
Introduction
Dro1/Ccdc80 has been identified as a tumor suppressor of colorectal, thyroid, and ovarian cancer [1–3]. Dro1/Ccdc80 suppresses anchorage independent growth [2], inhibits migration of cancer cells [4] and induces sensitization to detachment-induced apoptosis [2]. Down-regulation of Dro1/Ccdc80 has been shown in primary human colorectal, thyroid, and melanoma skin cancer [1, 2, 4, 5]. In ApcMin/+ mice ubiquitous inactivation of Dro1/Ccdc80 results in early death, a significant increase in the colonic tumor load, and the regular formation of adenocarcinoma in the colon [1]. Loss of DRO1/CCDC80 increases multiplicity of preneoplastic aberrant crypt foci and colonic tumors in carcinogen-induced colon carcinogenesis and promotes formation of colon adenocarcinoma during inflammation-driven carcinogenesis [6]. In colon tumors from ApcMin/+ mice loss of DRO1/CCDC80 induces ERK1/2 phosphorylation and leads to c-MYC oncogene activation [1]. The Dro1/Ccdc80 tumor suppressor function has been reviewed in detail in [7].
Tumors are composed of two distinct components, the tumor cells themselves and the surrounding microenvironment, also called the tumor stroma. The tumor stroma comprises capillaries, activated fibroblasts, nerves, basement membrane, extracellular matrix, and immune cells [8, 9]. Epithelial tumor cells and stromal cells, both closely interwoven within the tumor, interact by a complex and multifaceted paracrine cross-talk, which plays an integral part in tumor initiation, growth, and progression [8–11]. The stromal compartment is known to stimulate tumor cell proliferation and mediate evasion of tumor cells from apoptosis, promote continuous angiogenesis, and contribute to the invasive and metastatic process [11, 12]. Moreover, tumor stromal cells convey drug sensitivity as well as therapeutic resistance [13] and provide prognostic and response-predictive information [14].
To date, the tumor suppressor role of Dro1/Ccdc80 in vivo has always been addressed by ubiquitous gene inactivation in mice [1, 3, 6]. For the study of tumorigenesis this approach implicates Dro1/Ccdc80 deficiency in both the tumor parenchyma and the tumor microenvironment. The aim of the present study was to investigate whether Dro1/Ccdc80’s tumor suppressive function is tumor-cell-autonomous.
Results
Colon tumor development is unaffected by epithelial Dro1/Ccdc80
Ubiquitous inactivation of Dro1/Ccdc80 in ApcMin/+ mice results in early death, a dramatic increase in colon tumor number, and formation of adenocarcinoma in the colon [1]. To investigate whether tumor suppression by Dro1/Ccdc80 is tumor-cell-autonomous, we inactivated Dro1/Ccdc80 specifically in the intestinal epithelium of ApcMin/+ mice. Therefore, ApcMin/+ mice carrying floxed Dro1/Ccdc80 alleles were crossed to mice expressing Cre recombinase under control of the intestinal epithelium specific Villin promoter [15] to generate Dro1fl/fl;VillinCre+;ApcMin/+ mice and Dro1fl/fl;ApcMin/+ control littermates. Correct recombination on the Dro1/Ccdc80 locus by Cre recombinase in the intestinal epithelium was verified by PCR analysis (Supplementary Figure 1A). Survival was similar in Dro1fl/fl;VillinCre+;ApcMin/+ mice and Dro1fl/fl;ApcMin/+ controls (Figure 1A). Also, we found no differences in small intestinal as well as colon tumor number (Figure 1B) and tumor histology (Figure 1C). In both genotypes ~90% of colon tumors were benign adenoma and ~10% had progressed to intramucosal adenocarcinoma (Supplementary Table 1).
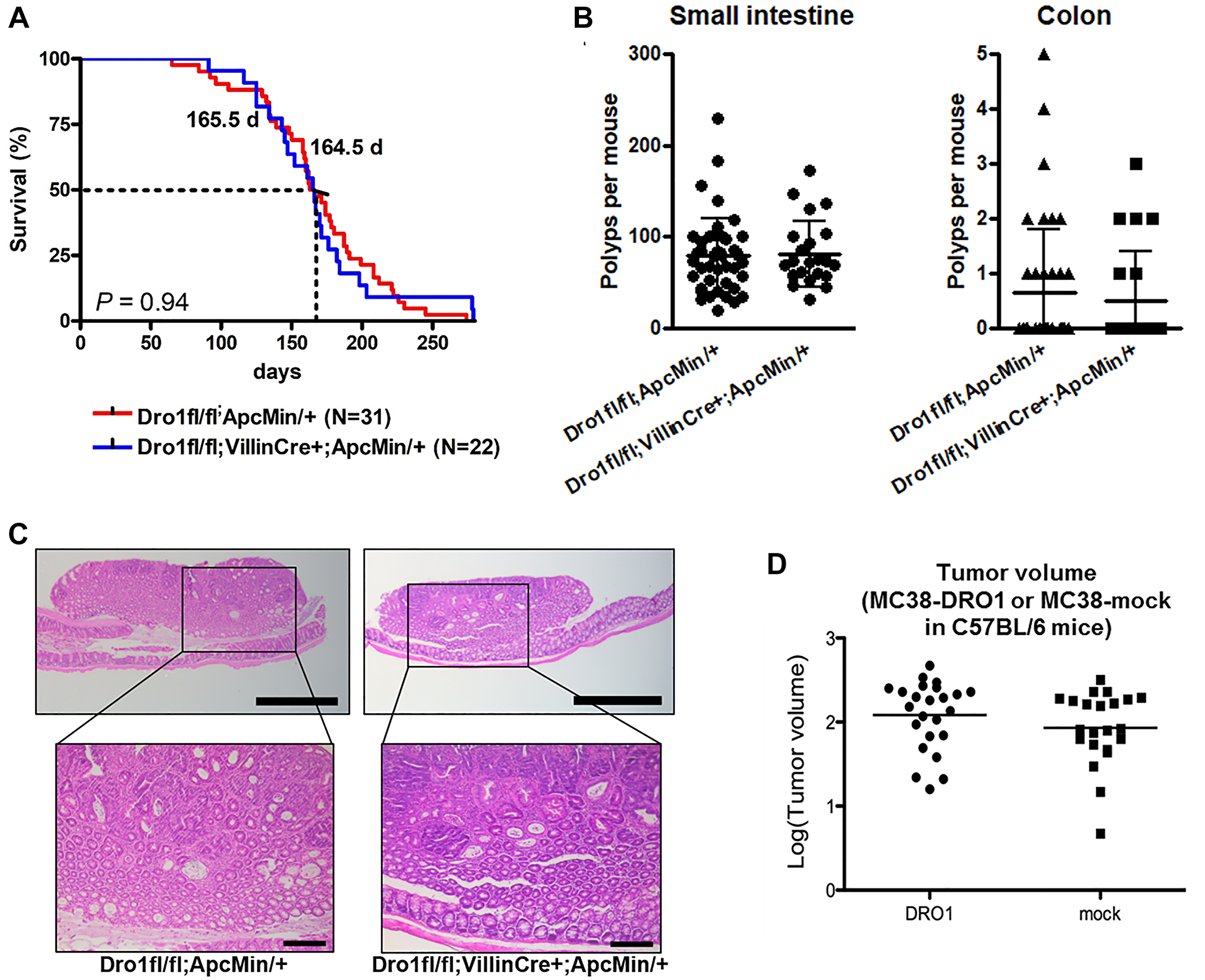
Figure 1: Colon tumor development is unaffected by epithelial Dro1/Ccdc80. (A) Survival of Dro1−/−;VillinCre+;ApcMin/+ (n = 22) and Dro1fl/fl;ApcMin/+ control (n = 31) mice. (B) Number of polyps per mouse in the small intestine and colon of moribund Dro1−/−;VillinCre+;ApcMin/+ (n = 22) and Dro1fl/fl;ApcMin/+ control (n = 42) mice. Error bars represent standard deviations. (C) Representative pictures of colon tumors from Dro1−/−;pVillinCre+;ApcMin/+ and ApcMin/+ control mice. H&E-staining. Scale bars, 500 μm and 200 μm. (D) Tumor volume of MC38-DRO1 and MC38-mock colorectal cancer cells subcutaneously injected into C57BL/6 mice (n = 24 tumors for MC38-DRO1 and n = 21 tumors for MC38-mock).
To further verify the initial observation of non-tumor-cell-autonomous tumor suppression by Dro1/Ccdc80, MC38 colorectal cancer cells reexpressing Dro1/Ccdc80 (MC38-DRO1) and MC38-mock cells were injected subcutaneously into C57BL/6 mice. Consistent with our observations in ApcMin/+ mice, growth (Figure 1D) and histology (Supplementary Figure 1B) of xenograft tumors were unaffected by Dro1/Ccdc80 expression in tumor cells.
Host Dro1/Ccdc80 suppresses growth of xenograft tumors
To study the effect of microenvironmental Dro1/Ccdc80 on colon tumorigenesis, we injected parental MC38 colorectal cancer cells subcutaneously into Dro1−/− and Dro1+/+ control mice. Growth of MC38 xenograft tumors was significantly increased in Dro1−/− mice compared to Dro1+/+ controls (Figure 2A). However, tumor incidence was unchanged by host Dro1/Ccdc80 inactivation (88% in Dro1−/− vs. 83% in Dro1+/+ mice). Similar results were obtained using B16 melanoma cells (Figure 2B; B16 tumor incidence: 100% in both Dro1−/− and Dro1+/+ mice). Moreover, when injecting B16 melanoma cells survival was dramatically reduced in the Dro1−/− group due to the fast and aggressive tumor growth (Figure 2C). Histopathological evaluation of MC38 and B16 xenograft tumors revealed no morphologic differences between tumors from Dro1−/− and Dro1+/+ control mice (Supplementary Figure 2A and 2B).
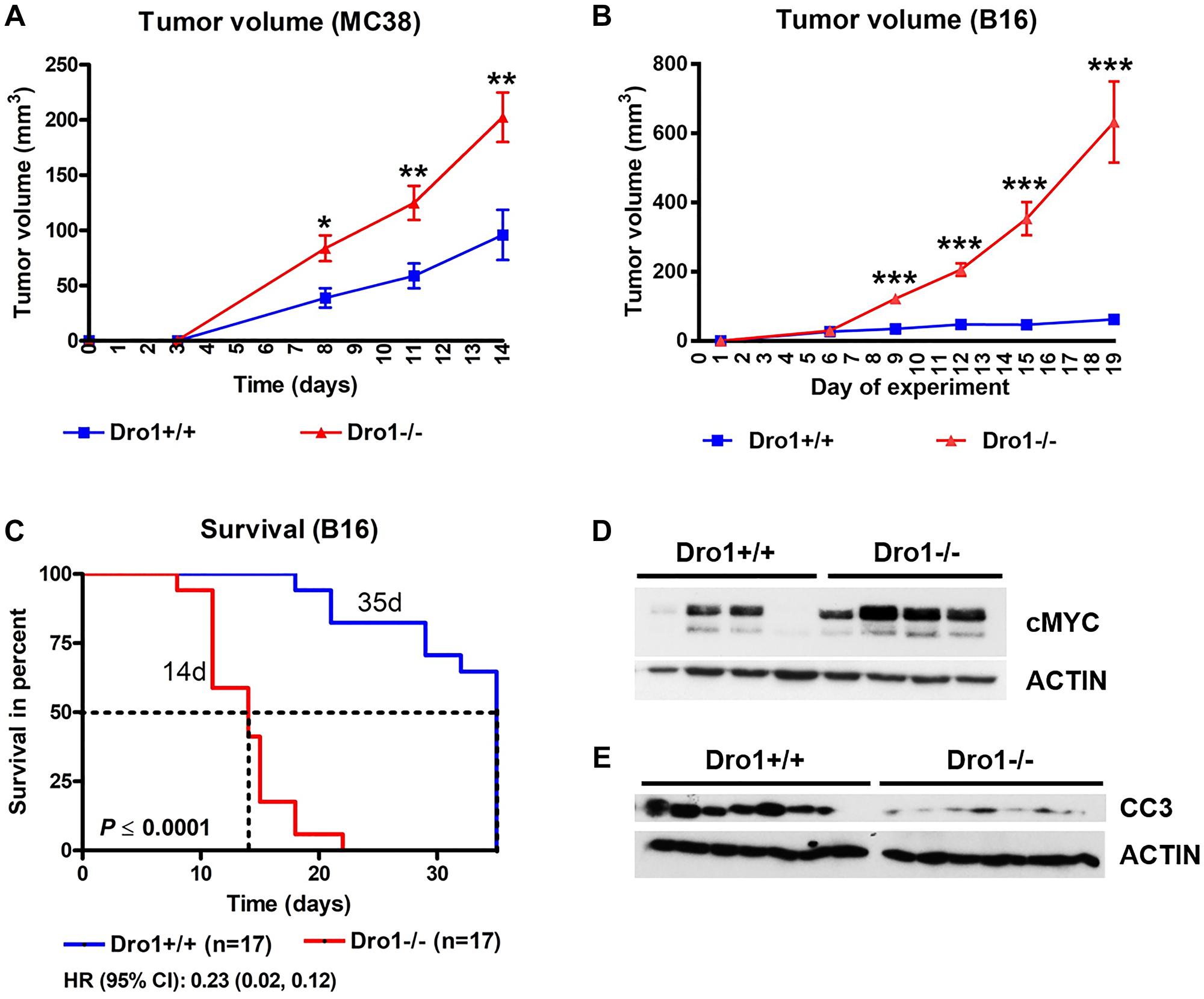
Figure 2: Host Dro1/Ccdc80 suppresses growth of xenograft tumors. (A) Tumor growth of parental MC38 colorectal cancer cells subcutaneously injected into Dro1-/- and Dro1+/+ control mice (n = 21 tumors from 12 Dro1−/− mice and n = 13 tumors from 8 Dro1+/+ control mice). (B) Tumor growth of B16 melanoma cells subcutaneously injected into Dro1−/− and Dro1+/+ control mice (n = 36 tumors from 18 Dro1−/− mice and n = 31 tumors from 18 Dro1+/+ control mice). (C) Survival of Dro1−/− and Dro1+/+ control mice after subcutaneous injection of B16 melanoma cells (n = 17/group). (D, E) Immunoblotting for indicated proteins on whole protein lysates from B16 xenograft tumors from Dro1−/− and Dro1+/+ control mice. Error bars represent standard deviations. *p < 0.05; **p < 0.01; ***p < 0.001.
Previously we have shown activation of both pERK1/2 and oncogenic c-MYC in colon tumors from ApcMin/+ mice upon ubiquitous inactivation of Dro1/Ccdc80 [1]. To investigate whether host DRO1/CCDC80 deficiency also impacts pERK1/2 and c-MYC status, immunoblot analysis of xenograft tumors was performed. No significant changes of pERK1/2 protein level were observed in B16 xenograft tumors upon microenvironmental DRO1/CCDC80 loss (Supplementary Figure 2C). However, consistent with our findings in ApcMin/+ mice, c-MYC oncogene was up-regulated in xenograft tumors from Dro1−/− mice compared to Dro1+/+ controls (Figure 2D). Moreover, depletion of host DRO1/CCDC80 resulted in reduced apoptosis in B16 tumors, as demonstrated by immunoblot analysis for cleaved-caspase-3 (Figure 2E).
Stromal DRO1/CCDC80 promotes apoptosis of cancer cells
Since loss of host DRO1/CCDC80 strongly decreased cleavage of caspase-3 in B16 xenograft tumors, we investigated the effect of Dro1/Ccdc80 inactivation in stromal cells on apoptosis of B16 cancer cells in vitro. Therefore, primary stromal cells (PSC) were generated from B16 xenograft tumors from Dro1−/− and Dro1+/+ control mice. B16 melanoma cells were treated with Dro1−/− and Dro1+/+ PSC conditioned medium (CM), respectively, and applied to UVB radiation for induction of apoptosis. Consistent with the in vivo findings, Dro1−/− PSC CM significantly reduced caspases-3/7 activity in B16 melanoma cells compared to Dro1+/+ control PSC CM (Figure 3A).
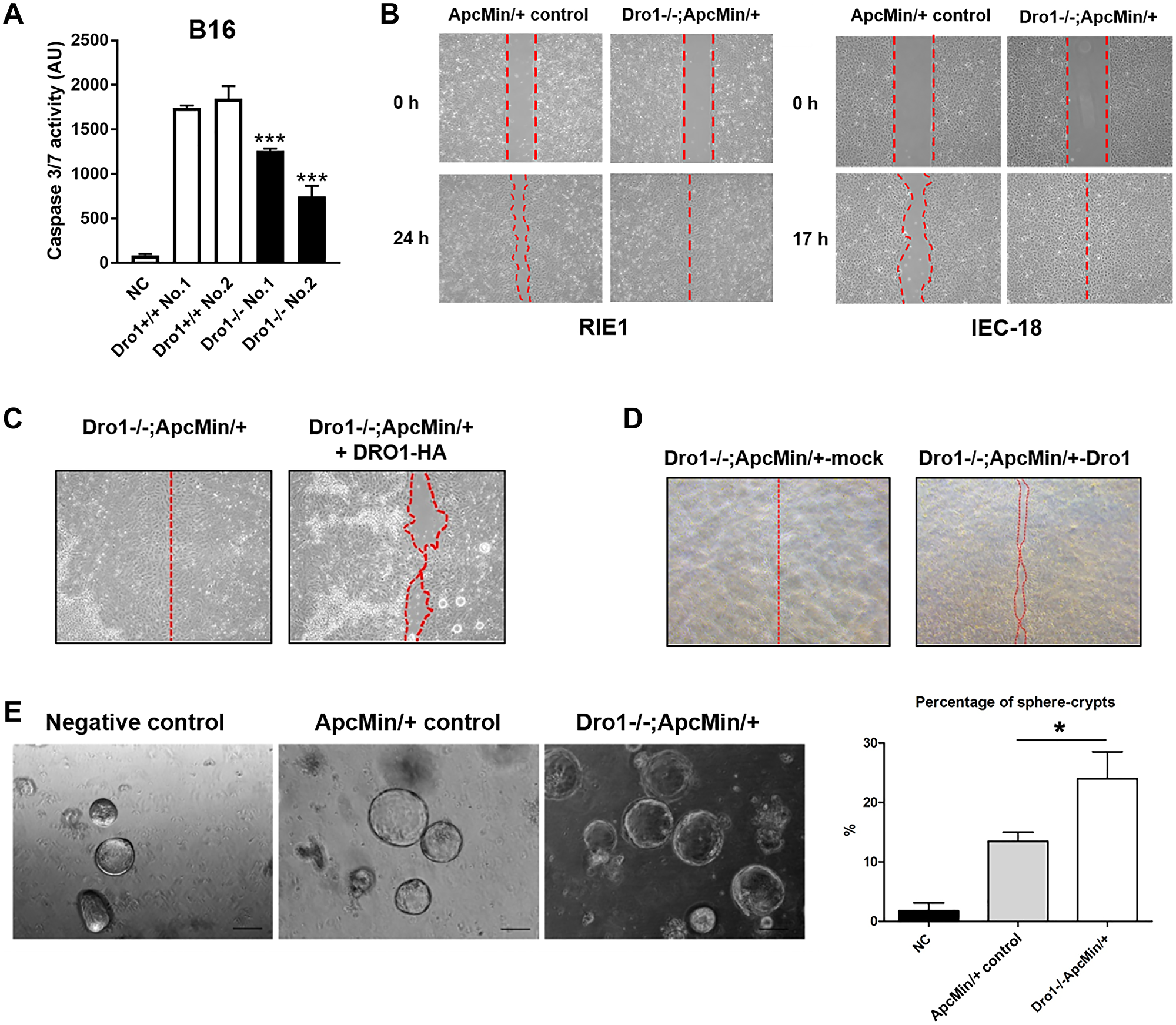
Figure 3: Inactivation of Dro1/Ccdc80 in stromal cells inhibits cancer cell apoptosis and promotes migration and sphere crypt formation of intestinal epithelial cells. (A) Caspase 3/7 activity in B16 melanoma cells 27 hours after induction of apoptosis by UVB radiation. B16 cells were treated with conditioned medium from primary stromal cells generated from B16 xenograft tumors from Dro1−/− and Dro1+/+ control mice. For negative control (NC) medium was used. Measurement was performed in triplicates. (B) Wound scratch assays. RIE1 and IEC-18 intestinal epithelial cells were treated with conditioned medium from primary stromal cells generated from the tumor-free colon of 5-week-old ApcMin/+ control and Dro1−/−;ApcMin/+ mice. (C) Wound scratch assay. Migration of RIE1 cells treated with conditioned medium from primary stromal cells generated from the tumor-free colon of 5-week-old Dro1−/−;ApcMin/+ mice. In the Dro1−/−;ApcMin/++DRO1-HA group the conditioned medium was supplemented with DRO1/CCDC80-HA. (D) Wound scratch assay. RIE1 cells were treated with conditioned medium from Dro1−/−;ApcMin/+-mock and Dro1−/−;ApcMin/+-DRO1 primary stromal cells. Dro1−/−;ApcMin/+ primary stromal cells were generated from the tumor-free colon of 5-week-old Dro1−/−;ApcMin/+ mice. (E) Sphere crypt assay. Small intestinal crypts were treated with conditioned medium from primary stromal cells generated from tumor-free colon of 5-week-old ApcMin/+ control and Dro1−/−;ApcMin/+ mice. Representative pictures of sphere crypts are shown (Scale bars, 100 μm). Percentage of sphere crypts was determined. Error bars represent standard deviations. *p < 0.05; ***p < 0.001.
Stromal DRO1/CCDC80 inhibits migration of intestinal epithelial cells
To study the effect of stromal Dro1/Ccdc80 on intestinal epithelial cells, PSC were generated from tumor-free colon from 5-week-old Dro1−/−;ApcMin/+ and ApcMin/+ control mice. Microscopic examination of PSC revealed no morphological differences upon DRO1/CCDC80 loss (data not shown).
Dro1−/−;ApcMin/+ PSC CM had no effect on cellular proliferation of both RIE1 intestinal epithelial and MC38 colorectal cancer cells compared to ApcMin/+ control CM (Supplementary Figure 3). Consistently, loss of DRO1/CCDC80 in PSC did not affect the growth of three-dimensional tumor spheroids generated by aggregation of MC38 colorectal cancer cells and PSC (Supplementary Figure 4A). Similar results were obtained using B16 melanoma cells (Supplementary Figure 4B).
Also, no effect of stromal Dro1/Ccdc80 on apoptosis of intestinal epithelial cells was observed as demonstrated by unchanged caspase-3/7 activity in RIE1 cells after treatment with PSC CM and UVB radiation (Supplementary Figure 5).
Dro1/Ccdc80 has been demonstrated to inhibit melanoma cancer cell migration [4]. To analyze the effect of stromal DRO1/CCDC80 on epithelial cell migration wound scratch assays were performed. Dro1−/−;ApcMin/+ PSC CM increased migration of both RIE1 and IEC18 intestinal epithelial cells compared to ApcMin/+ control CM (Figure 3B). To determine whether DRO1/CCDC80 protein inhibits migration of intestinal epithelial cells Dro1−/−;ApcMin/+ PSC CM was supplemented with DRO1/CCDC80 isolated from CM of SW480 Dro1/Ccdc80 over-expressing cells. Addition of DRO1/CCDC80 to the Dro1-/-;ApcMin/+ PSC CM inhibited migration of RIE1 cells compared to unmodified Dro1−/−;ApcMin/+ PSC CM (Figure 3C). Consistently, re-expression of Dro1/Ccdc80 in Dro1−/−;ApcMin/+ PSC resulted in reduced migration of RIE1 cells when treated with the PSC CM (Figure 3D). In contrast, Dro1−/−;ApcMin/+ PSC CM did not affect invasion of both RIE1 and MC38 cells into a basement coated membrane compared to ApcMin/+ control PSC CM (Supplementary Figure 6).
Ubiquitous inactivation of Dro1/Ccdc80 leads to increased phosphorylation of ERK1/2 and oncogenic c-MYC activation in colon tumors from ApcMin/+ mice [1]. Moreover, in the present study, we found loss of host DRO1/CCDC80 to increase c-MYC protein levels in xenograft tumors (Figure 2D). To investigate whether stromal inactivation of Dro1/Ccdc80 influences MAP kinase signaling and c-MYC levels in intestinal epithelial cells in vitro, RIE1 cells were treated with PSC CM and immunoblot analysis was performed. No significant differences in pERK1/2, ERK1/2, pP70S6K, P70S6K, and c-MYC protein levels were observed in RIE1 cells when treated with the CM from Dro1−/−;ApcMin/+ and ApcMin/+ control PSC, respectively (Supplementary Figure 7).
Stromal DRO1/CCDC80 affects formation of intestinal organoids
Epithelial loss of DRO1/CCDC80 had no effect on colon tumor development (Figure 1A–1D). To further investigate the role of epithelial Dro1/Ccdc80 in colon tumorigenesis, specifically in stemness, we established epithelial organoid cultures from colon tumors that were harvested from Dro1−/−;ApcMin/+ and ApcMin/+ control mice. In vitro-generated Dro1−/−;ApcMin/+ and ApcMin/+ control colon tumor organoids were growing as cystic structures (Supplementary Figure 8A). Diameter of Dro1−/−;ApcMin/+ colon tumor organoids was significantly decreased when compared to ApcMin/+ control organoids (207.2 ± 13.85 μm vs. 265.4 ± 17.88 μm; p = 0.0119; Supplementary Figure 8B).
Next we tested the effect of microenvironmental Dro1/Ccdc80 on the formation of intestinal organoids. Therefore, wildtype small intestinal crypts were treated with Dro1-/-;ApcMin/+ and ApcMin/+ PSC CM, respectively. The percentage of developing spheroides was significantly increased in the Dro1−/−;ApcMin/+ PSC group compared to the ApcMin/+ control group (Figure 3E).
Down-regulation of Dro1/Ccdc80 in the stromal tumor compartment
Dro1/Ccdc80 has been demonstrated to be expressed in various tissues including small intestine and colon [1, 2, 16]. Since former expression analyses utilized RNA isolated from whole small intestinal and colonic tissue samples, we now investigated expression of Dro1/Ccdc80 separately in the epithelial and stromal compartment. In small intestinal crypts from C57BL/6 mice and in scratched colon epithelium from ApcMin/+ mice Dro1/Ccdc80 expression was very low (Figure 4A and 4B). On the contrary, when the stromal compartment was studied, Dro1/Ccdc80 mRNA level was abundant in C57BL/6 small intestinal stromal cells, mouse embryonic fibroblasts, and murine gastric cancer associated fibroblasts (Figure 4A). Remarkably, in cancer associated fibroblasts Dro1/Ccdc80 expression was highly down-regulated compared to normal intestinal stromal cells (Figure 4A). Consistently, Dro1/Ccdc80 expression was strongly reduced in PSC isolated from colon tumors from moribund ApcMin/+ mice compared to PSC generated from tumor-free colon from 5-week-old ApcMin/+ mice (Figure 4B).
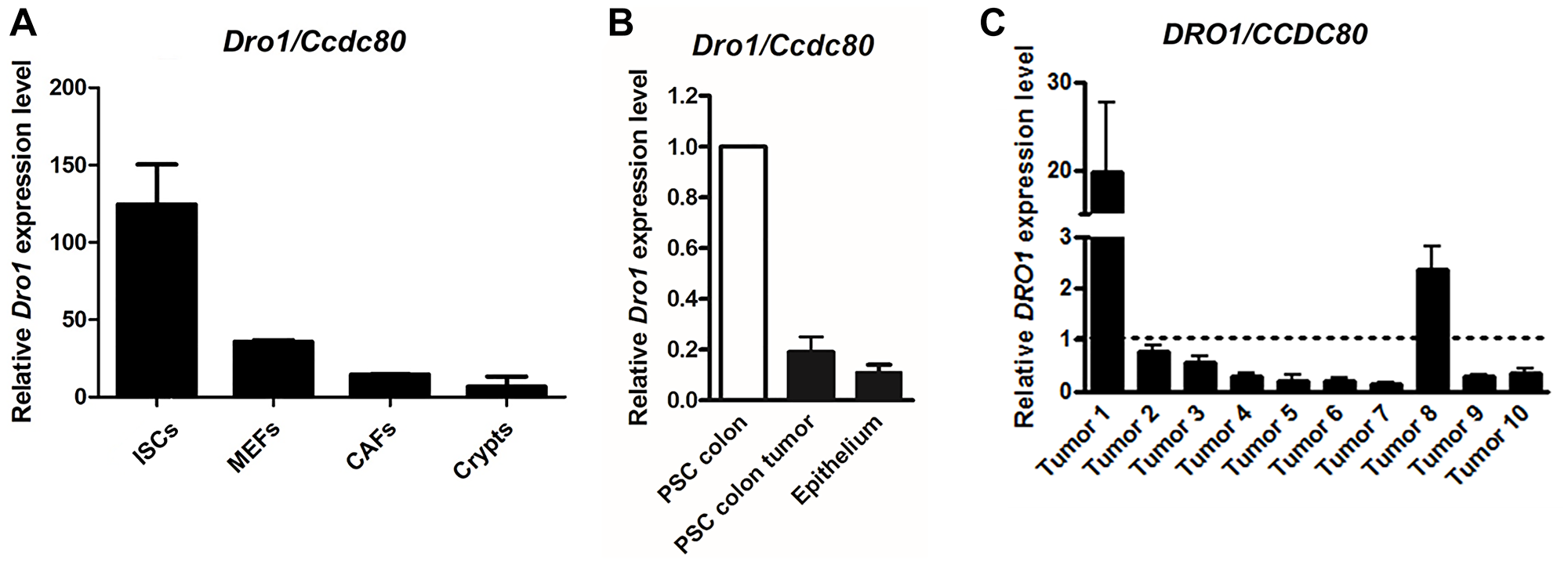
Figure 4: DRO1/CCDC80 is down-regulated in the stromal tumor compartment. (A) Relative Dro1/Ccdc80 mRNA expression in C57BL/6 mouse small intestinal stromal cells (ISCs), mouse embryonic fibroblasts (MEFs; day 18 p.c.), mouse gastric cancer associated fibroblasts (CAFs), and mouse small intestinal crypts (Crypts). (B) Relative Dro1/Ccdc80 mRNA expression in primary stromal cells generated from tumor-free colon from 5-week-old ApcMin/+ mice and from colon tumors from moribund ApcMin/+ mice and in scratched colon epithelium from 5-week-old ApcMin/+ mice. DRO1/CCDC80 expression in PSC from colon tumor and from epithelium is represented relative to expression in normal PSC (set to 1). (C) Relative DRO1/CCDC80 mRNA expression in microdissected human primary tumor stroma from colorectal carcinoma specimens compared to microdissected normal colorectal connective tissue. Matched pairs of tumor stroma and normal adjacent colorectal stroma from 10 patients were analyzed. DRO1/CCDC80 expression in colorectal carcinoma stroma is represented relative to expression in normal stroma (set to 1, see dotted line). Error bars represent standard deviations.
Previously, we have demonstrated DRO1/CCDC80 to be down-regulated in the majority of primary human colorectal carcinoma specimens [1, 2]. Since Dro1/Ccdc80 mRNA level was found to be reduced in murine cancer associated fibroblasts, we investigated expression of DRO1/CCDC80 in micro-dissected stroma from human primary colorectal cancer compared to expression in adjacent normal intestinal stroma. Microenvironmental down-regulation of DRO1/CCDC80 was found in 80% of human colorectal cancer specimens (Figure 4C).
DISCUSSION
In the present study we demonstrate that tumor suppression by Dro1/Ccdc80 is not tumor-cell-autonomous but is mediated by the tumor microenvironment. Colon tumor development in ApcMin/+ mice as well as formation of xenograft tumors was unaffected by Dro1/Ccdc80 expression in cancer cells, suggesting epithelial derived DRO1/CCDC80 to be dispensable for intestinal tissue homeostasis. In contrast, mutational inactivation of host Dro1/Ccdc80 highly promotes growth of xenograft tumors, suggesting a strong tumor suppressive role for microenvironmental Dro1/Ccdc80.
In B16 xenograft tumors inactivation of host Dro1/Ccdc80 resulted in dramatically reduced cleavage of caspase-3. Consistently, conditioned medium from Dro1/Ccdc80 deficient PSC from B16 xenograft tumors significantly inhibited caspase-3/7 activity in apoptosis induced B16 cancer cells. Thus, our data point to microenvironmental Dro1/Ccdc80 as a promoter of cellular apoptosis. Evasion from apoptosis is a prominent hallmark of cancer cells and fundamental for cancer development [17]. Previously, we have found Dro1/Ccdc80 to sensitize cancer cells to various apoptotic stimuli [2, 18]. In particular, DRO1/CCDC80 sensitizes cells to receptor-mediated apoptosis [2, 18]. Moreover, DRO1/CCDC80 significantly impairs cell survival under anchorage independent growth conditions by induction of detachment induced apoptosis (anoikis) [2]. In mammary carcinoma cells carrying an oncogenic activation of AIB1, inhibition of apoptosis has been shown to be at least partly mediated by repression of DRO1/CCDC80 expression [19]. Also, mouse embryonic fibroblasts generated from Dro1/Ccdc80 null mice are less sensitive to apoptotic stimuli with respect to mouse embryonic fibroblasts generated from wildtype mice [3].
In contrast to our findings in B16 melanoma cells, CM from ApcMin/+ Dro1/Ccdc80 knockout PSC did not affect activity of caspase-3/7 in apoptosis induced intestinal epithelial cells, suggesting no modulatory role for DRO1/CCDC80 in apoptosis in the intestinal epithelium. Consistently, we have found no effect of ubiquitous inactivation of Dro1/Ccdc80 on the rate of cellular apoptosis in the intestinal epithelium and in colon tumors from ApcMin/+ mice [1]. Moreover, we could observe no influence of ubiquitous DRO1/CCDC80 loss on the apoptotic rate in carcinogen-induced colonic neoplastic lesions [6]. Thus, Dro1/Ccdc80’s function in cellular apoptosis might be tissue dependent. Future studies are needed to further elucidate the role of Dro1/Ccdc80 in the apoptotic process during cancer development.
Activation of the c-MYC oncogene is a common molecular hallmark of many cancers contributing to both tumor initiation and progression [20]. We found depletion of host DRO1/CCDC80 to result in c-MYC oncogenic activation in xenograft tumors, suggesting microenvironmental loss of DRO1/CCDC80 to drive carcinogenesis by c-MYC. Consistently, we demonstrated ubiquitous inactivation of Dro1/Ccdc80 to implicate increased c-MYC protein levels in the colonic epithelium and in colon tumors from ApcMin/+ mice [1]. However, treatment of RIE1 cells with CM from Dro1/Ccdc80 knockout PSC had no impact on c-MYC protein level. In contrast to tumor tissue, RIE1 cells are non-tumorigenic intestinal epithelial cells [21]. Thus, the effect of DRO1/CCDC80 loss in stromal cells on c-MYC activation in epithelial cells might depend on second pro-tumorigenic mutational hits in epithelial cells. Similarly, despite Dro1/Ccdc80’s strong tumor suppressive function in ApcMin/+ mice as well as chemically-induced colon carcinogenesis [1, 6], ubiquitous loss of DRO1/CCDC80 alone was insufficient to induce spontaneous colonic tumor development in C57BL/6 mice [1].
Our results show inactivation of Dro1/Ccdc80 in stromal cells to stimulate migration of intestinal epithelial cells, indicating an important role for microenvironmental Dro1/Ccdc80 as regulator of epithelial cell motility. Previously, Dro1/Ccdc80 has also been shown to inhibit melanoma cancer cell migration [4]. Modulation of migratory properties of epithelial cells is implicated in many physiological processes, such as embryogenesis and wound healing, but also in cancer development [22]. During cancer progression, the tumor stroma has been shown to play a pivotal role in the control of migration, invasion, and finally metastasis of cancer cells by secretion of a plethora of molecules, including chemokines, cytokines, and growth factors [23]. Thus, loss of Dro1/Ccdc80 function in the tumor stroma might promote cancer progression by creating a permissive environment for cancer cell migration. Interestingly, we could observe no effect of stromal Dro1/Ccdc80 inactivation on invasive properties of intestinal epithelial cells, suggesting Dro1/Ccdc80 to control cell migration but not cell invasiveness.
Our data also indicate DRO1/CCDC80 to be an extracellular matrix (ECM) protein since the pro-migratory effect of Dro1/Ccdc80 loss in stromal cells on intestinal epithelial cells could be partly reversed by supplementation with DRO1/CCDC80 protein. A crucial role in cancer progression has been attributed to the ECM, which regulates cell behavior, tissue homeostasis, cell adhesion, and motility [22, 23]. Previously, DRO1/CCDC80 has been shown to be a secreted protein in various cell types [16, 24–26] and it has been identified as an ECM protein involved in matrix assembly and ECM molecule binding [25]. Moreover, DRO1/CCDC80 has been shown to be implicated in cell adhesiveness [25, 27]. Adhesion of chick lens cells and various tumor cell lines has been demonstrated to be mediated by DRO1/CCDC80 through heparin sulfate proteoglycan [27]. During chicken lens development extracellular DRO1/CCDC80 promotes lens cell adhesion and migration by activation of FGF signaling [27]. In contrast, we could observe no effect of Dro1/Ccdc80 loss in stromal cells on FGF signaling in intestinal epithelial cells as demonstrated by unchanged pERK1/2 levels. Our data support the hypothesis of DRO1/CCDC80 being an extracellular matrix protein that modifies epithelial cell migration, however, the molecular mechanisms by which DRO1/CCDC80 inhibits intestinal epithelial cell migration have to be elucidated.
Dro1/Ccdc80 inactivation did not improve growth of epithelial organoids derived from ApcMin/+ colon tumors, providing further evidence that the tumor suppressive function of DRO1/CCDC80 is not tumor-cell-autonomous but dictated by extrinsic cues provided by the local tissue microenvironment. In contrast, stromal Dro1/Ccdc80 inactivation increased sphere crypt forming ability of small intestinal organoids. Since intestinal organoids, known as mini-gut cultures, are maintained in vitro due to the presence of stem cells [28], we speculate that stromal cell-derived DRO1/CCDC80 may negatively regulate Lgr5+ stem cells, or Cnx43+ progenitors [29]. Since loss of stromal DRO1/CCDC80 facilitates migration of intestinal epithelial cells, stromal DRO1/CCDC80 might probably regulate transition from a stationary cancer stem cell into a migrating cancer stem cell during the multistep progression of colorectal cancer. Future studies are required to further investigate Dro1/Ccdc80’s role in cancer stemness.
We found Dro1/Ccdc80 to be significantly down-regulated in ApcMin/+ colon tumor primary stromal cells and in microdissected stroma from human colorectal cancer compared to normal, non-tumor stroma. Moreover, Dro1/Ccdc80 expression was reduced in gastric cancer associated fibroblasts with respect to normal intestinal stromal cells. Alterations in the tumor microenvironment are supposed to contribute to cancer initiation, progression, invasion, and metastasis [8–12]. Dramatic changes in gene expression patterns have been demonstrated for all cell types of the tumor microenvironment [30]. Several studies have shown genetic alterations in tumor-associated stromal cells, including somatic mutations in key tumor suppressor genes [31–39]. Frequent mutations in TP53 have been found in the stromal compartment of breast cancer and colorectal carcinoma [34–36]. PTEN has been demonstrated to be mutated in stromal cells of breast cancer [34] and somatic alterations in the GT198 tumor suppressor have been found in ovarian tumor stromal cells of various types of human ovarian cancer [40]. For breast and prostate cancer changes in microenvironmental gene expression patterns have also been shown to be at least partly due to distinct epigenetic modifications [39, 41, 42]. The data from the present study suggest microenvironmental loss of Dro1/Ccdc80’s tumor suppressive function to be an important event during carcinogenesis.
In summary, we identify Dro1/Ccdc80 as tumor suppressor in the tumor microenvironment. DRO1/CCDC80 in the stromal compartment strongly inhibited tumor growth, facilitated apoptosis in cancer cells, and reduced epithelial cell migration. Moreover, stromal DRO1/CCDC80 restrained the formation of epithelial organoids, indicating a possible role for Dro1/Ccdc80 in stemness. Our study provides new insights into the complex interaction between epithelial cells and their microenvironment and contributes to the understanding of cancer development. Future studies are needed to further elucidate the molecular mechanisms underlying Dro1/Ccdc80’s microenvironmental tumor suppressive function and to better characterize its role in human cancer.
Materials and Methods
Animals
Dro1−/− mice were generated as described previously [1]. Mice on the Dro1fl/fl background (in the following referred to as Dro1+/+ mice) were used as controls.
ApcMin/+ mice were purchased from the Jackson Laboratory and maintained on a C57BL/6J background. Mice were inspected on a daily basis and sacrificed when moribund.
Animals were housed under specific pathogen free conditions in a closed barrier system. All mice had access to water and to the same standard rodent diet (V1534, Ssniff, Soest, Germany) ad libitum. Experiments were carried out in accordance with the German Animal Welfare Act and with permission of the Government of Upper Bavaria (AZ55.2-1-54-2532-25-11 and AZ55.2-1-54-2532-48-2015).
PCR
Mice were genotyped by PCR analysis of genomic DNA from tail tip samples as described previously [1]. For purification of total RNA from microdissected FFPE tissue sections from human colorectal carcinoma specimens and from normal colon stroma, the RNeasy FFPE kit (Qiagen, Germany) was used. For quantitative RT-PCR RNA cleanup was performed using the RNeasy Mini kit (Qiagen, Germany). Genomic DNA was removed from the RNA preparation using DNase I, Amplification Grade (Invitrogen, USA). First-strand cDNA was generated using the SuperScript First Strand cDNA Synthesis System (Invitrogen, USA). Primer sequences for quantitative RT-PCR are displayed in Supplementary Table 2.
Tumor scoring and histology
Mice were sacrificed by cervical dislocation. The small intestine was rinsed with PBS, cut into 3 equal segments and each intestinal section was placed on a piece of filter paper, opened longitudinally, laid open and fixed in 4% buffered formaldehyde solution. Tumor number and their maximum diameter were determined under a dissecting microscope at 10× magnification. The colon and rectum were scored as “colon”. A quantity of small intestinal lesions and all colonic tumors sized ≥ 2 mm in diameter were resected including adjacent normal tissue, dehydrated and embedded in paraffin, 4 μm tissue sections cut in parallel with the mucosal surface and stained with hematoxylin and eosin (H&E). Histopathologic analysis of neoplastic lesions was performed in a blinded manner using standard criteria according to the classification of human adenomas of the colon and the assessment of the degree of dysplasia. The diagnosis intramucosal adenocarcinoma was made for lesions with high grade dysplasia/intraepithelial neoplasia (IEN) in combination with focal invasion of the lamina propria mucosae, cytologic features such as cribriform architecture with intraluminal accumulation of tumor and inflammatory cell debris (dirty necrosis) and desmoplastic stomal reaction. Adenocarcinomas invading through the lamina muscularis mucosae into the tela submucosa were classified as invasive adenocarcinoma [43]. Invasive adenocarcinoma and intramucosal adenocarcinoma were summarized under the diagnosis adenocarcinoma.
For xenograft experiments mice were sacrificed by cervical dislocation, the tumors excised, fixed in 4% buffered formaldehyde solution, and dehydrated and embedded in paraffin. 4 μm tissue sections were cut and stained with H&E. Histopathologic analysis of neoplastic lesions was performed in a blinded manner.
Xenograft experiments
Mice were injected subcutaneously with 106 tumor cells (MC38, B16) in 100 μl PBS. Each mouse received two injections (right and left flank). Mice were inspected every day and size of xenograft tumors was measured twice a week. Mice were sacrificed when one of the following endpoints was reached: tumor diameter ≥ 1.5 cm; penetration of tumor through the skin; moribund state. Tumors were dissected and the tumor weight determined.
Transfection of MC38 cells
MC38 cells were transfected with plasmids pCDNA3-DRO1-HA [2] and pCDNA3-mock for control. Transfection was performed as described before [44].
Immunoblotting
Protein lysates from mouse tissue samples were generated using M-PER mammalian extraction reagent (Thermo Scientific, USA) and separated by electrophoresis in discontinuous SDS-polyacrylamide gels. The following antibodies were used for immuno detection: pERK1/2 (Cell Signaling, USA), ERK1/2 (Cell Signaling, USA), c-MYC (Cell Signaling, USA), Cleaved-Caspase-3 (Cell Signaling, USA), pP70S6K (Cell Signaling, USA), P70S6K (Cell Signaling, USA), and anti-ACTIN (MP Biomedicals, USA).
Isolation of primary stromal cells
For generation of primary stromal cells from tumor-free whole colon samples, the colon from 5-week-old mice was cleaned from fecal debris and cut into 2–3 mm3 pieces and washed with Hank’s Balanced Salt Solution (HBSS, Life Technologies, USA). For generation of primary stromal cells from tumors, the tumors were excised, and cut into 2–3 mm3 pieces and washed with Hank’s Balanced Salt Solution (HBSS, Life Technologies, USA). The tissue pieces (colon/colon tumors) were incubated with 1 mM dithiothreitol (DTT, Sigma). After that, tissue pieces were incubated in 1 mM EDTA, followed by washing with HBSS. Then, incubation in 1 mM EDTA was repeated, and tissue pieces were washed with HBSS, followed by incubation in 1 mg/ml collagenase type I solution (Sigma). After washing in HBSS, tissue pieces were spun down and cultured in 10 cm Petri dishes in primary stromal cell medium (PSC medium) composed of: RPMI1640 (Life Technologies), 10% FBS, 1% penicillin/streptomycin (10,000 Units/ml penicillin; 10,000 μg/ml streptomycin; Life Technologies) and 100 μg/ml Normocin (Invivogen). Primary stromal cells migrated out of the tissue fragments and adhered to the plates.
In primary stromal cell cultures absence of epithelial cells was verified by immunofluorescence analysis for E-Cadherin. Primary stromal cells were also stained for α-SMA. For immunofluorescence primary stromal cells were grown on CELLview slides (Greiner Bio-One, Germany). Cells were fixed with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100 (Sigma). First antibodies were diluted 1: 100 for mouse-anti-E-Cadherin (BD) and 1: 200 for rabbit-anti-α-SMA (ACTA2, proteintech). Second antibodies were donkey anti-mouse IgG (H+L) highly cross-adsorbed antibody conjugated with Alexa Fluor 488 (Invitrogen, USA) for E-Cadherin and donkey anti-rabbit IgG antibody conjugated with Alexa Fluor 647 (Jackson ImmunoResearch) for α-SMA diluted 1: 1000. Slides were mounted using VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, USA). Fluorescence microscopy was performed on a Leica microscope.
Apoptosis assay (caspase-3/7 activity)
10.000 cells (RIE1, B16) were seeded per well of a 96-well-plate and incubated with conditioned medium from primary stromal cells for 24 h and subsequently applied to 450 mJ/cm2 UVB radiation [medisun HF-144 (Schulze & Böhm, Germany)] for induction of apoptosis. For negative control medium [DMEM (Sigma) containing 10% FCS and 1% penicillin/streptomycine] was used. After UVB radiation cells were again incubated with primary stromal cell conditioned medium or medium. 27 h after UVB radiation activity of caspase-3/7 was measured using the Apo-ONE Homogeneous caspase-3/7 assay (Promega, USA) according to the manufacturer’s manual. Experiments were performed in triplicates.
Proliferation assay
2.000 cells (RIE1, MC38) were seeded per well of a 96-well-plate and incubated with conditioned medium from primary stromal cells for the duration of the assay. Proliferation assays were performed using the Cell Proliferation ELISA, BrdU (colorimetric) (Sigma) according to the manufacturer’s manual. Experiments were performed in triplicates.
Generation of three-dimensional tumor spheroids
Tumor spheroids were generated by magnetic levitation [45] using the NanoShuttle™-PL system (Nano3D Biosciences, Inc., USA) according to the manufacturer’s manual. For labeling cells were incubated with NanoShuttle™-PL solution (1 μl/10.000 cells) in DMEM medium containing 0.5% FCS and 1% penicillin/streptomycin for 12 h. 125.000 labeled cancer cells (MC38, B16) and 125.000 labeled primary stromal cells per well were seeded in a 24-well cell-repellent surface plate (Greiner Bio-One, Germany). A levitating magnetic device (Nano3D Biosciences, Inc., USA) was put on top of the plate overnight. The next day, the levitating magnetic device was exchanged for a concentrating magnetic device (Nano3D Biosciences, Inc., USA) placed on the bottom of the plate and incubated overnight. The next day, the concentrating magnetic device was exchanged for the levitating magnetic device. Medium was exchanged twice a week using the concentrating magnetic device. Growth of spheroids was monitored every day under a light microscope. Pictures were taken with a Leica microscope (Type 11090137002). Size of spheroids was calculated from two-dimensional pictures using the Leica Application Suite V4.5 software. Experiments were performed in duplicates.
Invasion assay
Invasion assays were performed by co-cultivation in a Boyden chamber using CultreCoat Medium BME Cell Invasion Assay (R&D Systems, USA) according to the manufacturer’s manual. 25.000 epithelial cells (RIE1, MC38) per well were seeded in the upper, basement coated chamber. 15.000 primary stromal cells were seeded in the lower chamber. Experiments were performed in 8 replicates.
Wound scratch assay
Migration assays were performed in cell inserts having two separate wells divided by an insert (ibidi). After removal of the insert a defined ~500 μm cell free gap is left behind as migration area. For cultivation cell inserts were placed in 35 mm μ-dishes (ibidi). On day one 40.000 epithelial cells (RIE1, IEC-18) were seeded in DEMEM (Sigma) containing 10% FCS and 1% penicillin/streptomycine in each well of the two-well insert. On day two cultivation medium was changed to DEMEM (Sigma) containing 0.5% FCS and 1% penicillin/streptomycin. On day three cultivation medium was exchanged for conditioned medium of primary stromal cells and the insert was removed for start of cell migration. Pictures were taken at the time of removal of the insert (defined as 0 h of migration assay) and 17–24 h after removal of the insert with a Leica microscope (Type 11090137002). Experiments were performed in duplicates.
Purification of DRO1/CCDC80
DRO1/CCDC80 was purified from conditioned medium of SW480-Dro1-HA overexpressing cells [2]. For control experiments conditioned medium from SW480-mock cells was used [2]. SW480-Dro1-HA and SW480-mock cells were grown confluent in 10 cm cell culture dishes in DMEM (Sigma) culture medium containing 10% FCS and 1% penicillin/streptomycine. DRO1/CCDC80-HA expression in SW480 cells was induced by addition of Doxycycline (10 μg/ml) to the medium for 18 h. Anti-HA antibody (1: 16; Abcam) was preincubated with 50 μl Sepharose beads (Invitrogen, USA) in 250 μl PBS for 1 h and subsequently mixed with the conditioned medium. After washing with PBS a pH-shift was induced by addition of 0.1 M glycine (pH 3.0). Neutralisation was induced by treatment with 1M Tris (pH 9.0). pH-shift and neutralization was repeated to increase DRO1/CCDC80 yield. For migration experiments DRO1/CCDC80 isolated from 10 ml SW480-DRO1-HA conditioned medium was divided equally between two migration culture plates.
Colon tumor organoids
Colon tumors were harvested, washed with ice-cold Hank’s Balanced Salt Solution (Life Technologies) and cut into small pieces. Tissue fragments were washed with the washing solution: 10% FBS (Life Technologies) in PBS (Life Technologies); followed by incubation with 2 mM EDTA. Tissue fragments were firstly washed with the washing solution and then with the crypt basal medium (CBM; Supplementary Table 3). After that, tissue fragments were incubated with collagenase type I (1 mg/ml), washed with the washing solution, pelleted, mixed with Matrigel (BD Biosciences) and seeded in a 24-well plate. Just after the isolation, tumor crypts were cultured in a medium composed of 12.5% Wnt conditioned medium and 87.5% crypt complete medium (CCM; Supplementary Table 4), afterwards CCM was used for the culture. Wnt conditioned medium was collected from the supernatant of cultured L-Wnt3a cell line.
Sphere crypt assay
Small intestinal crypts were isolated as previously described [28, 46]. Briefly, the small intestine was harvested from a wildtype mouse and cut into small pieces. After several washing steps, tissue fragments were incubated with 2 mM EDTA and then passed through cell strainer 70 μm. The flow-through fraction, which contained crypts, was pelleted and then Matrigel (BD Biosciences) was added. Crypts resuspended in Matrigel were plated in a 24-well plate and cultured in CCM medium (Supplementary Table 4). For the sphere crypt assay, on day 0 crypts were seeded in a new 24-well plate and incubated with conditioned medium derived from colonic primary stromal cells. On day 1, sphere crypts were quantified. Sphere crypts were defined as non-budding, round crypts composed of a thin epithelial layer. For each experimental group at least 1000 organoids were quantified.
Cell line origin and authentication
Cell lines were obtained from Thermo Fischer Scientific (MC38, 1/2014), ATCC (IEC-18, 3/2014) and DSMZ (B16, 1/2014). Cell lines were tested and authenticated by generation of DNA profiles of eight highly polymorphic locations of Short Tandem Repeats (STRs) using a human nonaplex PCR system. In addition, the samples were tested for the presence of mitochondrial DNA sequences from rat, Syrien and Chinese Hamster. Finally, the samples were subjected to DNA Barcoding in order to identify the animal species. For mouse strain specificity murine STR technology was applied. Cell line authentication was performed by Leibniz-Institut DSMZ GmbH, Germany.
Statistical analysis
To display the time to tumor mortality Kaplan-Meier survival curves were used and logrank statistics was employed to test for differences between genotype groups. To analyze significance of differences, two-tailed Student’s t-test or two-tailed Mann Whitney U test were performed (GraphPad Prism). Multiple comparisons were performed using 1-way ANOVA (GraphPad Prism). P values < 0.05 were considered to be statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This research was supported by a grant of the German Research Foundation (GR4873/1-1) to JIC and a grant of the Wilhelm Sander-Stiftung (2013.084.1) to MRS and FTK. AP received the Leonhard-Lorenz-Stiftung Award (Technical University of Munich). The authors gratefully acknowledge Ingrid Renner-Müller and Petra Renner for help with animal care. Special thanks to Anna Ronja Binder for help with PSC isolation.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Grill JI, Neumann J, Herbst A, Hiltwein F, Ofner A, Marschall MK, Wolf E, Kirchner T, Göke B, Schneider MR, Kolligs FT. DRO1 inactivation drives colorectal carcinogenesis in ApcMin/+ mice. Mol Cancer Res. 2014; 12:1655–62. https://doi.org/10.1158/1541-7786.MCR-14-0205-T. [PubMed].
2. Bommer GT, Jäger C, Dürr EM, Baehs S, Eichhorst ST, Brabletz T, Hu G, Fröhlich T, Arnold G, Kress DC, Göke B, Fearon ER, Kolligs FT. DRO1, a gene down-regulated by oncogenes, mediates growth inhibition in colon and pancreatic cancer cells. J Biol Chem. 2005; 280:7962–75. https://doi.org/10.1074/jbc.M412593200. [PubMed].
3. Leone V, Ferraro A, Schepis F, Federico A, Sepe R, Arra C, Langella C, Palma G, De Lorenzo C, Troncone G, Masciullo V, Scambia G, Fusco A, Pallante P. The cl2/dro1/ccdc80 null mice develop thyroid and ovarian neoplasias. Cancer Lett. 2015; 357:535–41. https://doi.org/10.1016/j.canlet.2014.12.010. [PubMed].
4. Pei G, Lan Y, Lu W, Ji L, Hua ZC. The function of FAK/CCDC80/E-cadherin pathway in the regulation of B16F10 cell migration. Oncol Lett. 2018; 16:4761–67. https://doi.org/10.3892/ol.2018.9159. [PubMed].
5. Visconti R, Schepis F, Iuliano R, Pierantoni GM, Zhang L, Carlomagno F, Battaglia C, Martelli ML, Trapasso F, Santoro M, Fusco A. Cloning and molecular characterization of a novel gene strongly induced by the adenovirus E1A gene in rat thyroid cells. Oncogene. 2003; 22:1087–97. https://doi.org/10.1038/sj.onc.1206194. [PubMed].
6. Grill JI, Neumann J, Ofner A, Marschall MK, Zierahn H, Herbst A, Wolf E, Kolligs FT. Dro1/Ccdc80 inactivation promotes AOM/DSS-induced colorectal carcinogenesis and aggravates colitis by DSS in mice. Carcinogenesis. 2018; 39:1176–84. https://doi.org/10.1093/carcin/bgy077. [PubMed].
7. Grill JI, Kolligs FT. DRO1/CCDC80: a Novel Tumor Suppressor of Colorectal Carcinogenesis. Curr Colorectal Cancer Rep. 2015; 11:200–08. https://doi.org/10.1007/s11888-015-0276-3.
8. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016; 16:582–98. https://doi.org/10.1038/nrc.2016.73. [PubMed].
9. Quante M, Varga J, Wang TC, Greten FR. The gastrointestinal tumor microenvironment. Gastroenterology. 2013; 145:63–78. https://doi.org/10.1053/j.gastro.2013.03.052. [PubMed].
10. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006; 6:392–401. https://doi.org/10.1038/nrc1877. [PubMed].
11. Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev. 2009; 19:67–73. https://doi.org/10.1016/j.gde.2009.01.003. [PubMed].
12. Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010; 316:1324–31. https://doi.org/10.1016/j.yexcr.2010.02.045. [PubMed].
13. Ostman A. The tumor microenvironment controls drug sensitivity. Nat Med. 2012; 18:1332–34. https://doi.org/10.1038/nm.2938. [PubMed].
14. Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014; 25:61–68. https://doi.org/10.1016/j.semcancer.2014.02.006. [PubMed].
15. Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002; 277:33275–83. https://doi.org/10.1074/jbc.M204935200. [PubMed].
16. Tremblay F, Revett T, Huard C, Zhang Y, Tobin JF, Martinez RV, Gimeno RE. Bidirectional modulation of adipogenesis by the secreted protein Ccdc80/DRO1/URB. J Biol Chem. 2009; 284:8136–47. https://doi.org/10.1074/jbc.M809535200. [PubMed].
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013. [PubMed].
18. Herbst A, Bayer C, Wypior C, Kolligs FT. DRO1 sensitizes colorectal cancer cells to receptor-mediated apoptosis. Oncol Lett. 2011; 2:981–84. https://doi.org/10.3892/ol.2011.356. [PubMed].
19. Ferragud J, Avivar-Valderas A, Pla A, De Las Rivas J, Font de Mora J. Transcriptional repression of the tumor suppressor DRO1 by AIB1. FEBS Lett. 2011; 585:3041–46. https://doi.org/10.1016/j.febslet.2011.08.025. [PubMed].
20. Dang CV. MYC on the path to cancer. Cell. 2012; 149:22–35. https://doi.org/10.1016/j.cell.2012.03.003. [PubMed].
21. Blay J, Brown KD. Characterization of an epithelioid cell line derived from rat small intestine: demonstration of cytokeratin filaments. Cell Biol Int Rep. 1984; 8:551–60. [PubMed].
22. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003; 3:362–74. https://doi.org/10.1038/nrc1075. [PubMed].
23. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009; 9:239–52. https://doi.org/10.1038/nrc2618. [PubMed].
24. Song X, Tanaka H, Ohta K. Multiple roles of Equarin during lens development. Dev Growth Differ. 2014; 56:199–205. https://doi.org/10.1111/dgd.12121. [PubMed].
25. Manabe R, Tsutsui K, Yamada T, Kimura M, Nakano I, Shimono C, Sanzen N, Furutani Y, Fukuda T, Oguri Y, Shimamoto K, Kiyozumi D, Sato Y, et al. Transcriptome-based systematic identification of extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008; 105:12849–54. https://doi.org/10.1073/pnas.0803640105. [PubMed].
26. Okada T, Nishizawa H, Kurata A, Tamba S, Sonoda M, Yasui A, Kuroda Y, Hibuse T, Maeda N, Kihara S, Hadama T, Tobita K, Akamatsu S, et al. URB is abundantly expressed in adipose tissue and dysregulated in obesity. Biochem Biophys Res Commun. 2008; 367:370–76. https://doi.org/10.1016/j.bbrc.2007.12.164. [PubMed].
27. Song X, Sato Y, Sekiguchi K, Tanaka H, Ohta K. Equarin is involved in cell adhesion by means of heparan sulfate proteoglycan during lens development. Dev Dyn. 2013; 242:23–29. https://doi.org/10.1002/dvdy.23902. [PubMed].
28. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009; 459:262–65. https://doi.org/10.1038/nature07935. [PubMed].
29. Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F, Monteyne D, Pérez-Morga D, Vassart G, Garcia MI. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 2013; 5:421–32. https://doi.org/10.1016/j.celrep.2013.09.005. [PubMed].
30. Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004; 6:17–32. https://doi.org/10.1016/j.ccr.2004.06.010. [PubMed].
31. Lakhani SR, Chaggar R, Davies S, Jones C, Collins N, Odel C, Stratton MR, O'Hare MJ. Genetic alterations in 'normal' luminal and myoepithelial cells of the breast. J Pathol. 1999; 189:496–503. https://doi.org/10.1002/(SICI)1096-9896(199912)189:4<496::AID-PATH485>3.0.CO;2-D. [PubMed].
32. Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000; 60:2562–66. [PubMed].
33. Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. JAMA. 2007; 297:2103–11. https://doi.org/10.1001/jama.297.19.2103. [PubMed].
34. Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002; 32:355–57. https://doi.org/10.1038/ng1013. [PubMed].
35. Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, Eng C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007; 357:2543–51. https://doi.org/10.1056/NEJMoa071825. [PubMed].
36. Wernert N, Löcherbach C, Wellmann A, Behrens P, Hügel A. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001; 21:2259–64. [PubMed].
37. Weber F, Xu Y, Zhang L, Patocs A, Shen L, Platzer P, Eng C. Microenvironmental genomic alterations and clinicopathological behavior in head and neck squamous cell carcinoma. JAMA. 2007; 297:187–95. https://doi.org/10.1001/jama.297.2.187. [PubMed].
38. Moinfar F, Beham A, Friedrich G, Deutsch A, Hrzenjak A, Luschin G, Tavassoli FA. Macro-environment of breast carcinoma: frequent genetic alterations in the normal appearing skins of patients with breast cancer. Mod Pathol. 2008; 21:639–46. https://doi.org/10.1038/modpathol.2008.28. [PubMed].
39. Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005; 37:899–905. https://doi.org/10.1038/ng1596. [PubMed].
40. Peng M, Zhang H, Jaafar L, Risinger JI, Huang S, Mivechi NF, Ko L. Human ovarian cancer stroma contains luteinized theca cells harboring tumor suppressor gene GT198 mutations. J Biol Chem. 2013; 288:33387–97. https://doi.org/10.1074/jbc.M113.485581. [PubMed].
41. Fiegl H, Millinger S, Goebel G, Müller-Holzner E, Marth C, Laird PW, Widschwendter M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006; 66:29–33. https://doi.org/10.1158/0008-5472.CAN-05-2508. [PubMed].
42. Hanson JA, Gillespie JW, Grover A, Tangrea MA, Chuaqui RF, Emmert-Buck MR, Tangrea JA, Libutti SK, Linehan WM, Woodson KG. Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst. 2006; 98:255–61. https://doi.org/10.1093/jnci/djj051. [PubMed].
43. Washington MK, Powell AE, Sullivan R, Sundberg JP, Wright N, Coffey RJ, Dove WF. Pathology of rodent models of intestinal cancer: progress report and recommendations. Gastroenterology. 2013; 144:705–17. https://doi.org/10.1053/j.gastro.2013.01.067. [PubMed].
44. Grill JI, Neumann J, Herbst A, Ofner A, Hiltwein F, Marschall MK, Zierahn H, Wolf E, Schneider MR, Kolligs FT. Loss of DRO1/CCDC80 results in obesity and promotes adipocyte differentiation. Mol Cell Endocrinol. 2017; 439:286–96. https://doi.org/10.1016/j.mce.2016.09.014. [PubMed].
45. Haisler WL, Timm DM, Gage JA, Tseng H, Killian TC, Souza GR. Three-dimensional cell culturing by magnetic levitation. Nat Protoc. 2013; 8:1940–49. https://doi.org/10.1038/nprot.2013.125. [PubMed].
46. Pastula A, Quante M. Isolation and 3-dimensional Culture of Primary Murine Intestinal Epithelial Cells. Bio Protoc. 2014; 4:e1125. https://doi.org/10.21769/BioProtoc.1125.