
Introduction
The proto-oncogene MET encodes for a receptor tyrosine kinase which is a ubiquitously expressed cell surface receptor consisting of an extracellular alpha-chain disulfide-bonded to a membrane spanning beta-chain [1, 2]. The binding of the receptor to its ligand, the extracellular hepatocyte growth factor/scatter factor (HGF), induces dimerization of the receptor and triggers a conformational change [3, 4]. As a consequence, the activity of MET tyrosine kinase is activated and can lead to effects on cell growth, mortality, survival, invasion and angiogenesis [5–7]. Dysregulations in the signaling pathway were first investigated in 1990 as a possible cause of lung cancer [8]. Activation of the MET signaling pathway resulting from MET amplification or splice site alterations in MET exon 14 is associated with lung cancer growth and metastasis [9, 10]. In addition, MET amplification correlate with shorter overall survival in non-small cell lung cancer patients and, therefore, has prognostic value [11–13].
Common alterations are MET exon 14 splice site mutations and gene amplifications [14]. MET alterations are detected in 3–4% of lung adenocarcinomas and MET amplifications in 1–6% [15, 16]. In particular, patients diagnosed with lung adenocarcinoma and MET amplification showed acquired resistances when treated with epidermal growth factor receptor (EGFR) inhibitors like Gefitinib or Erlotinib, regardless of EGFR mutation status [17, 18]. In one study, resistance to EGFR inhibitors was found in 21% of lung cancer patients. Each of the affected patients displayed a MET amplification [19].
For these reasons, MET is considered a potentially targetable oncogenic driver [20, 21]. The tyrosine kinase inhibitors (TKIs) for MET amplifications are currently in the clinical stage of approval and show promising therapeutic results. For example, Capmatinib was recently approved by the U.S. Food and Drug Administration (FDA) as the first drug targeting specific mutations in advanced lung cancer [16, 22, 23]. It was developed to interrupt the signaling pathway by selectively binding to MET in order to prevent phosphorylation [24]. In addition, Crizotinib, a multikinase inhibitor used in lung cancer patients with ALK or ROS1 translocations, showed a beneficial effect in patients with MET alterations [25, 26].
The detection of MET amplification can be performed by several techniques such as polymerase chain reaction (PCR), southern blot, immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) [27–29]. It is very important to reliably detect MET changes. The standard analytical method for the detection of MET amplifications is FISH. However, in recent years, NGS has become a daily routine in molecular diagnostics and offers new opportunities to analyze MET alterations [30]. Despite the important therapeutic significance of MET alterations, there are currently no defined cut-off values for NGS.
The goal of this project was to find a way to analyze MET amplifications by NGS by defining cut-off values. The results of the NGS pipeline (n = 20) were compared to the gold standard FISH.
Results
Patient collective
The mean age was 64 years, ranging from 37 to 90 years. The cohort consisted of 10 men (50%) and 10 women (50%). 18 (90%) lung carcinomas and two cancers (10%) of unknown primary were included in the study. All samples were formalin-fixed paraffin embedded (FFPE) tumor tissues.
The patient collective was compiled based on the NGS result, wherein the selection criterion for the samples was the presence of a MET amplification and an overall sufficient sequencing quality, based on the quality parameters specified by the analysis software (QCI-A), to avoid false positive cases. Cases without MET amplification were also selected as controls. Data from a total of 20 patients were analyzed by NGS. 17 (85%) had a MET amplification by NGS and one (5%) a MET exon 14 skipping event while two (10%) were classified as wild type (Table 1).
Table 1: Characteristics of the patient collective including primary tumor site and NGS results of MET amplification according to determination via the nNGM-v1 panel, QIAGEN
| Study-ID | Primary tumor site | MET Status (NGS) | TC [%] | FC | CN 100% | CN 50% | CN 25% | CN 10% | p-value | CNMV 10–100% (NGS) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Lung | 2a | 40 | |||||||
| 2 | Lung | 1 | 40 | 1.51 | 3.02 | 4.03 | 6.06 | 12.16 | 1.24e-12 | 6.32 |
| 3 | Lung | 1 | 55 | 3.18 | 6.35 | 10.70 | 19.41 | 45.52 | 0 | 20.50 |
| 4 | Lung | 1 | 60 | 2.32 | 4.64 | 7.29 | 12.57 | 28.44 | 3.09e-12 | 13.24 |
| 5 | Lung | 1 | 70 | 2.49 | 4.98 | 7.96 | 13.92 | 31.79 | 4.0e-9 | 14.66 |
| 6 | Lung | 2 | 40 | |||||||
| 7 | Unknown | 1 | 70 | 1.48 | 2.96 | 3.91 | 5.82 | 11.55 | 2.14e-5 | 6.06 |
| 8 | Lung | 1 | 50 | 1.44 | 2.87 | 3.74 | 5.49 | 10.71 | 8.69e-5 | 5.70 |
| 9 | Lung | 1 | 50 | 1.60 | 3.20 | 4.40 | 6.79 | 13.98 | 4.07e-6 | 7.09 |
| 10 | Lung | 1 | 50 | 2.20 | 4.40 | 6.80 | 11.60 | 26.01 | 0 | 12.20 |
| 11 | Lung | 1 | 30 | 1.46 | 2.92 | 3.84 | 5.67 | 11.18 | 1.47e-3 | 5.90 |
| 12b | Lung | 1 | 80 | 3.50 | 6.99 | 11.98 | 21.97 | 51.92 | 3.18e-6 | 23.22 |
| 13 | Lung | 1 | 50 | 1.79 | 3.58 | 5.16 | 8.32 | 17.79 | 1.9e-6 | 8.71 |
| 14 | Unknown | 1 | 60 | 1.56 | 3.13 | 4.26 | 6.51 | 13.28 | 0.01 | 6.80 |
| 15 | Lung | 1 | 30 | 1.74 | 3.48 | 4.96 | 7.91 | 16.78 | 2.95e-11 | 8.28 |
| 16 | Lung | 1 | 20 | 1.51 | 3.03 | 4.05 | 6.10 | 12.26 | 1.26e-3 | 6.36 |
| 17 | Lung | 1 | 60 | 1.66 | 3.33 | 4.65 | 7.31 | 15.27 | 0.01 | 7.64 |
| 18 | Lung | 1 | 70 | 1.61 | 3.23 | 4.46 | 6.91 | 14.28 | 1.75e-10 | 7.22 |
| 19 | Lung | 1 | 80 | 2.61 | 5.22 | 8.44 | 14.88 | 34.20 | 5.68e-10 | 15.69 |
| 20 | Lung | 2 | 40 | |||||||
| Mean value | 1.86 | 3.72 | 5.43 | 8.87 | 19.17 | |||||
| Max | 3.50 | 6.99 | 11.98 | 21.97 | 51.92 | |||||
| Min | 1.44 | 2.87 | 3.74 | 5.49 | 10.71 |
All samples were subsequently assessed by FISH in order to proof the amplification status (Table 2).
Table 2: FISH results
| Study-ID | MV MET/nuclei | MV CEN7/nuclei | Ratio | Result |
|---|---|---|---|---|
| 1 | 2.97 | 1.88 | 1.50 | Negative |
| 2 | 2.42 | 2.01 | 1.20 | Negative |
| 3 | cluster | High-level (not countable) | ||
| 4 | 3.18 | 2.97 | 1.07 | Negative |
| 5 | 3.45 | 3.03 | 1.14 | Negative |
| 6 | 2.03 | 1.82 | 1.12 | Negative |
| 7 | 2.03 | 2.02 | 1.00 | Negative |
| 8 | 2.03 | 1.65 | 1.23 | Negative |
| 9 | 1.95 | 1.83 | 1.07 | Negative |
| 10 | 3.75 | 2.32 | 1.62 | Negative |
| 11 | 2.82 | 2.63 | 1.07 | Negative |
| 12 | 3.45 | 3.80 | 0.91 | Negative |
| 13 | 2.72 | 2.65 | 1.03 | Negative |
| 14 | 2.45 | 2.15 | 1.14 | Negative |
| 15 | 2.25 | 2.00 | 1.12 | Negative |
| 16 | 1.92 | 2.05 | 0.94 | Negative |
| 17 | 3.47 | 3.72 | 0.93 | Negative |
| 18 | 3.88 | 2.41 | 1.61 | Negative |
| 19 | 2.63 | 2.15 | 1.22 | Negative |
| 20 | 3.02 | 2.92 | 1.03 | Negative |
| Mean value | 2.76 | 2.42 | 1.15 | |
| Max | 3.88 | 3.80 | 1.62 | |
| Min | 1.92 | 1.65 | 0.91 |
MET alterations using NGS
QIAGEN calculates the values fold change (FC), copy number (CN) and p-value in the analysis of amplifications. These were considered in conjunction with the tumor cell content (TC) of the tissue samples for evaluation. The average FC of the 17 patients that were tested positive for MET amplification was 1.86, with a maximum value of 3.5 and a minimum value of 1.44. The copy number (100%) ranges from 6.99 to 2.87 with an average value of 3.72 (Table 1).
MET alterations using FISH
The evaluation of the FISH analysis was done by fluorescence microscopy. 20 tumor cell nuclei were counted in three separate areas; i.e., a total of 60 counts per patient. The ratio and mean value per cell (MV) for MET and CEN7 were calculated according to the criteria of Schildhaus et al. [31]. The results are shown in Table 2. 19 samples (95%) were negative. One sample (5%) showed a high-level/cluster amplification (Figure 1). No intermediate amplified cases were observed.
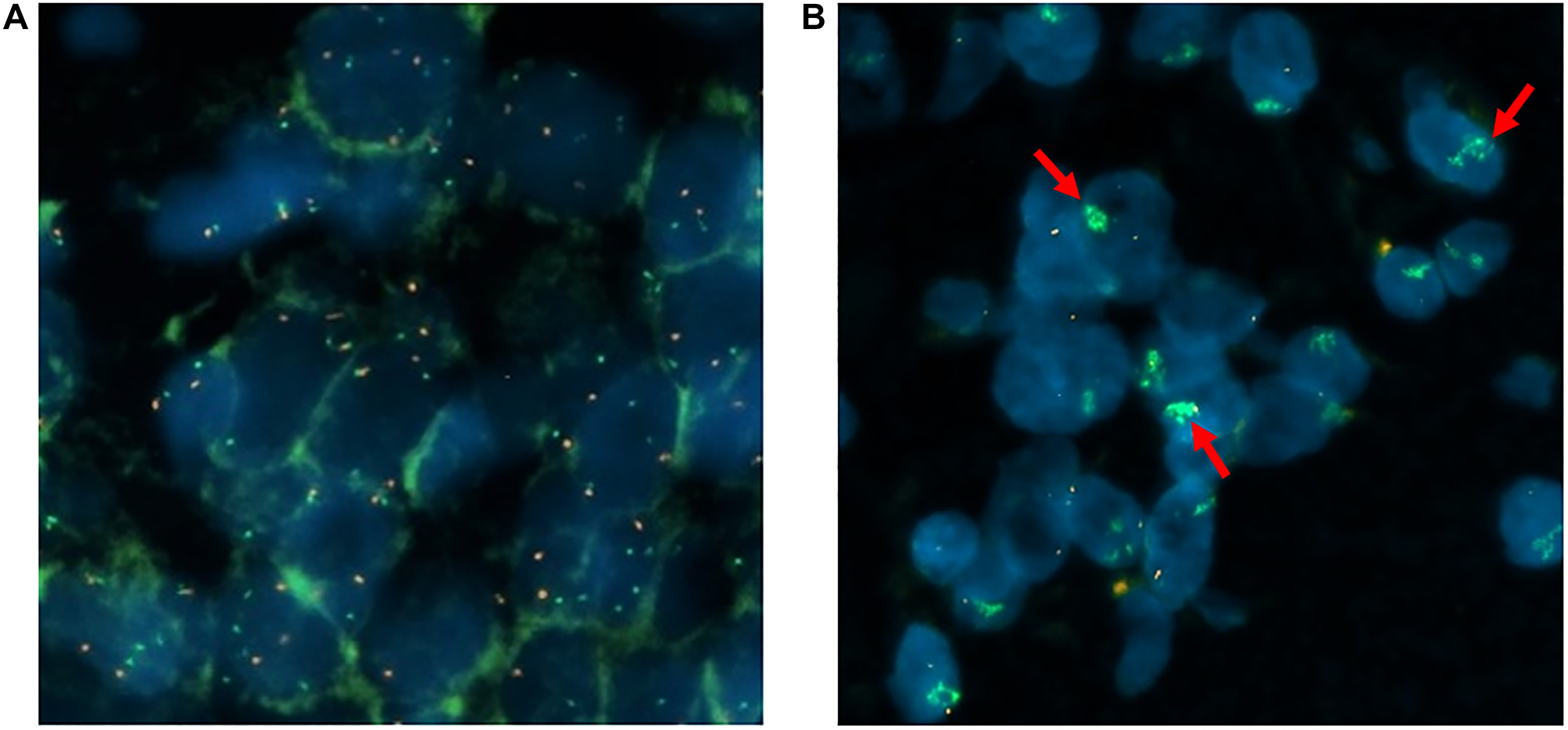
Figure 1: Representation of individual tumor cell nuclei (MET/CEN7 probe and DAPI). The MET gene is identified with green signals and the centromere of chromosome 7 is the reference gene marked in red: wild type (A), cluster amplification (B).
Comparison of NGS (QIAGEN workflow) and FISH analyses
Comparing the results of the two detection methods, the high-level MET amplification detected by FISH analysis showed agreement with the result of the NGS analysis. NGS analysis of 20 preselected specimens diagnosed a total of 17 MET amplification positive samples and one with a MET exon 14 skipping event. Two of the samples showed an FC ≥ 3 (sample 3 and 12). By FISH analysis, a high-level amplification was also detected in sample 3. Sample 12 had a reduced sequencing quality in the NGS analysis, which could be the reason for the discrepancy between the FISH and NGS result. Furthermore, it is not possible to detect exon skipping events by FISH analysis, so the MET exon 14 skipping event (a) identified by NGS in sample 1 could not be confirmed (Table 3).
Table 3: Comparison of NGS and FISH results regarding MET amplification
| Study-ID | TC [%] | MET Amplification Status (FISH) | MET Amplification Status (NGS) | FC | CN 100% | CN 50% | CN 25% | CN 10% | CNMV 10–100% (NGS) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 40 | 2a | 2b | ||||||
| 2 | 40 | 2 | 1 | 1.51 | 3.02 | 4.03 | 6.06 | 12.16 | 6.32 |
| 3 | 55 | 1 | 1 | 3.18 | 6.35 | 10.70 | 19.41 | 45.52 | 20.50 |
| 4 | 60 | 2 | 1 | 2.32 | 4.64 | 7.29 | 12.57 | 28.44 | 13.24 |
| 5 | 70 | 2 | 1 | 2.49 | 4.98 | 7.96 | 13.92 | 31.79 | 14.66 |
| 6 | 40 | 2 | 2 | ||||||
| 7 | 70 | 2 | 1 | 1.48 | 2.96 | 3.91 | 5.82 | 11.55 | 6.06 |
| 8 | 50 | 2 | 1 | 1.44 | 2.87 | 3.74 | 5.49 | 10.71 | 5.70 |
| 9 | 50 | 2 | 1 | 1.60 | 3.20 | 4.40 | 6.79 | 13.98 | 7.09 |
| 10 | 50 | 2 | 1 | 2.20 | 4.40 | 6.80 | 11.60 | 26.01 | 12.20 |
| 11 | 30 | 2 | 1 | 1.46 | 2.92 | 3.84 | 5.67 | 11.18 | 5.90 |
| 12 | 80 | 2 | 1c | 3.50 | 6.99 | 11.98 | 21.97 | 51.92 | 23.22 |
| 13 | 50 | 2 | 1 | 1.79 | 3.58 | 5.16 | 8.32 | 17.79 | 8.71 |
| 14 | 60 | 2 | 1 | 1.56 | 3.13 | 4.26 | 6.51 | 13.28 | 6.80 |
| 15 | 30 | 2 | 1 | 1.74 | 3.48 | 4.96 | 7.91 | 16.78 | 8.28 |
| 16 | 20 | 2 | 1 | 1.51 | 3.03 | 4.05 | 6.10 | 12.26 | 6.36 |
| 17 | 60 | 2 | 1 | 1.66 | 3.33 | 4.65 | 7.31 | 15.27 | 7.64 |
| 18 | 70 | 2 | 1 | 1.61 | 3.23 | 4.46 | 6.91 | 14.28 | 7.22 |
| 19 | 80 | 2 | 1 | 2.61 | 5.22 | 8.44 | 14.88 | 34.20 | 15.69 |
| 20 | 40 | 2 | 2 |
All MET wild type cases determined by FISH had a FC by NGS ranging from 1.44 to 2.61 and a CNMV 10–100% ranging from 5.70 to 15.69 (Figure 2). With the exception of the outlier sample 12 (Δ), which shows a FC of 3.50 and a CNMV 10–100% of 23.22. The only sample (sample 3, †) that was identified as MET amplified by FISH presents a FC of 3.18 and a CNMV 10–100% of 20.55 (Figure 2).
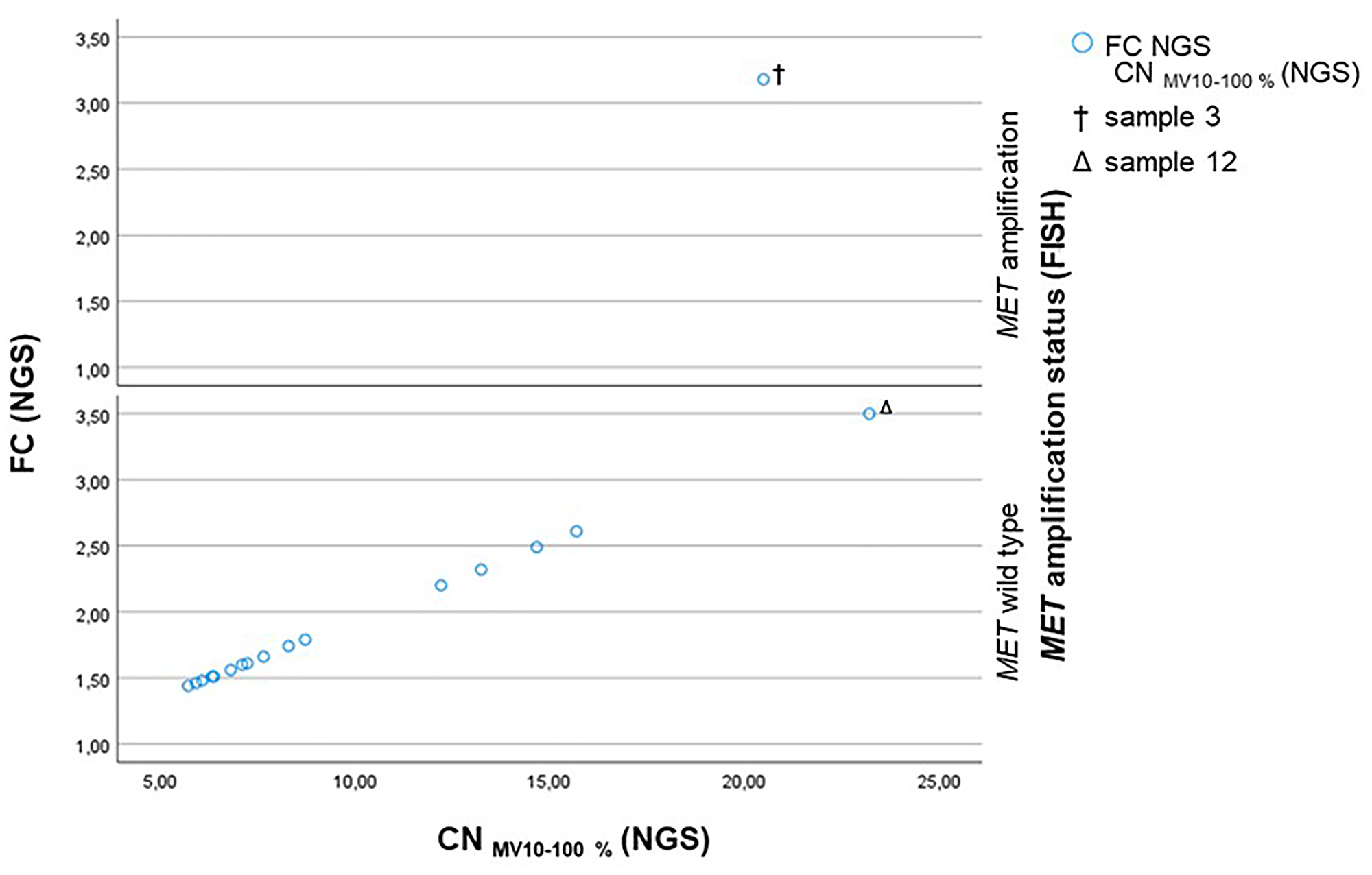
Figure 2: Scatterplot showing the CNMV 10–100% (NGS) plotted against the FC (NGS) subdivided into the MET amplification status assessed by FISH. Sample 3 (†) is MET amplified by FISH and shows a CNMV 10–100% of 20.55 and a FC of 3.18. Sample 12 (Δ) is determined as MET wild type by FISH and has a CN of 23.22 and a FC of 3.50 due to a low sequencing quality.
Analytical sensitivity and specificity
For the calculation of the analytical sensitivity and specificity, FISH analysis was assumed to be the gold standard. Thus, considering NGS positive cases with a FC of ≥ 3.0 and a CNMV 10–100% value of ≥ 20.0 as well as sufficient NGS sequencing quality results in both an analytical sensitivity (Equation 1) and an analytical specificity (Equation 2) of 1.
DISCUSSION
The aim of this study was to verify MET amplifications detected by NGS. Therefore, 20 samples with known amplification status (17 amplified cases and 3 non-amplified cases), were additionally checked by FISH.
FISH analysis is the current gold standard and the most widely used method for the detection of increased numbers of gene copies, such as MET amplification, with recommended evaluation criteria. For FISH analysis it is recommended to count a total of 50–100 cell nuclei, depending on the present tumor cell content [31]. In this study 60 tumor cell nuclei were counted per sample.
According to Schildhaus et al. FISH analysis enables differentiation between low-level, intermediate-level and high-level amplifications [31], however the therapeutically relevant cut-off value is still a matter of debate [32].
In NGS analysis, low tumor cell content and the sequencing quality influence the amplification results. Thus, false positive or false negative MET cases have to be kept in mind. It is not possible to differentiate between artifacts and real gene amplifications via sequencing, as the NGS methodology is only optimized for the detection of fusions and mutations. Amplifications are part of the panel but the sensitivity is limited. The detection of low- or intermediate-level gene amplifications by NGS is still matter of controversy.
In this study no low- or intermediate amplification was observed. Currently, most tumor samples are analyzed by NGS in routine diagnostics. Statistically, only 4% of lung adenocarcinomas show a MET amplification detected by FISH and are potential candidates for a targeted therapy [33].
Of the 20 preselected test samples, negative FISH results were consistent with negative NGS results. This fact supports the conclusion that a negative NGS result with a sufficient sequencing quality will strongly predict a negative FISH analysis. Sample 3, which was classified as high-level amplification via NGS (FC 3.18, CNMV 10–100% 20.5, sufficient sequencing quality), showed a positive FISH analysis, too. This therapeutically relevant mutation could be considered for study inclusion and treatment with TKIs. The discrepancy between the results for sample 12 by NGS as compared to FISH can be explained by a decreased NGS sequencing quality. In summary, a cut-off value of ≥ 3.0 and a CNMV 10–100% value of ≥ 20.0 can be recommended in the context of sufficient sequencing quality. This is further supported by the fact that (when positive cases with FC≥3.0 and CNMV 10–100% ≥ 20.0 are considered) an analytical sensitivity and specificity of 1 each can then be calculated, but still some more test samples should be run to finally determine the sensitivity and specificity.
In conclusion, MET alterations appear to be a promising therapeutic target. For this reason, it is important to be able to detect alterations with a high degree of accuracy based on a defined cut-off value. We recommend to use a combination of NGS and FISH for the detection of MET amplifications. The exon-14-skipping case (sample 1) in the MET gene cannot be detected by FISH, but can be detected by NGS analysis. It can be concluded that amplifications can be verified via FISH sufficiently. With this method each tumor sample can be evaluated microscopically in real time on the basis of the signals per tumor cell but you have to keep in mind overlapping cells to avoid a false increased MET signal counting. For NGS, a mixture of tumor, normal, necrotic and inflammatory cells are analyzed, which may reduce validity of the final results. Thus, these methods of molecular pathology complement each other and should be standard in every cancer center. In addition, the cut-off values of the NGS analyses should be specifically defined and validated in each lab depending on the NGS workflow. The following test algorithm is proposed for the detection of MET amplifications (Figure 3).
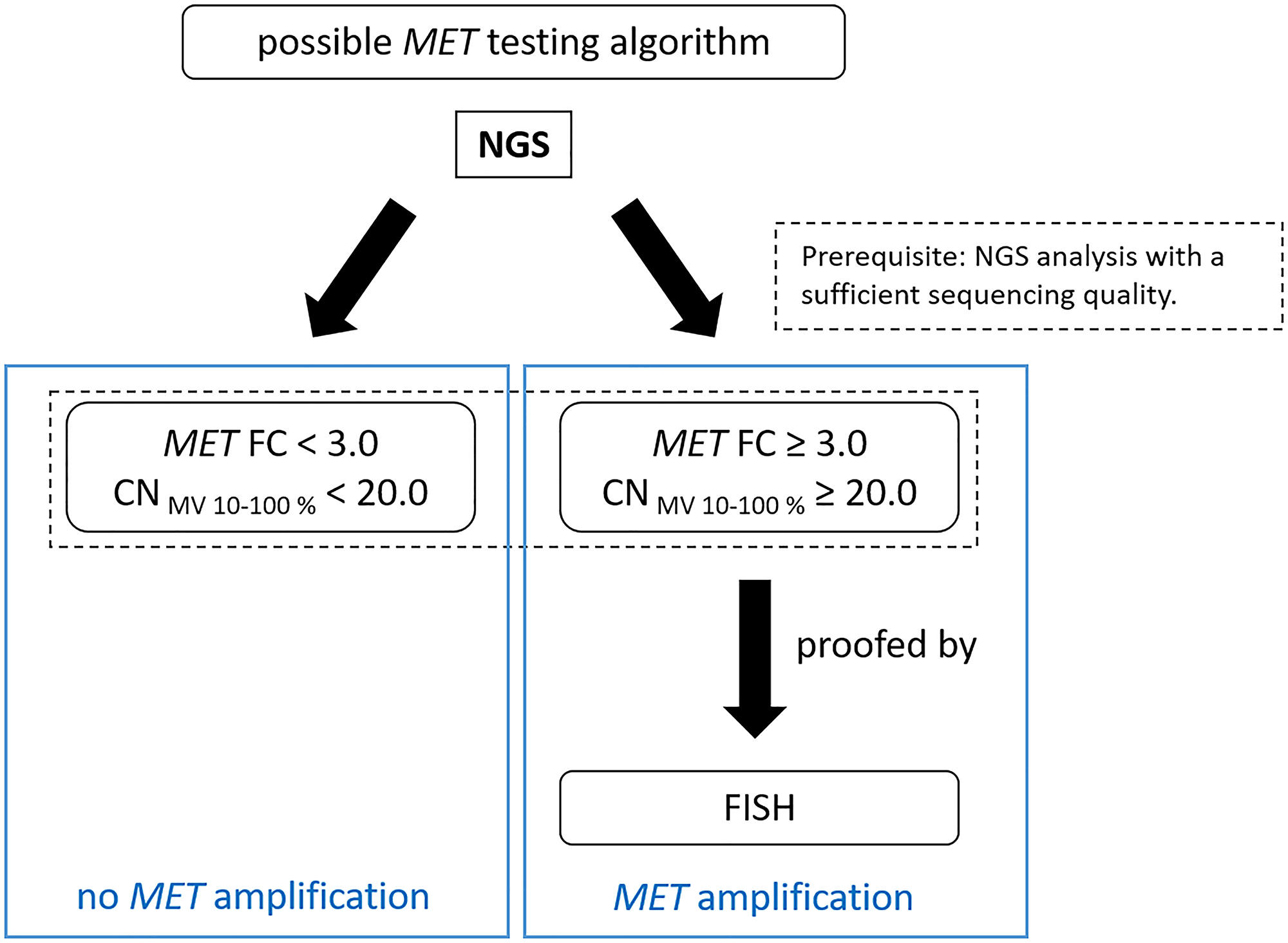
Figure 3: Suggested testing workflow for the detection of MET amplifications by QIAGEN workflow.
NGS analysis of the samples is performed: If the FC is ≥ 3 and the CNMV 10–100% value is ≥ 20, an additional analysis by FISH should follow to confirm MET gene amplification. If the FC is < 3 and the CNMV 10–100% value is < 20, MET is considered not amplified. Sufficient sequencing quality is a prerequisite for the NGS based testing algorithm.
Immunohistochemical analysis with recombinant Anti-Met (c-Met) antibody [SP44] – C-terminal (Abcam) as well as a review of the literature did not provide evidence of the validity of MET antibodies for the purposes of prescreening for MET alterations [34, 35].
In the next step we are going to increase the cohort size, including cases with negative, low-level, intermediate-level and high-level MET amplification. Additionally, copy number arrays (OncoScan Arrays; Thermo Fisher Scientific) as well as another NGS panel (Oncomine Comprehensive v3; Thermo Fisher Scientific) will be used to validate the results. Further clinical data are needed to define clinically relevant MET alterations and to establish a testing algorithm for routine diagnostics.
Materials and Methods
NGS
The samples were previously analyzed using NGS. After macrodissection of the tumor tissue, DNA was isolated using the Maxwell® RSC FFPE Plus DNA Kit (Promega). The library preparation was performed according to standard laboratory instructions and sequenced using the custom-made targeted resequencing panel (nNGM version 1.0) and the Gene Reader (QIAGEN) workflow. The results were analyzed with the QIAGEN bioinformatics software packages QCI-A and QCI-I. The cases were selected with respect to the quality parameters specified by the analysis software (QCI-A). Sequencing was performed on Gene Reader (QIAGEN) with a resulting sequence coverage of >100×. The detailed parameters of the bioinformatics NGS workflow for the detection of CNVs were set in the QCI-A software according to Table 4.
Table 4: CNVs detection configuration (workflow settings configured in the QCI-A software)
| Percentile used for fold-change calculation | 75 |
|---|---|
| Maximum combined p-value for amplification | 0.05 |
| Minimum fold change for amplification, absolute value | 1.40 |
| Maximum combined p-value for deletion | 0.05 |
| Minimum fold change for deletion, absolute value | 1.40 |
FISH
MET amplification status was assessed using ZytoLight® FISH-Tissue Implementation Kit (ZytoVision®) with a dual-color FISH probe set (ZytoLight® SPEC MET/CEN7) targeting MET and CEN7 on formalin-fixed, paraffin-embedded tissue sections. The ZytoBrite Hybridizer (Zytomed) was used for hybridization following the ZytoLight® protocol. The analysis was made using fluorescence microscopy (Carl Zeiss Microscopy GmbH).
FISH evaluation
Evaluation of the FISH analysis was performed and modified according to the criteria of Schildhaus et al. [31] (Table 5).
Table 5: FISH evaluation sheet modified according to Schildhaus et al. [31]
| Not amplified | None of the criteria below is fulfilled |
|---|---|
| high-level amplification | MET/CEN7 ratio ≥ 2.0 Average MET gene copy number per cell of ≥ 6.0 copies ≥ 10% of tumor cells containing ≥ 15 MET signals |
| intermediate level amplification | ≥ 50% of cells containing ≥ 5 MET signals Criteria for high-level amplification are not fulfilled |
| low level amplification | ≥ 40% of tumor cells showing ≥ 4 MET signals Criteria for high-level amplification |
Calculation of the analytical sensitivity and specificity
To calculate the analytical sensitivity and specificity, FISH analysis was assumed to be the gold standard. The calculation was performed according to the criteria of the “Deutsche Akkreditierungsstelle” (DakkS) [36].
Analytical sensitivity is a measure of the rate of true positive cases. It thus describes the ratio of correctly positive results to the total number of positive results.
The analytical specificity describes the rate of correctly negative results. It is a measure of the ratio of correct negative results to the total number of negative results.
ACKNOWLEDGMENTS
Tumor samples and patient data used in this study were provided by the Senckenberg Biobank, University Cancer Center Frankfurt-Marburg (UCT Frankfurt-Marburg). Written informed consent was obtained from all patients and the study was approved by the institutional Review Boards of the UCT and the Ethical Committee at the University Hospital Frankfurt (project-numbers: STO-2-2018).
CONFLICTS OF INTEREST
Peter J. Wild: Advisory role and honoraria (institutional or personal): AstraZeneca, Janssen, Roche, Astellas, Bayer, Bristol-Myers Squibb, Janssen, Novartis, Thermo Fisher Scientific, MSD, QIAGEN, Molecular Health, Sophia Genetics. Melanie Demes: Advisory role and honoraria (institutional or personal): Amgen, AstraZeneca, Bayer, Diaceutics, Biocartis, Sophia Genetics.
References
1. Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature. 1989; 339:155–56. https://doi.org/10.1038/339155a0. [PubMed].
2. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A. 1987; 84:6379–83. https://doi.org/10.1073/pnas.84.18.6379. [PubMed].
3. Califano R, Morgillo F, De Mello RA, Mountzios G. Role of mesenchymal-epithelial transition amplification in resistance to anti-epidermal growth factor receptor agents. Ann Transl Med. 2015; 3:81. https://doi.org/10.3978/j.issn.2305-5839.2015.03.44. [PubMed].
4. Baldanzi G, Graziani A. Physiological Signaling and Structure of the HGF Receptor MET. Biomedicines. 2014; 3:1–31. https://doi.org/10.3390/biomedicines3010001. [PubMed].
5. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003; 4:915–25. https://doi.org/10.1038/nrm1261. [PubMed].
6. Weidner KM, Behrens J, Vandekerckhove J, Birchmeier W. Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J Cell Biol. 1990; 111:2097–108. https://doi.org/10.1083/jcb.111.5.2097. [PubMed].
7. Sonnenberg E, Meyer D, Weidner KM, Birchmeier C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol. 1993; 123:223–35. https://doi.org/10.1083/jcb.123.1.223. [PubMed].
8. Rosell R, Chaib I, Santarpia M. Targeting MET amplification in EGFR-mutant non-small-cell lung cancer. Lancet Respir Med. 2020; 8:1068–70. https://doi.org/10.1016/S2213-2600(20)30171-5. [PubMed].
9. Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, Ahn MJ, Vansteenkiste JF, Su WC, Felip E, Chia V, Glaser S, Pultar P, et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J Clin Oncol. 2018; 36:3101–09. https://doi.org/10.1200/JCO.2018.77.7326. [PubMed].
10. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol. 2017; 12:15–26. https://doi.org/10.1016/j.jtho.2016.10.014. [PubMed].
11. Jin Y, Sun PL, Kim H, Seo AN, Jheon S, Lee CT, Chung JH. MET gene copy number gain is an independent poor prognostic marker in Korean stage I lung adenocarcinomas. Ann Surg Oncol. 2014; 21:621–28. https://doi.org/10.1245/s10434-013-3355-1. [PubMed].
12. Tanaka A, Sueoka-Aragane N, Nakamura T, Takeda Y, Mitsuoka M, Yamasaki F, Hayashi S, Sueoka E, Kimura S. Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer. 2012; 75:89–94. https://doi.org/10.1016/j.lungcan.2011.06.004. [PubMed].
13. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M, Toschi L, Finocchiaro G, Destro A, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009; 27:1667–74. https://doi.org/10.1200/JCO.2008.19.1635. [PubMed].
14. Garinet S, Laurent-Puig P, Blons H, Oudart JB. Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies? J Clin Med. 2018; 7:144. https://doi.org/10.3390/jcm7060144. [PubMed].
15. Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017; 3:148–53. https://doi.org/10.1016/j.cdtm.2017.06.002. [PubMed].
16. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, Tan DSW, Hida T, de Jonge M, Orlov SV, Smit EF, Souquet PJ, Vansteenkiste J, et al, and GEOMETRY mono-1 Investigators. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N Engl J Med. 2020; 383:944–57. https://doi.org/10.1056/NEJMoa2002787. [PubMed].
17. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–43. https://doi.org/10.1126/science.1141478. [PubMed].
18. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007; 104:20932–37. https://doi.org/10.1073/pnas.0710370104. [PubMed].
19. Robinson KW, Sandler AB. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist. 2013; 18:115–22. https://doi.org/10.1634/theoncologist.2012-0262. [PubMed].
20. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003; 22:309–25. https://doi.org/10.1023/a:1023768811842. [PubMed].
21. Gherardi E, Youles ME, Miguel RN, Blundell TL, Iamele L, Gough J, Bandyopadhyay A, Hartmann G, Butler PJ. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A. 2003; 100:12039–44. https://doi.org/10.1073/pnas.2034936100. [PubMed].
22. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, Tan DSW, Hida T, De Jonge MJ, Orlov SV, Smit EF, Souquet PJ, Vansteenkiste JF, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J Clin Oncol. 2019; 37:9004.
23. Novartis. Novartis announces MET inhibitor capmatinib (INC280), the first potential treatment for METΔex14 mutated advanced non-small cell lung cancer, granted priority FDA review | Novartis, https://www.novartis.com/news/media-releases/novartis-announces-met-inhibitor-capmatinib-inc280-first-potential-treatment-metex14-mutated-advanced-non-small-cell-lung-cancer-granted-priority-fda-review. (2020, accessed 14 November 2020).
24. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, Tan DS, Hida T, de Jonge MJA, Orlov SV, Smit EF, Souquet PJ, Vansteenkiste JF, et al. Results of the GEOMETRY mono-1 phase II study for evaluation of the MET inhibitor capmatinib (INC280) in patients (pts) with METΔex14 mutated advanced non-small cell lung cancer (NSCLC). Ann Oncol. 2018; 29:viii741–42. https://doi.org/10.1093/annonc/mdy424.090.
25. Drilon A, Clark JW, Weiss J, Ou SI, Camidge DR, Solomon BJ, Otterson GA, Villaruz LC, Riely GJ, Heist RS, Awad MM, Shapiro GI, Satouchi M, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020; 26:47–51. https://doi.org/10.1038/s41591-019-0716-8. [PubMed].
26. Nwizu T, Kanteti R, Kawada I, Rolle C, Vokes EE, Salgia R. Crizotinib (PF02341066) as a ALK /MET inhibitor- Special Emphasis as a Therapeutic Drug Against Lung Cancer. Drugs Future. 2011; 36:91–99. https://doi.org/10.1358/dof.2011.036.02.1584112. [PubMed].
27. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009; 4:5–11. https://doi.org/10.1097/JTO.0b013e3181913e0e. [PubMed].
28. Wasson JC, Brodeur GM. Molecular analysis of gene amplification in tumors. Curr Protoc Hum Genet. 2001; Chapter 10:Unit 10.5.1–10.5.18. https://doi.org/10.1002/0471142905.hg1005s02. [PubMed].
29. Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, Govindan R, Patel T, Orlov SV, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013; 31:4105–14. https://doi.org/10.1200/JCO.2012.47.4189. [PubMed].
30. Clavé S, Salido M, Rocha P, Hardy-Werbin M, Gibert J, Riera X, Weingartner E, Cerqueira G, Nichol D, Simmons J, Taus Á, Pijuan L, Bellosillo B, Arriola E. Identification of MET gene amplifications using next-generation sequencing in non-small cell lung cancer patients. Ann Oncol. 2019; 30:v800. https://doi.org/10.1093/annonc/mdz269.009.
31. Schildhaus HU, Schultheis AM, Rüschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, Schulte W, Ko YD, Schlesinger A, Bos M, Gardizi M, Engel-Riedel W, Brockmann M, et al. MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. 2015; 21:907–15. https://doi.org/10.1158/1078-0432.CCR-14-0450. [PubMed].
32. Eijkelenboom A, Tops BBJ, van den Berg A, van den Brule AJC, Dinjens WNM, Dubbink HJ, Ter Elst A, Geurts-Giele WRR, Groenen PJTA, Groenendijk FH, Heideman DAM, Huibers MMH, Huijsmans CJJ, et al. Recommendations for the clinical interpretation and reporting of copy number gains using gene panel NGS analysis in routine diagnostics. Virchows Arch. 2019; 474:673–80. https://doi.org/10.1007/s00428-019-02555-3. [PubMed].
33. Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015; 4:36–54. https://doi.org/10.3978/j.issn.2218-6751.2014.05.01. [PubMed].
34. Guo R, Berry LD, Aisner DL, Sheren J, Boyle T, Bunn PA Jr, Johnson BE, Kwiatkowski DJ, Drilon A, Sholl LM, Kris MG. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J Thorac Oncol. 2019; 14:1666–71. https://doi.org/10.1016/j.jtho.2019.06.009. [PubMed].
35. Moench S. Immunohistochemistry to Assess MET Status Is a Poor Screen for Actionable MET Alterations in Lung Adenocarcinoma. Cancer Therapy Advisor. 9 July 2019. https://www.cancertherapyadvisor.com/home/cancer-topics/lung-cancer/lung-cancer-met-status-immunohistochemistry-poor-screening.
36. DAkkS. Leitfaden des Sektorkomitees Pathologie/Neuropathologie für die Validierung von Untersuchungsverfahren in der Molekularpathologie: 71 SD 4037 | Revision: 1.1 2016. https://www.dakks.de/files/Dokumentensuche/Dateien/71%20SD%204%20037_Leitfaden%20Validierung%20MolPath_20161004_v1.1.pdf.