
INTRODUCTION
Despite recent clinical advancements, the treatment of advanced melanoma continues to represent a significant challenge. Historically, the 5-year survival rate for patients with stage IV disease was approximately 6% [1–3]. However, agents introduced in recent years have substantially improved the outlook for patients with melanoma. For example, BRAF or MEK inhibitors such as vemurafenib, dabrafenib or trametinib, which are indicated in the approximately 50–60% of patients with melanoma harboring a BRAFV600E/K mutation, are associated with high response rates (~20–80%), prolonged progression-free survival (PFS) (5–9 months), and improved overall survival (OS) [4–7]. Unfortunately, most if not all patients receiving BRAF or MEK inhibitors eventually develop resistant tumors leading to disease progression [4, 6, 8–11].
In contrast to these kinase inhibitors, a second major advance in clinical therapeutics came with the development of ipilimumab and tremelimumab; monoclonal antibodies that can induce an antitumor immune response by blocking the checkpoint molecule cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) [12, 13]. Although these anti-CTLA-4 antibodies have modest response rates in the range of 10% [12, 13], ipilimumab significantly improves OS, with a subset of patients experiencing long-term survival benefit [14]. In a phase III trial, tremelimumab was not associated with an improvement in OS [13], and tremelimumab is not currently approved for the treatment of melanoma. Across clinical trials, survival for ipilimumab-treated patients begins to separate from those patients treated in control arms at around 4–6 months, and improved survival rates are seen at 1, 2, and 3 years [12, 14, 15] (Table 1 [4, 7, 10, 12, 13, 16–25]). Further, in aggregating data for patients treated with ipilimumab, it appears that there may be a plateau in survival at approximately 3 years. Thereafter, patients who remain alive at 3 years may experience a persistent long-term survival benefit, including some patients who have been followed for up to 10 years [14, 26]. While BRAF inhibitors also provide improved OS over chemotherapy, similar long-term follow up is not yet available with these agents. It is possible that the long-term effect seen with ipilimumab is unique to immunotherapeutic approaches, as similar long-term survival in a subset of patients has been previously reported with interleukin (IL)-2 therapy [27]. These observations suggest that in some patients treated with immunotherapy, cancer can be kept in check for an extended period of time, which may be a consequence of an effective and ongoing immune response. The next generation of checkpoint inhibitors, used either as single agents or in combination regimens, offers the promise of extending clinical benefits to a larger number of patients.
Table 1: Mechanism of action of anticancer agents in melanoma and association with response patterns and safety profile [4, 7, 10, 12, 13, 16–25]
Type |
Examples |
Mechanism of Action |
Antitumor Activity |
Toxicities |
Reference(s) |
Chemotherapy |
Dacarbazine |
Induces DNA damage and death of dividing cells |
Directly cytotoxic effects cause tumor regression or non-progression |
Off-target effects responsible for neutropenia, thrombocytopenia, and leukopenia |
|
Targeted agents (e.g. BRAF and MEK kinase inhibitors) |
Vemurafenib, dabrafenib, trametinib |
Inhibit mutated signaling pathway (BRAF/MAPK) driving melanoma proliferation |
Directly antiproliferative effects lead to tumor regression or non-progression |
Effects on wild type BRAF and CRAF likely responsible for skin toxicities |
|
CTLA-4 checkpoint inhibitors |
Ipilimumab, tremelimumaba |
Inhibit CTLA-4-mediated T-cell inhibition; increases T-cell proliferation; depletion/inhibition of regulatory T cells |
Reactivated antitumor immune response can lead to immediate or delayed tumor regression or non-progression |
T-cell activation and proliferation can lead to immunologic AEs |
|
PD-1 checkpoint inhibitors |
Pembrolizumab, nivolumaba, pidilizumaba |
Inhibit PD-1:PD-L1– and PD-1:PD-L2– mediated T-cell inhibition |
Restored antitumor immune response can lead to immediate or delayed tumor regression or non-progression |
T-cell activation can lead to immunologic AEs |
|
PD-L1 checkpoint inhibitors |
MPDL3280Aa, MEDI4736a, MSB0010718C a |
Inhibit PD-1:PD-L1–mediated T-cell inhibition |
Restored antitumor immune response can lead to immediate or delayed tumor regression or non-progression |
T-cell activation can lead to immunologic AEs |
aNot FDA-approved for the treatment of patients with melanoma at the time of writing.
Mechanisms of action of immune checkpoint inhibitors
The goal of immunotherapy is to elicit or enhance antitumor immune responses. Whereas BRAF and MEK targeted agents specifically inhibit a node in the MAPK signaling pathway that can eventually be overcome by tumor mutation, cancer immunotherapy has the potential to induce the inherent capacity of the immune system to adapt to mutational tumor changes. Though cancer immunotherapy approaches have been pursued for decades and have been successful in some cases (e.g. IL-2 in melanoma), checkpoint inhibition and, in particular, PD-1/PD-L1 blockade, is the first strategy that is poised to impact the outcome in cancer patients on a broader scale.
Under physiologic conditions, both stimulatory and inhibitory pathways regulate the inflammatory immune response to pathogens and maintain tolerance to self-antigens. These are regulated by a diverse set of immune checkpoints, thereby protecting healthy tissues from damage [18]. These checkpoints can be co-opted by malignant tumors to dampen the immune response and evade destruction by the immune system [18]. The CTLA-4 and PD-1pathways have been the initial focus of anticancer agent development (Figure 1); agents targeting other pathways are also in development [18].
Generally, the CTLA-4 and PD-1 pathways operate at different stages of the immune response (Figure 1) [28]. CTLA-4 modulates the immune response early—at the time of T-cell activation by antigen presenting cells (APCs). T cells are activated by antigen presented on APC in the context of major histocompatibility complex (MHC) (signal 1), as well as co-stimulatory binding of CD28 to B7 (CD80/86) (signal 2). Upon T-cell activation, CTLA-4 is trafficked from the Golgi apparatus to the plasma membrane where it out-competes CD28 in binding to B7 ligands on APCs due to its much higher binding affinity. CTLA-4 binding to B7 ligands inhibits further T-cell activation, limiting the time for T-cell activity. In this way, the magnitude and duration of initial immune responses is physiologically controlled.
In the setting of cancer, inhibiting CTLA-4 may lead to continued activation of a larger number of T cells, resulting in more effective antitumor responses (Figure 2A). Preclinical evidence suggests that anti-CTLA-4 antibody can also deplete or inhibit regulatory T cells present in the tumor [19, 29]. CTLA-4 blockade has the potential to activate T cells that are specific for a wide range of antigens, including self-antigens, or deplete regulatory T cells that control autoreactive effector T cells, which may explain the autoimmune-like toxicities observed with CTLA-4 blockade.
In contrast to the effect of CTLA-4 on early T-cell activation, the PD-1 pathway appears to impact the T-cell response at the (later) effector stage (Figure 1). PD-1 is upregulated on T cells after persistent antigen exposure, typically in response to chronic infections or tumors. PD-L1 and PD-L2, the ligands for PD-1, can be expressed by tumor cells, as well as several other hematopoietic and non-hematopoietic cell types. Expression of PD-L1 and PD-L2 is induced by inflammatory cytokines, predominately interferon-γ [30]. In tumors, oncogenic signaling pathways can also upregulate PD-L1 expression [31]. When PD-1 binds its ligand, the T cell receives an inhibitory signal. Over time, chronic inhibition via PD-1:PD-L1 in tumor leads to T-cell anergy and blockade of a productive antitumor immune response [32].
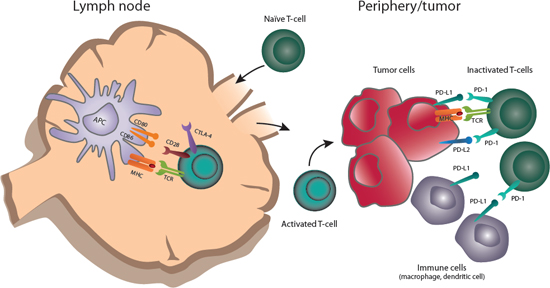
Figure 1: Role of CTLA-4 and PD-1 in antitumor immune responses. Naïve T cells are primed by antigens presented by APCs in the context of MHC (signal 1), as well as co-stimulatory binding of CD28 to B7 (CD80/86) (signal 2). T cells upregulate CTLA-4 shortly after activation. Ligation of CTLA-4 with CD80 or CD86 limits T-cell activation and proliferation. Activated T cells traffic to the periphery and encounter tumor antigens at the tumor site. PD-1 is upregulated on T cells after prolonged activation; binding to PD-1 ligands (PD-L1 or PD-L2) expressed by tumor or other immune cells, including macrophages and dendritic cells, causes T-cell activation and dampens an ongoing antitumor immune response.
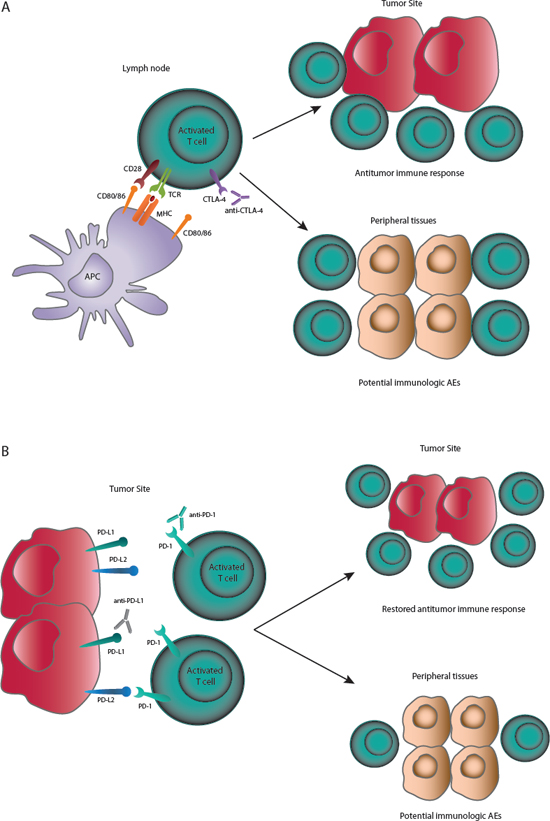
Figure 2: (A) CTLA-4 checkpoint inhibition. CTLA-4 inhibition prevents early deactivation of T cells responding to tumor antigens presented by APCs. Activated T cells can migrate to the tumor site and mount effective antitumor immune responses. Activation of T cells with cross-reactivity to host antigens may cause immunologic AEs. (B) PD-1 and PD-L1 checkpoint inhibition. PD-1 checkpoint inhibitors will prevent PD-1:PD-L1– and PD-1:PD-L2–mediated deactivation of T cells. PD-L1 checkpoint inhibitors will prevent PD-1:PD-L1– mediated deactivation of T cells. PD-1 pathway inhibition can restore antitumor immune responses directly at the tumor site and also facilitate T-cell activation in lymph nodes or other sites. Activation of T cells with cross-reactivity to host antigens may cause immunologic AEs.
Expression of PD-1 ligands is a mechanism for tumors to evade antitumor immune responses [30]. In many tumor types, tumor infiltrating lymphocytes (TILs) have been observed in both primary and metastatic specimens, indicating immune recognition of cancer cells. The presence of these TILs has been associated with improved outcomes in several cancers, including melanoma and renal cell carcinoma [33, 34]. Blockade of the PD-1/PD-L1 axis may restore the activity of TILs that have become quiescent as a result of PD-L1 ligation (Figure 2B).
Whereas CTLA-4 is widely expressed by T cells, PD-1 is expressed by activated T cells that have developed an “exhausted” or near-anergic state. PD-1 pathway blockade, in contrast to CTLA-4 blockade, may thus enhance activation of T cells with greater selectivity for tumors. However, human tumors can also express PD-L2, and this ligand has been found to be a negative prognostic factor in some cancers [30, 35]. PD-1 has a higher affinity for PD-L2 than for PD-L1, although whether this leads to differences in signaling or T-cell functions is not known [35]. PD-L2 is expressed in a number of tissues, most notably the lung, where it dampens the immune response to self-antigens and prevents autoimmunity [36]. It has been postulated that for selective blockade of PD-1:PD-L1 binding, keeping PD-1:PD-L2 interactions intact could offer a benefit in terms of reduced toxicities [37]. However, to date, a differential toxicity profile has not been observed between anti-PD-1 and anti-PD-L1 antibodies, and no direct comparisons from randomized trials are available.
Monoclonal antibodies directed at PD-1 (nivolumab, pembrolizumab, pidilizumab [CT-011]) are designed to prevent PD-1 from binding both PD-L1 and PD-L2 (Table 1 [4, 7, 10, 12, 13, 16–25]). PD-L1 inhibitors (MEDI4736, MPDL3280A, and MSB0010718C) block PD-1:PD-L1 binding, but not PD-1:PD-L2 binding. PD-L1 blockade also inhibits the binding of PD-L1 to CD80, which is expressed on activated T cells. The implication of this additional effect is not clear, as the role of PD-L1:CD80 interaction is not fully understood [18]. Pembrolizumab was approved by the United States Food and Drug Administration (FDA) in September 2014 for the treatment of patients with unresectable or metastatic melanoma with disease progression following ipilimumab and, if BRAFV600 mutant, a BRAF inhibitor. Nivolumab was approved in Japan for the treatment of patients with unresectable melanoma in July 2014. The other PD-1 and PD-L1 directed agents are currently in Phase I–III clinical trials in multiple tumor types.
Clinical response patterns
Rapid and dramatic responses have been observed with oncogene-directed treatments, such as BRAF inhibitors in melanoma and epidermal growth factor receptor (EGFR) inhibitors in non-small cell lung cancer. These agents specifically inhibit oncogenic signaling pathways. However, response duration with these agents has been modest. For example, the median PFS of BRAF inhibitors in BRAFV600E/K mutant melanoma ranges from 5–8 months [4, 8, 10, 11]. With dual BRAF and MEK blockade, PFS is longer, approximately 9 months [6, 9].
Checkpoint inhibitors display a range of response patterns, which may reflect the complexities of inducing a tumor-directed immune response and the individuality of a patient's immune system and tumor. Response kinetics may also depend on which pathway is inhibited. In theory, a patient with extensive tumor infiltration of PD-1-expressing T cells could have a rapid response with a PD-1 pathway inhibitor. In contrast, a patient with low numbers of pre-existing tumor-specific T cells could have a delayed or no response to PD-1 or CTLA-4 pathway blockade. Late or delayed responses occurring months to years after treatment initiation have been described with checkpoint inhibitors [17, 38–40]. In the case of a delayed response, tumor size may initially increase—as a result of either true tumor growth or increased tumor volume due to infiltration by immune cells—prior to subsequent tumor regression. As such, PFS based on traditionally used response criteria, such as RECIST, may not be the most appropriate efficacy measure with immunotherapies. To guide clinical practice, expert opinion has suggested the use of modified response criteria for immunotherapies, termed immune-related response criteria (irRC). These response criteria take into account the potential for increase in tumor size or number of lesions prior to declaration of progressive disease (Table 2 [12, 17, 21, 38, 40–46]). It has been suggested that RECIST may underestimate the benefit of PD-1 inhibitors in approximately 10% of patients relative to irRC [41].
In clinical trials of ipilimumab, responses predominately occurred by 12 weeks, though there was a subset of patients with delayed response [47]. Further, some patients had an initial partial response that later converted to a complete response; the average time to reach a complete response was 30 months [47]. At the FDA-approved dose of pembrolizumab (2 mg/kg every 3 weeks), an open-label phase I trial reported an ORR of 26% in patients with ipilimumab-refractory advanced melanoma. The majority of patients had a response by the time of first assessment at 3 months (12 weeks); the median time to response was 3 months (range: 2.8 to 9) [48]. In a phase III trial of nivolumab (3 mg/kg, every 2 weeks) in patients with untreated metastatic melanoma, the ORR was 40% (versus 14% with dacarbazine). The median time to response with nivolumab was 2.1 months (range: 1.2 to 7.6), similar to that of dacarbazine (median 2.1 months; range: 1.8 to 3.6) [46].
In addition to delayed responses, treatment with checkpoint inhibition has been associated with durable, long-lasting responses, even after discontinuing therapy in some patients. In the phase I trial of nivolumab, the vmedian duration of responses was 99 weeks (range, 17+ to 117+ weeks), and 56% (19/34) of responses were ongoing at the time of last analysis (range of follow-up of responding patients, approximately 32–144 weeks, median follow up not disclosed) [20]. With shorter follow-up in the pembrolizumab trial, the median response duration had not been reached (range, 6+ to 76+ weeks), and 88% of responses were ongoing (range of follow-up of responding patients, approximately 18–88 weeks, median follow up not disclosed) [40]. Many patients who have discontinued immunotherapy (either anti-PD-1 or anti-CTLA-4) for reasons other than disease progression had persistent responses, indicative of a sustained antitumor immune response. In the phase 1 trial of nivolumab, 52% (11/21) of responding patients who discontinued therapy early had responses lasting ≥ 24 weeks off therapy [20]. Of these 11 patients, 7 (64%) had ongoing responses at the time of analysis.
Table 2: Select immunologic adverse events reported in patients with melanoma receiving checkpoint inhibitors [12, 17, 21, 38, 40–46]
Type |
Examples |
Frequency (All Grades) |
Frequency (Grade ≥ 3) |
Typical Timing of First Occurrencea |
Treatment Approaches for Grade ≥ 3 AEsb |
Typical Time to Resolution (Grades ≥ 2) |
Dermatologic |
Rash, pruritus, vitiligo |
21–50% |
≤ 4% |
2–3 weeks |
Dermatologist evaluation; drug interruption or discontinuation and systemic corticosteroids |
≤ 3 months |
Gastrointestinal |
Diarrhea, colitis |
Ipilimumab: 30–35% PD-1 inhibitors: 17–20% |
Ipilimumab: 5–8% PD-1 inhibitors: ≤ 2% |
5–6 weeks |
Gastrointestinal consultation; colonoscopy may be considered; evaluation to rule out infection; drug discontinuation and systemic corticosteroids with≥30 day taper; infliximabc |
≤ 3 months |
Endocrine |
Hypothyroidism, hyperthyroidism, hypophysitis, adrenal insufficiency |
4–13% |
≤ 1% |
8–9 weeks |
Endocrinologist evaluation; drug interruption or discontinuation; systemic corticosteroids and/or hormone replacement therapy |
> 4 months; may be irreversible |
Hepatic |
Elevated ALT, elevated AST |
2–9% |
≤ 2% |
6–8 weeks |
Drug discontinuation and systemic corticosteroids; mycophenolate mofetil or other immunosuppressants per local guidelines |
≤ 3 months |
Pulmonary |
Pneumonitis |
≤ 4% |
≤ 2% |
6–12 weeksd |
Drug interruption or discontinuation and systemic corticosteroids; additional immunosuppressant therapy as needede |
> 4 months in one patientd |
Ocular |
Conjunctivitis, scleritis, uveitis, Graves' ophthalmopathy |
≤ 1% |
< 1% |
d |
Drug interruption or discontinuation and topical or systemic corticosteroids; ophthalmologist consultation as needed |
≤ 3 monthsd |
Neurologic |
Myopathy, Guillain-Barré syndrome, myasthenia gravis |
< 1% |
< 1% |
d |
Neurologist evaluation; drug discontinuation and systemic corticosteroids; IVIG or other immunosuppressants per local guidelines |
d |
aAfter treatment initiation. Individual patient experiences will vary.
bWith the exception of endocrinopathies, add prophylactic antibiotics for opportunistic infections. Patients on IV steroids may be switched to an equivalent dose of oral corticosteroids (e.g. prednisone) at start of tapering or earlier, once sustained clinical improvement is observed. The lower bioavailability of oral corticosteroids should be taken into account.
cUnless contraindicated; should not be used in cases of perforation or sepsis.
dInformation is limited due to small numbers of cases.
eInfliximab, cyclophosphamide, IVIG, or mycophenolate mofetil.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Survival with checkpoint inhibitors
In clinical practice, ipilimumab is administered at 3 mg/kg every 3 weeks for a total of 4 doses, based on the approved FDA label [49]. There is speculation in the field as to whether a higher dose (10 mg/kg) or the possibility of maintenance administration (every 12 weeks after the initial 4 doses) could influence the activity of the drug, and clinical trials are ongoing to investigate these possibilities. Nevertheless, the approved regimen has led to > 5-year survival in some heavily pretreated patients [14, 26, 47]. The durability and adaptability of tumor responses among patients who discontinued nivolumab and pembrolizumab therapy may be similar to that observed with ipilimumab, and may also explain partial or complete responses observed in patients who received PD-1 inhibitors after ipilimumab [40, 50].
Survival data with PD-1 inhibitors are promising. Significantly improved survival was seen with nivolumab versus dacarbazine in patients with previously untreated metastatic melanoma in a randomized phase III trial [46]. The 1-year survival rate was 73% with nivolumab and 42% with dacarbazine, and median PFS was 5.1 months and 2.2 months, respectively. In the phase I trial of pembrolizumab, median OS was not reached at the time of analysis, and 1-year and 18-month survival rates were 69% and 62%, respectively. In this study, 48% of patients had received ≥ 2 prior therapies [40]. Median OS was 17.3 months for all patients treated in the nivolumab phase I trial, 62% of whom had received ≥ 2 prior therapies, and 1-, 2- and 3-year survival rates were 63%, 48%, and 41%, respectively [20].
Safety profile with checkpoint inhibitors
Safety with monotherapy
Related to the immune mechanism of antitumor activity, checkpoint inhibitors are associated with immune-related adverse events (irAEs). While different from those of targeted agents and chemotherapy, the safety profiles of checkpoint inhibitors are typically manageable and tolerable for most patients. Since these therapies induce tumor regression by stimulation of immune responses, side effects may be caused by activating potentially self-reactive T cells (Figure 2A and 2B). One exception to this is hypophysitis, which is reported in about 4% of patients receiving ipilimumab, and is attributed to ectopic expression of CTLA-4 in the pituitary gland, leading to ipilimumab binding to endocrine cells, complement fixation, and inflammation [51].
In the phase III study of ipilimumab monotherapy compared with the gp100 vaccine or ipiliumumab plus gp100 vaccine, the incidence of grade 3–4 adverse events (AEs) in the ipilimumab monotherapy arm was 24% [12]. In comparison, the incidence of treatment-related grade 3–4 AEs in the phase I nivolumab and pembrolizumab trials were 22% and 12%, respectively [21, 40]. In the phase III nivolumab trial, reported rates of treatment-related grade 3–4 AEs were 12% with nivolumab and 18% with dacarbazine [46]. The most commonly-reported treatment-related AE with ipilimumab is fatigue, while the most common clinically significant immunologic AEs include diarrhea/colitis, rash/pruritus, endocrinopathies, and hepatitis (Table 2 [12, 17, 21, 35, 40–46]). While fatigue is also common with PD-1 inhibitors, diarrhea may be less common as compared with ipilimumab (Table 2 [12, 17, 21, 38, 40–46]). In contrast, pneumonitis, though infrequent, may be more common with PD-1 pathway inhibitors than with ipilimumab. The rate of grade 3–4 pneumonitis was 2% or less in trials of PD-1 pathway inhibitors [22, 52, 53]. However, pneumonitis was the cause of death in 3 patients with cancer who received nivolumab; no deaths occurred in patients with melanoma [21].
Most irAEs with checkpoint inhibitors occur during the first 2 to 6 months of treatment, but can occur anytime, even after treatment discontinuation [21, 42, 45] (Table 2 [12, 17, 21, 38, 40–46]). The general timing and grade of irAEs with ipilimumab have been described by Weber and colleagues (Figure 3 [38]). However, whether PD-1 pathway inhibitors have similar AE kinetics is not currently clear. With chemotherapies, toxicities can be cumulative, but this may not be the case with immunotherapies. In patients who received up to 2 years of nivolumab treatment, there was no evidence of increasing toxicity with increased drug exposure [21]. Furthermore, no maximum tolerated dose was reached in the phase I studies of nivolumab or pembrolizumab [54, 55].
Safety with checkpoint blockade combination therapy
Preclinical animal studies suggest that dual checkpoint blockade with anti-CTLA-4, anti-PD-1, and anti-PD-L1 antibodies leads to stronger immune stimulation and enhanced antitumor activity [56]. In a phase I trial, concurrent checkpoint blockade with both anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab) antibodies showed increased efficacy over what has been observed with single agent therapy. The ORR with combination therapy was 42% across all tested doses, versus 11% and 32% reported in trials of ipilimumab and nivolumab monotherapy, respectively [12, 20, 57], and 42% of patients had ≥ 80% tumor reduction at 36 weeks [57]. Preliminary survival rates at 1 and 2 years were 85% and 79%, respectively, for the combination regimen, which compared favorably with the reported 1- and 2-year rates of 46% and 24% for ipilimumab and 63% and 48% for nivolumab [12, 20, 57].
Combining CTLA-4 and PD-1 pathway blockade resulted in a higher incidence of AEs compared with single agent therapy, whereby the types of AEs were similar to what has been observed with ipilimumab or nivolumab alone. Sixty-two percent of patients with concurrent ipilimumab plus nivolumab therapy experienced a grade 3–4 treatment-related AE, and 23% of patients discontinued therapy due to a treatment-related AE [57]. The majority of grade 3–4 treatment-related AEs were laboratory abnormalities, including elevated aspartate aminotransferase (13%), lipase (13%), alanine aminotransferase (11%), creatinine (6%), and diarrhea (6%) [50]. Multiple ongoing studies are evaluating different checkpoint combinations and doses to optimize the risk/benefit profile of dual checkpoint blockade [23].
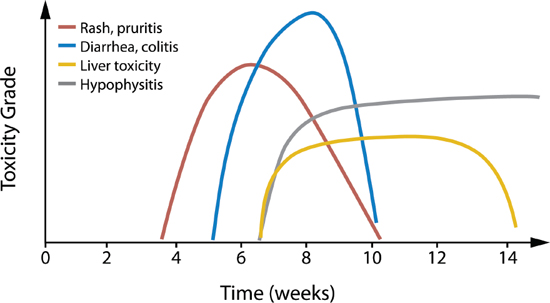
Figure 3: Kinetics of irAEs in ipilimumab-treated patients. The overall approximate timing and relative grade of the most common irAEs in ipilimumab-treated melanoma patients is depicted. Individual patient experiences vary. Reprinted with permission. © 2012 American Society of Clinical Oncology. All rights reserved. Weber JS et al: J Clin Oncol. 30 (21), 2012: 2691–2697 [38].
Since pembrolizumab is approved for patients with progression after ipilimumab therapy, and other PD-1 targeting agents are likely to be approved soon, patients who have previously received ipilimumab may subsequently receive a different checkpoint inhibitor. To date, trials of PD-1 pathway inhibitors in patients who previously received ipilimumab have not shown an increased risk of select irAEs [40, 44, 48, 58]. In one study, patients who had previously experienced grade 3 irAEs with ipilimumab did not experience similar AEs when treated with nivolumab [44]. In a trial of pembrolizumab in patients who were ipilimumab-naïve or who had previously received ipilimumab (on average 33–34 weeks prior; range, 4–248 weeks), the rates of grade 3–4 treatment-related AEs and the rates of patients discontinuing due to AEs were similar between ipilimumab-naïve and ipilimumab-treated patients: 14% versus 11% and 4% versus 5%, respectively [40, 48].
The trial investigating concurrent nivolumab plus ipilimumab also evaluated nivolumab following progressive disease and lack of grade 3–4 AEs in patients who had received ipilimumab in the previous 4–12 weeks [50]. In this cohort, 18% of patients experienced a grade 3–4 treatment-related AE and 9% of patients discontinued due to a treatment-related AE. As patients who had previously experienced high grade AEs with ipilimumab were excluded, the study did not assess the safety of sequential therapy in this patient population. While initial evidence suggests that PD-1 pathway checkpoint inhibitors can be used safely in patients with prior ipilimumab treatment, no data yet exists to describe the experience of patients treated with anti-CTLA-4 antibody following anti-PD-1 antibody. This is an area of ongoing investigation that will be of particular importance in future clinical practice.
Several fully-accrued trials are further investigating ipilimumab plus nivolumab in patients with advanced melanoma, and an expanded access program is now available [23]. A phase II study is evaluating the safety of sequential use of ipilimumab after nivolumab (four 3-week cycles of ipilimumab, followed by nivolumab every 2 weeks until progression or unacceptable toxicity) versus nivolumab after ipilimumab (six 2-week cycles of nivolumab followed by four 3-week cycles of ipilimumab followed by nivolumab every 2 weeks until progression or unacceptable toxicity) [NCT01783938]. A phase III study is evaluating nivolumab alone or with ipilimumab versus ipilimumab in patients with previously untreated advanced melanoma; the primary endpoint is OS [NCT01844505]. Another phase II study has a similar design, without the nivolumab alone group, and has ORR as the primary read-out [NCT01927419]. Other combinations of a CTLA-4-targeted agent plus a PD-1 pathway inhibitor are also being evaluated in phase I trials, including ipilimumab plus pembrolizumab or MPDL3280A, and tremelimumab plus MEDI4736 [23].
Strategies for managing AEs with checkpoint inhibitors
Detailed treatment algorithms and recommendations are available for the approved agent ipilimumab and nivolumab (Table 2 [12, 17, 21, 38, 40–46]). Patients should be examined for potential irAEs at each visit, and prompt work-up of suspected AEs should be performed to minimize the risk of worsening. Since asymptomatic grade 3–4 elevations in aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) have been noted in several studies of checkpoint inhibitors, clinical trials required that certain laboratory tests be performed at regular intervals, including those evaluating liver, renal, and thyroid functions [21, 38, 43, 45].
A particular AE to note with checkpoint inhibitors is diarrhea, which is due an inflammatory immune response, not off-target drug effects, as with chemotherapy or targeted agents [45]. Therefore, diarrhea induced by checkpoint blockade is treated with corticosteroids or other immunosuppressant(s) (Table 2 [12, 17, 21, 38, 40–46]). Similarly, most irAEs can be effectively managed with corticosteroid treatment; however, a prolonged taper is often required for complete resolution. General treatment strategies for irAEs are as follows:
Grade 1–2 AEs are treated symptomatically, with increased frequency of monitoring.
Grade 1–2 AEs that remain persistent or become more symptomatic should be managed similarly to grade 3–4 AEs.
Grade 3–4 AEs should be treated with corticosteroids and tapered over 4 or more weeks [38, 42, 45, 46].
Endocrine disorders with checkpoint inhibitors have been managed with hormone replacement, which may or may not be permanent. Prolonged exposure to corticosteroid therapy, possibly to manage irAEs, may also lead to adrenal insufficiency and hypogonadism. Each of these supportive measures should be taken into consideration during the assessment of endocrinopathies [45]. Long-term exposure to corticosteroids can lead to infection, with opportunistic infections and gastrointestinal irritation [59]. Therefore, prophylactic antibiosis and gastric acid suppression may be indicated in patients requiring extended steroid tapers. The use of corticosteroids to manage irAEs does not appear to negatively impact the efficacy of checkpoint inhibitors, as tumor response duration appears to be unaffected in patients requiring this intervention [45]. In many patients, checkpoint therapy can be restarted after successful resolution of the AE [42, 48, 54]. However, patients who experience severe AEs should permanently discontinue treatment [38, 42, 45, 49].
Patient selection
The identification of a selection marker for treatment, such as a BRAF mutation in melanoma, offers the ability to prospectively identify patients more likely to benefit from certain therapies. While ORRs with targeted therapies are high, not all patients are eligible. Initial evidence suggests that checkpoint inhibition may be more broadly applicable than targeted therapy. In trials of patients with melanoma being treated with checkpoint inhibitors, responses have been observed in patients with and without BRAF mutations, brain metastases, or prior treatment [12, 20, 40, 45, 57, 60]. As a potential biomarker of response, a rise in absolute lymphocyte count at 3, 7 or 12 weeks of ipilimumab treatment has been correlated with improved survival [61–64]. Studies have also shown a correlation between an increased eosinophil count—either at baseline or a rise between the second and third ipilimumab infusions—and improved survival [61, 65]. Also, an exploratory study found that pembrolizumab-treated patients with smaller baseline tumor size (≤ 90 mm) had higher responses and improved OS at 1 year as compared with patients with larger baseline tumors. However, patients with larger tumors also derived benefit from pembrolizumab [66]. Predictors of toxicity are also being evaluated with checkpoint inhibitors. For example, IL-17 levels at 7 weeks of treatment with ipilimumab predicted colitis [67]. Additionally, patients with a history of autoimmune disease may be more at risk for development of immunologic AEs with checkpoint inhibitors and were excluded from clinical trials [43, 50, 54].
Many trials are now investigating whether PD-L1 expression by tumors can be used as a predictive biomarker of response to PD-1 pathway inhibitors. Initial results suggest that response rates with PD-1 inhibitor monotherapy may be higher in patients with PD-L1-positive melanoma versus PD-L1 negative melanoma [20, 46, 57, 68]. However, in nearly all studies, responses were also seen in patients with negative or low PD-L1 tumor expression. It is likely that PD-L1 expression will be associated with an improved response rate with single agent PD-1 inhibition. Nevertheless, tumor PD-L1 expression will not discriminate against patients unable to benefit. In patients receiving concurrent CTLA-4 and PD-1 pathway inhibitors, high proportions of patients responded, regardless of baseline PD-L1 status (ORR: 57% for PD-L1 positive; 35% for PD-L1 negative) [57]. Therefore, tumor PD-L1 expression may not be useful as a prognostic biomarker for patients receiving combination regimens. Further evaluation of potential biomarkers is needed, with one option being evaluation of the inflamed versus non-inflamed tumor phenotype [69, 70].
CONCLUSION
Checkpoint inhibitors, such as ipilimumab, anti-PD-1, and anti-PD-L1 antibodies, have emerged as new treatment modalities for patients with melanoma, and likely various other cancers. For a subset of patients treated with checkpoint inhibitors, durable clinical responses lasting many years may be possible. Immunotherapy combinations have shown increased efficacy and toxicity compared with monotherapy; however, to date, most toxicities have been manageable. Clinical trials are underway to examine various combinations and sequencing of ipilimumab and PD-1 pathway blockers, and it remains to be seen if sequential administration of immuno-oncology agents will be as efficacious or exhibit an improved (or worsened) toxicity profile. As such, treatment of patients with combinations or sequential approaches will require the close attention of clinicians for the development of immune-related toxicities. The understanding that immunologic AEs are caused by uncontrolled off-target immune responses, and therefore frequently require active treatment with steroids, is critical for clinicians to effectively manage patients receiving these therapies.
ACKNOWLEDGMENTS
The authors take full responsibility for the content of this publication and confirm that it reflects their viewpoint and expertise. Professional medical writing assistance was provided by Britt Anderson, PhD and professional editing assistance was provided by Karin McGlynn at StemScientific and was funded by Bristol-Myers Squibb. Bristol-Myers Squibb generated the concept for this Review Article; however, the authors developed the content. Bristol-Myers Squibb reviewed a draft for medical accuracy only. Neither Bristol-Myers Squibb nor StemScientific influenced the content of the manuscript, nor did the authors receive financial compensation for authoring the manuscript.
Conflict of interest statement
Dr. Luke reports consultancy and travel from Amgen, Bayer, and Genentech, and clinical trial support to his institution from EMD Serono, GlaxoSmithKline, and Novartis.
Dr. Ott reports consultancy from Bristol-Myers Squibb and clinical trial support to his institution from ARMO BioSciences, Bristol-Myers Squibb, MedImmune, and Merck.
REFERENCES
1. Barth A, Wanek LA, Morton DL. Prognostic factors in 1,521 melanoma patients with distant metastases. J Am Coll Surg. 1995; 181:193–201.
2. Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000; 18:3782–3793.
3. Unger JM, Flaherty LE, Liu PY, Albain KS, Sondak VK. Gender and other survival predictors in patients with metastatic melanoma on Southwest Oncology Group trials. Cancer. 2001; 91:1148–1155.
4. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–2516.
5. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367:107–114.
6. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, et al. Combined BRAF, and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694–1703.
7. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380:358–365.
8. Ravnan MC, Matalka MS. Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma. Clin Ther. 2012; 34:1474–1486.
9. Long GV, Stroyakovsky DL, Gogas H, Levchencko E, de Braud F, Larkin JMG, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion-Sileni V, Lebbe C, Mandalà M, et al. COMBI-d: A randomized, double-blinded, phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma. J Clin Oncol. 2014; 32:abstr 9011^. http://meetinglibrary.asco.org/content/128598-144. (Accessed Jul 1, 2014).
10. Luke JJ, Hodi FS. Ipilimumab, vemurafenib, dabrafenib, and trametinib: synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist. 2013; 18:717–725.
11. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012; 366:707–714.
12. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711–723.
13. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, Lorigan P, Kendra KL, Maio M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013; 31:616–622.
14. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. Eur J Cancer. 2013; 49:abstr 4LBA. http://eccamsterdam2013.ecco-org.eu/Scientific-Programme/Abstract-search.aspx. (Accessed Jul 1, 2014).
15. Robert C, Thomas L, Bondarenko I, O'Day S, M D JW, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364:2517–2526.
16. Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De VL. Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res. 2007; 56:275–287.
17. Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, Humphrey R, Hodi FS. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009; 15:7412–7420.
18. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–264.
19. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009; 206:1717–1725.
20. Hodi FS, Sznol M, Kluger HM, McDermott DF, Carvajal RD, Lawrence DP, Topalian SL, Atkins MB, Powderly JD, Sharfman WH, Puzanov I, Smith DC, Leming PD, et al. Long-term survival of ipilimumab-naïve patients (pts) with advanced melanoma (MEL) treated with nivolumab (anti-PD-1, BMS-936558; ONO-4538) in a phase I trial. J Clin Oncol. 2014; 32:abstr 9002. http://meetinglibrary.asco.org/content/125578-144. (Accessed Jul 1, 2014).
21. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014; 32:1020–1030.
22. Atkins MB, Kudchadkar RR, Sznol M, McDermott DF, Lotem M, Schachter J, Wolchok JD, Urba WJ, Kuzel T, Schuchter LM, Slingluff CL, Ernstoff MS, Fay JW, et al. Pidilizumab in metastatic melanoma: results from a multicenter Phase, II, open-label, randomized trial. J Clin Oncol. 2014; 32:abstr 9001. http://meetinglibrary.asco.org/content/131463-144. (Accessed Jul 1, 2014).
23. ClinicalTrials.gov. 2014; http://clinicaltrials.gov. (Accessed Jul 7, 2014).
24. Hamid O, Sosman JA, Lawrence DP, Sullivan RJ, Ibrahim N, Kluger HM, Boasberg PD, Flaherty K, Hwu P, Ballinger M, Mokatrin A, Kowanetz M, Chen DS, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013; 31:abstr 9010. http://meetinglibrary.asco.org/content/115916–132. (Accessed Aug 14, 2014).
25. Segal NH, Antonia SJ, Brahmer JR, Maio M, Blake-Haskins A, Li X, Vasselli J, Ibrahim RA, Lutzky J, Khleif S. Preliminary data from a multi-arm expansion study of MEDI4736, an anti-PD-L1 antibody. J Clin Oncol. 2014; 32:abstr 3002. http://meetinglibrary.asco.org/content/134136-144. (Accessed Jul 1, 2014).
26. Lebbé C, Weber JS, Maio M, Neyns B, Harmankaya K, Hamid O, O'Day SJ, Konto C, Cykowski L, McHenry MB, Wolchok JD. Survival follow-up and ipilimumab retreatment for patients with advanced melanoma who received ipilimumab in prior phase II studies. Ann Oncol. 2014; (Epub ahead of print). pii: mdu441.
27. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999; 17:2105–2116.
28. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008; 224:166–182.
29. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013; 1:32–42.
30. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008; 8:467–477.
31. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B, Ribas A. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014; 20:3446–3457.
32. Wherry EJ. T cell exhaustion. Nat Immunol. 2011; 12:492–499.
33. Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012; 72:1070–1080.
34. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12:298–306.
35. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer-induced immune suppression. Clin Dev Immunol. 2012; 2012:656340.
36. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008; 26:677–704.
37. Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy—inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012; 18:6580–6587.
38. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012; 30:2691–2697.
39. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, Xu H, Yao S, Pons A, Chen L, Pardoll DM, Brahmer JR, Topalian SL. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013; 19:462–468.
40. Ribas A, Hodi FS, Kefford R, Hamid O, Daud A, Wolchok JD, Hwu WJ, Gangadhar TC, Patnaik A, Joshua AM, Hersey P, Weber JS, Dronca RS, et al. Efficacy and safety of the anti-PD-1 monoclonal antibody MK-3475 in 411 patients (pts) with melanoma (MEL). J Clin Oncol. 2014; 32:abstr LBA9000^. http://meetinglibrary.asco.org/content/133842-144. (Accessed Jul 1, 2014).
41. Hodi FS, Ribas A, Daud A, Hamid O, Robert C, Kefford R, Hwu WJ, Gangadhar TC, Joshua AM, Hersey P, Weber JS, Dronca RS, Perrone AM, et al. Evaluation of immune-related response criteria (irRC) in patients (pts) with advanced melanoma (MEL) treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014; 32:abstr 3006. http://meetinglibrary.asco.org/content/134449-144. (Accessed Jul 1, 2014).
42. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013; 18:733–743.
43. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013; 369:134–144.
44. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, Kroeger J, Eysmans C, Sarnaik AA, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013; 31:4311–4318.
45. Tarhini A. Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy: the underlying mechanisms and clinical management. Scientifica (Cairo). 2013; 2013:857519.
46. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2014; Nov 16; [Epub ahead of print].
47. Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, Levy CL, Rosenberg SA, Phan GQ. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012; 18:2039–2047.
48. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384:1109–1117.
49. Yervoy® (ipilimumab) prescribing information. Bristol-Myers Squibb Company (Princeton, NJ, USA: 2013. http://packageinserts.bms.com/pi/pi_yervoy.pdf. (Accessed Jul 1, 2014).
50. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369:122–133.
51. Iwama S, De RA, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Transl Med. 2014; 6:230ra45.
52. Topalian SL, Sznol M, Brahmer JR, McDermott DF, Smith DC, Gettinger SN, Taube JM, Drake CG, Pardoll DM, Powderly JD, Carvajal RD, Sosman JA, Atkins MD, et al. Nivolumab in patients with advanced solid tumors: survival and long-term safety in a phase I trial. J Clin Oncol. 2013; 31:abstr 3002^. http://meetinglibrary.asco.org/content/83737. (Accessed Jul 1, 2014).
53. Garon EB, Leighl NB, Rizvi NA, Blumenschein GR, Balmanoukian AS, Eder JP, Goldman JW, Hui R, Soria JC, Gangadhar TC, Sun JM, Patnaik A, Gubens MA, et al. Safety and clinical activity of MK-3475 in previously treated patients (pts) with non-small cell lung cancer (NSCLC). J Clin Oncol. 2014; 32:abstr 8020. http://meetinglibrary.asco.org/content/133339-144. (Accessed 1 Jul 2014).
54. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–2454.
55. Patnaik A, Kang SP, Tolcher AW, Rasco DW, Papadopoulos KP, Beeram M, Drengler R, Chen C, Smith L, Perez C, Gergich K, Lehnert M. Phase I study of MK-3475 (anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. J Clin Oncol. 2012; 30:abstr 2512. http://meetinglibrary.asco.org/content/100724-114. (Accessed Jul 1, 2014).
56. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013; 19:4917–4924.
57. Sznol M, Kluger HM, Callahan MK, Postow MA, Gordon RA, Segal NH, Rizvi NA, Lesokhin AM, Atkins MB, Kirkwood JM, Burke MM, Ralabate AL, Rivera AL, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL). J Clin Oncol. 2014; 32:abstr LBA9003. http://meetinglibrary.asco.org/content/126008-144. (Accessed Jul 1, 2014).
58. Hamid O, Robert C, Ribas A, Wolchok JD, Hodi FS, Kefford R, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Hersey P, Weber JS, Joseph RW, et al. Randomized comparison of two doses of the anti-PD-1 monoclonal antibody MK-3475 for ipilimumab-refractory (IPI-R) and IPI-naive (IPI-N) melanoma (MEL). J Clin Oncol. 2014; 32:abstr 3000. http://meetinglibrary.asco.org/content/134788-144. (Accessed Jul 1, 2014).
59. Kyi C, Hellmann MD, Wolchok JD, Chapman PB, Postow MA. Opportunistic infections in patients treated with immunotherapy for cancer. J Immunother Cancer. 2014; 2:19.
60. Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, Chin K, Canetta R, Humphrey R. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010; 37:533–546.
61. Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, Roy S, Eggermont AM, Routier E, Robert C. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013; 24:1697–1703.
62. Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, Chapman PB, Schwartz GK, Allison JP, Wolchok JD. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010; 116:1767–1775.
63. Postow MA, Yuan J, Panageas K, Bogatch K, Callahan M, Cheng M, Schroeder SEA, Kendle RF, Harding JJ, Dickson MA, D'Angelo SP, Carvajal RD, Schwartz GK, et al. Evaluation of the absolute lymphocyte count as a biomarker for melanoma patients treated with the commercially available dose of ipilimumab (3 mg/kg). J Clin Oncol. 2012; 30:abstr 8575. http://meetinglibrary.asco.org/content/98956-114. (Accessed Dec 8, 2014).
64. Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, Esposito A, Paone M, Palla M, Cavalcanti E, Sandomenico F, Petrillo A, Botti G, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014; 63:675–683.
65. Schindler K, Harmankaya K, Postow MA, Frantal S, Bello D, Ariyan CE, Michielin OA, Hoeller C, Pehamberger H, Wolchok JD. Pretreatment levels of absolute and relative eosinophil count to improve overall survival (OS) in patients with metastatic melanoma under treatment with ipilimumab, an anti CTLA-4 antibody. J Clin Oncol. 2013; 31:abstr 9024. http://meetinglibrary.asco.org/content/118098–132. (Accessed Dec 1, 2014).
66. Joseph RW, Elassaiss-Schaap J, Wolchok JD, Joshua AM, Ribas A, Hodi FS, Hamid O, Robert C, Daud A, Hwu WJ, Kefford R, Hersey P, Weber JS, et al. Baseline tumor size as an independent prognostic factor for overall survival in patients with metastatic melanoma treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014; 32:abstr 3015. http://meetinglibrary.asco.org/content/134724144. (Accessed Jul 1, 2014).
67. Callahan MK, Yang A, Tandon S, Xu Y, Subudhi SK, Roman RA, Heine AI, Pogoriler E, Kuk D, Panageas K, Yuan JD, Allison JP, Wolchok JD. Evaluation of serum IL-17 levels during ipilimumab therapy: Correlation with colitis. J Clin Oncol. 2011; 29:abstr 2505. http://meetinglibrary.asco.org/content/79357-102. (Accessed Aug 20, 2014).
68. Kefford R, Ribas A, Hamid O, Robert C, Daud A, Wolchok JD, Joshua AM, Hodi FS, Gangadhar TC, Hersey P, Weber JS, Dronca RS, Patnaik A, et al. Clinical efficacy and correlation with tumor PD-L1 expression in patients (pts) with melanoma (MEL) treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014; 32:abstr 3005. http://meetinglibrary.asco.org/content/130007-144. (Accessed Jul 1, 2014).
69. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol. 2011; 23:286–292.
70. Spranger S, Gajewski T. Rational combinations of immunotherapeutics that target discrete pathways. J Immunother Cancer. 2013; 1:16.