
Introduction
Ovarian cancer is a lethal and common gynecological malignancy, with 80% of patients being diagnosed at an advanced stage of disease [1]. Fewer than half of patients survive beyond five years after diagnosis due to the prevalence of aggressive high-grade serous carcinomas and lack of accurate early detection methods. As with many other cancers, much of what we know about ovarian cancer relates to genetic abnormalities that give cell growth or survival advantages. Mutations in genes such as TP53, PTEN, KRAS, and Rb1 are considered major driver mutations in many cancers and are in part responsible for establishing ovarian tumors [2–4]. However, some of these mutations, such as KRAS, are conspicuously absent in the most lethal form of OC, high grade serous ovarian cancer (HGSOC). A possible explanation for this observation is the presence of other cancer-specific adaptations that are independent of DNA mutation.
Research has shown that increased lipid uptake supports the high energy needs of growing malignant cells, with alterations in lipid metabolic genes often already present in early stages of ovarian cancer and becoming more prevalent with disease progression [5]. Lipid metabolism is critical to the growth and proliferation of all eukaryotic cells. Lipid metabolism refers to the synthesis, catabolism, and uptake of lipids from the surrounding environment. The metabolism of lipid molecules is important for normal cell biology and is critical to the development of pathological conditions, such as cancer.
Lipids have a dynamic role in the context of tumorigenesis in the ovaries, and they are involved in supporting cancer cell growth and suppressing the immune response. Lipid intermediates in ascites and fat-containing cells of the omentum have been shown to negatively affect the function of T-lymphocytes, which could inhibit the anti-tumor activity of the immune system [6–9]. Additionally, cancer cell survival and spread has been linked to increased levels of lipogenic enzymes. For instance, fatty acid synthase (FASN) and stearoyl-CoA desaturase (SCD1) are elevated in high-grade, metastatic ovarian tumors and lead to elevated levels of unsaturated fatty acids [5, 10, 11]. This increase in lipogenic enzymes, and their products (e.g., unsaturated fatty acid) often correlates with poor patient outcomes. Therefore, it is of interest to examine the impact of alterations in lipid metabolism to gain a better understanding of ways to subvert the molecular pathogenesis seen in ovarian cancer. The current review presents a logical argument for developing more approaches to therapeutically target the lipogenic pathways in cancer cells to improve patient outcomes.
LIPID DYSREGULATION IN CANCER
As stated earlier, lipids play important roles in biological processes in eukaryotic cells. Abnormal lipid homeostasis is pathognomonic with several diseases such as metabolic syndrome, obesity, diabetes, liver steatosis, cardiovascular disease, and cancer. Rapidly dividing cancer cells produce significantly more fatty acids and sterols (for energy and increased membrane synthesis) compared to non-transformed cells. In addition, cancer cells derive nearly 95% of their saturated and mono-unsaturated fatty acids de novo, even in the presence of adequate dietary lipids. Furthermore, studies have suggested that cancer cells also utilize lipolysis (the breakdown of fatty acid) to provide additional raw materials for cellular energetic demands [12–14]. This combination of metabolic alterations has led some researchers to suggest that tumorigenesis is the consequence of epithelial cells capitalizing on an overabundance of lipids in the environment (Figure 1) [15, 16].
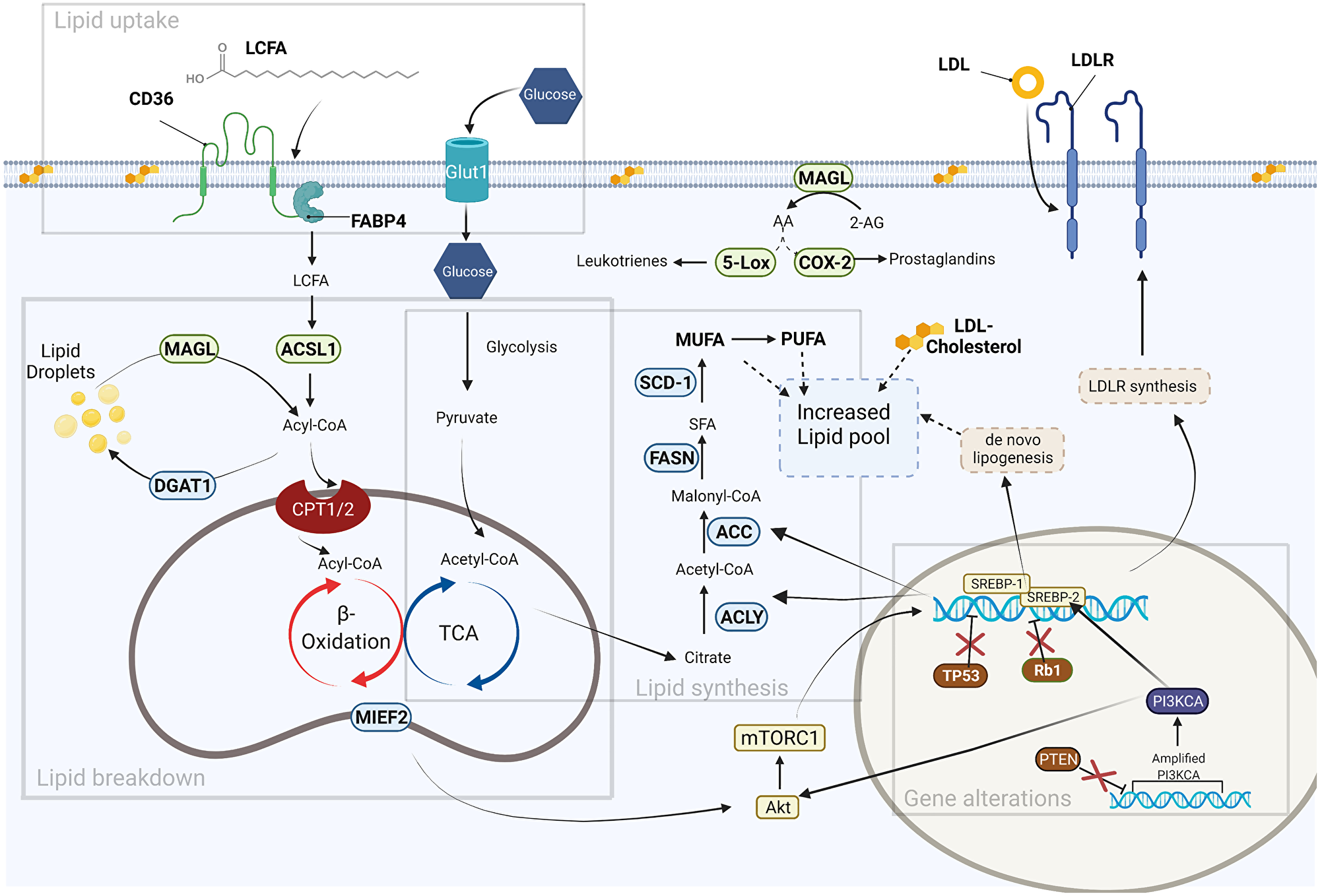
Figure 1: Fatty acid metabolism in cancer. Gene alterations: Several gene alterations (mutations, amplifications, deletions) contribute to increased production of lipogenic genes either directly via transcription regulation, or indirectly by loss of repressors. Lipid uptake: Fatty acids (FAs) are obtained via de novo lipogenesis and exogenous uptake. FA translocase CD36 is responsible for the exogenous uptake of FAs from the surrounding microenvironment. These FAs can be converted to triacylglycerols (DGAT1) and stored in lipid droplets or used in generation of acetyl-CoA through β-oxidation. Lipid Synthesis: Glucose is a major carbon source for de novo lipogenesis. Pyruvate derived from glucose contributes the substrate for several lipogenic enzymes (ACLY, ACC, FASN, SCD-1) leading to an increased lipid pool. Lipid breakdown: Lipid droplets are mobilized by lipase enzymes (MAGL) to provide energy for cancer cell growth and secondary bioactive lipids that modify the tumor microenvironment. Several promising lipid-targeting anti-cancer therapies are based on disrupting the lipid metabolic pathways (enzymes, receptors, and bioactive lipids) that are shown in this figure. Created with https://biorender.com.
In transformed cells, lipid metabolism, along with several other biosynthetic pathways, is increased to keep pace with the energetic demands of the rapidly proliferating cancer cells. For many years, researchers focused almost exclusively on glucose metabolism, but more recently other aspects of metabolism have come into the spotlight as well, including amino acid and lipid metabolism [17, 18]. Cells with high metabolic demands often experience alterations in key genes leading to increased expression of genes related to lipid synthesis [14, 19–21]. Interestingly, genes related to lipogenesis are not often mutated in cancer, leading many to believe that lipid alterations in cancer are a compensatory effect in response to other driver mutations. In support of this idea, many cancers, including OC, are known to possess “driver mutations” in genes that regulate the expression and activity of many lipogenic genes to the benefit of growing cells [22]. For instance, major driver mutations associated with ovarian carcinoma development are in genes encoding for PTEN, TP53, and Rb1, [4]. Mutations in these genes lie upstream of several highly regulated enzymes involved in various pathways that converge on lipid metabolism.
MUTATIONS IN GENES THAT AFFECT LIPOGENIC ENZYMES INVOLVED IN OVARIAN CANCER
As therapeutic interests in lipid metabolism of cancer have increased, an increasing number of studies have highlighted how some of these enzymes are involved in affecting the characteristics of ovarian tumors. As mentioned earlier, alterations in lipid metabolism appear to stem from effects of mutations in genes upstream of the lipogenic pathway. However, these mutations substantially impact the overall metabolic conditions within the cell and provide a boost to lipid production and turnover. What begins to come clear is that the presence of excess lipids provides growth advantages to transformed cells that should be considered when developing therapeutic approaches.
PTEN
Phosphatase and tensin (PTEN) homolog is a multi-functional tumor suppressor found in nearly every cell of the body. Its deletion on Chromosome 10 is associated with oncogenesis. As a tumor suppressor, PTEN functions as a negative regulator of another enzyme associated with oncogenesis, PI3KCA [23]. PI3KCA is rarely mutated in serous OC, but the PI3KCA gene is often amplified leading to increased activation of Akt and mTORC1 signaling (a major control center for cellular metabolism) and activation of sterol response element binding proteins (SREBP1 and SREBP2) [24–27]. SREBP1 is the master regulator of lipid biosynthesis and controls the expression levels of many biosynthetic genes such as fatty acid synthase (FASN), SREBP2, acetyl CoA carboxylase (ACC), ATP citrate lyase (ACLY), HMG-CoA carboxylase (HMGC), HMG-CoA reductase (HMGCR), low density lipoprotein receptor (LDLR), and many others [24]. Taken together, loss of PTEN via gene deletion in cancer cells can explain a fair amount of increased lipogenesis.
TP53
In addition to the tumor suppressor PTEN, Tumor protein 53 (TP53) is often downregulated in several models of cancer [2, 28, 29]. TP53 mutations are almost ubiquitous in HGSOC, and yet deletion of TP53 alone does not lead to the transformation of ovarian surface epithelial cells. This strongly suggests that p53 is not a unique driver of tumorigenesis but instead works in concert with other mutations [4]. This idea is supported by evidence in HGSOC tumors with TP53 mutations, roughly 67% of which also contain mutations in the Rb1 gene. In models of ovarian cancer, it has been estimated that 5–8 genetic mutations are needed to establish oncogenic growth. This suggests that loss of functional TP53 and Rb1 synergize in HGSOC to promote tumorigenesis.
RB1
Retinoblastoma protein (Rb1) is a cell cycle checkpoint regulator that controls the cell’s transition from G1 to S-phase. In cancer development Rb1 is considered a tumor suppressor and therefore its deletion is closely correlated with the development of several malignancies, in particular retinoblastoma [30–32]. Rb1 is involved in several metabolic pathways including autophagy, glycolysis, oxidative phosphorylation, and mitochondrial biogenesis [31]. Studies investigating the cellular role of Rb1 show that Rb1 deletion leads to increased production of lipids, especially fatty acids, as cells enter the cell cycle. This lipogenic effect is mediated by increased binding of the E2F transcription factor to the promoter of target genes like SREBP1. Increased expression and activity of SREBP1 leads to increased expression of stearoyl-CoA desaturase (SCD1) and fatty acid elongase 6 (ELOVL6). The activity of SCD1, and to a lesser extent ELOVL6 are responsible for the increased levels of unsaturated fatty acids present in cancer cells [30].
LIPOGENIC ENZYMES DYSREGULATED IN OVARIAN CANCER
Stearoyl CoA desaturase (SCD1)
It is important to understand that increased lipogenesis and uptake can be potentially toxic to cells. When cellular lipid stores decline, they signal the activation of the transcription factor SREBP1. This leads to corresponding increases in the expression of fatty acid synthetic genes including FASN, ACC, ACLY, and LDLR. The production of fatty acids potentiates the transformation of acetyl-CoA into saturated fatty acids, palmitate, and stearate. However, high levels of saturated fat are detrimental to membrane integrity and are therefore toxic to cells This is called lipotoxicity [33, 34]. Lipotoxicity occurs when fatty acid or sterol accumulation exceeds the ability of the cell to package them into triacylglycerols and sterol esters [35]. In cancer cells, as fatty acid synthesis increases, the conversion of saturated fat to unsaturated fatty acids also increases due to the activity of stearoyl-CoA desaturase (SCD1) [36].
SCD1 is a delta-9 desaturase that introduces a single double bond into the 9th position of stearic and palmitic acid (making oleic and palmitoyl oleic acid, respectively) [37]. SCD1-mediated production of unsaturated fatty acids is essential to produce cell membranes and phospholipids as unsaturated lipids are required for proper membrane function [38, 39]. Many studies have demonstrated that SCD1 levels are elevated in cancer compared to normal cells [40–42]. Accordingly, SCD1 has been proposed as a therapeutic target [11, 38, 43–47]. In preclinical studies, pharmacological inhibition of SCD1 blocks cell proliferation when exogenous lipids are limited [48]. This suggests that cancer cell dependence on lipids needs to be thwarted both at the synthesis and uptake stages to have an integrative anti-cancer effect. Although clinical data in this regard is still limited, there is evidence that populations of ovarian cancer stem cells are heavily reliant on SCD1 activity [11].
Cancer stem cells are regarded as being responsible for much of the proliferative capacity in tumors as well as resistance to therapy. In single cell analysis, lipid desaturation markers were elevated in cancer stem cells when compared to non-stem cancer cells [11]. Furthermore, analyses of cell proliferation and migratory capacity were shown to be largely supported by the presence of unsaturated fatty acids. Taken together, it appears that the presence of unsaturated fatty acid contributes significantly to the functions of cancer cells that are associated with poor disease outcomes.
ATP-Citrate lyase (ACLY)
It is not only the latter steps in fatty acid generation that are altered in OC. Key initiating enzymes are also altered to some extent as well. ATP-Citrate Lyase (ACLY), an upstream regulator of fatty acid synthesis, is responsible for converting 6-carbon molecules (glucose and glutamine) into molecules of oxaloacetate and acetyl-CoA. ACLY links sugar- or glycol-metabolism to lipid metabolism, illustrating the important relationship between elevated glucose uptake and lipid metabolism in cancer cells [49]. Increased ACLY levels are beneficial to OC cell fitness [2]. Correspondingly, targeting ACLY with interfering RNAs decreases the proliferative capacity of ovarian cancer cells, demonstrating its critical role in supporting cancer cell growth [50].
Acetyl CoA carboxylase (ACC)
Continuing along the fatty acid synthetic pathway, Acetyl-CoA is similarly elevated in OC. Acetyl-CoA Carboxylase is the enzyme responsible for the first committed step in fatty acid synthesis, catalyzing the carboxylation of cytosolic acetyl-CoA to form malonyl-CoA. ACC activity is regulated during post-translational phosphorylation by adenosine monophosphate kinase (AMPK). In ovarian cancer, inhibition of ACC with the allosteric inhibitor TOFA induces G0/G1 cell cycle arrest and apoptosis [51]. AMPK-ACC pathways in cancer cells are regulated by lysophosphatidic acid (LPA), a bioactive lipid-like growth factor mediator, which is found in ascites of ovarian cancer patients at high levels. Increased activity of ACC, along with fatty acid synthase, drives de novo lipid production. Studies examining LPA-mediated mechanisms in ovarian cancer reveal the role of LPA in activating AMPK-ACC cascades, resulting in an increase of de novo lipogenesis [52]. There is even evidence that increased ACC activity in response to upstream metabolic signals (increased AMPK activity) is partly responsible for the resistance of ovarian cancer cells to chemotherapy [53].
Fatty acid synthase (FASN)
In support of the central role lipid production plays in cancer cell survival, inhibition of SREBP1 has been demonstrated to prevent the growth of ovarian cancer in xenograft models [26]. In a more focused analysis, targeting the final enzyme of the canonical fatty acid biosynthetic pathway, fatty acid synthase (FASN), showed very similar results. FASN is essential for lipid synthesis and uses acetyl CoA derived from glucose to synthesize palmitate and other fatty acids used in lipid signaling, cell proliferation, and triglyceride storage [54]. Early studies that monitored FASN expression in primary prostate cancers showed that FASN expression was detected in 57% of 99 primary prostate cancers, which corresponded to decreased disease-free survival in patients [55]. This is corroborated by another study that showed FASN functions as an oncogene when expressed in excessive amounts [56]. Increased levels of FASN have been linked with decreased patient survival, increased disease recurrence, and increased invasive capacity of cancer in patients [19, 57].
More recent work suggests that FASN expression is associated with worse outcomes in cancers, including ovarian cancer. [55, 56, 58, 59]. This observation is likely linked to the association of increased FASN with resistance to cytotoxic stress induced by chemo- radiotherapy, leading to poorer patient outcomes. Several studies have demonstrated that inhibition of FASN prevents the growth of the immunosuppressive phenotype associated with a variety of cancers including ovarian cancer, further implicating its critical role in tumorigenesis [10, 17, 60–62]. Inhibitors of FASN have demonstrated efficacy as anticancer therapies based on the results of studies using cell lines and ovarian tumor mouse xenograft preclinical models [50, 51, 63].
Mitochondrial elongation factor 2 (MIEF2)
Even outside of the canonical lipogenic/ fatty acid biosynthetic pathway there are enzymes whose activities affect lipogenic properties of cancer cells. One such molecule is Mitochondrial elongation factor (MIEF2), a regulator of mitochondrial fission. In the context of ovarian cancer, high expression of this protein is predictive of a poor prognosis. It has been shown that knockdown of MIEF2 decreases levels of free fatty acid, triglycerides, and cholesterol in ovarian cancer cell lines; the converse has also been observed [64]. The increase in lipogenic genes caused by increased expression of MIEF2 appears to be the result of increased activation of the ROS/Akt/mTOR pathway, which results in increased SREBP1 and SREBP2 mRNA levels. As discussed earlier, increased expression of SREBP1 ultimately results in increased levels of lipids and preferred growth conditions in the affected cells.
Diacylglycerol O-Acyltransferase 1 (DGAT1)
As cells generate increasing levels of lipid molecules even as they are converted to less problematic unsaturated species, they still must be exported or stored. Storage comes in the form of di- and triacylglycerols. Along with SCD1, the protein Diacylglycerol O-Acyltransferase 1 (DGAT1) helps with the toxification of lipid. DGAT1 is responsible for catalyzing the conversion of diacylglycerol and fatty acyl-CoA to triacylglycerol. DGAT1 is overexpressed in ovarian cancer and correlates with poor survival of patients. In fact, the levels of DGAT1 are positively associated with ovarian tumor growth [65]; the larger the tumor, the more DGAT1 will be produced, likely supporting the storage of ever-increasing amounts of lipid. Due to its role in aiding lipid storage, DGAT1 activity is regulated in part by the availability of glucose or glycolytic activity of the cell, as glycolysis provides the raw material that will eventually become new acyl-CoA molecules.
ENZYMES INVOLVED IN LIPID DEGRADATION IN OVARIAN CANCER
Not only is the ability to generate large amounts of lipid a benefit to cancer cells, breakdown and conversion of lipid is also critical to tumor development and growth. Several studies have shown that enzymes involved in lipid degradation are elevated in cancer cells when compared to non-transformed cells [22, 64, 66, 67]. In some cases, enzymes can be involved in both the biosynthesis and degeneration of lipids such as ACSL1 [67].
Acyl-CoA synthetase long chain family member 1 (ACSL1)
Acyl-CoA Synthetase Long Chain family member 1 is an isozyme of the long chain fatty acid coenzyme A ligase family. ACSL1 converts free long chain fatty acids into fatty acyl CoA esters and plays a key role in lipid biosynthesis and fatty acid degradation during beta-oxidation. Highly metastatic ovarian cancer cells have a distinct lipid profile as compared to less metastatic cells [66]. There is a notable increase in phospholipids, in particular phosphatidylcholine in these cells. ACSL1 overexpression in non-metastatic ovarian cancer cells increases their metastatic dissemination in xenograft models, indicating that ACSL1 activity can drive metastasis.
Arachidonate 5-Lipoxygenase (5-LOX)
Arachidonate 5-Lipoxygenase (5-LOX) is an enzyme responsible for catalyzing the synthesis of bioactive, proinflammatory lipids known as leukotrienes from the polyunsaturated fatty acid arachidonic acid. 5-LOX is highly expressed in ovarian cancer cells and correlates with poor prognosis in patients [5]. 5-LOX-derived leukotrienes are associated with increased cell migration and metalloproteinase expression, leading to increased metastasis [68]. 5-LOX also promotes recruitment of pro-inflammatory tumor associated macrophages (TAMs) into hypoxic regions of the tumor. Increased TAMs density within the tumor is associated with metastasis and tumor stage. Furthermore, it was determined that metabolites of 5-LOX helped promote a positive feedback loop in which the hypoxic environment helped recruit more TAMs, which in turn promoted 5-LOX activity. Similarly, expression of leukotriene receptors (Leukotriene B4 receptor B2) has also been demonstrated to correlate with poor clinical outcomes in ovarian cancer patients [69]. The presence of inflammatory leukotrienes driven by the expression of 5-LOX and its receptors may be a treatment target worth further investigation in ovarian cancer patients.
Cyclooxygenase-2 (COX-2)
Cyclooxygenase-2 is another rate-limiting enzyme involved in the metabolism of the polyunsaturated fatty acid, arachidonic acid, into bioactive prostaglandins. In ovarian cancer, COX-2 expression is increased leading to increased presence of its product, the Prostaglandin E2 (PGE2) [5, 70]. The synergistic presence of COX2 and PGE2 has been implicated in promoting the expression of the pro-angiogenic cytokine vascular endothelial growth factor (VEGF) [5, 68]. Several studies have demonstrated that COX2 expression also promotes metastasis by increasing the expression of metalloproteinase enzymes that degrade the extracellular matrix surrounding tumor cells [71]. There is evidence that celecoxib, a selective inhibitor of COX-2, successfully inhibits growth and induces apoptosis in ovarian cancer cells [72].
Carnitine palmitoyl transferase
Carnitine Palmitoyl Transferase (CPT) is responsible for helping cells adapt to low glucose conditions by switching to beta oxidation [73, 74]. CPT helps convert long chain fatty acids in the cell into acyl chains that are then subjected to beta oxidation in the mitochondria. CPT expression is high in ovarian cancer, which facilitates the use of the beta oxidation pathway in these cells, simultaneously with glycolysis [75]. This gives ovarian cancer cells a significant growth and proliferation advantage. The effect of the genetic ablation of CPT1 dramatically alters beta oxidation and induces cell cycle arrest and p21-mediated apoptosis, demonstrating the importance of CPT1 to cancer survival [75].
Monoacylglycerol lipase (MAGL)
Monoacylglycerol lipase (MAGL) is responsible for catalyzing the decomposition of monoacylglycerol into free fatty acids and glycerol. This effectively increases the level of fatty acid within the cell. The expression of MAGL is elevated in ovarian cancer [5]. Elevated MAGL is associated with increased epithelial-mesenchymal-transition (EMT) potential in cancer cells [76].
LIPID UPTAKE IN CANCER
CD36
The uptake of lipids from the circulation is a complementary means by which cells maintain internal stores of fatty acids and sterols. An essential protein involved in the uptake of lipids in cells is the scavenger receptor, or CD36, a fatty acid translocase [77]. CD36 is an 88-kDa transmembrane glycoprotein expressed in numerous cell types including macrophages, endothelial cells, and adipocytes. CD36 translocates from the cytoplasm to the plasma membrane, where it binds to low density lipoproteins and transports them across the membrane, which is the functional mechanism whereby CD36 contributes to the total lipid content of cells. Recent studies have shown that CD36 expression is increased in solid tumors of the breast, stomach, and ovary [7, 78, 79]. Increased CD36 expression levels likely occur in response to the increased energy demands on the cell, similar to what occurs in other metabolic pathways.
CD36 plays a role in the initiation of cancer and is correlated with poor prognosis in melanoma and breast cancer [77]. Inhibition and knockdown of CD36 have deleterious effects on cellular proliferation in many cancers, rendering CD36 a diagnostic biomarker and a potential target for therapy. The reason for this potent anti-cancer effect in CD36 inhibition is linked to inactivation of Wnt/Beta-catenin, a major driver of oncogenic cell growth in cancer cells [80]. If CD36 levels are reduced, then the growth signals provided by the Wnt pathway are substantially inhibited as well.
Fatty acid binding protein 4
Fatty acid binding protein 4 (FABP4) helps to promote the uptake of long chain fatty acids into cells [5, 81]. FABP4 overexpression has been reported in many cancers including ovarian cancer. FABP4 has the potential to predict the presence of residual disease in ovarian cancer [2]. Immunohistochemical-based FABP4 expression appears to be enriched in areas along the carcinoma cell/adipocyte junction, likely owing to its role in transporting lipids into cells that require additional forms of energy [82]. Cancer cell dependence on readily accessible sources of fatty acids is associated with FABP4 becoming an attractive therapeutic target. Studies targeting FABP4 have shown that FABP4-inhibited cancer cells have decreased aggressiveness (less metastasis) and have an increased sensitivity to carboplatin therapy [8, 81].
Low density lipoprotein receptor (LDLR)
Low density lipoprotein receptor (LDLR) is a membrane protein that transports cholesterol into cells in the periphery, away from the liver [83]. LDLR is an SREBP responsive-gene, and its expression is increased along with many of the lipogenic genes. SREBP2 and LDLR expression is often elevated in chemoresistant cells in ovarian cancer [84]. Accordingly, elevated LDLR expression in ovarian cancer is correlated with a poor response to platinum-based drugs. Conversely, knockdown of LDLR increases sensitivity to platinum-based therapies [85].
LIPID METABOLISM IN OVARIAN CANCER STEM CELLS
Tumor relapse and resistance to conventional chemo- and radiotherapies leading to fatal metastatic disease has been associated with the development of cancer stem cells (CSCs) [86]. CSCs play a role in treatment failures and cancer progression through their interaction with other cells and molecules in the tumor microenvironment [87]. The importance of the microenvironment can be seen in the interaction between ovarian cancer cells and their stroma through the regulation of ascitic fluid contents in stromal cells and growth processes. Within the microenvironment, cancer-associated fibroblasts, a major cell population in the stroma, have been found to enhance the generation of ovarian CSCs and cause angiogenesis by inducing the secretion of vascular endothelial growth factors (VEGF) [11, 88].
CSCs also accumulate excess lipids and cholesterol inside lipid droplets [11]. Cancer cells, like adipocytes, store excess energy in lipid droplets that can be broken down into free fatty acids. This formation occurs by lipogenic enzymes which are highly expressed in cancer cells and include acetyl-CoA carboxylase, fatty acid synthase, and ATP citrate lyase.
Tumor growth is dependent on active angiogenesis, therefore, vascular reduction results in tumor inhibition in non-adipose tissues [89]. Analysis of Raman scattered imaging and mass spectrometry of lipids shows significant increases in unsaturated lipid levels within ovarian cancer stem cells compared to non-stem cells [11]. Therefore, this provides evidence that metabolic alterations in lipogenesis with the use of increased unsaturated lipids could be a metabolic marker for ovarian cancer stem cells. These alterations in intracellular lipid content develop from the utilization of extracellular lipids or by de novo synthesis.
Ovarian cancer stem cells also can differentiate into other cell types which can further contribute to angiogenesis, progression, and metastasis [84]. De novo adipocytes in adipose and tumor tissue differentiate from mesenchymal stem cells and can be influenced by external factors from the tumor microenvironment to enable the storage of excessive levels of lipid content. The formation of remodeled adipocytes, due to cancer cell invasion, results in tumor tissue growth by engulfment of adipocyte clusters [89].
ROLE OF LIPID METABOLISM IN ANTI-CANCER DRUG RESISTANCE
Ovarian cancer is among the diseases wherein excess adiposity has a causal role [90, 91]. Results from a recent large-scale study conducted by Si et al. to identify the risk factors associated with ovarian cancer indicated that body mass index (BMI), body fat percentage, body fat mass, and basal metabolic rate are significantly associated with ovarian cancer [92]. Based on data from a cohort of 13,222 women diagnosed with ovarian cancer in the United Kingdom with >20 years’ follow-up for ovarian cancer incidence and cause-specific mortality, BMI was identified as a modifiable means of improving survival [93].
One way that BMI can be modified is by adhering to a ketogenic diet consisting of low levels of carbohydrates and high levels of fat to create a metabolic state of ketosis. With ketogenic diets, by increasing insulin sensitivity and restricting carbohydrate intake, adipose tissue is selectively decreased while maintaining lean mass [94]. A study of 30 women with polycystic ovary syndrome demonstrated that adherence to a ketogenic diet resulted in reduced central obesity [95]. Consumption of low levels of carbohydrates resulted in preferential loss of fat mass from metabolically harmful adipose deposits, whereas a diet with high levels of carbohydrates resulted in a re-partitioning of lean mass to fat mass. Supporting the effectiveness of diet modification strategies in improving outcomes for women with ovarian cancer, results from the Women’s Health Initiative Randomized Controlled Dietary Modification Trial indicated that a low-fat diet may be associated with beneficial health outcomes [96].
Obesity has a causal role in the anti-cancer drug response of patients with ovarian cancer. More specifically, adipocytes and lipid metabolism play complex roles in modulating anti-cancer drug resistance, which is one of the most significant challenges to successful ovarian cancer treatment. Although the dependence of cancer cells on glycolysis for energy production has been studied extensively [97], less is known about the roles of adipocytes and alterations in lipid metabolic programming in the context of the growth, metastasis, and drug responses of cancer cells. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth [98]. Adipocyte-rich environments support tumor growth via several mechanisms. In ovarian cancer, metastatic cells are home to omental adipose tissue which contain high concentrations of triglycerides. Free fatty acids are generated from the hydrolysis of these free fatty acids which metastatic ovarian cancer cells uptake and utilize as energy sources. Adipocytes secrete factors that increase ovarian cancer cell resistance against chemotherapeutic drugs by Akt pathway activation, and the Akt pathway has been demonstrated to mediate the anti-apoptotic activity of adipocytes [53]. Additionally, arachidonic acid, a polyunsaturated fatty acid present in cell membrane phospholipids, is capable of activating Akt and inhibiting cisplatin-induced apoptosis [89]. The level of Akt activation is positively correlated with the chemo-protective effect of arachidonic acid.
Among the potential mechanisms that underlie poor anti-cancer drug response in obese cancer patients are adipose hypoxia, altered pharmacokinetics, increased ATP production, altered microbiota, the production of tumor-promoting growth factors and cytokines, and the generation of drug-resistant cancer stem cells [89, 99]. The low-grade hypoxia which occurs in adipose tissues stimulates angiogenesis, inhibits macrophage migration and pre-adipocyte differentiation, increases fibrosis, and suppresses immune cell recruitment [100]. Based on the results from studies using pancreatic cancer cells, Harbuzariu et al. proposed that elevated levels of leptin in obese individuals could protect cells from chemotherapy-induced apoptosis [101]. Obesity could enhance fibrosis in tumors by facilitating interactions between pro-inflammatory and fibrotic pathways, consequently impeding tumor drug delivery [102].
Lipids and cholesterol have critical roles in the proliferation of cancer cells. In these highly proliferative cells, lipid catabolism occurs via the fatty acid ß-oxidation (FAO) pathway, which involves exogenous and endogenous fatty acids [22]. In nutrient- and oxygen-depleted environmental conditions, cancer cells exhibit an increased dependence on FAO [103]. It therefore follows that therapeutic strategies intended to modulate lipid-associated metabolism in cancer cells must be designed to avoid adverse effects on normal metabolic functions.
Ovarian cancer cells utilize lipid metabolism in the ascites or omental microenvironment during metastatic progression through AMPK/ACC/FASN-mediated lipogenesis and AMPK/TAK1/NF-κB signaling pathways. Chen et al. demonstrated that targeting lipid metabolism and/or suppressing TAK1/NF-κB signaling is an effective therapeutic strategy to prevent and treat peritoneal metastases in ovarian cancer cells [104, 105]. Cholesterol-lowering drugs such as statins target lipid metabolism. Statins have been shown to induce the apoptosis of ovarian cancer cells [106]. Lovastatin was utilized in a clinical trial in combination with the chemotherapeutic agent Paclitaxel to improve the standard-of-care for patients with refractory or relapsed ovarian cancer [107]. Another targeted strategy for altering lipid metabolism in ovarian cancer is the knockdown of ceramide transport protein (CERT) [108]. Knockdown of CERT causes the accumulation of ceramide in the ER, increasing ER stress, and sensitizing ovarian cancer cells to Paclitaxel treatment [105].
LIPIDS AS DIAGNOSTIC OR PROGNOSTIC BIOMARKERS
Analytical methods including imaging techniques and mass spectrometry have enabled the determination of fatty acid composition and intra-tumor lipid class spatial distribution toward the development of lipid and lipid metabolism-associated biomarkers [109, 110]. For example, breast tumors and epithelial ovarian cancer cells have been shown to exhibit increased levels of membrane-associated phosphatidylcholine and phosphatidylethanolamine [111–113]. These phospholipids have potential roles in improving clinical diagnosis as well as the identification of new therapeutic targets.
Promising lipid-targeting anti-cancer therapies are based on disrupting lipid metabolic pathways by targeting enzymes, receptors, or bioactive lipids to consequently stimulate tumor regression and prevent metastasis. Components of altered metabolic lipid pathways that could function as potential prognostic biomarkers or therapeutic targets to prevent the growth of cancer cells or to overcome chemotherapy resistance include cholesterol [106], fatty acid synthase [114–117], autotaxin [96] ceramide [118], and CERT [108]. These and other lipids and lipid metabolism-associated biomarkers are summarized in Table 1.
Table 1: Classification of lipid and lipid metabolism-associated biomarkers
| Biomarker | Classification | Biomarker Class | References |
|---|---|---|---|
| Cholesterol | Bioactive lipid mediator | Prognostic | [106] |
| Fatty acid synthase | Key metabolic enzyme | Prognostic | [114–116] |
| Autotaxin | LPA-producing enzyme | Prognostic | [117] |
| Ceramide | Bioactive lipid (metastasis-suppressor lipid) | Prognostic | [118] |
| Ceramide transport protein | Lipid-transfer protein | Prognostic | [108] |
| PI3, RGS, ADORA3, CH25H, CCDC80, PTGER3, MATK, KLRB1, CCL19, CXCL9, and CXCL10 | Lipid metabolism related genes | Prognostic | [131] |
| Lysophosphatidic acid | Bioactive lipid mediator | Diagnostic | [120, 121] |
| Sulfatides | Sulfoglycolipid | Diagnostic | [122] |
| Phospholipase A2 | Phospholipid cleaving enzyme | Diagnostic | [113, 123] |
| Lysophospholipids | Bioactive lipid molecule | Diagnostic | [124] |
| CD36 | Fatty acid receptor | Diagnostic | [7] |
The phospholipid composition of cancer cells as determined by lipidomic profiling could aid the differentiation between low- and high-grade tumors and malignant vs. benign cells [119]. Potential lipid ovarian cancer diagnostic markers include lysophosphatidic acid [117, 118], sulfatides [122], phospholipase A2 [113, 123], and lysophospholipids [124]. The results from a nested case-control study within the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial using tandem mass spectrometry analysis of serum indicated that five arachidonic acid/linoleic acid/alpha-linoleic acid metabolites were positively associated with ovarian cancer risk: 8-HETE, 12,13-DHOME, 13-HODE, 9-HODE, and 9,12,13-THOME [125].
Ovarian cancer cells co-cultured with primary human omental adipocytes have a high expression level of the fatty acid receptor CD36 in the plasma membrane, which facilitates exogenous fatty acid uptake [7]. Thus, it has been proposed that inhibiting CD36 could be an effective treatment strategy against ovarian cancer metastasis.
Using Raman scattering imaging of single living cells and mass spectrometry analysis of extracted lipids, Li et al. observed significantly increased levels of unsaturated lipids in ovarian cancer stem cells compared to non-cancer stem cells [11]. The results of their study demonstrated that since enhanced lipid unsaturation is a metabolic marker for ovarian cancer stem cells, it can function as a target for cancer stem cell-specific therapy.
Tebbe et al. demonstrated that Metformin limits the adipocyte tumor-promoting effect on ovarian cancer by inhibiting adipocyte-mediated ovarian cancer cell proliferation, migration, expression of cancer-associated genes and bio-energetic changes [126]. Acetyl-CoA carboxylase is the target of the drug 5-tetradecyloxy-2-furoic acid (TOFA), which was found to be cytotoxic to COC1 and COC1/DDP ovarian cancer cell lines and inhibit COC1/DDP cell growth in ovarian tumor xenografts [126]. However, Pouyafar et al. demonstrated that TOFA-induced lipolysis inhibition is not as effective as glycolysis inhibition in preventing ovarian cancer stem cell differentiation into endothelial-like cells [13].
Anti-cancer drugs that perturb the cholesterol content of cell membranes can be employed to hinder lipid raft-dependent cell survival or cell death pathways. Methyl-ß-cyclodextrin (MCD) is an example of one such drug that depletes the cholesterol content of cell membranes and inhibits ovarian cancer growth while avoiding acute systemic cytotoxicity [127]. MCD has been shown to increase the efficacy of the estrogen-modulating anti-cancer drug, tamoxifen [128].
As an example of taking advantage of cancer cells’ increased dependence on saturated fatty acids, the delivery of drugs to tumors has been enhanced by loading drugs into liposomes enriched in saturated phosphatidylcholine [129]. Among the pharmaceutical benefits of liposomes are their low toxicity and ability to improve the biopharmaceutical features and therapeutic index of drugs, consequently increasing efficacy and reducing side effects [130].
Lipids also have roles as prognostic ovarian cancer biomarkers. Zheng et al. recently developed and validated an 11-gene prognostic model for serous ovarian carcinomas based on a lipid metabolism expression profile [131]. Using RNA-seq data from The Cancer Genome Atlas and Gene Expression Omnibus databases, a multi-gene prognosis model was established which consists of the following lipid metabolism-related genes: PI3, RGS, ADORA3, CH25H, CCDC80, PTGER3, MATK, KLRB1, CCL19, CXCL9, and CXCL10. This model could be utilized as a novel approach for a molecular diagnostic test to assess the prognosis and potential risk factors for patients with ovarian cancer.
FUTURE DIRECTIONS
Ovarian cancer and its predominant subtype, high grade serous ovarian cancer, present a significant cancer burden globally. As with many other types of cancer, patients with ovarian cancer have a lower likelihood of disease-free survival if they are diagnosed with advanced disease. Although the survival rates for several forms of solid tumors have improved over the last 30 years, survival rates for patients with ovarian cancer have not changed substantially in this period. Therefore, it is of critical importance to identify new ways to diagnose and treat ovarian cancer to improve outcomes.
Like many other malignancies, ovarian cancer proliferation appears to benefit from increased levels of lipids not only in the tumor microenvironment but also in the transformed cells themselves, making lipid metabolism an attractive target for therapy. Understanding how lipid accumulation benefits the initiation and growth of ovarian cancer cells will help in determining new molecular pathways critical to tumorigenesis.
In this review, we presented evidence from the literature supporting the case that lipid metabolism in ovarian cancer cells is a crucial metabolic process that should be further investigated to improve treatment efficacy for patients with ovarian cancer. Interestingly, several lines of evidence have demonstrated that lipids (fatty acids, sterols, sphingolipids) are important mediators of classical oncogenic signaling, yet it remains unclear the extent to which the function of lipids in non-cancerous cells differs from non-transformed cells. Indeed, several novel drugs targeting various aspects of lipid metabolism pathways are being developed, which can provide new strategies for ovarian cancer treatment.
Abbreviations
ACC: Acetyl CoA carboxylase; ACSL1: Acyl-CoA Synthetase Long Chain family member 1; AMPK: Adenosine monophosphate kinase; 5-LOX: Arachidonate 5-Lipoxygenase; ACLY: ATP citrate lyase; BMI: Body mass index; CSCs: Cancer stem cells; CPT: Carnitine Palmitoyl Transferase; CERT: Ceramide transport protein; COX-2: Cyclooxygenase-2; DGAT1: Diacylglycerol O-Acyltransferase 1; DHOME: Dihydroxyoctadecenoic acid; FABP4: Fatty acid binding protein 4; FASN: Fatty acid synthase; HMGC: HMG-CoA carboxylase; HMGCR: HMG-CoA reductase; HETE: Hydroxyeicosatetraenoic acid; HODE: Hydroxyoctadecadienoic acid; THOME: Trihydroxyoctadecenoic acid; LDLR: Low density lipoprotein receptor; MCD: Methyl-ß-cyclodextrin; MIEF2: Mitochondrial elongation factor; MAGL: Monoacylglycerol lipase; OC: Ovarian cancer; ELOVL6: Fatty acid elongase 6; PGE2: Prostaglandin E2; PTEN: Phosphatase and tensin; Rb1: Retinoblastoma protein; SCD1: Stearoyl-CoA desaturase; SREBP1 and 2: Sterol response element binding protein 1 and 2; TP53: Tumor protein 53; KRAS: Kirsten Rat Sarcoma Viral Oncogene Homolog; HGSOC: High grade serous ovarian cancer.
ACKNOWLEDGMENTS AND FUNDING
GES and SNT acknowledge funding from the National Institutes of Health’s National Center for Advancing Translational Sciences, grant UL1TR002494. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health’s National Center for Advancing Translational Sciences. SNT acknowledges funding from the V Foundation for Cancer Research and startup funds provided by the University of Minnesota Department of Laboratory Medicine and Pathology.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018; 68:284–96. https://doi.org/10.3322/caac.21456. [PubMed].
2. Cole AJ, Dwight T, Gill AJ, Dickson KA, Zhu Y, Clarkson A, Gard GB, Maidens J, Valmadre S, Clifton-Bligh R, Marsh DJ. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016; 6:26191. https://doi.org/10.1038/srep26191. [PubMed].
3. Nakayama N, Nakayama K, Yeasmin S, Ishibashi M, Katagiri A, Iida K, Fukumoto M, Miyazaki K. KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br J Cancer. 2008; 99:2020–28. https://doi.org/10.1038/sj.bjc.6604783. [PubMed].
4. Yamulla RJ, Nalubola S, Flesken-Nikitin A, Nikitin AY, Schimenti JC. Most Commonly Mutated Genes in High-Grade Serous Ovarian Carcinoma Are Nonessential for Ovarian Surface Epithelial Stem Cell Transformation. Cell Rep. 2020; 32:108086. https://doi.org/10.1016/j.celrep.2020.108086. [PubMed].
5. Ji Z, Shen Y, Feng X, Kong Y, Shao Y, Meng J, Zhang X, Yang G. Deregulation of Lipid Metabolism: The Critical Factors in Ovarian Cancer. Front Oncol. 2020; 10:593017. https://doi.org/10.3389/fonc.2020.593017. [PubMed].
6. Dai L, Song K, Di W. Adipocytes: active facilitators in epithelial ovarian cancer progression? J Ovarian Res. 2020; 13:115. https://doi.org/10.1186/s13048-020-00718-4. [PubMed].
7. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, Yamada SD, Abumrad NA, Lengyel E. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018; 37:2285–301. https://doi.org/10.1038/s41388-017-0093-z. [PubMed].
8. Mukherjee A, Chiang CY, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O, Lengyel E. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res. 2020; 80:1748–61. https://doi.org/10.1158/0008-5472.CAN-19-1999. [PubMed].
9. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, Müller R. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol. 2017; 7:24. https://doi.org/10.3389/fonc.2017.00024. [PubMed].
10. Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front Immunol. 2018; 9:2927. https://doi.org/10.3389/fimmu.2018.02927. [PubMed].
11. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, Matei D, Cheng JX. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell. 2017; 20:303–14.e5. https://doi.org/10.1016/j.stem.2016.11.004. [PubMed].
12. Najt CP, Khan SA, Heden TD, Witthuhn BA, Perez M, Heier JL, Mead LE, Franklin MP, Karanja KK, Graham MJ, Mashek MT, Bernlohr DA, Parker L, et al. Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol Cell. 2020; 77:810–24.e8. https://doi.org/10.1016/j.molcel.2019.12.003. [PubMed].
13. Pouyafar A, Heydarabad MZ, Abdolalizadeh J, Zade JA, Rahbarghazi R, Talebi M. Modulation of lipolysis and glycolysis pathways in cancer stem cells changed multipotentiality and differentiation capacity toward endothelial lineage. Cell Biosci. 2019; 9:30. https://doi.org/10.1186/s13578-019-0293-z. [PubMed].
14. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013; 52:585–89. https://doi.org/10.1016/j.plipres.2013.08.005. [PubMed].
15. Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013; 6:1353–63. https://doi.org/10.1242/dmm.011338. [PubMed].
16. Kuo CY, Ann DK. When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun (Lond). 2018; 38:47. https://doi.org/10.1186/s40880-018-0317-9. [PubMed].
17. Stoiber K, Nagło O, Pernpeintner C, Zhang S, Koeberle A, Ulrich M, Werz O, Müller R, Zahler S, Lohmüller T, Feldmann J, Braig S. Targeting de novo lipogenesis as a novel approach in anti-cancer therapy. Br J Cancer. 2018; 118:43–51. https://doi.org/10.1038/bjc.2017.374. [PubMed].
18. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of Amino Acids in Cancer. Front Cell Dev Biol. 2021; 8:603837. https://doi.org/10.3389/fcell.2020.603837. [PubMed].
19. Dang Q, Chen YA, Hsieh JT. The dysfunctional lipids in prostate cancer. Am J Clin Exp Urol. 2019; 7:273–80. [PubMed].
20. Ferro M, Terracciano D, Buonerba C, Lucarelli G, Bottero D, Perdonà S, Autorino R, Serino A, Cantiello F, Damiano R, Andras I, De Placido S, Di Lorenzo G, et al. The emerging role of obesity, diet and lipid metabolism in prostate cancer. Future Oncol. 2017; 13:285–93. https://doi.org/10.2217/fon-2016-0217. [PubMed].
21. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020; 122:4–22. https://doi.org/10.1038/s41416-019-0650-z. [PubMed].
22. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016; 5:e189. https://doi.org/10.1038/oncsis.2015.49. [PubMed].
23. Martins FC, Couturier DL, Paterson A, Karnezis AN, Chow C, Nazeran TM, Odunsi A, Gentry-Maharaj A, Vrvilo A, Hein A, Talhouk A, Osorio A, Hartkopf AD, et al. Clinical and pathological associations of PTEN expression in ovarian cancer: a multicentre study from the Ovarian Tumour Tissue Analysis Consortium. Br J Cancer. 2020; 123:793–802. https://doi.org/10.1038/s41416-020-0900-0. [PubMed].
24. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM, Menon AV, Webster KA, Ng C, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018; 50:206–18. https://doi.org/10.1038/s41588-017-0027-2. [PubMed].
25. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. 2012; 10:133–42. https://doi.org/10.1158/1541-7786.MCR-11-0206. [PubMed].
26. Nie LY, Lu QT, Li WH, Yang N, Dongol S, Zhang X, Jiang J. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol Rep. 2013; 30:1346–54. https://doi.org/10.3892/or.2013.2575. [PubMed].
27. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016; 35:1250–60. https://doi.org/10.1038/onc.2015.179. [PubMed].
28. Wu JH, Guo JP, Shi J, Wang H, Li LL, Guo B, Liu DX, Cao Q, Yuan ZY. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol. 2017; 23:2308–17. https://doi.org/10.3748/wjg.v23.i13.2308. [PubMed].
29. Lasham A, Moloney S, Hale T, Homer C, Zhang YF, Murison JG, Braithwaite AW, Watson J. The Y-box-binding protein, YB1, is a potential negative regulator of the p53 tumor suppressor. J Biol Chem. 2003; 278:35516–23. https://doi.org/10.1074/jbc.M303920200. [PubMed].
30. Muranaka H, Hayashi A, Minami K, Kitajima S, Kohno S, Nishimoto Y, Nagatani N, Suzuki M, Kulathunga LAN, Sasaki N, Okada N, Matsuzaka T, Shimano H, et al. A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis. 2017; 6:e350. https://doi.org/10.1038/oncsis.2017.51. [PubMed].
31. Linn P, Kohno S, Sheng J, Kulathunga N, Yu H, Zhang Z, Voon D, Watanabe Y, Takahashi C. Targeting RB1 Loss in Cancers. Cancers (Basel). 2021; 13:3737. https://doi.org/10.3390/cancers13153737. [PubMed].
32. Lefèvre SH, Vogt N, Dutrillaux AM, Chauveinc L, Stoppa-Lyonnet D, Doz F, Desjardins L, Dutrillaux B, Chevillard S, Malfoy B. Genome instability in secondary solid tumors developing after radiotherapy of bilateral retinoblastoma. Oncogene. 2001; 20:8092–99. https://doi.org/10.1038/sj.onc.1205009. [PubMed].
33. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003; 100:3077–82. https://doi.org/10.1073/pnas.0630588100. [PubMed].
34. Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003; 14:281–87. https://doi.org/10.1097/00041433-200306000-00008. [PubMed].
35. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019; 20:137–55. https://doi.org/10.1038/s41580-018-0085-z. [PubMed].
36. Schaffer JE. Lipotoxicity: Many Roads to Cell Dysfunction and Cell Death: Introduction to a Thematic Review Series. J Lipid Res. 2016; 57:1327–28. https://doi.org/10.1194/jlr.E069880. [PubMed].
37. Koeberle A, Löser K, Thürmer M. Stearoyl-CoA desaturase-1 and adaptive stress signaling. Biochim Biophys Acta. 2016; 1861:1719–26. https://doi.org/10.1016/j.bbalip.2016.08.009. [PubMed].
38. Nishizawa S, Sumi H, Satoh Y, Yamamoto Y, Kitazawa S, Honda K, Araki H, Kakoi K, Imamura K, Sasaki M, Miyahisa I, Satomi Y, Nishigaki R, et al. In vitro and in vivo antitumor activities of T-3764518, a novel and orally available small molecule stearoyl-CoA desaturase 1 inhibitor. Eur J Pharmacol. 2017; 807:21–31. https://doi.org/10.1016/j.ejphar.2017.03.064. [PubMed].
39. Ntambi JM. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. J Lipid Res. 1999; 40:1549–58. [PubMed].
40. Falvella FS, Pascale RM, Gariboldi M, Manenti G, De Miglio MR, Simile MM, Dragani TA, Feo F. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis. 2002; 23:1933–36. https://doi.org/10.1093/carcin/23.11.1933. [PubMed].
41. Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avancès C, Allory Y, de la Taille A, Culine S, Blancou H, Cristol JP, Michel F, Sardet C, Fajas L. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther. 2010; 9:1740–54. https://doi.org/10.1158/1535-7163.MCT-09-1064. [PubMed].
42. von Roemeling CA, Marlow LA, Pinkerton AB, Crist A, Miller J, Tun HW, Smallridge RC, Copland JA. Aberrant lipid metabolism in anaplastic thyroid carcinoma reveals stearoyl CoA desaturase 1 as a novel therapeutic target. J Clin Endocrinol Metab. 2015; 100:E697–709. https://doi.org/10.1210/jc.2014-2764. [PubMed].
43. Huang J, Fan XX, He J, Pan H, Li RZ, Huang L, Jiang Z, Yao XJ, Liu L, Leung EL, He JX. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget. 2016; 7:39970–79. https://doi.org/10.18632/oncotarget.9461. [PubMed].
44. Iida T, Ubukata M, Mitani I, Nakagawa Y, Maeda K, Imai H, Ogoshi Y, Hotta T, Sakata S, Sano R, Morinaga H, Negoro T, Oshida S, et al. Discovery of potent liver-selective stearoyl-CoA desaturase-1 (SCD1) inhibitors, thiazole-4-acetic acid derivatives, for the treatment of diabetes, hepatic steatosis, and obesity. Eur J Med Chem. 2018; 158:832–52. https://doi.org/10.1016/j.ejmech.2018.09.003. [PubMed].
45. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, Sorrentino G, Chianese G, Taglialatela-Scafati O, Trisciuoglio D, Del Bufalo D, Di Martile M, Di Napoli A, et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene. 2017; 36:4573–84. https://doi.org/10.1038/onc.2017.75. [PubMed].
46. Noto A, Raffa S, De Vitis C, Roscilli G, Malpicci D, Coluccia P, Di Napoli A, Ricci A, Giovagnoli MR, Aurisicchio L, Torrisi MR, Ciliberto G, Mancini R. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013; 4:e947. https://doi.org/10.1038/cddis.2013.444. [PubMed].
47. von Roemeling CA, Caulfield TR, Marlow L, Bok I, Wen J, Miller JL, Hughes R, Hazlehurst L, Pinkerton AB, Radisky DC, Tun HW, Kim YSB, Lane AL, Copland JA. Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy. Oncotarget. 2017; 9:3–20. https://doi.org/10.18632/oncotarget.21545. [PubMed].
48. von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, Copland JA. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res. 2013; 19:2368–80. https://doi.org/10.1158/1078-0432.CCR-12-3249. [PubMed].
49. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018; 157:1276–91. https://doi.org/10.1016/j.ejmech.2018.09.001. [PubMed].
50. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005; 8:311–21. https://doi.org/10.1016/j.ccr.2005.09.008. [PubMed].
51. Li S, Qiu L, Wu B, Shen H, Zhu J, Zhou L, Gu L, Di W. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol Med Rep. 2013; 8:373–78. https://doi.org/10.3892/mmr.2013.1505. [PubMed].
52. Mukherjee A, Wu J, Barbour S, Fang X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J Biol Chem. 2012; 287:24990–5000. https://doi.org/10.1074/jbc.M112.340083. [PubMed].
53. Yang J, Zaman MM, Vlasakov I, Roy R, Huang L, Martin CR, Freedman SD, Serhan CN, Moses MA. Adipocytes promote ovarian cancer chemoresistance. Sci Rep. 2019; 9:13316. https://doi.org/10.1038/s41598-019-49649-1. [PubMed].
54. Shah US, Dhir R, Gollin SM, Chandran UR, Lewis D, Acquafondata M, Pflug BR. Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma. Hum Pathol. 2006; 37:401–9. https://doi.org/10.1016/j.humpath.2005.11.022. [PubMed].
55. Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. 1996; 27:917–21. https://doi.org/10.1016/s0046-8177(96)90218-x. [PubMed].
56. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009; 101:519–32. https://doi.org/10.1093/jnci/djp030. [PubMed].
57. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006; 66:5977–80. https://doi.org/10.1158/0008-5472.CAN-05-4673. [PubMed].
58. Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X, Liu J, Jiang X, Shen Y, Zhang L, Ren M, Huang P. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol. 2015; 32:391. https://doi.org/10.1007/s12032-014-0391-z. [PubMed].
59. Myers JS, von Lersner AK, Sang QX. Proteomic Upregulation of Fatty Acid Synthase and Fatty Acid Binding Protein 5 and Identification of Cancer- and Race-Specific Pathway Associations in Human Prostate Cancer Tissues. J Cancer. 2016; 7:1452–64. https://doi.org/10.7150/jca.15860. [PubMed].
60. Menendez JA, Lupu R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets. 2017; 21:1001–16. https://doi.org/10.1080/14728222.2017.1381087. [PubMed].
61. Lupu R, Menendez JA. Targeting fatty acid synthase in breast and endometrial cancer: An alternative to selective estrogen receptor modulators? Endocrinology. 2006; 147:4056–66. https://doi.org/10.1210/en.2006-0486. [PubMed].
62. Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, Waki A, Tsuji AB, Sogawa C, Wakizaka H, Fukumura T, Yoshii H, Fujibayashi Y, et al. Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PLoS One. 2013; 8:e64570. https://doi.org/10.1371/journal.pone.0064570. [PubMed].
63. Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010; 6:551–62. https://doi.org/10.2217/fon.10.11. [PubMed].
64. Zhao S, Cheng L, Shi Y, Li J, Yun Q, Yang H. MIEF2 reprograms lipid metabolism to drive progression of ovarian cancer through ROS/AKT/mTOR signaling pathway. Cell Death Dis. 2021; 12:18. https://doi.org/10.1038/s41419-020-03336-6. [PubMed].
65. Xia L, Wang Y, Cai S, Xu M. DGAT1 Expression Promotes Ovarian Cancer Progression and Is Associated with Poor Prognosis. J Immunol Res. 2021; 2021:6636791. https://doi.org/10.1155/2021/6636791. [PubMed].
66. Zhang Q, Zhou W, Yu S, Ju Y, To SKY, Wong AST, Jiao Y, Poon TCW, Tam KY, Lee LTO. Metabolic reprogramming of ovarian cancer involves ACSL1-mediated metastasis stimulation through upregulated protein myristoylation. Oncogene. 2021; 40:97–111. https://doi.org/10.1038/s41388-020-01516-4. [PubMed].
67. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, Fang X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018; 435:92–100. https://doi.org/10.1016/j.canlet.2018.08.006. [PubMed].
68. Wen Z, Liu H, Li M, Li B, Gao W, Shao Q, Fan B, Zhao F, Wang Q, Xie Q, Yang Y, Yu J, Qu X. Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene. 2015; 34:1241–52. https://doi.org/10.1038/onc.2014.85. [PubMed].
69. Rocconi RP, Kirby TO, Seitz RS, Beck R, Straughn JM Jr, Alvarez RD, Huh WK. Lipoxygenase pathway receptor expression in ovarian cancer. Reprod Sci. 2008; 15:321–26. https://doi.org/10.1177/1933719108316390. [PubMed].
70. Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010; 62:233–44. https://doi.org/10.1016/s1734-1140(10)70262-0. [PubMed].
71. Gu P, Su Y, Guo S, Teng L, Xu Y, Qi J, Gong H, Cai Y. Over-expression of COX-2 induces human ovarian cancer cells (CAOV-3) viability, migration and proliferation in association with PI3-k/Akt activation. Cancer Invest. 2008; 26:822–29. https://doi.org/10.1080/07357900801941860. [PubMed].
72. Wang YP, Wang QY, Li CH, Li XW. COX-2 inhibition by celecoxib in epithelial ovarian cancer attenuates E-cadherin suppression through reduced Snail nuclear translocation. Chem Biol Interact. 2018; 292:24–29. https://doi.org/10.1016/j.cbi.2018.06.020. [PubMed].
73. Adeva-Andany MM, Calvo-Castro I, Fernández-Fernández C, Donapetry-García C, Pedre-Piñeiro AM. Significance of l-carnitine for human health. IUBMB Life. 2017; 69:578–94. https://doi.org/10.1002/iub.1646. [PubMed].
74. Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004; 25:495–520. https://doi.org/10.1016/j.mam.2004.06.004. [PubMed].
75. Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, Zhang Y, Spiegel S, Idowu MO, Fang X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016; 7:3832–46. https://doi.org/10.18632/oncotarget.6757. [PubMed].
76. Gonzalez-Guerrico AM, Espinoza I, Schroeder B, Park CH, Kvp CM, Khurana A, Corominas-Faja B, Cuyàs E, Alarcón T, Kleer C, Menendez JA, Lupu R. Suppression of endogenous lipogenesis induces reversion of the malignant phenotype and normalized differentiation in breast cancer. Oncotarget. 2016; 7:71151–68. https://doi.org/10.18632/oncotarget.9463. [PubMed].
77. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescós C, Di Croce L, Benitah SA. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017; 541:41–45. https://doi.org/10.1038/nature20791. [PubMed].
78. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S, Chen D, Li W, Shi Y, Gao X, Zhang H, Zhang Z, Du W, et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics. 2019; 9:5359–73. https://doi.org/10.7150/thno.34024. [PubMed].
79. Feng WW, Bang S, Kurokawa M. CD36: a key mediator of resistance to HER2 inhibitors in breast cancer. Mol Cell Oncol. 2020; 7:1715766. https://doi.org/10.1080/23723556.2020.1715766. [PubMed].
80. Huangfu N, Xu Z, Zheng W, Wang Y, Cheng J, Chen X. LncRNA MALAT1 regulates oxLDL-induced CD36 expression via activating β-catenin. Biochem Biophys Res Commun. 2018; 495:2111–17. https://doi.org/10.1016/j.bbrc.2017.12.086. [PubMed].
81. Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, Bayraktar E, Nagaraja AS, Mangala LS, Zhang X, Haemmerle M, Hu W, Rodriguez-Aguayo C, et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun. 2018; 9:2923. https://doi.org/10.1038/s41467-018-04987-y. [PubMed].
82. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011; 17:1498–503. https://doi.org/10.1038/nm.2492. [PubMed].
83. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009; 29:431–38. https://doi.org/10.1161/ATVBAHA.108.179564. [PubMed].
84. Zheng L, Li L, Lu Y, Jiang F, Yang XA. SREBP2 contributes to cisplatin resistance in ovarian cancer cells. Exp Biol Med (Maywood). 2018; 243:655–62. https://doi.org/10.1177/1535370218760283. [PubMed].
85. Chang WC, Wang HC, Cheng WC, Yang JC, Chung WM, Ho YP, Chen L, Hung YC, Ma WL. LDLR-mediated lipidome-transcriptome reprogramming in cisplatin insensitivity. Endocr Relat Cancer. 2020; 27:81–95. https://doi.org/10.1530/ERC-19-0095. [PubMed].
86. Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, Silasi DA, Steffensen KD, Waldstrom M, Visintin I, Mor G. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009; 8:158–66. https://doi.org/10.4161/cc.8.1.7533. [PubMed].
87. Varas-Godoy M, Rice G, Illanes SE. The Crosstalk between Ovarian Cancer Stem Cell Niche and the Tumor Microenvironment. Stem Cells Int. 2017; 2017:5263974. https://doi.org/10.1155/2017/5263974. [PubMed].
88. Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013; 437:397–402. https://doi.org/10.1016/j.bbrc.2013.06.089. [PubMed].
89. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019; 129:3006–17. https://doi.org/10.1172/JCI127201. [PubMed].
90. De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013; 2013:291546. https://doi.org/10.1155/2013/291546. [PubMed].
91. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004; 4:579–91. https://doi.org/10.1038/nrc1408. [PubMed].
92. Si S, Li J, Tewara MA, Li H, Liu X, Li Y, Chen X, Liu C, Yuan T, Li W, Wang B, Xue F. Identifying causality, genetic correlation, priority and pathways of large-scale complex exposures of breast and ovarian cancers. Br J Cancer. 2021; 125:1570–81. https://doi.org/10.1038/s41416-021-01576-7. [PubMed].
93. Gaitskell K, Hermon C, Barnes I, Pirie K, Floud S, Green J, Beral V, Reeves GK, and Million Women Study Collaborators. Ovarian cancer survival by stage, histotype, and pre-diagnostic lifestyle factors, in the prospective UK Million Women Study. Cancer Epidemiol. 2022; 76:102074. https://doi.org/10.1016/j.canep.2021.102074. [PubMed].
94. Goss AM, Chandler-Laney PC, Ovalle F, Goree LL, Azziz R, Desmond RA, Wright Bates G, Gower BA. Effects of a eucaloric reduced-carbohydrate diet on body composition and fat distribution in women with PCOS. Metabolism. 2014; 63:1257–64. https://doi.org/10.1016/j.metabol.2014.07.007. [PubMed].
95. Cohen CW, Fontaine KR, Arend RC, Alvarez RD, Leath CA III, Huh WK, Bevis KS, Kim KH, Straughn JM Jr, Gower BA. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J Nutr. 2018; 148:1253–60. https://doi.org/10.1093/jn/nxy119. [PubMed].
96. Bós AM, Howard BV, Beresford SA, Urban N, Tinker LF, Waters H, Bós AJ, Chlebowski R, Ennis JM. Cost-effectiveness analysis of a low-fat diet in the prevention of breast and ovarian cancer. J Am Diet Assoc. 2011; 111:56–66. https://doi.org/10.1016/j.jada.2010.10.011. [PubMed].
97. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33. https://doi.org/10.1126/science.1160809. [PubMed].
98. Kurelac I, Umesh Ganesh N, Iorio M, Porcelli AM, Gasparre G. The multifaceted effects of metformin on tumor microenvironment. Semin Cell Dev Biol. 2020; 98:90–97. https://doi.org/10.1016/j.semcdb.2019.05.010. [PubMed].
99. Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu-Farha M. The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis. 2016; 15:108. https://doi.org/10.1186/s12944-016-0278-4. [PubMed].
100. Engin A. Adipose Tissue Hypoxia in Obesity and Its Impact on Preadipocytes and Macrophages: Hypoxia Hypothesis. Adv Exp Med Biol. 2017; 960:305–26. https://doi.org/10.1007/978-3-319-48382-5_13. [PubMed].
101. Harbuzariu A, Gonzalez-Perez RR. Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget. 2018; 9:18239–53. https://doi.org/10.18632/oncotarget.24435. [PubMed].
102. Cascetta P, Cavaliere A, Piro G, Torroni L, Santoro R, Tortora G, Melisi D, Carbone C. Pancreatic Cancer and Obesity: Molecular Mechanisms of Cell Transformation and Chemoresistance. Int J Mol Sci. 2018; 19:3331. https://doi.org/10.3390/ijms19113331. [PubMed].
103. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013; 110:8882–87. https://doi.org/10.1073/pnas.1307237110. [PubMed].
104. Chen RR, Yung MMH, Xuan Y, Zhan S, Leung LL, Liang RR, Leung THY, Yang H, Xu D, Sharma R, Chan KKL, Ngu SF, Ngan HYS, Chan DW. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun Biol. 2019; 2:281. https://doi.org/10.1038/s42003-019-0508-1. [PubMed].
105. Pyragius CE, Fuller M, Ricciardelli C, Oehler MK. Aberrant lipid metabolism: an emerging diagnostic and therapeutic target in ovarian cancer. Int J Mol Sci. 2013; 14:7742–56. https://doi.org/10.3390/ijms14047742. [PubMed].
106. Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010; 10:103. https://doi.org/10.1186/1471-2407-10-103. [PubMed].
107. A Phase II Study of Interaction of Lovastatin and Paclitaxel For Patients With Refractory or Relapsed Ovarian Cancer. https://clinicaltrials.gov. Available 2022 Feb 12, from https://clinicaltrials.gov/ct2/show/NCT00585052.
108. Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, Brenton JD, Downward J, Nicke B. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007; 11:498–512. https://doi.org/10.1016/j.ccr.2007.04.011. [PubMed].
109. Le TT, Yue S, Cheng JX. Shedding new light on lipid biology with coherent anti-Stokes Raman scattering microscopy. J Lipid Res. 2010; 51:3091–102. https://doi.org/10.1194/jlr.R008730. [PubMed].
110. Li J, Cheng JX. Direct visualization of de novo lipogenesis in single living cells. Sci Rep. 2014; 4:6807. https://doi.org/10.1038/srep06807. [PubMed].
111. Guenther S, Muirhead LJ, Speller AV, Golf O, Strittmatter N, Ramakrishnan R, Goldin RD, Jones E, Veselkov K, Nicholson J, Darzi A, Takats Z. Spatially resolved metabolic phenotyping of breast cancer by desorption electrospray ionization mass spectrometry. Cancer Res. 2015; 75:1828–37. https://doi.org/10.1158/0008-5472.CAN-14-2258. [PubMed].
112. Hilvo M, Denkert C, Lehtinen L, Müller B, Brockmöller S, Seppänen-Laakso T, Budczies J, Bucher E, Yetukuri L, Castillo S, Berg E, Nygren H, Sysi-Aho M, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011; 71:3236–45. https://doi.org/10.1158/0008-5472.CAN-10-3894. [PubMed].
113. Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, Venturini E, Glunde K, Bhujwalla ZM, Mezzanzanica D, Canevari S, Podo F. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010; 70:2126–35. https://doi.org/10.1158/0008-5472.CAN-09-3833. [PubMed].
114. Tomek K, Wagner R, Varga F, Singer CF, Karlic H, Grunt TW. Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Mol Cancer Res. 2011; 9:1767–79. https://doi.org/10.1158/1541-7786.MCR-10-0467. [PubMed].
115. Ueda SM, Yap KL, Davidson B, Tian Y, Murthy V, Wang TL, Visvanathan K, Kuhajda FP, Bristow RE, Zhang H, Shih IeM. Expression of Fatty Acid Synthase Depends on NAC1 and Is Associated with Recurrent Ovarian Serous Carcinomas. J Oncol. 2010; 2010:285191. https://doi.org/10.1155/2010/285191. [PubMed].
116. Zhou W, Han WF, Landree LE, Thupari JN, Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, El Meskini R, Ronnett GV, Townsend CA, Kuhajda FP. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007; 67:2964–71. https://doi.org/10.1158/0008-5472.CAN-06-3439. [PubMed].
117. Vidot S, Witham J, Agarwal R, Greenhough S, Bamrah HS, Tigyi GJ, Kaye SB, Richardson A. Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells. Cell Signal. 2010; 22:926–35. https://doi.org/10.1016/j.cellsig.2010.01.017. [PubMed].
118. van Vlerken LE, Duan Z, Seiden MV, Amiji MM. Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer Res. 2007; 67:4843–50. https://doi.org/10.1158/0008-5472.CAN-06-1648. [PubMed].
119. Eberlin LS, Gabay M, Fan AC, Gouw AM, Tibshirani RJ, Felsher DW, Zare RN. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc Natl Acad Sci U S A. 2014; 111:10450–55. https://doi.org/10.1073/pnas.1409778111. [PubMed].
120. De La Franier B, Thompson M. Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors (Basel). 2020; 10:13. https://doi.org/10.3390/bios10020013. [PubMed].
121. Zhang YJ, Cao LY, Fu ZZ, Wang YJ, Wang GX, Gu T. Clinical significance of plasma lysophosphatidic acid levels in the differential diagnosis of ovarian cancer. J Cancer Res Ther. 2015; 11:375–80. https://doi.org/10.4103/0973-1482.157335. [PubMed].
122. Liu Y, Chen Y, Momin A, Shaner R, Wang E, Bowen NJ, Matyunina LV, Walker LD, McDonald JF, Sullards MC, Merrill AH Jr. Elevation of sulfatides in ovarian cancer: an integrated transcriptomic and lipidomic analysis including tissue-imaging mass spectrometry. Mol Cancer. 2010; 9:186. https://doi.org/10.1186/1476-4598-9-186. [PubMed].
123. Cai Q, Zhao Z, Antalis C, Yan L, Del Priore G, Hamed AH, Stehman FB, Schilder JM, Xu Y. Elevated and secreted phospholipase A2 activities as new potential therapeutic targets in human epithelial ovarian cancer. FASEB J. 2012; 26:3306–20. https://doi.org/10.1096/fj.12-207597. [PubMed].
124. Sutphen R, Xu Y, Wilbanks GD, Fiorica J, Grendys EC Jr, LaPolla JP, Arango H, Hoffman MS, Martino M, Wakeley K, Griffin D, Blanco RW, Cantor AB, et al. Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2004; 13:1185–91. [PubMed].
125. Hada M, Edin ML, Hartge P, Lih FB, Wentzensen N, Zeldin DC, Trabert B. Prediagnostic Serum Levels of Fatty Acid Metabolites and Risk of Ovarian Cancer in the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial. Cancer Epidemiol Biomarkers Prev. 2019; 28:189–97. https://doi.org/10.1158/1055-9965.EPI-18-0392. [PubMed].
126. Tebbe C, Chhina J, Dar SA, Sarigiannis K, Giri S, Munkarah AR, Rattan R. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget. 2014; 5:4746–64. https://doi.org/10.18632/oncotarget.2012. [PubMed].
127. Grosse PY, Bressolle F, Pinguet F. Antiproliferative effect of methyl-beta-cyclodextrin in vitro and in human tumour xenografted athymic nude mice. Br J Cancer. 1998; 78:1165–69. https://doi.org/10.1038/bjc.1998.648. [PubMed].
128. Mohammad N, Malvi P, Meena AS, Singh SV, Chaube B, Vannuruswamy G, Kulkarni MJ, Bhat MK. Cholesterol depletion by methyl-β-cyclodextrin augments tamoxifen induced cell death by enhancing its uptake in melanoma. Mol Cancer. 2014; 13:204. https://doi.org/10.1186/1476-4598-13-204. [PubMed].
129. Graeser R, Bornmann C, Esser N, Ziroli V, Jantscheff P, Unger C, Hopt UT, Schaechtele C, von Dobschuetz E, Massing U. Antimetastatic effects of liposomal gemcitabine and empty liposomes in an orthotopic mouse model of pancreatic cancer. Pancreas. 2009; 38:330–37. https://doi.org/10.1097/MPA.0b013e31819436e6. [PubMed].
130. Gentile E, Cilurzo F, Di Marzio L, Carafa M, Ventura CA, Wolfram J, Paolino D, Celia C. Liposomal chemotherapeutics. Future Oncol. 2013; 9:1849–59. https://doi.org/10.2217/fon.13.146. [PubMed].
131. Zheng M, Mullikin H, Hester A, Czogalla B, Heidegger H, Vilsmaier T, Vattai A, Chelariu-Raicu A, Jeschke U, Trillsch F, Mahner S, Kaltofen T. Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile. Int J Mol Sci. 2020; 21:9169. https://doi.org/10.3390/ijms21239169. [PubMed].