
Introduction
Familial/hereditary cancer syndromes (FCS) are an important component of overall cancer incidence, therefore family history of cancer is a key factor in determining overall cancer risk and prognosis [1–3]. FCS is of particular concern in Saudi Arabia, which has one of the highest rates of consanguinity worldwide [4–7]. Cancer increased in the Kingdom of Saudi Arabia by 136% between 1999 and 2015 [4].
An estimated 20% of all cancer patients in Saudi Arabia have a positive family history of cancer [5–8] and are therefore likely to carry mutant alleles. Where a mutation is carried in one allele, damage to the second, wild type allele at the same locus can result in the loss of a checkpoint to cancer development [9]. The clustering of cancer in families may be due to shared environmental exposures and/or inherited genetic factors, including complex interactions between the two; hereditary causes alone account for ~5% of regional cancer cases [8].
The most common forms of cancer in the population are breast and colorectal cancer, with respective prevalences of 53% and 51% [10], both of which are associated with FCS. Although breast cancer incidence is lower than in most Western populations, it is steadily climbing, and is expected to eventually plateau at an ASR comparable to Western countries as lifestyles change over time [11]. Age at onset is lower than is typical globally, and women are likely to present at a later stage, with poorer prognoses.
Colorectal cancer incidence in Saudi Arabia was estimated at 14.4% in 2020, with a higher than typical level of early onset CRC [12]. A 2020 study of colorectal cancer (CRC) in Saudi Arabia found that colon cancer patients with a corresponding family history had a statistically significant higher risk of mortality than colon cancer patients with no family history of CRC [5]. CRC can be familial when caused by familiar adenomatous polyposis (FAP), or by Lynch syndrome, which appears to be more common in this population than in Western countries [12].
As the prevalence of FCS and cancer-related mortality in the Saudi population appears to be increasing [6, 7, 13–18], an understanding of which pathogenic variants (PVs) are prevalent in the Saudi population has practical value. As approximately 40% of all marriages in Saudi Arabia are between relatives [19, 20], it is important to inform related individuals who wish to marry about the risk to future offspring of inheriting cancer-associated alleles from both parents. Moreover, knowing which variants in which genes contribute to the cancer incidence in Saudi populations would allow both for genetic screening and for physicians to recommend preventative measures, including surgical interventions or simply regular physical or radiological screening, to those at high risk due to inheritance of mutations. It could also be appropriate for relatives who have not inherited variants associated with familial cancer to reduce their frequency of screening via procedures such as colonoscopy or mammography, which have associated medical risks [2, 10]. However, to reap these benefits, the exact variant(s) being transmitted in the family must be known, and this can require screening multiple genes to find relevant PVs.
Recent advances in the field of medical genomics and, in particular in the use of next-generation sequencing (NGS), have opened new avenues for a better understanding of the underlying genetic risk factors for cancer [13–21]. NGS gene panels allow for the screening of multiple cancer-associated genes simultaneously, making them a cost-effective and time-saving way to detect familial variants [13, 16]. In particular, they detect inherited mutations in individuals who might not meet the criteria of international screening standards, and, in individuals who have inherited a familial cancer-associated mutation in one gene, multi-gene panels detect variants in additional genes that may increase the cancer risk for those individuals [16, 22]. Furthermore, using an NGS gene panel approach has been shown to detect cancer-associated germline mutations in genes not generally associated with the type of cancer that runs in the family, which would have been missed if only the genes associated with the particular cancer type had been sequenced [16].
There are many studies which have screened for germline mutations in populations with few or no Arab individuals [16, 23]. However, since the prevalence of individual mutations may vary across populations, it is important to determine the prevalence of cancer-associated mutations within specific populations [14, 24, 25]. Previous studies using gene panels to screen for cancer-associated mutations have typically involved either large but non-Arab-specific populations [15, 23, 26], or Arab-specific but small-sized populations [14].
Studies specific to Arab populations have typically screened for tumor-specific rather than broad germline mutations [14, 15, 26, 27], or have focused on one gene or a small number of genes associated with one type of cancer [28–44]. These show that Arab cohorts differ greatly in pathogenic variant prevalence from Western cohorts, and from each other. A meta-analysis of ovarian cancer cohorts across 22 Arab-predominant countries, has revealed eight ovarian-cancer linked mutations apparently unique to Arab populations, six of which were only observed in Saudi Arabia, with the most commonly observed mutations being in BRCA1 (77% of patients) [45].
In a Lebanese study, fewer than 6% of patients with early onset or familial breast cancer carried a known BRCA1 or BRCA2 PV, suggesting that alternative variants drive breast cancer incidence in this population. A total of 12% carried a VUS, with 26% having one haplotype featuring the same seven variants, and the haplotypes observed in Lebanese patients varied significantly from those observed in Tunisian and Algerian patients [46].
Two previous case studies have demonstrated links between ‘rare’ variants and familial cancer in Arab populations. In the first, the extremely rare TP53 missense variant, c.799C > T (p. Arg267Trp) was found in a 2-year-old Saudi proband diagnosed with choroid plexus carcinoma (CPC), and six first- and second-degree relatives [47]. This family comprised seven of the eight known individuals with this mutation in the Saudi population, six of which will be analyzed in the current study. The authors also observed this variant in an additional, unrelated individual with colon, breast and ovarian cancer, indicating the potential relevance of this variant to multiple types of cancer in this population. A second study reports on a 5-year-old female, whose data is also included in this study, with glioblastoma multiforme and constitutional mismatch repair-deficiency due to an MSH6 homozygous c.1883G>A mutation, who continued to experience an exceptional and durable response, 9 months later, to the immune checkpoint inhibitor (ICPI) nivolumab [48]. To the authors’ knowledge, this was the first report in the Arab world of a durable response to ICPIs in a pediatric glioblastoma patient. There is minimal other literature on specific cancer-related genetic variants in the Saudi population, which reinforces the importance of the current study.
In this study, candidates were screened using a commercial NGS panel kit comprising 30 whole or partial gene loci, for which variants have previously been reportedly associated with the development of breast, ovarian, colorectal, melanoma, pancreatic, prostate, uterine and stomach cancers based on the existing literature. This kit has been previously trialed as a means of capturing potential PVs at a population level in Nigeria and the Caribbean, and in identifying rare variants in cancer patients who have tested negative for common cancer variants [35–38]. Our results not only have an immediate clinical impact for the patients screened, but also provide important knowledge about population-specific genetic cancer risk factors that can guide future interventional programs, to prevent the development or progression of familial cancers, in Saudi Arabia and in Arab populations around the world.
Results
A total of 313 subjects were eligible for screening for sequence variants in 30 genes. For three of these patients, sequencing failed. The results of two patients had previously been recorded in another study, avoiding retesting. Within the remaining cohort, there were 310 patients and relatives (188 males and 122 females), with a mean age of 35.0 yrs (sd = 15.6 yrs), ranging from 6 months to 75 years (Supplementary Table 1). The mean age at cancer diagnosis (n = 32), was 39.1 ± 12.0 yrs.
We defined three subgroups of participants among the 313 subjects; these included 110 index patients with cancer, 143 of their family members, and 57 individuals without cancer. This latter group was designated low-risk for the purposes of the analysis, based on having no family history of cancer. The majority of these individuals were patients of the hospital for non-cancerous conditions, which justified their testing (Table 1, Figure 1).
Table 1: Subject characteristics
| N | % | |
|---|---|---|
| Relationship to Proband* | ||
| Self | 110 | 35.48 |
| Mother | 20 | 6.45 |
| Father | 15 | 4.52 |
| Son | 18 | 5.81 |
| Daughter | 18 | 5.81 |
| Sister | 33 | 10.6 |
| Brother | 19 | 6.13 |
| Granddaughter | 3 | 0.97 |
| Grandson | 2 | 0.65 |
| Maternal Aunt | 1 | 0.32 |
| Maternal Uncle | 1 | 0.32 |
| Paternal Aunt | 3 | 0.97 |
| Paternal Uncle | 2 | 0.65 |
| Nephew | 2 | 0.65 |
| Niece | 5 | 1.61 |
| Cousin | 1 | 0.32 |
| No family history of cancer | 57 | 18.39 |
| Gender | ||
| Male | 188 | 60.65% |
| Female | 122 | 39.35% |
| History of cancer | ||
| Yes | 126 | 40.65% |
| No | 184 | 59.35% |
| Age (yrs) | ||
| <20 | 55 | 17.74% |
| 20–39 | 141 | 45.48% |
| 40–60 | 89 | 28.71% |
| >60 | 20 | 6.45% |
| Missing | 5 | 1.61% |
| Total | ||
| All subjects | 310 | 100% |
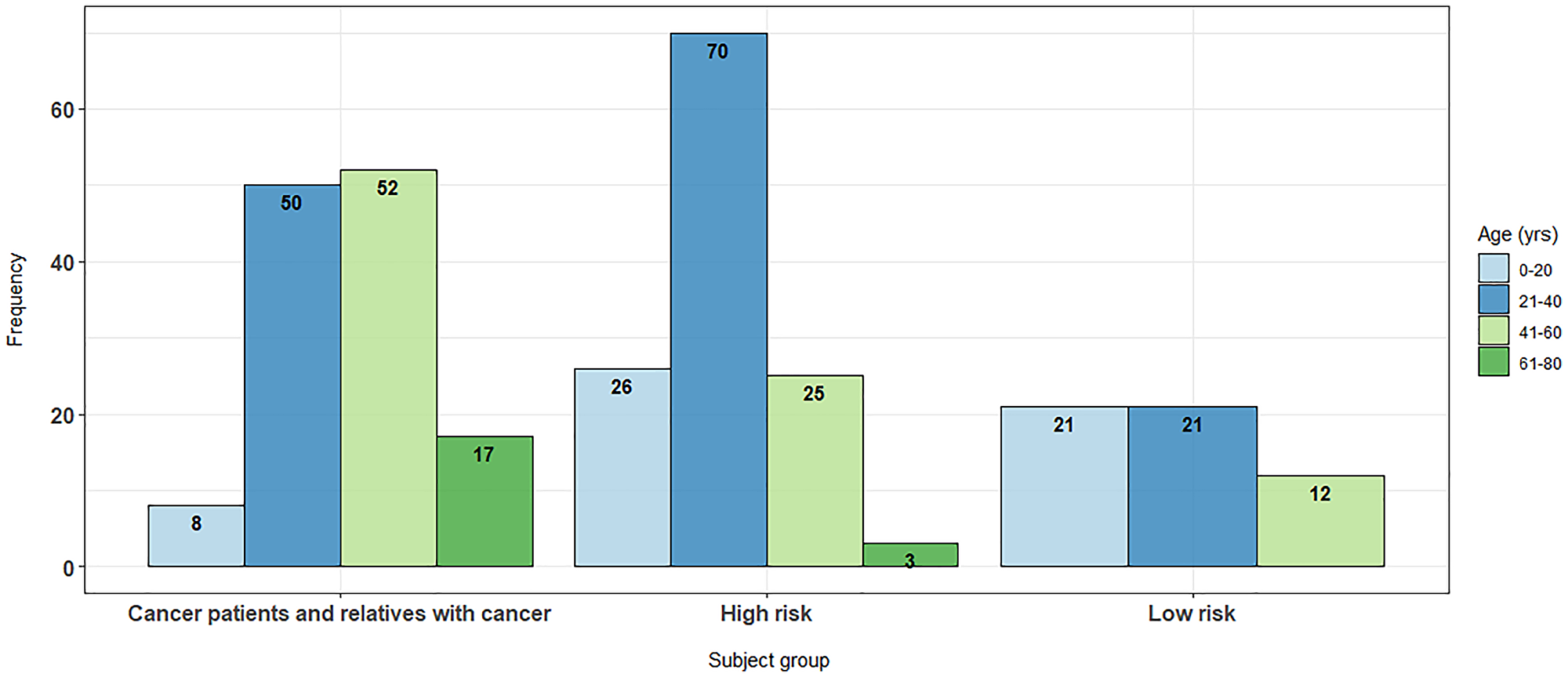
Figure 1: Age distributions across cohort subgroups.
Sixteen of the 143 family members of cancer patients (11.2%) had also been diagnosed with cancer at any time prior to this study, and are therefore grouped with the ‘index’ cancer patients for the purpose of the analysis. These diagnosed relatives and our index cancer patients are collectively referred to within the analysis as ‘persons with a history of cancer’.
Family members of cancer patients, who had never prior to this study been diagnosed with any cancer, are designated separately within the analyses as ‘high risk’ individuals. We consider them to be at higher risk of developing cancers in the future than individuals with no family history of cancer.
The relatives of index patients tended to be their descendants, therefore the age distributions of patients varied significantly between these groups, with high-risk individuals and non-cancer patients both typically being younger than the index cancer patients (Figure 1; p < 0.001). Due to ethical considerations and the relatedness of the subjects it was not feasible to resample by age or to prospectively recruit to address the age skew.
Across all groups, there were more female than male participants, although the distribution of gender was similar across groups (p > 0.05). Information on consanguinity was only available for 27 of the patients, 12 of which reported consanguinity.
Pathogenic variants
We identified germline PVs and likely PVs in the following genes: APC, ATM, BRCA1, BRCA2, BRIP1, CDKN2A (in both p14ARF and p16INK open reading frames), CHEK2, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, and TP53 (Supplementary Tables 2 and 3). Variants of undetermined significance (VUS) were observed in the following genes: BRCA2, APC, ATM, PALB2, MSH2, MLH1, MSH6, CHEK2, CDKN2A, TP53, NBN, PMS2, RAD51D, BRIP1, BMPR1A, and CDH1.
Roughly half of the 123 unique variants observed were singleton variants (n = 67, Supplementary Table 3). However, we identified 13 frequent variants occurring in five or more subjects, 2 of which are considered likely PVs (APC c.3920T>A and TP53 c.799C>T), and six of which have been previously classified as PVs (BRCA1 c.5123C>A, MSH2 c.1964del, MUTYH c.544C>T and c.734G>A, PMS2 c.1376C>G and c.1606C>T). PMS2 c.1376C>G was the most frequent Lynch syndrome-associated variant in a survey of Saudi CRC patients [39].
The proportions of persons with a history of cancer (including index patients and diagnosed relatives), high-risk participants (undiagnosed relatives of index patients) and low-risk participants (non-cancer patients with no patient history), with PVs, likely PVs and VUS, were determined and compared (Table 2, Figure 2). In total, there were 123 individual genetic variants found or reported among the 310 subjects (Supplementary Table 3), including 34 PVs, 7 likely PVs and 82 VUS. Considering the numbers of patients with and without PVs, likely PVs and VUS, there was no significant difference in the numbers of subjects with and without PVs (p > 0.05, Fisher’s exact test). Similarly, there was no significant difference in the proportion of patients with likely PVs across groups, or in the prevalence of VUS between groups (p > 0.05). However, the likelihood of homozygosity at one or more PV or likely PV was significantly higher in persons with cancer (n = 10 of 126, 7.9%) based on an exact test for a multinomial distribution (p LLR<0.05) when compared to one case of homozygosity at a PV/likely PV among the low-risk participants and only one case among the high-risk participants.
Table 2: Individuals with PVs, likely PVs and/or VUS
| Number of patients with different variant classifications | Cancer patients (n = 110) and family members with cancer (n = 16) n = 126 (%) | High Risk Individuals n = 125 (%) | Low Risk Individuals n = 57 (%) |
|---|---|---|---|
| Pathogenic variant(s) (PVs) | 36 (28.3) | 31 (24.8) | 12 (21.1) |
| Likely PV(s), no PVs | 13 (10.3) | 20 (16.0) | 5 (8.8) |
| VUS only | 29 (23.0) | 26 (20.8) | 17 (29.8) |
| Negative for all tested variants | 48 (38.1) | 48 (38.4) | 21 (36.8) |
| Not tested- syndicated data | 0 | 0 | 2 (3.5) |
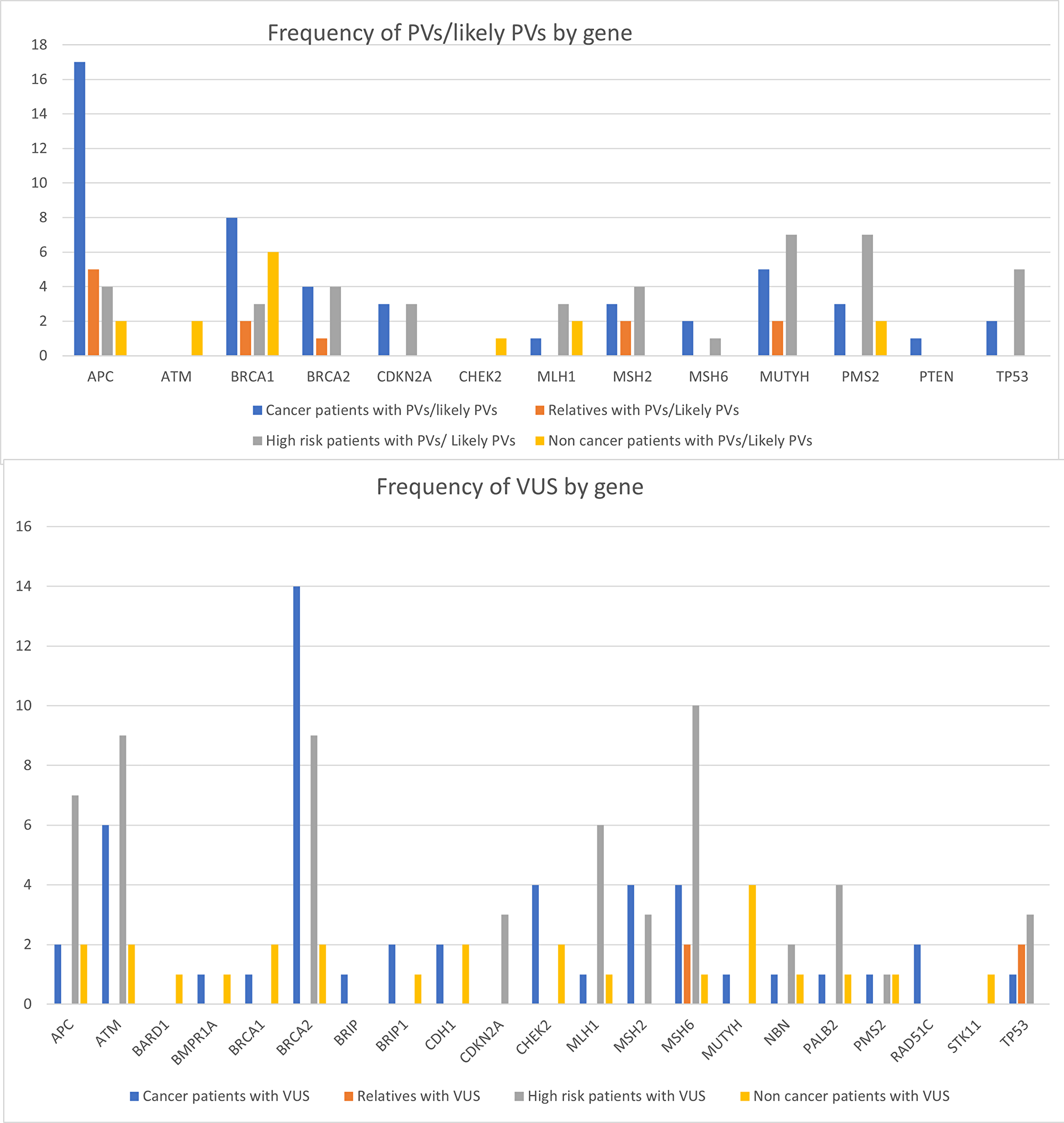
Figure 2: Frequencies of PVs and likely PVs per gene, by subject group. A list of all variants discovered in each patient group, along with any cancer types diagnosed in patients with these variants, can be found in the Supplementary Tables 1 and 4.
There were 13 instances in which patients were homozygous at SNP loci, ten of which involved index cancer patients. Nine of these loci were PVs and three were likely PVs (Supplementary Tables 2, 3 and Supplementary Figures). There was one instance of a duplication in MSH6 in a colon cancer patient, and three patients with duplications of BRCA2 c.5557 within the same family, two of which reported breast cancer. Fifteen subjects had deletions within single exons, and seven patients had deletions spanning multiple exons in BRCA1, APC, ATM, CHEK2 or MSH2. We observed a large chromosomal rearrangement of chromosome 9 removing CDKN2A (p.14ARF) in a bone marrow cancer patient (Supplementary Tables 1 and 3), and six out of 18 unique indel events were observed in multiple members of a family.
Genes of interest
PVs in the APC gene were significantly more frequent in cancer patients than in the other groups and were observed at least twice as frequently in cancer patients than PVs in any other gene within the panel (p LLR = 0.0003). Other PVs appeared to be normally distributed (p > 0.05, Figure 2), although the overall numbers of subjects with most PVs were small enough that a likelihood ratio version of the exact test for a multinomial had to be used, so it is not surprising that no association was detected. BRCA2 variants were also significantly more frequent in persons with cancer (p = 9.82e−08), with a notable enrichment of BRCA2 VUS in these patients (Figure 2). We anticipate that some of the VUS flagged within this panel may prove pathogenic or to be linked to PVs if investigated in larger Arab population samples.
Associations with cancer
Despite their approximately proportionate overall distribution, the various PVs do not have the same impact across all types of cancer, so it is important to consider the association of each individual PV with the various types of cancer observed. In order to detect any associations between individual variants and cancer occurrence, the observed cancers were grouped by the affected organ (or organ system). APC PVs/likely PVs were found in 20 of the 126 participants with cancer (15.9%). The most common types of cancer observed in patients with APC PVs/ likely PVs were colon cancer (n = 9), rectal cancer (n = 5) and breast cancer (n = 4). APC PVs/ likely PVs were also observed in 21 of 125 high-risk patients (16.8%) and 7 of 57 ‘low-risk’ participants with no family history of cancer (12.3%). The most common APC variant was likely PV c.3920T>A, occurring in 39 subjects, this was observed with MSH2 Val655Aspfs*30 in 5 subjects, all from one family, and with MSH6 variants in 7 cases, which again were all from the same family. There was a significant association of the APC c.3920T>A; p.Ile1307Lys variant with colorectal cancers, i.e. colon, rectal, and sigmoid cancers, as well as polyposis (p-value = 0.03).
Of the 19 persons with cancer carrying BRCA2 variants, eight had breast cancer, seven of which lacked BRCA1 variants. Four patients carrying BRCA2 variants had rectal cancer, and three had colon cancer. All PVs observed within BRCA2 were deletion or duplication events which significantly disrupt gene function (c.3170_3174del, c.4787del, c.5557dup), these appeared only in cancer patients and high-risk patients, with 5 of 7 BRCA2 PV carriers affected. Carriers were significantly more likely to have breast cancer than non-carriers (p = 8e−06), with 50% of carriers having breast cancer across four unrelated families (Supplementary Table 3). The most frequent BRCA2 variant in this sample was the previously considered VUS c.122C>T, although carriers with cancer did not report breast cancer. No individual BRCA1 variants were significantly associated with cancer or breast cancer specifically within this sample, most likely due to sample size.
Other statistically significant associations included an association of TP53 c.868C>T; p.Arg290Cys with multiple colon polyposis in this population. There were also 21 less common variants appearing 3 or more times whose risk association with cancer could not be quantified because they only appeared in cancer and/or high-risk subjects with a family history of cancer, and many unique variants (Supplementary Table 3). For example, there were 13 subjects with MSH6 c.733A>T, which is considered a VUS, all of whom had cancer or were high-risk subjects. These frequent variants, unique to cancer and high-risk patients, which should be noted as potential PVs, included TP53 c.799C>T, APC c.3183_3187delACAAA, ATM c.1516G>T, BRCA2 c.3170_3174del, c.122C>T, c.5909C>T and c.5557dup, CDKN2A c.238C>T, MSH2 c.1964del, MSH6 c.733A>T deletion of the whole of exon 16 of MLH1, MUTYH c.544C>T and c.734G>A, and PALB2 c.1102A>T.
For persons with cancer, eight cancer types were most commonly observed. The most common primary cancer types were colorectal cancers, including colon, rectal, and sigmoid cancers, as well as polyposis (n = 60; 47.2% of persons with cancer), breast cancers (n = 34; 27.6%), and ovarian cancer (n = 5; 3.9%) (Figure 3). Some patients had more than one of these eight cancer types on primary presentation or had a different secondary cancer type (Supplementary Data 1).
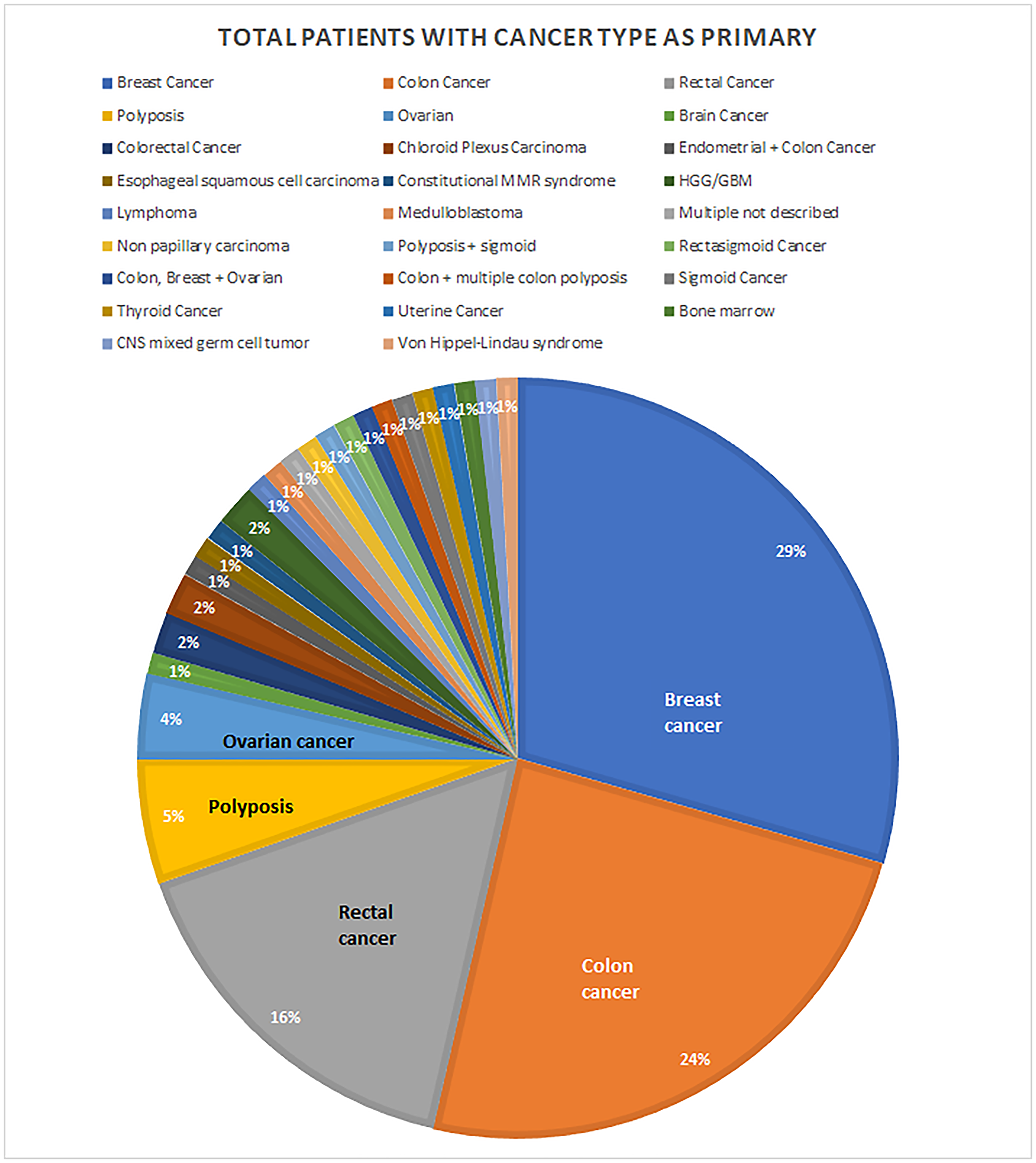
Figure 3: Locations of primary cancers. Some patients have reported multiple cancer types on primary presentation, each patient is counted once.
A total of 107 participants, 49 (45.8%) of which had a history of cancer, had one or more PV or likely PV. Out of the remaining 203 participants, who were negative for the PVs and likely PVs included in the panel, 72 (35.5%) had VUS, and 40.3% (n = 29) of those patients with VUS had some form of cancer. This suggests the influence of one or more VUS that have not been previously categorized as pathogenic, but which have pathogenic activity in this Arab population.
A correlation analysis of variants showed 14 distinct pairs of variants were significantly correlated (frequently appearing together), even if the genes involved were on different chromosomes and therefore unlinked. This may be because several of the study participants were grouped into families, including some in which one or both parents had multiple variants; additionally, some variants are likely to be present in both parents due to the high degree of consanguinity in Saudi Arabia. Thus, the parents had an increased chance of passing on certain variants to their children. Of the 14 significant correlations, six involved PVs and a further three involved likely PVs (Supplementary Table 4). Notably, APC c.3920T>A was significantly linked to MSH2 c.1964del (p = 1.8e-07) and MSH6 c.733A>T (p = 0.02).
DISCUSSION
Recent advances in the field of medical genetics have allowed a much deeper understanding of the underlying genetic risk factors for cancer. In particular, cancer screening utilizing germline genetic sequencing of panels of cancer susceptibility genes has become a powerful tool to identify potential underlying familial genetic variants and their associations with elevated cancer risks [40]. For example, studies within the Israeli Ashkenazi Jewish population, which is well-characterized, have illuminated the existence of three founder mutations that are estimated to account for up to 30% of early-onset breast cancer and 60% of ovarian cancer in this population [41, 42]. The discovery of these mutations has fundamentally changed the public health management of early-onset cancer in this population, which includes the recommendation of genetic testing for all individuals of Ashkenazi Jewish descent as part of early cancer detection and prevention [41]. There may be genetic variants associated with cancer in many ethnic or regional populations, such as people of Arab descent, that do not appear be associated with cancer, or that show lower association with cancer, in the European populations normally recruited for these types of association studies [14].
This observational study investigated familial cancer biomarkers in 110 cancer patients, their family members, and unrelated patients with other conditions. Roughly a quarter (24.4%, n = 31) of the family members of the patients with cancer who had not yet received any cancer diagnosis (n = 127), and 8 of 16 relatives with cancer themselves (50%), had at least one PV as defined based on PV effects demonstrated in non-Arab-specific populations (Supplementary Table 1). These data demonstrate a sufficient presence of genetic variants with the potential to increase cancer risk in the Saudi population to justify prioritizing genetic surveillance in Saudi Arabia. However, it must be acknowledged that PVs can have low penetrance, and often do not lead to disease onset [43].
The disproportionately high percentage of individuals with variants demonstrated in other settings to be pathogenic, combined with the higher number of homozygotes for PVs or likely PVs, in index patients and family members with cancer than would be expected from a proportional distribution, indicates that, despite the variant classifications being based on studies of non-Arab specific populations, they do apply, at large, to people of Middle Eastern descent.
The roughly proportionate numbers of cancer patients, family members with cancer and high-risk relatives carrying one or more VUS indicates that some of these variants are benign in those of Arab ethnicity. We should therefore not make any assumptions that each variant within our panel is relevant, or that the panel of potential variants is complete. While the overall categorization of variant types based on non-Arab populations applies to the study population, individual variants not believed to play a role in cancer occurrence in one population may exhibit such a role in another population [14]. Additionally, the frequencies of VUS, likely PVs and PVs can depend on the ancestry of the population [24]. Therefore, all variants found in the Arab, mostly Saudi Arabian, subjects of this study were statistically analyzed for their potential association with cancer in general Arab populations.
Where associations observed have previously been specifically linked to hereditary cancers in other settings, this leads us to believe that we are correct in considering them familial. Over half of all subjects had one or more variant in the APC gene, the most common of which was APC c.3920T>A; p.Ile1307Lys, which was significantly associated with colon cancer (p = 0.03predict). This is considered a low penetrance VUS in most settings [16], but it has been demonstrably associated with colorectal tumor risk in the Israeli Ashkenazi Jewish population [42]. APC c.3920T>A; p.Ile1307Lys was observed in 18 of 126 cancer cases (14.3%) and 10 of 67 colorectal cancer cases (14.9%), compared to 4 of 57 individuals without cancer or family history of cancer (7.0%). We propose that APC c.3920T>A; p.Ile1307Lys may act as a pathogenic variant in this population, and may prove a useful marker for colon cancer risk in Arab populations.
Multiple variants may impact the same signaling pathways to create a higher likelihood of cancer if appearing together. For example, reduced expression of MUTYH and TP53 mutation, both of which are associate with diffuse-type histology, have been shown to be associated in adenocarcinomas of gastric cardia patients. In this cohort, APC c.3920T>A; p.Ile1307Lys was linked to MSH2 c.1964del; p.Val655Aspfs*30 and MSH6 c.733A>T; p.Ile245Leu. All three of these variants have been tentatively associated with Lynch syndrome. MSH2 c.1964de has previously been identified in Lynch syndrome patients with colorectal tumors within Saudi Arabia [29].
It is our intention that our findings influence future screening protocols. We note with interest the diversity of BRCA1 and BRCA2 variants within our cancer patient group, and the enrichment of BRCA2 VUS in cancer patients. Whilst no specific variants dominated the pool, in general terms BRCA2 variants were associated with cancer and BRCA2 confirmed PVs were strongly associated with breast cancer collectively (p = 8e−06). Only in one instance did a breast cancer patient have both BRCA1 and BRCA2 variants. Despite identifying five ovarian cancer patients, there was no crossover between the variants we identified in our patients with cancer and the five most common BRCA1 variants identified in the meta-analysis by Younes and Zayid [45], nor any carriers of the key Ashkenazi ‘founder’ BRCA1 and BRCA2 variants [41, 42].
The BRCA1 variant c5530delC, which has been flagged as unique among Saudi Arabian ovarian cancer patients [45], and one of the most common variants observed in another Saudi Arabian ovarian cancer cohort by Agha et al. [49], was present in one subject with ovarian cancer, with each patient carrying a different variant (3 in BRCA1, 1 in APC, 1 in TP53, see Supplementary Table 3). We note a similar overall incidence of BRCA1 mutation in association with ovarian cancer between this study and that of Agha et al. (60% vs. 77%), although our ovarian cancer patient pool is small. Screening for a diverse panel of BRCA1 and BRCA2 variants may be necessary to protect women in Arab populations who have a known family history of breast and/or ovarian cancer.
As APC c.3920T>A; p.Ile1307Lys was found to be associated with a higher occurrence of CRC (including colon, rectal and sigmoid cancers, as well as polyposis; p-value = 0.0257), the unexpected frequency of the variant in low risk individuals may indicate a greater predisposition for, or prevalence of, CRC in the general Saudi population than has been previously detected. Asymptomatic individuals with this variant may need to be followed up by an oncologist, as they may develop cancer in the future. A larger study might reveal the need for national screening for this variant in any Saudi individuals who have a family history of CRC, and this variant may function as a CRC-associated marker in the Saudi population. Until further investigation can be completed into the specific impact of frequent and rare APC and BRCA2 variants in Arab cohorts, all patients carrying either APC or BRCA2 variants should be considered at elevated risk of CRC and breast cancer and be offered screening accordingly.
Whilst frequent variants are excellent candidates for future screening panels, rarer variants are important to acknowledge. TP53 c.868C>T; p.Arg290Cys was significantly associated with multiple colon polyposis in this population, despite not having been previously linked to any specific cancer form. In our study population, TP53 c.868C>T; p.Arg290Cys is carried by five members of the same family, two of which have polyposis and one of which may have polyposis. The family do not share any other variant detected in this gene panel.
There were many variants appearing 3 or more times only in cancer and/or high-risk subjects which should be considered to be potential PVs (TP53 c.799C>, APC c.3183_3187delACAAA, ATM c.1516G>T, BRCA2 c.3170_3174del, c.122C>T, c.5909C>T and c.5557dup, CDKN2A c.238C>T, MSH2 c.1964del, MSH6 c.733A>T deletion of the whole of exon 16 of MLH1, MUTYH c.544C>T and c.734G>A, and PALB2 c.1102A>T). All TP53 c.799C > T carriers were those patients syndicated from a previous family case study [44], with no additional carriers of this variant identified among our patient cohort or their family members.
Limitations of this study included limited sample size. However, this is still the largest prospective study of its type in this population. Another limitation of the study is the uneven distribution of ages between various subject and control groups, involving a skewing towards older individuals in patients with cancer. A longitudinal follow-up studies of these high-risk relatives, low-risk individuals, or controls with variants should reveal or strengthen any such associations over time. The limited NGS panel was intended to be used as preliminary screening tool, therefore we recommended a more comprehensive panel to be used in negative high-risk cases (Figure 4). Finally, the cancer negative patients screened via panel test are not fully representative of a healthy population, as each had some clinical symptoms or family history prompting their initial panel testing for other purposes.
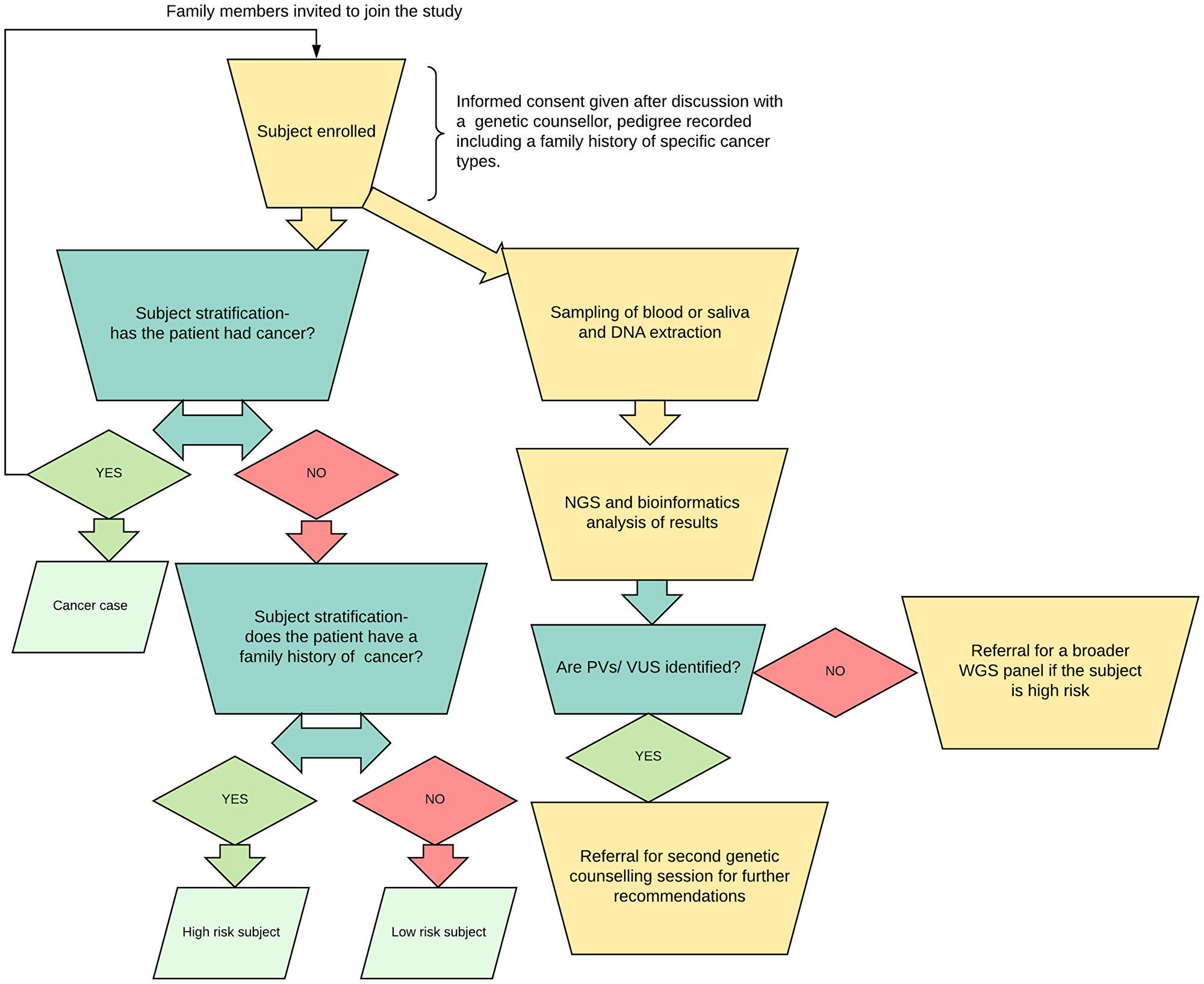
Figure 4: Workflow diagram. Subjects were stratified into three groups, their saliva samples were processed and sent for next generation sequencing. Family history was defined, in order to apply to all cancer subgroups, as having one or more relatives (parents, grandparents etc.) with any type of cancer. A breakdown of the relationships to the cancer patient subject demographic data for all subjects are available in Supplementary Table 1.
These results indicate that, in general, the categorization of PVs based on their effects on non-Arab populations may produce an incomplete picture of variants with relevance for a Saudi population. Some PVs common in Caucasian populations be less frequent or less likely to predict cancer onset, and some VUS that occur at low frequency in other reported populations may play a primary role in cancer risk in Arab populations.
In conclusion, this study is one of the first to report the prevalence of inherited cancer genetic variants in a cohort from the Arab world. Our study gives critical first insights into the genetic variants associated with overall cancer risk in this specific population, and specific forms including CRC/Lynch syndrome and breast cancer. Whilst a larger population level study is still needed, we demonstrate that multigene NGS panel testing may serve as non-invasive diagnostic and cost-effective tool to predict familial cancer risk at the pre-clinical stage, allowing targeted screening and enabling early intervention.
Materials and Methods
Subjects
The study cohort was recruited from the King Fahad Medical City (KFMC) clinical departments and from other medical centers throughout Saudi Arabia, and therefore represents a geographically diverse set of patients. KFMC is a major tertiary referral center in Middle East with ~1200 beds and 8 specialized centers including a comprehensive cancer center. Cancer patients diagnosed with breast, ovarian, colorectal, brain, thyroid, melanoma, pancreatic, prostate, uterine, or stomach cancer, and their family members, were invited to participate in the study. All of the subjects were referred to the Familial Genetic Counseling Clinic for a preliminary meeting, and again if relevant following sequencing, to be informed of any mutations relevant to their health. The majority of participants were Saudi Arabian nationals, and a small minority represented other Arab nationalities (Table 1). Although race and ethnicity data were not available, an estimated 90% of Saudi Arabian nationals are Arab (CIA, 2022), therefore we assume our cohort to be predominantly Arab.
The panel results of two of the included patients, neither or which had a history of cancer, were syndicated from another clinic within the same medical group (Supplementary Table 1).
Study procedures
Enrolled subjects were stratified into groups, and their DNA samples were obtained and processed, after seeing a certified genetic counselor in a cancer genetics clinic and obtaining informed consent (Figure 4).
Next generation sequencing
Target enrichment was carried out according to Agilent’s SureSelect method (v1.7), followed by sequencing via Illumina’s NextSeq 500 or NovaSeq600 (paired-end 150 bp, High Output kit), conducted at Color® Genomics Laboratory, to analyze the panel of 30 genes featured in the cancer susceptibility gene panel tool kit from Color® Genomics Laboratory (APC, ATM, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A [p14ARF and p16INK4a], CHEK2, EPCAM, GREM1, MITF, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, POLE, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53), in which mutations have been associated with an elevated risk for breast, ovarian, colorectal, brain, thyroid, melanoma, pancreatic, prostate, uterine, or stomach cancers (Supplementary Table 2). The majority of these genes were assessed for variants within all coding exons with +/− 20 bp flanking each exon. This panel kit should broadly capture gene variants reported to be associated with a variety of cancers, and has been applied across diverse research settings to characterize cancers and to determine patient risk [35–38, 50, 51]. The methodology was followed by our team as recommended by the provider, including quality control checks incorporated to ensure proper sample identification and efficiency of DNA isolation, library preparation and target capture. In addition, each sequencing test contained two fully-characterized positive controls. The Color Test for hereditary cancer has been developed in compliance with the Clinical Laboratory Improvements Act of 1988.
Bioinformatics analysis
The bioinformatics analysis of sequence data followed a previously published pipeline [37, 38]. Copy number variations were detected using dedicated internally developed algorithms for read depth analysis and split-read alignment detection. Variants were classified according to the standards and guidelines for sequence variant interpretation of the American College of Medical Genetics and Genomics (ACMG) [37, 38, 52] into pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign categories. All variants were evaluated by a board-certified medical geneticist or pathologist. Variants classified as pathogenic or likely pathogenic were confirmed through secondary technology (Sanger sequencing, array comparative genomic hybridization, or multiplex-ligation dependent probe amplification) before reporting.
Statistical analysis
All statistical analyses were conducted using R statistical software v4.1.2 and R package XNomial (function”xmulti”) v1.0.4. Study participants were classified according to whether they had only PVs, likely PVs, VUS (with variants reported to have conflicting interpretations of pathogenicity considered as VUS), or various combinations of these three variant types. Variant classifications were assigned to the genetic variants found in subjects following the criteria of the ACMG, based on characteristics described either on the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) or in the literature, although we apply these criteria tentatively to this Arab-specific study population. Statistical tests were conducted as described in the Supplementary Methods.
Abbreviations
NGS: next-generation sequencing; PV: pathogenic variant; FCS: familial/hereditary cancer syndromes; CRC: colorectal cancer; CPC: choroid plexus carcinoma; ICPI: immune checkpoint inhibitor; KFMC: King Fahad Medical City; VUS: variant of uncertain significance.
ACKNOWLEDGMENTS
This study was supported by King Fahad Medical City (RFA17-17). We would like to thank all volunteers in this study, including genetics counselors Balsam AlMaarak (2018–2019) and Lamia AlSubaie (2017–2018).
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
Ethical statement and consent
We declare that informed consent was obtained from all participants in adherence with the Declaration of Helsinki and the KFMC IRB and Research Advisory Committees (RAC) rules and regulations under the following approved project (KFMC IRB 16.310). All protocols are carried out in accordance with relevant guidelines and regulations. Approval was obtained from the KFMC ethics committee. Participants gave informed consent to participate in the study before taking part.
FUNDING
This study was supported by King Fahad Medical City MA and MAA (RFA17-17). The funding body was not involved in any stage of the study and had no role in the design of the study, the collection, analysis, and interpretation of data or the writing of the manuscript.
References
1. Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg. 2010; 44:293–311. https://doi.org/10.1016/j.yasu.2010.05.011. [PubMed].
2. National Cancer Institute (USA). Cancer Genetics Overview (PDQ®)–Health Professional Version. 2006. https://www.cancer.gov/about-cancer/causes-prevention/genetics/overview-pdq.
3. Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer. 1996; 77:2318–24. https://doi.org/10.1002/(SICI)1097-0142(19960601)77:11<2318::AID-CNCR21>3.0.CO;2-Z. [PubMed].
4. Jazieh AR, Da’ar OB, Alkaiyat M, Zaatreh YA, Saad AA, Bustami R, Alrujaib M, Alkattan K. Cancer Incidence Trends From 1999 to 2015 And Contributions Of Various Cancer Types To The Overall Burden: Projections To 2030 And Extrapolation Of Economic Burden In Saudi Arabia. Cancer Manag Res. 2019; 11:9665–74. https://doi.org/10.2147/CMAR.S222667. [PubMed].
5. Azzam N, AlRuthia Y, Alharbi O, Aljebreen A, Almadi M, Alarfaj M, Alsaleh K, Almasoud A, Alsharidah M, Alseneidi S, Alali F, Alalwan M. Predictors of Survival Among Colorectal Cancer Patients in a Low Incidence Area. Cancer Manag Res. 2020; 12:451–59. https://doi.org/10.2147/CMAR.S233215. [PubMed].
6. Chaudhri E, Fathi W, Hussain F, Hashmi SK. The Increasing Trends in Cases of the Most Common Cancers in Saudi Arabia. J Epidemiol Glob Health. 2020; 10:258–62. https://doi.org/10.2991/jegh.k.200515.001. [PubMed].
7. Abedalthagafi MS. Precision medicine of monogenic disorders: Lessons learned from the Saudi human genome. Front Biosci (Landmark Ed). 2019; 24:870–89. https://doi.org/10.2741/4757. [PubMed].
8. AlHarthi FS, Qari A, Edress A, Abedalthagafi M. Familial/inherited cancer syndrome: a focus on the highly consanguineous Arab population. NPJ Genom Med. 2020; 5:3. https://doi.org/10.1038/s41525-019-0110-y. [PubMed].
9. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008; 359:2143–53. https://doi.org/10.1056/NEJMra0802968. [PubMed].
10. Alqahtani WS, Almufareh NA, Domiaty DM, Albasher G, Alduwish MA, Alkhalaf H, Almuzzaini B, Al-Marshidy SS, Alfraihi R, Elasbali AM, Ahmed HG, Almutlaq BA. Epidemiology of cancer in Saudi Arabia thru 2010-2019: a systematic review with constrained meta-analysis. AIMS Public Health. 2020; 7:679–96. https://doi.org/10.3934/publichealth.2020053. [PubMed].
11. Basudan AM. Breast Cancer Incidence Patterns in the Saudi Female Population: A 17-Year Retrospective Analysis. Medicina (Kaunas). 2022; 58:1617. https://doi.org/10.3390/medicina58111617. [PubMed].
12. Zacharakis G, Almasoud A, Aldossari K. Colorectal cancer screening challenges in Saudi Arabia. A comprehensive review article. Archives of Medical Science - Civilization Diseases. 2022; 7:e24–32. https://doi.org/10.5114/amscd.2022.119965.
13. Woodward ER, Green K, Burghel GJ, Bulman M, Clancy T, Lalloo F, Schlecht H, Wallace AJ, Evans DG. 30 year experience of index case identification and outcomes of cascade testing in high-risk breast and colorectal cancer predisposition genes. Eur J Hum Genet. 2022; 30:413–19. https://doi.org/10.1038/s41431-021-01011-8. [PubMed].
14. Barakeh DH, Aljelaify R, Bashawri Y, Almutairi A, Alqubaishi F, Alnamnakani M, Almubarak L, Al Naeem A, Almushawah F, Alrashed M, Abedalthagafi M. Landscape of somatic mutations in breast cancer: new opportunities for targeted therapies in Saudi Arabian patients. Oncotarget. 2021; 12:686–97. https://doi.org/10.18632/oncotarget.27909. [PubMed].
15. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017; 23:703–13. https://doi.org/10.1038/nm.4333. [PubMed].
16. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, Goldberg RM, Paskett E, Shields PG, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017; 3:464–71. https://doi.org/10.1001/jamaoncol.2016.5194. [PubMed].
17. Alabdulkarim B, Hassanain M, Bokhari A, AlSaif A, Alkarji H. Age distribution and outcomes in patients undergoing breast cancer resection in Saudi Arabia. A single-institute study. Saudi Med J. 2018; 39:464–69. https://doi.org/10.15537/smj.2018.5.21993. [PubMed].
18. Mosli MH, Al-Ahwal MS. Colorectal cancer in the Kingdom of Saudi Arabia: need for screening. Asian Pac J Cancer Prev. 2012; 13:3809–13. https://doi.org/10.7314/apjcp.2012.13.8.3809. [PubMed].
19. Albanghali MA. Prevalence of Consanguineous Marriage among Saudi Citizens of Albaha, a Cross-Sectional Study. Int J Environ Res Public Health. 2023; 20:3767. https://doi.org/10.3390/ijerph20043767. [PubMed].
20. Mahboub SM, Alsaqabi AA, Allwimi NA, Aleissa DN, Al-Mubarak BA. Prevalence and pattern of consanguineous marriage among educated married individuals in Riyadh. J Biosoc Sci. 2020; 52:768–75. https://doi.org/10.1017/S0021932019000786. [PubMed].
21. Nagahashi M, Shimada Y, Ichikawa H, Kameyama H, Takabe K, Okuda S, Wakai T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019; 110:6–15. https://doi.org/10.1111/cas.13837. [PubMed].
22. Mandelker D, Zhang L, Kemel Y, Stadler ZK, Joseph V, Zehir A, Pradhan N, Arnold A, Walsh MF, Li Y, Balakrishnan AR, Syed A, Prasad M, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA. 2017; 318:825–35. https://doi.org/10.1001/jama.2017.11137. [PubMed].
23. Hu C, Hart SN, Gnanaolivu R, Huang H, Lee KY, Na J, Gao C, Lilyquist J, Yadav S, Boddicker NJ, Samara R, Klebba J, Ambrosone CB, et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med. 2021; 384:440–51. https://doi.org/10.1056/NEJMoa2005936. [PubMed].
24. Roberts ME, Susswein LR, Janice Cheng W, Carter NJ, Carter AC, Klein RT, Hruska KS, Marshall ML. Ancestry-specific hereditary cancer panel yields: Moving toward more personalized risk assessment. J Genet Couns. 2020; 29:598–606. https://doi.org/10.1002/jgc4.1257. [PubMed].
25. Liede A, Narod SA. Hereditary breast and ovarian cancer in Asia: genetic epidemiology of BRCA1 and BRCA2. Hum Mutat. 2002; 20:413–24. https://doi.org/10.1002/humu.10154. [PubMed].
26. Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, Wahlström C, Pooley KA, Parsons MT, Fortuno C, Wang Q, Bolla MK, Dennis J, et al. and Breast Cancer Association Consortium. Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med. 2021; 384:428–39. https://doi.org/10.1056/NEJMoa1913948. [PubMed].
27. Khoueiry P, Fakhri G, Akel R, El Assaad M, Mahfouz R, Khuri F, Chami H, Petersen J, Viet S, Davies G, Kadara H, Tfayli A. Deep targeted sequencing analysis of hot spot mutations in non-small cell lung cancer patients from the Middle Eastern population. J Thorac Dis. 2019; 11:2383–91. https://doi.org/10.21037/jtd.2019.05.74. [PubMed].
28. Siraj AK, Masoodi T, Bu R, Parvathareddy SK, Iqbal K, Azam S, Al-Rasheed M, Ajarim D, Tulbah A, Al-Dayel F, Al-Kuraya KS. Prevalence of germline TP53 mutation among early onset middle eastern breast cancer patients. Hered Cancer Clin Pract. 2021; 19:49. https://doi.org/10.1186/s13053-021-00206-w. [PubMed].
29. Siraj AK, Kumar Parvathareddy S, Pratheeshkumar P, Padmaja Divya S, Ahmed SO, Melosantos R, Begum R, Concepcion RMJ, Al-Sanea N, Ashari LH, Abduljabbar A, Al-Dayel F, Al-Kuraya KS. APC truncating mutations in Middle Eastern Population: Tankyrase inhibitor is an effective strategy to sensitize APC mutant CRC To 5-FU chemotherapy. Biomed Pharmacother. 2020; 121:109572. https://doi.org/10.1016/j.biopha.2019.109572. [PubMed].
30. Siraj AK, Masoodi T, Bu R, Pratheeshkumar P, Al-Sanea N, Ashari LH, Abduljabbar A, Alhomoud S, Al-Dayel F, Alkuraya FS, Al-Kuraya KS. MED12 is recurrently mutated in Middle Eastern colorectal cancer. Gut. 2018; 67:663–71. https://doi.org/10.1136/gutjnl-2016-313334. [PubMed].
31. Siraj AK, Prabhakaran S, Bavi P, Bu R, Beg S, Hazmi MA, Al-Rasheed M, Al-Assiri M, Sairafi R, Al-Dayel F, Al-Sanea N, Uddin S, Al-Kuraya KS. Prevalence of Lynch syndrome in a Middle Eastern population with colorectal cancer. Cancer. 2015; 121:1762–71. https://doi.org/10.1002/cncr.29288. [PubMed].
32. Siraj AK, Bu R, Prabhakaran S, Bavi P, Beg S, Al Hazmi M, Al-Rasheed M, Alobaisi K, Al-Dayel F, AlManea H, Al-Sanea N, Uddin S, Al-Kuraya KS. A very low incidence of BRAF mutations in Middle Eastern colorectal carcinoma. Mol Cancer. 2014; 13:168. https://doi.org/10.1186/1476-4598-13-168. [PubMed].
33. Karakas B, Colak D, Kaya N, Ghebeh H, Al-Qasem A, Hendrayani F, Toulimat M, Al-Tweigeri T, Park BH, Aboussekhra A. Prevalence of PIK3CA mutations and the SNP rs17849079 in Arab breast cancer patients. Cancer Biol Ther. 2013; 14:888–96. https://doi.org/10.4161/cbt.25945. [PubMed].
34. Al-Qasem AJ, Toulimat M, Eldali AM, Tulbah A, Al-Yousef N, Al-Daihan SK, Al-Tassan N, Al-Tweigeri T, Aboussekhra A. TP53 genetic alterations in Arab breast cancer patients: Novel mutations, pattern and distribution. Oncol Lett. 2011; 2:363–69. https://doi.org/10.3892/ol.2011.236. [PubMed].
35. Adejumo PO, Aniagwu TIG, Awolude OA, Oni AO, Ajayi OO, Fagbenle O, Ogungbade D, Kochheiser M, Ogundiran T, Olopade OI. Feasibility of genetic testing for cancer risk assessment programme in Nigeria. Ecancermedicalscience. 2021; 15:1283. https://doi.org/10.3332/ecancer.2021.1283. [PubMed].
36. George SHL, Donenberg T, Alexis C, DeGennaro V Jr, Dyer H, Yin S, Ali J, Butler R, Chin SN, Curling D, Lowe D, Lunn J, Turnquest T, et al. Gene Sequencing for Pathogenic Variants Among Adults With Breast and Ovarian Cancer in the Caribbean. JAMA Netw Open. 2021; 4:e210307. https://doi.org/10.1001/jamanetworkopen.2021.0307. [PubMed].
37. Neben CL, Zimmer AD, Stedden W, van den Akker J, O’Connor R, Chan RC, Chen E, Tan Z, Leon A, Ji J, Topper S, Zhou AY. Multi-Gene Panel Testing of 23,179 Individuals for Hereditary Cancer Risk Identifies Pathogenic Variant Carriers Missed by Current Genetic Testing Guidelines. J Mol Diagn. 2019; 21:646–57. https://doi.org/10.1016/j.jmoldx.2019.03.001. [PubMed].
38. Crawford B, Adams SB, Sittler T, van den Akker J, Chan S, Leitner O, Ryan L, Gil E, van ‘t Veer L. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat. 2017; 163:383–90. https://doi.org/10.1007/s10549-017-4181-0. [PubMed].
39. Siraj AK, Masoodi T, Bu R, Parvathareddy SK, Siraj S, Alassiri A, Al-Dayel F, Alkuraya FS, Al-Kuraya KS. The study of Lynch syndrome in a special population reveals a strong founder effect and an unusual mutational mechanism in familial adenomatous polyposis. Gut. 2020; 69:2048–49. https://doi.org/10.1136/gutjnl-2019-320511. [PubMed].
40. Raoof S, Kennedy CJ, Wallach DA, Bitton A, Green RC. Molecular cancer screening: in search of evidence. Nat Med. 2021; 27:1139–42. https://doi.org/10.1038/s41591-021-01431-5. [PubMed].
41. Levy-Lahad E, Catane R, Eisenberg S, Kaufman B, Hornreich G, Lishinsky E, Shohat M, Weber BL, Beller U, Lahad A, Halle D. Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. Am J Hum Genet. 1997; 60:1059–67. [PubMed].
42. Abeliovich D, Kaduri L, Lerer I, Weinberg N, Amir G, Sagi M, Zlotogora J, Heching N, Peretz T. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of early-onset breast cancer patients among Ashkenazi women. Am J Hum Genet. 1997; 60:505–14. [PubMed].
43. Forrest IS, Chaudhary K, Vy HMT, Petrazzini BO, Bafna S, Jordan DM, Rocheleau G, Loos RJF, Nadkarni GN, Cho JH, Do R. Population-Based Penetrance of Deleterious Clinical Variants. JAMA. 2022; 327:350–59. https://doi.org/10.1001/jama.2021.23686. [PubMed].
44. Zauber P, Bishop T, Taylor C, Sabbath-Solitare M, Marotta S, Tomlinson I. Colorectal tumors from APC*I1307K carriers principally harbor somatic APC mutations outside the A8 tract. PLoS One. 2014; 9:e84498. https://doi.org/10.1371/journal.pone.0084498. [PubMed].
45. Younes N, Zayed H. Genetic epidemiology of ovarian cancer in the 22 Arab countries: A systematic review. Gene. 2019; 684:154–64. https://doi.org/10.1016/j.gene.2018.10.044. [PubMed].
46. El Saghir NS, Zgheib NK, Assi HA, Khoury KE, Bidet Y, Jaber SM, Charara RN, Farhat RA, Kreidieh FY, Decousus S, Romero P, Nemer GM, Salem Z, et al. BRCA1 and BRCA2 mutations in ethnic Lebanese Arab women with high hereditary risk breast cancer. Oncologist. 2015; 20:357–64. https://doi.org/10.1634/theoncologist.2014-0364. [PubMed].
47. AlHarbi M, Ali Mobark N, AlMubarak L, Aljelaify R, AlSaeed M, Almutairi A, Alqubaishi F, Hussain ME, Balbaid AAO, Said Marie A, AlSubaie L, AlShieban S, alTassan N, et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist. 2018; 23:1401–6. https://doi.org/10.1634/theoncologist.2018-0163. [PubMed].
48. AlHarbi M, Mubarak N, AlMubarak L, Aljelaify R, AlSaeed M, Almutairi A, AlJabarat W, Alqubaishi F, Al-Subaie L, AlTassan N, Neben CL, Zhou AY, Abedalthagafi M. Rare TP53 variant associated with Li-Fraumeni syndrome exhibits variable penetrance in a Saudi family. NPJ Genom Med. 2018; 3:35. https://doi.org/10.1038/s41525-018-0074-3. [PubMed].
49. Agha N, Alshamsan B, Al-Farsi S, Ateya HA, Almugbel FA, Alotaibi HA, Omar A, Mohamed AS, Alharthy H, Elhassan T, Salem H, Alhusaini H. Assessing frequency and clinical outcomes of BRCA mutated ovarian cancer in Saudi women. BMC Cancer. 2022; 22:18. https://doi.org/10.1186/s12885-021-09123-6. [PubMed].
50. Adedokun B, Zheng Y, Ndom P, Gakwaya A, Makumbi T, Zhou AY, Yoshimatsu TF, Rodriguez A, Madduri RK, Foster IT, Sallam A, Olopade OI, Huo D. Prevalence of Inherited Mutations in Breast Cancer Predisposition Genes among Women in Uganda and Cameroon. Cancer Epidemiol Biomarkers Prev. 2020; 29:359–67. https://doi.org/10.1158/1055-9965.EPI-19-0506. [PubMed].
51. Bishop MR, Omeler-Fenaud SM, Huskey ALW, Merner ND. Gene panel screening for insight towards breast cancer susceptibility in different ethnicities. PLoS One. 2020; 15:e0238295. https://doi.org/10.1371/journal.pone.0238295. [PubMed].
52. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, and ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24. https://doi.org/10.1038/gim.2015.30. [PubMed].