
Introduction
A major challenge to overcome in cancer is to understand how cells adapt to survive to therapy. The classical Darwinian mechanism of resistance, in which random mutations permit natural selection, plays a role in cancer resistance, but is far from the only process involved. Mechanisms of tolerance generated through dynamics in cellular processes can also support therapy evasion. Therefore, the increased knowledge about the dynamics in several phenotypes in cancer cells in the last years has opened our eyes on the degree of these dynamics and their potential role in therapeutic resistance [1, 2]. Such fluctuation together with the occurrence of different level of expression memory along generations [3, 4] can contribute to heterogeneity found among related cells, and indicate that the heterogeneity of a tumor goes far beyond mutations.
So far, most studies of dynamics have focused on signaling pathways, expression of specific genes and modulation of cellular processes [5, 6], however, the most important omniphenotype in cancer is the number of live descendant of a cancer cell after a defined time, i.e., its fitness. Several studies have shown the improve on cell fitness due to phenotypic changes [7, 8], however the focus is usually populational and not on the dynamics of single cells. A very remarkable example is the impact of the flower isoforms on fitness. This transmembrane protein has four isoforms, two of them are related with a winner phenotype and the other two with a loser phenotype, thus being considered a ‘fitness fingerprint’ [9, 10]. The outcome depends on the expression of neighboring cells and other proteins involved in signaling [9, 11, 12]. Even though it is known that the flower protein expression is important to tumor growth and metastasis [9, 13], it is not known if the expression of the flower gene is stable or fluctuates over time in single cells. In this regard, the question remains whether a winner cell will produce descendants that are all winners, or will the formed clone be a mix of winner and looser cells in a similar proportion of the population of non-related cells?
The major challenge to measure the dynamic of fitness at single cells is that when a cell dies, the ability to get information about its future is lost, and when a cell divides, the cell is not the same anymore. To overcome this methodological barrier and address the dynamics in cancer cell fitness, we created the Dynamic Fitness Analysis (DynaFit) method [14]. The rationale of DynaFit is that if fitness does not change during the formation of colonies, the fitness level of the founding cell will be phenotocopied to all cells in the colony and therefore the comparison between colonies will reveal a variance in fitness similar to the variance of their founding cells. However, if fitness changes over time or with cell division, cells with diverse fitness levels will produce colonies that will be formed from high and low fitness cells and therefore these colonies will have a behavior much closer to the average fitness and therefore a variance in fitness among colonies much lower than the variance of their founding cells (Figure 1). As DynaFit takes into account the size of the colony, one can estimate the degree of dynamics in fitness.
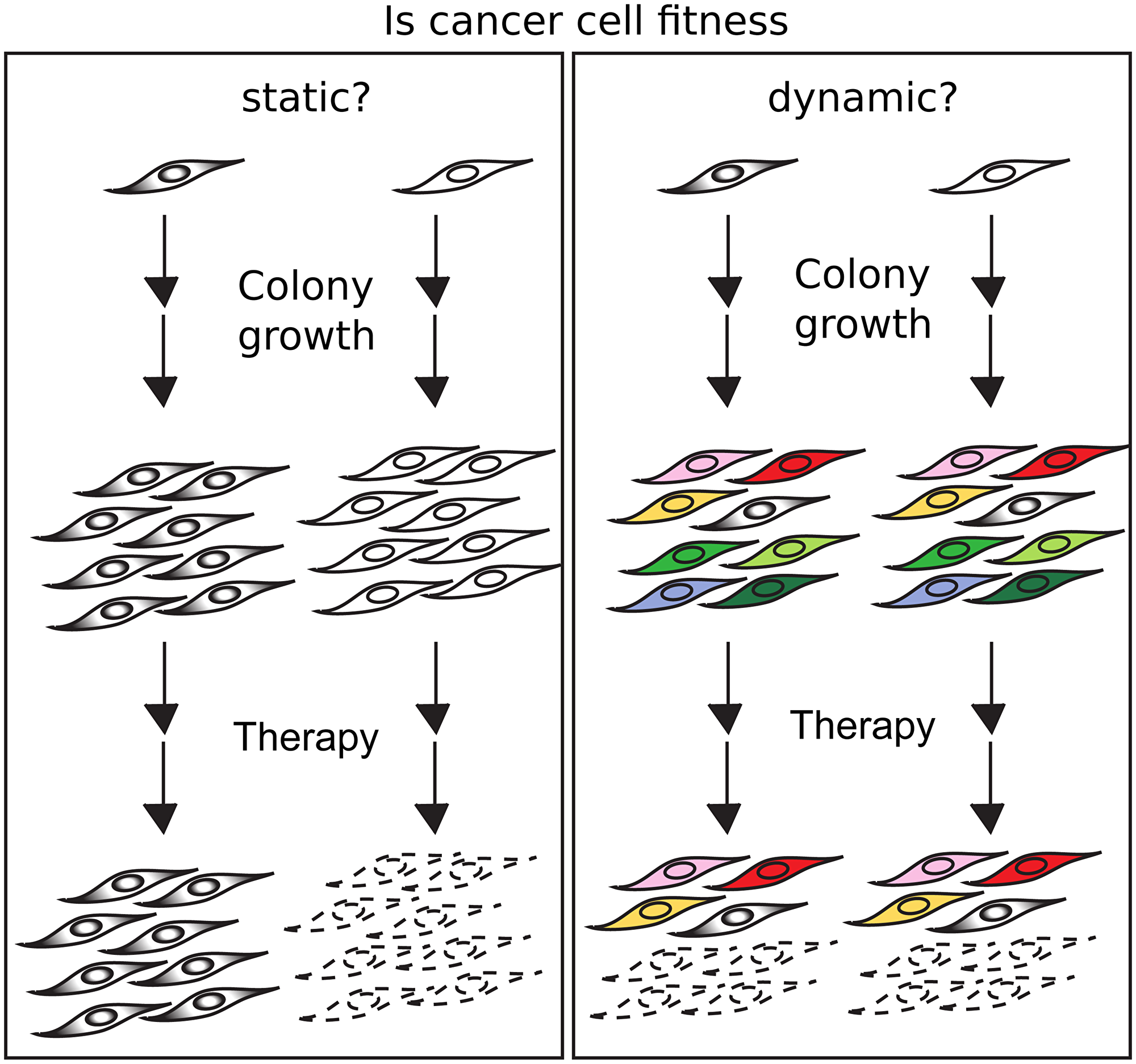
Figure 1: If fitness is conserved, cells with high and low fitness will generate colonies which will be resistant or sensitive to a therapeutic agent (left). If fitness changes with time and/or division, the colonies formed from high and low fitness cells will be similar, in which both colonies will undergo fractional killings in the presence of a therapeutic agent (right).
The results obtained with DynaFit clearly show that normal and cancer cells change their fitness after only a few generations both under normal growth condition or in the presence of chemotherapeutic agents [14]. This indicates that tolerance to this agents fluctuates and may thus contribute for the high rate of failure of treatment of tumors such as glioblastoma.
The origins of phenotypic fluctuations are diverse and far from understood. We observed that sister cells already have different levels of MAPK signaling levels and that cell cycle desynchronization and the induction of apoptosis and senescence becomes different among cells in colonies after a few divisions [14]. Others have also shown the contribution of cell cycle-dependent fluctuations [15], transcriptional noise [16, 17] and epigenetic modulation [18] on the variation of cells. Tolerance is a dynamic phenomenon caused by epigenetic reprograming [19, 20] which can either remain dynamic or be fixed in population through genetic assimilation [21] or genetic resistance [22]. We hypothesized that alterations in epigenetic pathways through epigenetic inhibitors could alter the dynamic of fitness. Indeed, cells treated with both histone deacetylase inhibitor and DNA methyltransferase inhibitor maintain a variance among colonies similar to the observed in single cells. Besides that, fractional killing was reduced in bigger colonies when they were treated with epigenetic inhibitors along with TMZ compared with colonies treated only with TMZ, indicating that when epigenetic mechanisms are blocked the sensitive phenotype is phenocopied through generations [14].
The strategy to freeze the level of tolerance of cells has the drawback of fixing both the good and the bad. The strategy of evolutionary trap could be employed to deal with cells with a stabilized tolerant state to a given drug [23]. In that case, the pressure of a drug will positively select a trail that confers an advantage, but also produce fitness trade-offs, that can be explored with a second therapeutic intervention. This concept has been demonstrated in relation to the aneuploid phenotype [24] and the antagonistic pleotropy between bromodomain and BCL-2 inhibitors in acute myeloid leukemia [25]. However, the practical applicability of the rationale of evolutionary trap in a therapeutic setting is still in the distant future.
The incorporation of dynamic processes in our knowledge of cancer biology is fundamental for basic and applied cancer research. Integrating Darwinian and Lamarckian evolutionary concepts in cancer cell adaptation to therapeutic pressure is key for moving forward in the comprehension on how cancer cells adapt to the challenges of therapy and is the only way to improve therapies available.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011; 146:633–44. https://doi.org/10.1016/j.cell.2011.07.026. [PubMed].
2. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Pätzold S, Villanueva J, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013; 23:811–25. https://doi.org/10.1016/j.ccr.2013.05.003. [PubMed].
3. Shaffer SM, Emert BL, Reyes Hueros RA, Cote C, Harmange G, Schaff DL, Sizemore AE, Gupte R, Torre E, Singh A, Bassett DS, Raj A. Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors. Cell. 2020; 182:947–59.e17. https://doi.org/10.1016/j.cell.2020.07.003. [PubMed].
4. Phillips NE, Mandic A, Omidi S, Naef F, Suter DM. Memory and relatedness of transcriptional activity in mammalian cell lineages. Nat Commun. 2019; 10:1208. https://doi.org/10.1038/s41467-019-09189-8. [PubMed].
5. Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016; 165:631–42. https://doi.org/10.1016/j.cell.2016.03.025. [PubMed].
6. Filippi-Chiela EC, Bueno e Silva MM, Thomé MP, Lenz G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy. 2015; 11:1099–113. https://doi.org/10.1080/15548627.2015.1009795. [PubMed].
7. McGuirk S, Audet-Delage Y, St-Pierre J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer. 2020; 6:49–61. https://doi.org/10.1016/j.trecan.2019.11.009. [PubMed].
8. Menon DR, Hammerlindl H, Torrano J, Schaider H, Fujita M. Epigenetics and metabolism at the crossroads of stress-induced plasticity, stemness and therapeutic resistance in cancer. Theranostics. 2020; 10:6261–77. https://doi.org/10.7150/thno.42523. [PubMed].
9. Madan E, Pelham CJ, Nagane M, Parker TM, Canas-Marques R, Fazio K, Shaik K, Yuan Y, Henriques V, Galzerano A, Yamashita T, Pinto MAF, Palma AM, et al. Flower isoforms promote competitive growth in cancer. Nature. 2019; 572:260–64. https://doi.org/10.1038/s41586-019-1429-3. [PubMed].
10. Madan E, Peixoto ML, Dimitrion P, Eubank TD, Yekelchyk M, Talukdar S, Fisher PB, Mi QS, Moreno E, Gogna R. Cell Competition Boosts Clonal Evolution and Hypoxic Selection in Cancer. Trends Cell Biol. 2020; 30:967–78. https://doi.org/10.1016/j.tcb.2020.10.002. [PubMed].
11. Merino MM, Rhiner C, Lopez-Gay JM, Buechel D, Hauert B, Moreno E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell. 2015; 160:461–76. https://doi.org/10.1016/j.cell.2014.12.017. [PubMed].
12. Gogna R, Shee K, Moreno E. Cell Competition During Growth and Regeneration. Annu Rev Genet. 2015; 49:697–718. https://doi.org/10.1146/annurev-genet-112414-055214. [PubMed].
13. Parker T, Madan E, Gupta K, Moreno E, Gogna R. Cell Competition Spurs Selection of Aggressive Cancer Cells. Trends Cancer. 2020; 6:732–36. https://doi.org/10.1016/j.trecan.2020.03.008. [PubMed].
14. Lenz LS, Faccioni JL, Bracco PA, Santos JAF, Pereira LC, Buss JH, Tamborindeguy MT, Torgo D, Monteiro T, Mantovani GB, Santo CN, Marcolin JC, Dalsin E, et al. Cancer Cell Fitness Is Dynamic. Cancer Res. 2021; 81:1040–51. https://doi.org/10.1158/0008-5472.CAN-20-2488. [PubMed].
15. Hsu CH, Altschuler SJ, Wu LF. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell. 2019; 178:361–73.e12. https://doi.org/10.1016/j.cell.2019.05.041. [PubMed].
16. Kannoly S, Gao T, Dey S, Wang IN, Singh A, Dennehy JJ. Optimum Threshold Minimizes Noise in Timing of Intracellular Events. iScience. 2020; 23:101186. https://doi.org/10.1016/j.isci.2020.101186. [PubMed].
17. Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002; 297:1183–86. https://doi.org/10.1126/science.1070919. [PubMed].
18. Meir Z, Mukamel Z, Chomsky E, Lifshitz A, Tanay A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat Genet. 2020; 52:709–718. https://doi.org/10.1038/s41588-020-0645-y. [PubMed].
19. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell. 2010; 141:69–80. https://doi.org/10.1016/j.cell.2010.02.027. [PubMed].
20. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, Eggan E, Anastopoulos IN, Vargas-Garcia CA, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017; 546:431–35. https://doi.org/10.1038/nature22794. [PubMed].
21. Salgia R, Kulkarni P. The Genetic/Non-genetic Duality of Drug ‘Resistance’ in Cancer. Trends in Cancer. Trends Cancer. 2018; 4:110–18. https://doi.org/10.1016/j.trecan.2018.01.001. [PubMed].
22. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016; 22:262–69. https://doi.org/10.1038/nm.4040. [PubMed].
23. Gatenby RA, Brown JS. Integrating evolutionary dynamics into cancer therapy. Nat Rev Clin Oncol. 2020; 17:675–86. https://doi.org/10.1038/s41571-020-0411-1. [PubMed].
24. Chen G, Mulla WA, Kucharavy A, Tsai HJ, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, Seidel CW, Berman J, Li R. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015; 160:771–84. https://doi.org/10.1016/j.cell.2015.01.026. [PubMed].
25. Lin KH, Rutter JC, Xie A, Pardieu B, Winn ET, Bello RD, Forget A, Itzykson R, Ahn YR, Dai Z, Sobhan RT, Anderson GR, Singleton KR, et al. Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap to target drug resistance in cancer. Nat Genet. 2020; 52:408–17. https://doi.org/10.1038/s41588-020-0590-9. [PubMed].