
Introduction
Cancer stem cells (CSCs) constitute a cancer cell subpopulation similar to the other stem cell types in terms of self-renewal and multilineage differentiation potential but drive tumor development besides heterogeneity and dissemination of cancer cells [1–9]. CSCs have been extensively studied, with some of these studies focusing on their identification and their origin from differentiated cancer cells due to microenvironment’s influence, which contributes to their heterogeneous phenotypes [6, 10].
Targeting CSCs for therapeutic purposes is a goal of the scientific community. Currently, cancer treatments target the bulk population of the tumor cells without identifying and targeting CSCs [11–14]. This inability of the contemporary therapeutic protocols is significant in treatment resistance and metastasis of cancer cells [15–19]. The significant problem in this regard is the lack of identification marker/s specific for CSCs. Bonnet and Dick were the first to report the existence of CSCs in the tumor in acute myeloid leukaemia (AML) samples [20]. They found that CD34+ CD38- subpopulation cells in leukemia were similar to normal hematopoietic stem cells (HSCs) and could initiate AML in immuno-deficient mice [20]. Since the publication of this report, CSCs have been extensively studied for specific markers but with little success [21, 22]. Nevertheless, CD34+CD38- expression differs between normal and CSCs and may be useful for the identification of CSCs [23, 24]. Receptors expressed in several cell types bind other molecules and be specific for normal, epithelial cancer cells, and CSCs [25, 26]. In the recent decades, scientists have often discussed the identification of CSCs and the use of a specific superficial target that can identify CSCs is the most commonly used approach [27–35].
The main characteristic of these antigens is the capacity to target an endogenous stem cell. However, a standard marker specific for CSCs has not yet been found, although several gene markers have been recently described for CSCs in different tumors, including brain, breast, blood, and lung [36–41]. One of the major problems in finding such a marker is that several markers are able to detect not only CSCs, but also non-tumor cells, which represents an obstacle in developing new therapies targeting CSCs [42–44].
Indeed, there are currently no markers able to distinguish between stem cells and CSCs. Thus far, the best markers identified are those of onco-fetal stem cells, which are absent in adult organs and present in cancer cells [45–48]. Most of the markers identifying stem cells are proteins or glycoproteins, i.e., CD15, an embryogenic target in glioblastoma [49–51]. These identification markers may be subjected to genetic modifications and identify specific CSCs subpopulations with different molecular characteristics. For example, the upregulated ATP-binding cassette (ABC) transporter G subfamily (ABCG), absence or presence of mutated NOTCH1, or different isotypes as observed for CD44v [33, 52–56].
Another essential characteristic of these markers is the variability in their localization as they may be on the cell membrane like CD133 or in the cytoplasm/nucleus like aldehyde dehydrogenase 1 (ALDH1), which is located in the cytoplasm and has been observed in several solid tumors as well as leukemia [57–61]. The localization and the characteristics of these markers reveal that CSCs may have different epigenetic and genetic alterations [62, 63]. This may be one of the most important reasons why scientists are still debating about possible CSCs markers in different solid tumors [45, 64]. Despite that, these aspects have been investigated in cell lines and experimental animals’ models for decades. Sullivan et al. (2010) described the role of ALDH as a possible marker for lung cancer stem cell, as ALDH+ cells in cancer cell lines, as well as those extracted from lung cancer tissue, for the properties that it showed as forming spheres in culture tumor cells lines as well as in cells extracted from lung cancer tissue [65]. However, currently there is no specific or standard marker in lung cancer cells that can define this subpopulation of cells, probably due to the complex localization of these markers and their epigenetic regulation.
Different markers for CSCs have been studied in the last recent decades, and scientists tried have attempted to improve the identification of this better this subpopulation by using a double or triple marker [15, 32, 33, 65–70]. Masciale et al. reported a novel subpopulation of cells expressing CD44+/epithelial cell adhesion molecule (EpCAM+) that was positively correlated with ALDH+ cells [33]. They compared two similar CSC populations. These significant data may form the basis to develop new targeted treatments to eliminate CSCs, which are considered to be one of the leading causes of tumour recurrence and progression [71–75].
A theory regarding the role of CSCs in cancer progression is based on the premise that tumor tissue is hierarchically organized into different types of cells wherein CSC subpopulation is at the top of this hierarchy [1, 28, 29], with the other levels consisting of more differentiated tumor cells or cells with a limited proliferative potential [30]. CSCs have many attributes, including quiescence, chemotherapeutic resistance, and slow cycling [74–76]. Another essential characteristic that places CSCs at the top of the tumor cell hierarchy is their unlimited proliferation potential, which allows them to repopulate the tumor even if bulk tumor cells have been removed [18, 77–87]. It is important to note that CSCs may represent a dynamic cellular state in which stem cell-like traits are acquired to mediate resistance and induce tumor dissemination [77, 86, 87]. The complexity of cell composition, which is the base of cancer heterogeneity, has been discussed for a long time due to the different mechanisms that are the cause of the cancerization process variability [89]. Moreover, the discovery of the plasticity of CSCs and the possibility of switching from stem to non-stem cells led to a more complex picture of the origin of tumor heterogeneity [88]. Peter Nowell was the first to describe the “clonal evolution theory,” defining cancer as a complex process resulting from the development of a single out-of-control cell with multiple cell mutations that result in the progression of the tumor, which is kept viable through the selection of the most aggressive clones [89]. He also hypothesized that the dominant clone cells showed the most substantial tumorigenic properties [89]. An opposing theory is based on the concept that CSCs are a group of cells endowed with a high self-renewal capacity that can set different phenotypes of tumorigenic cells [18, 88]. The CSC theory was the most impressive in solid tumors in the absence of another approach to distinguish between a tumorigenic and non-tumorigenic cell due to similarity in their kinetic properties and because cancer is hierarchically organized [21]. One of the main obstacles to proving the CSC model is the difficulty in identification and isolation of these cells [7, 33, 91]. In fact, scientists first tried to define a CSC model using xenografts for the testing and identification of markers, and isolation of tumor-initiating cells [91, 92]. This model showed the existence of highly tumorigenic cells but did not clarify the superficial markers that might be useful to define this subpopulation compared with non-CSCs. One of the most important aspects that require in-depth study in the future is the capacity of these cells to survive chemotherapy. In addition to these aspects demonstrated in an animal models, these cells’ self-renewal capacity, which helps the tumor grow, disseminate, and relapse remains a topic of intense interest in the scientific community. Although the CSC model alone is not supported enough to explain functional heterogeneity in cancer, scientists have recently considered the role of the tumor microenvironment (TME) as a significant factor in CSCs’ plasticity, especially in the process of turning from non-CSCs to CSCs [90, 92]. This mechanism seems to rely on cell-to-cell interactions within the tumor niche [77].
Furthermore, these connections between CSCs and other cells is the primary source of protection for this subpopulation during induction of cell transformation, tumor growth, and resistance to common oncological treatments. In this context, the TME seems to play a crucial role in tumor progression and metastasis by building a synergistic relationship with CSCs [6, 10, 16, 76, 93] (Figure 1).
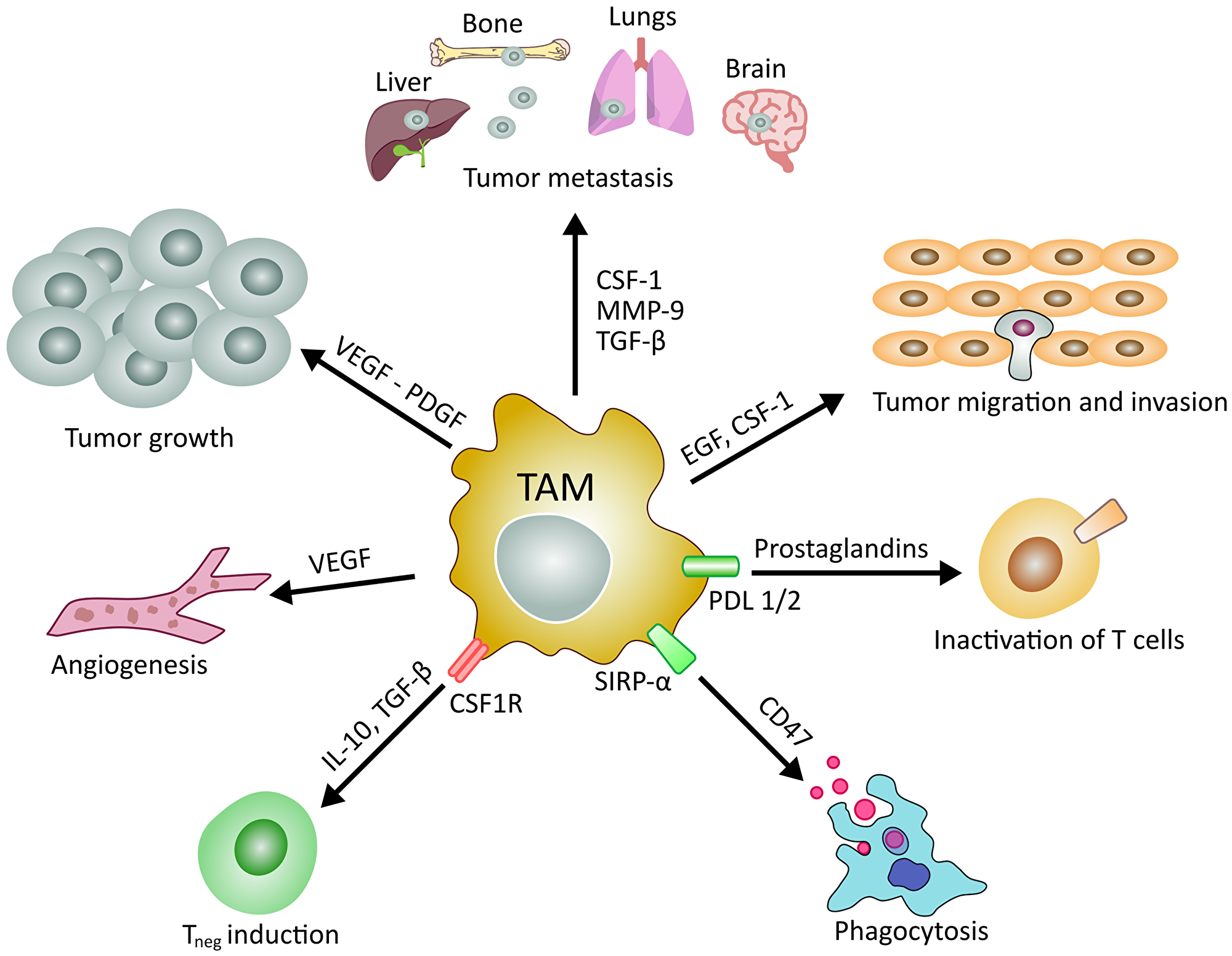
Figure 1: Main roles of tumor associated macrophages in cancer development and manteinance. TAMs and their released factors are involved in different processes controlling the evolution of cancer. Schematic representation of the role of TAMs in tumor growth, angiogenesis, invasion, metastasis and regulation of T lymphocytes.
CSCS ARE THE KEY DRIVERS OF TUMOR INITIATION AND PROGRESSION
The role of CSCs in carcinogenesis has not yet been well-defined [86, 87, 93, 94]. The process of tumor initiation involves the accumulation of mutations that facilitate the uncontrolled proliferation of tumor cells. Any mutation in these proliferation-promoting genes results in the failure of DNA-repair mechanisms [95, 96]. Additional mutations induce clonal selection, which selects more aggressive phenotypes. Unfortunately, these molecular alterations usually take place in the early stages of cancer that continue unabated in the absence of any treatment, as cancers are rarely identified in early stages [97]; thus, cancer grows and disseminate during this time unchecked [1, 98]. The most substantial support for metastasis comes from the TME, which provides favourable signals to support the metastatic cascade. The molecular interactions between cancer cells and the TME influence the tumor’s capacity to survive and evolve, developing more resistant and aggressive phenotypes [6, 10, 16, 76, 93]. This is the possible reason that cancer treatments are not able to reduce or stop tumor progression, especially in advanced stages. The tumour’s aggressiveness is primarily determined by the subpopulation of CSCs, as they are often resistant to therapies and can re-initiate tumor even if the bulk tumor cells are eliminated, resulting in tumor relapse [99].
The dualistic model explains this capacity to mediate tumour progression. By this model, CSCs may divide either symmetrically, giving rise to two identical CSCs or two differentiated cancer cells, or asymmetrically thus resulting in one CSC and one differentiated cancer cell. Only the complete extinction of the CSCs population would completely eradicate the tumor [100, 101]. Alternatively, if we consider the stochastic model in which each cell can be tumour-initiating, the path to eradicating the tumor is much more complicated [102]. Olmeda et al. put forth a tumor initiation model comprised of CSCs, differentiated cancer cells, and all the other cells within a tumour [103]. Each cell type in the tumour then creates an active link with the microenvironment. By this model, each cell subpopulation can actively interact with the microenvironment via diverse chemical pathways and physical interactions [103].
The CSC initiation process is stemness transcription factors dependent that drive the expression of genes not expressed in normal cells but is highly expressed in CSCs, especially during the initiation process [102–104]. Blanpain et al. demonstrated a vital role for the transcription factor sex-determining region Y-Box 2 (SOX2) in the melanoma initiation and progression [105, 106]. In this study, they noted that SOX2 was not present in normal skin but was clearly expressed at an early stage in tumor formation [106]. As these data, two now better understand these stemness genes and their potential targets during the control of tumor progression. In fact, in the case of melanoma, it has been thought that tumor initiation may be prevented by the deletion of the SOX2 gene and that the removal of SOX2-positive cells from established tumors may lead to regression. Potential treatments developed based on the study of stemness genes are very promising for controlling CSCs and, consequently, tumor resolution [106]. Future studies are required in many solid organs to identify the genes implicated in the mechanism regulating CSCs proliferation, tumor survival, and invasion [107].
MACROPHAGES AND TAMS
Macrophages are large specialized phagocytic cells that exist in tissues or at infection sites. They arise from monocytes in the bone marrow and perform different functions and roles in the microenvironments of normal and tumor tissue [108, 109]. Macrophages differentiate into classically activated subtypes: CD68 expressing M1 mainly involved in pro-inflammatory activities, and CD163 expressing M2, that promote anti-inflammatory processes. In tumors, tumor-associated macrophages (TAM) comprise up to 50% of the tumor mass, with M2 phenotype being most abundant in the TME [110–112]. The primary signals provided by TAMs include interleukin 4 (IL-4) and transforming growth factor-beta (TGF-β). TAMs play a key role in tumor initiation, development, and cancer cell propagation [113] (Figure 1).
Recent studies have demonstrated that high numbers of TAMs correlate with a poor clinical prognosis in lung tumors and gastric cancer, among other cancer types [114–118]. Another important aspect is the protective role of TAMs for tumors undergoing chemotherapy, which may impact chemotherapeutic resistance and consequent tumor relapse [119]. There is a general reception that TAMs decrease the effectiveness of chemotherapy while the presence of CD68+ and CD163+ cells correlates with a poor prognosis in esophageal and pancreatic cancers [120, 121].
Yang et al. have suggested that in breast tumors, TAMs are responsible for chemotherapy drug resistance via the interleukin 10 (IL-10)/signal transducer and activator of transcription 3 (STAT3)/B-cell lymphoma 2 (Bcl-2) signaling pathway [122]. TAMs are also correlated with tamoxifen resistance, which is used in endocrine therapy in postmenopausal patients with breast cancer [122]. Moreover, TAMs contribute to unfavorable outcomes during radiotherapy due to their capacity to modulate cancer cells’ response to therapy [123, 124]. In particular, macrophages are responsible for the side effects of radiotherapy [123–125]. These findings explain that irradiation, DNA damage, cell death, and hypoxia stimulate tumors to produce vascular endothelial growth factor (VEGF), stromal cell-derived factor 1α (SDF-1α), and colony-stimulating factor 1 (CSF-1), which are involved in the recruitment of macrophages to the tumor [122–126] (Figure 1). The recruitment of TAMs leads to the production of proangiogenic cytokines, which stimulate blood vessel formation [121, 122]. Irradiated macrophages can also promote cancer cell migration and tumor angiogenesis (Figure 1). Notably, it has been demonstrated that therapies targeted against TAMs can improve radiotherapy’s efficacy [127–130]; specifically, the inhibition of TAM recruitment can prevent tumor regrowth [127].
TUMOR ESCAPE AND TAMS
Immune system can recognize the presence of cancer cells and induce their rejection response. However, some specific phenotypes of cells manage to escape the vigilant immune surveillance to form tumors despite the presence of normal activated immune cells [131–133]. Immunoediting comprises all of the immune processes that lead to the control of tumor progression, including an important phase of immunosurveillance [134]. As a consequence of this tumor elimination phase, a specific subpopulation of CSCs is able to escape immune mechanisms to escape the immune response via, for example, the downregulation of antigen-presenting cells (APCs) [135, 136]. As the presentation of tumor antigens from human leukocyte antigen (HLA)-1 to T cells is essential for the recognition phase, the expression of HLA-1 on CSCs may decrease as reported by Di Tomaso et al. in a study on glioblastoma CSCs [137]. This downregulation of HLAs has also been observed in melanoma CSCs, in which abnormally high expression of HLA-II and low expression of the melanoma-associated antigens MART-1, ML-IAP, NY-ESO-1, and MAGE-A was observed [138, 139]. The antigen-processing machinery is defective in CSCs; thus, these highly specialized cells exhibit low immunogenicity [140]. Some CSCs, particularly in melanoma, modulate immune responses by expressing ABCB5 gene, conferring chemoresistance [141–143]. These mechanisms mentioned above describe the modulation of immune response via the induction of T-cell anergy and downregulation of cancer-associated antigens to escape immune-mediated tumor clearance [141–143].
Another mechanism that can influence immune cells presence and function is associated with TAMs [121]. TAMs are the predominant immune cells within tumors; consequently, they significantly impact the tumor initiation process and can affect T lymphocytes, natural killer (NK) cells, dendritic cells, neutrophils, and myeloid-derived suppressor cells (MDSCs) [144]. Also, TAMs express chemokines, i.e., CCL5, CCL22, and CCL20 and cytokines i.e., IL-10 and TGF-β, which recruit and activate regulatory T cells (Tregs), thus contributing to immunosuppression in the TME [145], and participate in tumor cells’ escape and tumorigenesis. In particular, TAMs can suppress the antitumor effects of tumor-infiltrating T cells and NK cells [148] and inhibit T cell function by releasing specific enzymes, such as nitric oxide synthase and arginase I [146, 147].
Besides, TAMs inhibit the cytotoxic functions of T cells, natural killer T cells, and NK cells through the expression of the ligands for the immune checkpoint receptors programmed cell death-1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) [121], which are highly expressed on the surface of CSCs in various cancer types, allowing CSCs to escape and avoid elimination by the immune cells [148].
Depending on their phenotype, macrophages have a dual role in cancer. M1 macrophages are involved in the earlier stages of neoplasia, while M2 macrophages are involved in tumor spreading [109]. DNA damage is the principal mechanism by which inflammation promotes tumorigenesis. TAM-generated free-radicals lead to DNA damage, causing alterations that predispose a cell to cancer. An example of this macrophage-mediated induction of tumorigenesis is Crohn’s disease, which significantly enhances the risk of colorectal cancer [149]. In the metastatic process, TAMs are responsible for local invasion and intravasation into the blood system, as well as homing to the pre-metastatic niche (by promoting EMT) [150, 151]. TAMs also foster both tumor growth and migration by producing a variety of chemokines, inflammatory agents, and growth factors. For example, CD68+HLA-DR+ TAMs in hepatocellular carcinoma (HCC) induce HCC cell migration via the nuclear factor kappa B (NF-κB)/ focal adhesion kinase pathway [109, 152]. One of the most exciting characteristics of TAMs is their capacity to promote angiogenesis and lymphangiogenesis that allow tumor growth and the spread of cells in the TME.
The formation of new blood vessels or lymphatic vessels provides support channels for neoplastic tissues. Massive angiogenesis contributes to poor prognosis in primary tumors. The principal contributors in angiogenesis are hypoxia, hyper-osmosis, and proangiogenic factors such as VEGF, TGF-β, cyclooxygenase 2, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), angiopoietins, and chemokines [152]. TAMs can also synthesize proteins associated with vascular endothelial cells, such as Wnt7b besides VEGF, thereby promoting the angiogenetic switch [153]. One of the significant angiogenesis-inducing factors, pro-matrix metalloproteinase-9 (proMMP-9), is supplied by TAMs to the TME [156]. MMP-9 plays a fundamental role in tumor angiogenesis and metastasis by activating the angiogenic switch that mediates the development and maintenance of distinct neovascular networks [154, 155] (Figure 1).
Cross-talk between CSCs and TAMs involves the recruitment of TAMs through vascularization and the release of chemokines by TAMs to preserve the quiescence of CSCs and modification of their antigens to escape from recruitment by immune cells. This crosstalk influences all the aspects of tumorigenesis to metastasis. Consequently, immunotherapies to fight cancer, such as checkpoint inhibitors or T-lymphocyte transfer strategies are emerging novel therapeutic strategies in the oncology field.
MECHANISMS LINKING CSCs AND TAMs
TAMs may constitute more than 50% of the tumor mass [109, 112, 156]. They promote tumor growth by inducing neoangiogenesis, supporting CSCs, and downregulating tumour-targeting immune cells’ number and function [125, 153–155]. Due to the significance of the tasks in which TAMs are involved, TAMs are increasingly becoming principal targets of novel therapeutic approaches, especially in the field of nanomedicine. It is now generally accepted that the M2 macrophages have an essential role in immunosuppression and trophic activity in response to Th2 cytokines [121, 156–159].
The roles, connections, and functions of the crosstalk between TAMs and CSCs have been studied in-depth during the recent past. [158–160]. The interactions may be direct or indirect, and the effects on CSCs include chemoresistance, preservation, and the capacity to differentiate [161, 162]. TAMs produce cytokines including milk fat globule epidermal growth factor 8 (MFG-E8); interleukin 6 (IL-6), which can activate STAT3; and the Hedgehog signaling pathway, which seems to be one of the causes of drug resistance [163, 164]. For example, in hepatocarcinoma, IL-6 promotes the expression of CD44+, inducing tumor development [165].
The role of IL-6 in the induction of TAM-mediated CSCs has been studied by the inhibition of IL-6 with the anti-IL6-R antibody tocilizumab, which was able to decrease the number of the tumor spheres and chemoresistant cells [166, 167]. IL-6 also plays a role in cancer by stimulating the conversion of non-stem cell into stem-like cells. In breast cancer, in vitro stimulation with IL-6 increases tumor mammospheres and CD44+/CD24+ breast CSCs. It is also known that IL-6 stimulates the conversion of non-stem cell into stem-like cells. In breast cancer, in vitro stimulation with IL-6 results in an increase in the number of tumor mammospheres and CD44+/CD24+ breast CSCs [168]. Substantial evidence demonstrates the important role of IL-6 in the niche microenvironment to guide the metastatic process in cancer [164, 165]. Moreover, IL-6 promotes stemness in osteosarcoma by upregulating the signalling pathway regulating EMT through phosphorylation of the STAT3. Further, IL-6 levels are associated with tumor growth and metastases [169–172]. The IL-6 release, together with CCL5 and IL-8, has been linked to the β-catenin/Wnt pathway, leading to the spread of CSCs [173, 174].
Another cytokine able to drive EMT in hepatoma cells is TGF-β1. This induction promotes the development of cancer stem-like characteristics, and it can be reduced or stopped by the depletion of TGF-β1 [175, 176]. A microarray from 96 patients diagnosed with pancreatic ductal adenocarcinoma showed CD44+CD133+ markers for CSCs and CD204+ for TAM, suggesting a possible connection between CSCs and TAMs in cancers, which also appears to be correlated with overall decreased disease-free survival [177]. Also, breast cancer CSCs appear to interact directly with TAMs through CD90/CD11b anchoring [178]. This interplay induces EphA4 receptor-mediated activation of both the nuclear factor- kappa B (NF-κB) and Src signaling pathways in CSCs. Cytokines and granulocyte-macrophage colony-stimulating factor (GM-CSF) are secreted by CSCs through a cascade of juxtracrine signals. However, the interactions between CSCs and TAMs may be facilitated by several components of the extracellular matrix (ECM). In particular, there are molecules like TGF-β1 responsible for EMT, which promotes the development of cells with cancer stem-like characteristics [167].
In leukemia, several cytokines, growth factors, and ECM proteins derived from mesenchymal stromal cells promote abnormal proliferation and dissemination of cancer cells [179]. Periostin (POSTN) is also involved in metastasis of different cancer types by promoting tumor niche formation, especially the CSC niche [179], and CCL2 was shown to increase the development of CSC in breast cancer by acting on cells in the TME [180, 181]. A recently identified pro-TME factor, Wnt-induced signaling protein 1 (WISP1) has been related with the promotion CSCs and TAMs’ survival of both in glioblastoma thus affecting the disease course [182, 183]. Moreover, in breast cancer, upregulation of hyaluronan synthase 2 (HAS2) induces the production of the hyaluronic component of the ECM, which is can promote TAMs-released platelet-derived growth factor-BB (PDGF-BB) [184]. In turn, PDGF-BB activates stromal cells, which induce CSCs’ self-renewal through the secretion of fibroblast growth factor 7 (FGF7) and FGF9. Inhibition of HAS2 by 4-methylumbelliferone blocks cancer development and reduces the incidence of recurrence [184]. This finding highlighted that inhibiting HAS2 can control the interactions between CSCs and TAMs. This could be considered very important in developing a new generation of therapeutic approaches targeting both of these cell populations [184].
TAMs also indirectly affect CSCs’ differentiation by NK cells by secreting IFN-γ [185, 186]. TAMs originate from monocytes upon their activation in the TME. The tumor actively recruits monocytes and favor the M1-to-M2 conversion, a local tumor-associated event that becomes more frequent with tumor development [112, 121, 156, 157, 187]. Efforts are underway to reprogram or inhibit the tumor-protective properties of TAMs, and develop potential strategies to increase the efficacy of conventional chemotherapy by combining it with macrophage-associated delivery of nano-drugs [121, 156, 157, 187]. The potential link between monocyte activation and macrophage conversion into TAMs, and the interactions between M2 macrophages and CSCs, are not well understood. It appears that CSCs should promote the conversion from M1 to M2, induce neo-vascularisation via VEGF release, and create CSCs niches through tissue repair pathways [188]. Molecular studies have demonstrated a crosstalk between TAMS and CSCs wherein TAMs release milk fat globule–EGF factor 8to activate the CSC-associated pathways STAT3 and Shh and amplifies the drug resistance and tumorigenicity of CSCs [189]. Indeed, the drug resistance of murine mammary CSCs was linked with the EGFR/STAT3/SOX2 signaling pathway by Yang et al., who reported paracrine activity established through a complex interplay between CSCs and TAMs [189–191].
In glioma, macrophages of the microglia and brain also produce high levels of TGF-β, thus rendering glioma stem-like cells (GSLCs) more invasive. This was accompanied by a substantial amount of MMP-9, a serine protease that contributed to the invasiveness of GSLCs [191–193]. However, there is insufficient data to identify all of the tumor-associated factors engaged in macrophages’ conversion to TAMs. The participation of M2 macrophages in tumor development is similar to their role in wound healing. The wound healing process has four programmed phases including hemostasis, inflammation, proliferation, and remodeling [194]. Wounds trigger mobilization of bone marrow MSCs and EPCs involved in neovascularization. These steps share similarities with tumorigenesis, in which CSCs initiate the formation of the primary tumor or metastatic nodes and, perhaps, play an essential role in the M1-to-M2 conversion. Therefore, tumorigenesis is often considered as a deviated natural healing process involving the participation of transformed stem cells (CSCs) and macrophages (TAMs) [109, 156]. CSCs release factors that attract macrophages and convert them into TAMs. CSF-1, a significant growth factor involved in this process, helps recruit macrophages to the tumor site, promoting tumor progression to malignancy. Inhibition of TAM recruitment by a CSF-1 may likely improve the ability of chemotherapeutic agents to reduce tumor progression and metastasis [109, 156, 195, 196]. Resting CSCs populate the hypoxic areas of tumor and get activated after chemotherapy-induced injury, when most peripheral cancer cells are eliminated. When macrophages are recruited to remove debris, they activate dormant CSCs. TAMs participate in reparative mechanisms after radiotherapy or antiangiogenic treatment [152–154]. Depending on various factors, they either enhance or antagonize the efficacy of radiotherapy or chemotherapy and immunotherapeutic agents such as tumor-targeting antibodies [67, 156].
One of the metastasis hypotheses suggests that metastasizing cells move to the peripheral niches occupied by CSCs [196]. According to this hypothesis, TAMs form cell hybrids with tumor cells and travel to distant sites to initiate metastases [197]. The theory of forming a hybrid cell was proposed in 2006 by John Pawelek. He explained this phenomenon as a fusion between a myeloid cell and a tumor cell, leading to a genomic hybridization [198]. This theory was further expanded to include fusion between macrophages and tumor cells in general and TAMs in particular.
It is pertinent to mention that the hybrid cells have a reduced proliferative ability as compared to the parental cell lines. According to the CSCs hypothesis, CSCs form spheroids that migrate out of the primary tumor site via the bloodstream or lymphatic circulation and undergo metastases in the niches with appropriate conditions. Macrophages associated with the repair of the injured lesions may serve as niche-forming cells attracting CSCs. Metastatic foci can be further supported by the mutual interaction of these two cell types and the acquisition of TAM characteristics by M2 macrophages to allow tumor growth [199].
The presentation of an antigen is a standard process by which the immune cells eliminate the abnormal cells. However, the hybrid cells have a reduced proliferative potential compared to their parental cell lines. According to the CSC-hypothesis, CSCs form spheroids that migrate out of the primary tumor site via the bloodstream or lymphatic circulation and form metastases in niches under conducive conditions. Macrophages associated with the repair of the injured lesions may serve as niche-forming cells attracting CSCs. Metastatic foci can be further supported by the mutual interaction of these two cell types and the acquisition of TAM characteristics by M2 macrophages to allow tumor growth [200–203].
As antigen presentation is an essential part of the immune response against tumor cells, immune cells such as CD4+ T helper cells and CD8+ cytotoxic T lymphocytes represent one of the most considered and studied processes to eliminate cancer cells. Experimental studies have been done on CSCs and their interactions with lymphocytes in the tumor. For example, in 2019, Masciale et al. described two interesting correlations between CSCs and tumor-infiltrating lymphocytes in NSCLC; between CD3+ T cells and CSCs and between CD8+ T cells and CSCs. These findings are useful for defining the antitumor effects of the cytotoxic CD8+ T cells, and the regulatory CD8+ T cells since CD8+ and CD3+ T cells support the host defense against cancer [5]. In several tumors, cytotoxic T lymphocytes are important predictors of outcomes, both for the disease progression and immunotherapy response [203–205].
CSCs from head and neck squamous cell carcinoma that were positive for CD44 have reduced MHC-I expression compared with the CD44-negative epithelial cancer cells [206]. Similarly, the expression of MHC is lower in CSCs in other cancers [207–210]. On the other hand, no differences in MHC-I expression between CSCs and epithelial cancer cells have been reported [211]. However, the downregulation APCs impede the targeting of CSCs by T cells. This aspect is an essential feature of CSCs in the TME [212]. The ability to regulate APC expression reduces the cell differentiation process and is generally caused by an epigenetic process, such as suppressing histones or DNA methylation. Using a drug-induced DNA demethylation protocol, upregulation of antigen-presenting cells in glioblastoma CSCs has been reported, although the expression remained less than in epithelial cancer cells. This is likely due to the distinct molecular mechanisms that cause different gene expression patterns [208]. However, despite the reduced expression of MHC-I, CSCs remain targets of the immune system, particularly NK cells.
In summary, equilibrium of various signals defines the destiny of CSCs and cancer epithelial cells, and their ability to eradicate or evade an immune response. APCs alone are generally not enough to stimulate T cells. Hence, in the absence of additional signals; T cells destroy themselves or remain inactive. This stimulation is derived from molecules on APCs, i.e., CD80 or CD86 that can bind with T cells. On the other hand, a mechanism of inhibition is PDL1 (B7-H1) presented on tumor cells or APCs. PDL1 binds PD1, inducing the apoptosis or inactivation of T cells. Overexpression of PDL1 is commonly used by cancer cells to inhibit T cells [213]. This aspect has been observed in many cancer types, including glioblastoma and head & neck squamous cell carcinoma [208, 214, 215], thus suggesting an interaction between CSCs and immune cells to have a meaningful impact on promoting or inhibiting T cells. In particular, high levels of PDL1 on CSCs may be interesting to investigate as possible CSC immunotargets [216].
FUTURE PERSPECTIVES IN CANCER REGARDING TAMs AND CSCs
Surgery is the gold-standard treatment only for cancer in early stages, whereas treatment of advanced cancer stages requires chemotherapy and radiotherapy alone or in combination [217, 218]. However, in many cases, all treatments (chemo-, radio-, and immunotherapy), fail to prevent cancer recurrence [219]. The primary cause of failure in cancer treatment is the emergence of drug resistance that promotes the tumor spreading [220]. Several clinical trials investigate the best cell target to fight cancer, including, but not limited to, CSCs, since CSCs are sustained by other cells, including TAMs (Table 1). Indeed, anti-macrophage drugs such as trabectedin [14, 221, 222], RG7155 (anti-CSF-1R) [223], and an anti-MIF (macrophage migration inhibitory factor) antibody have been developed. However, CSCs are still considered as the most important subpopulation of cells as a novel target due to their integral participation in cancer relapse [224–226]. A recent study has demonstrated that CSCs release cytokines in pro-tumor microenvironment through the generation of CD163+ macrophages like myeloid cells [227]. Moreover, targeting TAMs together with CSCs offer another possible option in treating pancreatic ductal adenocarcinoma to better control cancer progression and avoid tumor dissemination [228]. An important finding is the inhibition of phagocytosis by macrophages through the interaction between signal regulatory protein alpha (SIRPα) and CD47, specific for epithelial cancer cells [229, 230]. Weissman et al. used a monoclonal antibody to block in vitro the activity of CD47 for increasing the phagocytosis of the tumor cells leading to a reduction of the tumor growth in vivo [228, 229, 231–233].
Table 1: Clinical trials targeting CSCs
| Drug name | Mechanism | Condition or disease | NCT Number | Current Status |
|---|---|---|---|---|
| Vismodegib (GDC-0449) | Hedgehog Pathway Inhibitor | Ovarian Cancer | NCT00959647 | Completed |
| Hedgehog Pathway Inhibitor | Basal Cell Carcinoma | NCT00959647 | Completed | |
| Hedgehog Pathway Inhibitor | Metastatic Colorectal Cancer | NCT00959647 | Completed | |
| Sonidegib (LDE225) | Hedgehog Pathway Inhibitor | Medulloblastoma | NCT01708174 | Completed |
| BMS-833923 | Hedgehog Pathway Inhibitor | Leukemia | NCT02100371 | Completed |
| MK-0752 | Notch pathway inhibitors | Metastatic Breast Cancer | NCT00645333 | Completed |
| RO4929097 | Notch pathway inhibitors | Adenocarcinoma of the Pancreas | NCT01122901 | Terminated |
| Notch pathway inhibitors | Recurrent Adult Brain Tumor | NCT01122901 | Terminated | |
| Nirogacestat (PF-03084014) | Notch pathway inhibitors | Desmoid tumors/aggressive fibromatosis | NCT01981551 | Active, not recruiting |
| Crenigacestat (LY3039478) | Notch signaling pathway | Neoplasms | NCT01695005 | Completed |
| Notch signaling pathway | Lymphoma | NCT01695005 | Completed | |
| Demcizumab (OMP-21M18) | Notch pathway inhibitors | Non-Small Cell Lung Cancer | NCT01189968 | Completed |
| Ipafricept (OMP-54F28) | WNT pathway inhibitors | Stage IV Pancreatic Cancer | NCT02092363 | Completed |
| WNT pathway inhibitors | Pancreatic Cancer | NCT02050178 | Completed | |
| Vantictumab (OMP-18R5) | WNT pathway inhibitors | Metastatic breast cancer | NCT01973309 | Completed |
| PRI-724 | Wnt signaling pathway blocking | Advanced Solid Tumors | NCT01302405 | Terminated |
| AVID 200 | TGF-β inihibitors | Malignant solid tumor | NCT03834662 | Active, not recruiting |
| Fresolimumab (GC1008) | TGF-β inihibitors | Metastatic breast cancer | NCT01401062 | Completed |
| TGF-β inihibitors | Stage IA Non-Small Cell Lung Carcinoma | NCT02581787 | Recruiting | |
| NIS793 | TGF-β inihibitors | MPN (Myeloproliferative Neoplasms) | NCT02947165 | Active, not recruiting |
| TGF-β inihibitors | Lung cancer | NCT02947165 | Active, not recruiting | |
| TGF-β inihibitors | Hepatocellular Cancer | NCT02947165 | Active, not recruiting | |
| TGF-β inihibitors | Colorectal Cancer | NCT02947165 | Active, not recruiting | |
| TGF-β inihibitors | Pancreatic Cancer | NCT02947165 | Active, not recruiting | |
| Ruxolitinib | JAK inihibitors | Metastatic breast cancer | NCT01348490 | Completed |
| JAK inihibitors | Myeloproliferative neoplasms | NCT01348490 | Completed | |
| AZD4205 | JAK inihibitors | Advanced non-small cell lung cancer | NCT03450330 | Completed |
| SAR245409 | PI3K and mTOR inihibitors | Advanced or metastatic solid tumors | NCT01240460 | Completed |
| Matuzumab (EMD 72000) | EGFR inhibitors | Non small cell lung carcinoma | NCT00753246 | Completed |
Cioffi et al. have extended this concept in a pancreatic cancer model, describing a novel therapy that induced phagocytosis of CSCs [230]. This approach has been tested in pancreatic cancer with encouraging results and hence necessitates further studies must be performed in other solid tumors.
Another approach recently taken into consideration as a prospective approach in cancer therapies is nanomedicine. The term “nanos” refers to preparations with non-sized particles ranging in size between 1–100 nm in diameter [229–235]. Their main composition of such preparations may include lipids, proteins, or polymer [236, 237], and they may be used as a loader of drugs or genes or for diagnostic purposes [237, 238]. Ex vivo loading of patients’ cells, i.e., macrophages, monocytes, or MSCs, with a loaded nanodrug, are considered to be the most effective approach for nanodrug delivery without compromising the cell viability and mobility [239]. These drug-loaded cells can home-in to the tumor site or areas of inflammation [240] focusing on immunocytes and stem cells due to their intrinsic neoangiogenic capacity around inflammatory or tumor sites.
The cell-based targeted nanoparticles have been described in several studies and can adhere to antibodies, peptides, etc. [235, 239, 241, 242–255]. Their targets are generally membrane-bound and intracellular receptors [256, 257], or mitochondria [258]. The most complicated aspect of this interaction is the space between the target and the molecules that needs to be bound: This space must be no larger than a few nm [259]; otherwise, the interaction would not be effective. This aspect is easier to address in vitro. It may represent a problem in vivo, as the characteristics of the TME are more varied and are different from the media commonly used in vitro. It has been shown that the target of nanoparticles does not produce an increment of the particles able to bind cancer cells in vivo [260]. This aspect suggests that the target of cell-based nanoparticles in oncology has been over-considered, while the physical characteristics of nanoparticles must receive more focus [261]. In addition to these aspects, the microenvironment, particularly the cell-to-cell interactions and the cell-immune system interactions, should also be taken into consideration, as they may constitute a huge obstacle for the optimization of this approach against cancer. The synthesis of oral nanodrugs will likely be more useful than intravenous solutions, and this may represent the main factor in the clinical use of nanodrugs to treat cancer [262, 263]. However, several barriers are required to be overcome before interaction between nanoparticles and the components of the TME.
Interestingly, the intrinsic phagocytic activity of macrophages has been exploited to load anti-cancer nanodrugs. One of the first accepted therapies in this field involved the innovative delivery of bioactive proteins into macrophages to treat neurodegenerative diseases [264, 265]. This therapeutic approach is based on nanozymes, which can be rapidly internalized by monocyte-derived macrophages and released in an active form within 24 hours. In a mouse model of Parkinson’s disease, the injected monocyte-derived macrophages were able to home-in to the brain, and there was an overall reduction in oxidative stress. Macrophages loaded with a DNA plasmid encoding for catalase had a similar effect as exosomes secreted by the macrophages and attenuated oxidative stress in neurons [266]. This strategy exhibits promise in improving motor neuron functions in mouse models of Parkinson’s disease. These nano-carriers are particularly useful due to their ability to cross the blood–brain barrier (BBB) [267]. The same approach has been used for antiretroviral drugs against HIV, encapsulating them in biodegradable nanoparticles loaded ex vivo into monocyte-derived macrophages. These macrophages were able to deliver drugs across the BBB and inhibit HIV infection in the brain. More recently, given these promising effects, macrophage-loading with drugs has been extended to antiviral drugs such as ritonavir, indinavir, and efavirenz [268].
A novel approach to improving nanodrug capture efficiency by macrophages for anti-HIV therapy is being developed [269]. This approach uses a well-known chemoattractant for macrophages, N-formyl-methionyl-leucyl-phenylalanine peptide conjugated onto PEGylated nanoparticles. This approach was useful for nanodrug internalization by peritoneal macrophages, which are the primary HIV reservoirs. At molecular level, macrophage scavenger receptors are actively involved in capturing circulating nanodrugs [270]. These studies demonstrated the importance of using nanoparticles that can be captured by macrophage scavenger receptors.
Conclusions
In-depth understanding of interaction between TAMs and CSCs is needed to develop novel treatment strategies in future. In this direction, researchers have already reported the presence of CSCs in many solid tumors as the leading cause of cancer relapse and chemotherapeutic drug resistance. In addition to this subpopulation of cells, macrophages and other immune cells also participate in interactions that may aid or impede the fight against cancer. For this reason, the targeting TAMs offer a novel treatment option against cancer. The different therapeutic approaches developed to target TAMs include the depletion, blockade of monocyte/macrophage recruitment, reprogramming of TAMs into pro-inflammatory M1-like macrophages, and neutralizing the products of TAMs [271]. Although most TAM-targeting strategies are in the pre-clinical stages, several factors used for TAMs depletion have already been tested in clinical trials [271, 272]. However, the effects of these novel treatments targeting TAMs on checkpoint blockade-based immunotherapies must be further investigated [273]. We believe that targeting TAMs may trigger various stromal reactions in the tumor milieu that are difficult to predict, even if the variability from patient to patient is kept as a consideration. Targeting TAMs could not only inhibit the TME, but also renovate the tumor “soil” to build a tumor-suppressive microenvironment, thereby suppressing tumor development. This strategy may become an effective therapeutic intervention that may be used either alone or in combination with other therapeutic strategies to treat cancer [273].
In summary, generating new information about the interaction between TAMs and CSCs will be one of the most important challenges for the development of more effective targeted cancer therapies.
Abbreviations
TAM: tumor-associated macrophages; ALDH: aldehyde dehydrogenase; ARGI: arginase I; BBB: blood–brain barrier; CSCs: Cancer stem cells; COX-2: cyclooxygenase 2; CTLA-4: cytotoxic T lymphocyte-associated protein 4; EGF: epidermal growth factor; EMT: epithelial-to-mesenchymal transition; GSLCs: glioma stem-like cells; HCC: hepatocellular carcinoma; NOS: nitric oxide synthase; PDGF: platelet-derived growth factor; PD-1: receptors programmed cell death; TME: tumor microenvironment; Tregs: recruit regulatory T cells.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018; 25:20. https://doi.org/10.1186/s12929-018-0426-4. [PubMed].
2. Teng YD, Wang L, Kabatas S, Ulrich H, Zafonte RD. Cancer Stem Cells or Tumor Survival Cells? Stem Cells Dev. 2018; 27:1466– 78. https://doi.org/10.1089/scd.2018.0129. [PubMed].
3. Schulenburg A, Blatt K, Cerny-Reiterer S, Sadovnik I, Herrmann H, Marian B, Grunt TW, Zielinski CC, Valent P. Cancer stem cells in basic science and in translational oncology: can we translate into clinical application? J Hematol Oncol. 2015; 8:16. https://doi.org/10.1186/s13045-015-0113-9. [PubMed].
4. Aramini B, Masciale V, Haider KH. Defining lung cancer stem cells exosomal payload of miRNAs in clinical perspective. World J Stem Cells. 2020; 12:406–21. https://doi.org/10.4252/wjsc.v12.i6.406. [PubMed].
5. Masciale V, Grisendi G, Banchelli F, D’Amico R, Maiorana A, Sighinolfi P, Pinelli M, Lovati E, Stefani A, Morandi U, Dominici M, Aramini B. Correlating tumor-infiltrating lymphocytes and lung cancer stem cells: a cross-sectional study. Ann Transl Med. 2019; 7:619. https://doi.org/10.21037/atm.2019.11.27. [PubMed].
6. Masciale V, Grisendi G, Banchelli F, D’Amico R, Morandi U, Dominici M, Aramini B. Cancer Stem Cells and Their Microenvironment. In: Khawaja Husnain Haider (Editor), Stem Cells: World Scientific Publishing. 2011; 146–178. https://doi.org/10.1142/9789811205538_0006.
7. Masciale V, Grisendi G, Banchelli F, D’Amico R, Maiorana A, Sighinolfi P, Stefani A, Morandi U, Dominici M, Aramini B. Isolation and Identification of Cancer Stem-Like Cells in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Pilot Study. Front Oncol. 2019; 9:1394. https://doi.org/10.3389/fonc.2019.01394. [PubMed].
8. Masciale V, Grisendi G, Banchelli F, D’Amico R, Maiorana A, Morandi U, Dominici M, Aramini B. Cancer stem-neuroendocrine cells in an atypical carcinoid case report. Transl Lung Cancer Res. 2019; 8:1157–62. https://doi.org/10.21037/tlcr.2019.12.07. [PubMed].
9. Lathia JD, Liu H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target Oncol. 2017; 12:387–99. https://doi.org/10.1007/s11523-017-0508-3. [PubMed].
10. Osman A, Afify SM, Hassan G, Fu X, Seno A, Seno M. Revisiting Cancer Stem Cells as the Origin of Cancer-Associated Cells in the Tumor Microenvironment: A Hypothetical View from the Potential of iPSCs. Cancers (Basel). 2020; 12:879. https://doi.org/10.3390/cancers12040879. [PubMed].
11. Gao XM, Zhang R, Dong QZ, Qin LX. Properties and feasibility of using cancer stem cells in clinical cancer treatment. Cancer Biol Med. 2016; 13:489–95. https://doi.org/10.20892/j.issn.2095-3941.2016.0076. [PubMed].
12. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F, Cui H. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020; 5:8. https://doi.org/10.1038/s41392-020-0110-5. [PubMed].
13. Harrop R, O’Neill E, Stern PL. Cancer stem cell mobilization and therapeutic targeting of the 5T4 oncofetal antigen. Ther Adv Vaccines Immunother. 2019; 7:2515135518821623. https://doi.org/10.1177/2515135518821623. [PubMed].
14. Du FY, Zhou QF, Sun WJ, Chen GL. Targeting cancer stem cells in drug discovery: current state and future perspectives. World J Stem Cells. 2019; 11:398–420. https://doi.org/10.4252/wjsc.v11.i7.398. [PubMed].
15. Wu G, Song X, Liu J, Li S, Gao W, Qiu M, Yang C, Ma Y, Chen Y. Expression of CD44 and the survival in glioma: a meta-analysis. Biosci Rep. 2020; 40:BSR20200520. https://doi.org/10.1042/BSR20200520. [PubMed].
16. Sun Y. Translational horizons in the tumor microenvironment: harnessing breakthroughs and targeting cures. Med Res Rev. 2015; 35:408–36. https://doi.org/10.1002/med.21338. [PubMed].
17. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017; 23:1124–34. https://doi.org/10.1038/nm.4409. [PubMed].
18. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105–11. https://doi.org/10.1038/35102167. [PubMed].
19. Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016; 2016:1740936. https://doi.org/10.1155/2016/1740936. [PubMed].
20. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3:730–37. https://doi.org/10.1038/nm0797-730. [PubMed].
21. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012; 22:457–72. https://doi.org/10.1038/cr.2012.13. [PubMed].
22. Karsten U, Goletz S. What makes cancer stem cell markers different? Springerplus. 2013; 2:301. https://doi.org/10.1186/2193-1801-2-301. [PubMed].
23. Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol. 2013; Chapter 14:Unit 14.25. https://doi.org/10.1002/0471141755.ph1425s61. [PubMed].
24. Hamid AB, Petreaca RC. Secondary Resistant Mutations to Small Molecule Inhibitors in Cancer Cells. Cancers (Basel). 2020; 12:927. https://doi.org/10.3390/cancers12040927. [PubMed].
25. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014; 13:497–512. https://doi.org/10.1038/nrd4253. [PubMed].
26. Yadav AK, Desai NS. Cancer Stem Cells: Acquisition, Characteristics, Therapeutic Implications, Targeting Strategies and Future Prospects. Stem Cell Rev Rep. 2019; 15:331–55. https://doi.org/10.1007/s12015-019-09887-2. [PubMed].
27. Kim WT, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017; 50:285–98. https://doi.org/10.5483/bmbrep.2017.50.6.039. [PubMed].
28. Xia P. Surface markers of cancer stem cells in solid tumors. Curr Stem Cell Res Ther. 2014; 9:102–11. https://doi.org/10.2174/1574888x09666131217003709. [PubMed].
29. Feng L, Huang S, An G, Wang G, Gu S, Zhao X. Identification of new cancer stem cell markers and signaling pathways in HER-2-positive breast cancer by transcriptome sequencing. Int J Oncol. 2019; 55:1003–18. https://doi.org/10.3892/ijo.2019.4876. [PubMed].
30. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S, Kossatz-Boehlert U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol. 2020; 11:1280. https://doi.org/10.3389/fimmu.2020.01280. [PubMed].
31. Zakaria N, Yusoff NM, Zakaria Z, Lim MN, Baharuddin PJ, Fakiruddin KS, Yahaya B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer. 2015; 15:84. https://doi.org/10.1186/s12885-015-1086-3. [PubMed].
32. Satar NA, Fakiruddin KS, Lim MN, Mok PL, Zakaria N, Fakharuzi NA, Abd Rahman AZ, Zakaria Z, Yahaya BH, Baharuddin P. Novel triple-positive markers identified in human non-small cell lung cancer cell line with chemotherapy-resistant and putative cancer stem cell characteristics. Oncol Rep. 2018; 40:669–81. https://doi.org/10.3892/or.2018.6461. [PubMed].
33. Masciale V, Grisendi G, Banchelli F, D’Amico R, Maiorana A, Sighinolfi P, Stefani A, Morandi U, Dominici M, Aramini B. CD44+/EPCAM+ cells detect a subpopulation of ALDHhigh cells in human non-small cell lung cancer: A chance for targeting cancer stem cells? Oncotarget. 2020; 11:1545–55. https://doi.org/10.18632/oncotarget.27568. [PubMed].
34. Nusblat LM, Tanna S, Roth CM. Gene silencing of HIF-2α disrupts glioblastoma stem cell phenotype. Cancer Drug Resist. 2020; 3:199–208. https://doi.org/10.20517/cdr.2019.96. [PubMed].
35. Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. 2017; 7:13856. https://doi.org/10.1038/s41598-017-14364-2. [PubMed].
36. Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, Ji X, Liu W, Huang B, Luo W, Liu B, Lei Y, Du S, et al. Breast cancer development and progression: risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018; 5:77–106. https://doi.org/10.1016/j.gendis.2018.05.001. [PubMed].
37. Kong DS. Cancer stem cells in brain tumors and their lineage hierarchy. Int J Stem Cells. 2012; 5:12–15. https://doi.org/10.15283/ijsc.2012.5.1.12. [PubMed].
38. Prabavathy D, Swarnalatha Y, Ramadoss N. Lung cancer stem cells-origin, characteristics and therapy. Stem Cell Investig. 2018; 5:6. https://doi.org/10.21037/sci.2018.02.01. [PubMed].
39. Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, Liu F, Liu J, Dai B, Chen X, Wang F, Zeng F, Xu H, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2012; 22:248–58. https://doi.org/10.1038/cr.2011.109. [PubMed].
40. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367:645–48. https://doi.org/10.1038/367645a0. [PubMed].
41. Scioli MG, Storti G, D’Amico F, Gentile P, Fabbri G, Cervelli V, Orlandi A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers (Basel). 2019; 11:1021. https://doi.org/10.3390/cancers11071021. [PubMed].
42. Wang T, Shigdar S, Gantier MP, Hou Y, Wang L, Li Y, Shamaileh HA, Yin W, Zhou SF, Zhao X, Duan W. Cancer stem cell targeted therapy: progress amid controversies. Oncotarget. 2015; 6:44191–206. https://doi.org/10.18632/oncotarget.6176. [PubMed].
43. Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009; 8:806–23. https://doi.org/10.1038/nrd2137. [PubMed].
44. Liu G, David BT, Trawczynski M, Fessler RG. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev Rep. 2020; 16:3–32. https://doi.org/10.1007/s12015-019-09935-x. [PubMed].
45. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555–67. https://doi.org/10.1016/j.stem.2007.08.014. [PubMed].
46. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432:396–401. https://doi.org/10.1038/nature03128. [PubMed].
47. Lehmann C, Jobs G, Thomas M, Burtscher H, Kubbies M. Established breast cancer stem cell markers do not correlate with in vivo tumorigenicity of tumor-initiating cells. Int J Oncol. 2012; 41:1932–42. https://doi.org/10.3892/ijo.2012.1654. [PubMed].
48. Vikram R, Chou WC, Hung SC, Shen CY. Tumorigenic and Metastatic Role of CD44-/low/CD24-/low Cells in Luminal Breast Cancer. Cancers (Basel). 2020; 12:1239. https://doi.org/10.3390/cancers12051239. [PubMed].
49. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015; 29:1203–17. https://doi.org/10.1101/gad.261982.115. [PubMed].
50. Gimple RC, Bhargava S, Dixit D, Rich JN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019; 33:591–609. https://doi.org/10.1101/gad.324301.119. [PubMed].
51. Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, Febbo PG, Wechsler-Reya RJ. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009; 15:135–47. https://doi.org/10.1016/j.ccr.2008.12.016. [PubMed].
52. Kenney-Herbert E, Al-Mayhani T, Piccirillo SG, Fowler J, Spiteri I, Jones P, Watts C. CD15 Expression Does Not Identify a Phenotypically or Genetically Distinct Glioblastoma Population. Stem Cells Transl Med. 2015; 4:822–31. https://doi.org/10.5966/sctm.2014-0047. [PubMed].
53. Huls M, Russel FG, Masereeuw R. The role of ATP binding cassette transporters in tissue defense and organ regeneration. J Pharmacol Exp Ther. 2009; 328:3–9. https://doi.org/10.1124/jpet.107.132225. [PubMed].
54. Begicevic RR, Falasca M. ABC Transporters in Cancer Stem Cells: beyond Chemoresistance. Int J Mol Sci. 2017; 18:2362. https://doi.org/10.3390/ijms18112362. [PubMed].
55. Park JW, Jung KH, Byun Y, Lee JH, Moon SH, Cho YS, Lee KH. ATP-binding Cassette Transporters Substantially Reduce Estimates of ALDH-positive Cancer Cells based on Aldefluor and AldeRed588 Assays. Sci Rep. 2019; 9:6462. https://doi.org/10.1038/s41598-019-42954-9. [PubMed].
56. Hu J, Li G, Zhang P, Zhuang X, Hu G. A CD44v+ subpopulation of breast cancer stem-like cells with enhanced lung metastasis capacity. Cell Death Dis. 2017; 8:e2679. https://doi.org/10.1038/cddis.2017.72. [PubMed].
57. Fleischman AG. ALDH marks leukemia stem cell. Blood. 2012; 119:3376–77. https://doi.org/10.1182/blood-2012-02-406751. [PubMed].
58. Triviai I, Stübig T, Niebuhr B, Hussein K, Tsiftsoglou A, Fehse B, Stocking C, Kröger N. CD133 marks a stem cell population that drives human primary myelofibrosis. Haematologica. 2015; 100:768–79. https://doi.org/10.3324/haematol.2014.118463. [PubMed].
59. Fathi F, Rahbarghazi R, Movassaghpour AA, Rashidi MR. Detection of CD133-marked cancer stem cells by surface plasmon resonance: its application in leukemia patients. Biochim Biophys Acta Gen Subj. 2019; 1863:1575–82. https://doi.org/10.1016/j.bbagen.2019.06.009. [PubMed].
60. Hoang VT, Buss EC, Wang W, Hoffmann I, Raffel S, Zepeda-Moreno A, Baran N, Wuchter P, Eckstein V, Trumpp A, Jauch A, Ho AD, Lutz C. The rarity of ALDH(+) cells is the key to separation of normal versus leukemia stem cells by ALDH activity in AML patients. Int J Cancer. 2015; 137:525–36. https://doi.org/10.1002/ijc.29410. [PubMed].
61. Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, Galkin S, Collector MI, Perkins B, Levis MJ, Griffin CA, Sharkis SJ, Borowitz MJ, et al. A clinically relevant population of leukemic CD34(+) CD38(-) cells in acute myeloid leukemia. Blood. 2012; 119:3571–77. https://doi.org/10.1182/blood-2011-06-364182. [PubMed].
62. Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012; 6:620–36. https://doi.org/10.1016/j.molonc.2012.10.006. [PubMed].
63. Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017; 16:29. https://doi.org/10.1186/s12943-017-0596-9. [PubMed].
64. Zhao Y, Dong Q, Li J, Zhang K, Qin J, Zhao J, Sun Q, Wang Z, Wartmann T, Jauch KW, Nelson PJ, Qin L, Bruns C. Targeting cancer stem cells and their niche: perspectives for future therapeutic targets and strategies. Semin Cancer Biol. 2018; 53:139–55. https://doi.org/10.1016/j.semcancer.2018.08.002. [PubMed].
65. Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, Honorio S, Xie Y, Scaglioni PP, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010; 70:9937–48. https://doi.org/10.1158/0008-5472.CAN-10-0881. [PubMed].
66. Vassalli G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019; 2019:3904645. https://doi.org/10.1155/2019/3904645. [PubMed].
67. Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther. 2008; 3:237–46. https://doi.org/10.2174/157488808786734006. [PubMed].
68. Xu X, Chai S, Wang P, Zhang C, Yang Y, Yang Y, Wang K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015; 369:50–57. https://doi.org/10.1016/j.canlet.2015.08.018. [PubMed].
69. Toledo-Guzmán ME, Hernández MI, Gómez-Gallegos ÁA, Ortiz-Sánchez E. ALDH as a Stem Cell Marker in Solid Tumors. Curr Stem Cell Res Ther. 2019; 14:375–88. https://doi.org/10.2174/1574888X13666180810120012. [PubMed].
70. Wang L, Zuo X, Xie K, Wei D. The Role of CD44 and Cancer Stem Cells. Methods Mol Biol. 2018; 1692:31–42. https://doi.org/10.1007/978-1-4939-7401-6_3. [PubMed].
71. Yan Y, Zuo X, Wei D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. 2015; 4:1033–43. https://doi.org/10.5966/sctm.2015-0048. [PubMed].
72. Chang JC. Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016; 95:S20–25. https://doi.org/10.1097/MD.0000000000004766. [PubMed].
73. Li L, Li JC, Yang H, Zhang X, Liu LL, Li Y, Zeng TT, Zhu YH, Li XD, Li Y, Xie D, Fu L, Guan XY. Expansion of cancer stem cell pool initiates lung cancer recurrence before angiogenesis. Proc Natl Acad Sci U S A. 2018; 115:E8948–57. https://doi.org/10.1073/pnas.1806219115. [PubMed].
74. Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore). 2016; 95:S2–7. https://doi.org/10.1097/MD.0000000000004764. [PubMed].
75. Gasch C, Ffrench B, O’Leary JJ, Gallagher MF. Catching moving targets: cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol Cancer. 2017; 16:43. https://doi.org/10.1186/s12943-017-0601-3. [PubMed].
76. Aponte PM, Caicedo A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017; 2017:5619472. https://doi.org/10.1155/2017/5619472. [PubMed].
77. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015; 16:225–38. https://doi.org/10.1016/j.stem.2015.02.015. [PubMed].
78. Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015; 16:239–53. https://doi.org/10.1016/j.stem.2015.02.019. [PubMed].
79. He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol. 2009; 25:377–406. https://doi.org/10.1146/annurev.cellbio.042308.113248. [PubMed].
80. Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008; 132:598–611. https://doi.org/10.1016/j.cell.2008.01.038. [PubMed].
81. Rezza A, Wang Z, Sennett R, Qiao W, Wang D, Heitman N, Mok KW, Clavel C, Yi R, Zandstra P, Ma’ayan A, Rendl M. Signaling Networks among Stem Cell Precursors, Transit-Amplifying Progenitors, and their Niche in Developing Hair Follicles. Cell Rep. 2016; 14:3001–18. https://doi.org/10.1016/j.celrep.2016.02.078. [PubMed].
82. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014; 54:716–27. https://doi.org/10.1016/j.molcel.2014.05.015. [PubMed].
83. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8:97–106. https://doi.org/10.1038/nrclinonc.2010.196. [PubMed].
84. McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH, Daggett J Jr, Cullen K, Kantoff E, et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A. 2012; 109:E2939–48. https://doi.org/10.1073/pnas.1206400109. [PubMed].
85. Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, Miele L. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010; 16:3141–52. https://doi.org/10.1158/1078-0432.CCR-09-2823. [PubMed].
86. Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012; 198:281–93. https://doi.org/10.1083/jcb.201202014. [PubMed].
87. Agliano A, Calvo A, Box C. The challenge of targeting cancer stem cells to halt metastasis. Semin Cancer Biol. 2017; 44:25–42. https://doi.org/10.1016/j.semcancer.2017.03.003. [PubMed].
88. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017; 16:41. https://doi.org/10.1186/s12943-017-0600-4. [PubMed].
89. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–28. https://doi.org/10.1126/science. [PubMed].
90. Cruz MH, Sidén A, Calaf GM, Delwar ZM, Yakisich JS. The stemness phenotype model. ISRN Oncol. 2012; 2012:392647. https://doi.org/10.5402/2012/392647. [PubMed].
91. Pantic I. Cancer stem cell hypotheses: impact on modern molecular physiology and pharmacology research. J Biosci. 2011; 36:957–61. https://doi.org/10.1007/s12038-011-9155-5. [PubMed].
92. Welte Y, Adjaye J, Lehrach HR, Regenbrecht CR. Cancer stem cells in solid tumors: elusive or illusive? Cell Commun Signal. 2010; 8:6. https://doi.org/10.1186/1478-811X-8-6. [PubMed].
93. Sun HR, Wang S, Yan SC, Zhang Y, Nelson PJ, Jia HL, Qin LX, Dong QZ. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front Oncol. 2019; 9:1104. https://doi.org/10.3389/fonc.2019.01104. [PubMed].
94. Jiang W, Peng J, Zhang Y, Cho WC, Jin K. The implications of cancer stem cells for cancer therapy. Int J Mol Sci. 2012; 13:16636–57. https://doi.org/10.3390/ijms131216636. [PubMed].
95. Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet. 2015; 6:157. https://doi.org/10.3389/fgene.2015.00157. [PubMed].
96. Adjiri A. Tracing the path of cancer initiation: the AA protein-based model for cancer genesis. BMC Cancer. 2018; 18:831. https://doi.org/10.1186/s12885-018-4739-1. [PubMed].
97. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016; 8:a019505. https://doi.org/10.1101/cshperspect.a019505. [PubMed].
98. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019; 234:8381–95. https://doi.org/10.1002/jcp.27740. [PubMed].
99. De Angelis ML, Francescangeli F, La Torre F, Zeuner A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front Oncol. 2019; 9:626. https://doi.org/10.3389/fonc.2019.00626. [PubMed].
100. Olmeda F, Ben Amar M. Clonal pattern dynamics in tumor: the concept of cancer stem cells. Sci Rep. 2019; 9:15607. https://doi.org/10.1038/s41598-019-51575-1. [PubMed].
101. Hale JS, Li M, Lathia JD. The malignant social network: cell-cell adhesion and communication in cancer stem cells. Cell Adh Migr. 2012; 6:346–55. https://doi.org/10.4161/cam.21294. [PubMed].
102. Badri H, Leder K. Optimal treatment and stochastic modeling of heterogeneous tumors. Biol Direct. 2016; 11:40. https://doi.org/10.1186/s13062-016-0142-5. [PubMed].
103. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014; 1840:2506–19. https://doi.org/10.1016/j.bbagen.2014.01.010. [PubMed].
104. Liu A, Yu X, Liu S. Pluripotency transcription factors and cancer stem cells: small genes make a big difference. Chin J Cancer. 2013; 32:483–87. https://doi.org/10.5732/cjc.012.10282. [PubMed].
105. Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004; 118:635–648. https://doi.org/10.1016/j.cell.2004.08.012. [PubMed].
106. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, Brohée S, Salmon I, Dubois C, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014; 511:246–50. https://doi.org/10.1038/nature13305. [PubMed].
107. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013; 501:328–37. https://doi.org/10.1038/nature12624. [PubMed].
108. Gutknecht MF, Bouton AH. Functional significance of mononuclear phagocyte populations generated through adult hematopoiesis. J Leukoc Biol. 2014; 96:969–80. https://doi.org/10.1189/jlb.1RI0414-195R. [PubMed].
109. Guo Q, Jin Z, Yuan Y, Liu R, Xu T, Wei H, Xu X, He S, Chen S, Shi Z, Hou W, Hua B. Corrigendum to “New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy”. J Immunol Res. 2018; 2018:6728474. https://doi.org/10.1155/2018/6728474. [PubMed].
110. Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: phenotypical vs. Functional Differentiation. Front Immunol. 2014; 5:514. https://doi.org/10.3389/fimmu.2014.00514. [PubMed].
111. Zheng X, Turkowski K, Mora J, Brüne B, Seeger W, Weigert A, Savai R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017; 8:48436–52. https://doi.org/10.18632/oncotarget.17061. [PubMed].
112. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014; 6:1670–90. https://doi.org/10.3390/cancers6031670. [PubMed].
113. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019; 12:76. https://doi.org/10.1186/s13045-019-0760-3. [PubMed].
114. Räihä MR, Puolakkainen PA. Tumor-associated macrophages (TAMs) as biomarkers for gastric cancer: A review. Chronic Dis Transl Med. 2018; 4:156–63. https://doi.org/10.1016/j.cdtm.2018.07.001. [PubMed].
115. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017; 14:399–416. https://doi.org/10.1038/nrclinonc.2016.217. [PubMed].
116. Lin CN, Wang CJ, Chao YJ, Lai MD, Shan YS. The significance of the co-existence of osteopontin and tumor-associated macrophages in gastric cancer progression. BMC Cancer. 2015; 15:128. https://doi.org/10.1186/s12885-015-1114-3. [PubMed].
117. Wang XL, Jiang JT, Wu CP. Prognostic significance of tumor-associated macrophage infiltration in gastric cancer: a meta-analysis. Genet Mol Res. 2016; 15. https://doi.org/10.4238/gmr15049040. [PubMed].
118. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp Ther Med. 2019; 18:4490–98. https://doi.org/10.3892/etm.2019.8068. [PubMed].
119. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci. 2017; 18:2574. https://doi.org/10.3390/ijms18122574. [PubMed].
120. Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology. 2019; 8:1596004. https://doi.org/10.1080/2162402X.2019.1596004. [PubMed].
121. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014; 41:49–61. https://doi.org/10.1016/j.immuni.2014.06.010. [PubMed].
122. Yang C, He L, He P, Liu Y, Wang W, He Y, Du Y, Gao F. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol. 2015; 32:352. https://doi.org/10.1007/s12032-014-0352-6. [PubMed].
123. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012; 9:193–99. https://doi.org/10.7150/ijms.3635. [PubMed].
124. Genard G, Lucas S, Michiels C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front Immunol. 2017; 8:828. https://doi.org/10.3389/fimmu.2017.00828. [PubMed].
125. Anfray C, Ummarino A, Andón FT, Allavena P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells. 2019; 9:E46. https://doi.org/10.3390/cells9010046. [PubMed].
126. Pinto TA, Pinto LM, Cardoso PA, Monteiro C, Pinto TM, Maia FA, Castro P, Figueira R, Monteiro A, Marques M, Mareel M, Dos Santos SG, Seruca R, et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci Rep. 2016; 6:18765. https://doi.org/10.1038/srep18765. [PubMed].
127. Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautès-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: anticancer radioimmunotherapy. Oncoimmunology. 2013; 2:e25595. https://doi.org/10.4161/onci.25595. [PubMed].
128. Sulli G, Di Micco R, d'Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012; 12:709–20. https://doi.org/10.1038/nrc3344. [PubMed].
129. Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010; 31:363–72. https://doi.org/10.1007/s13277-010-0042-8. [PubMed].
130. Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. 2014; 4:1. https://doi.org/10.3389/fonc.2014.00001. [PubMed].
131. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018; 32:1267–84. https://doi.org/10.1101/gad.314617.118. [PubMed].
132. Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020 May 27. https://doi.org/10.1038/s41417-020-0183-x. [Epub ahead of print]. [PubMed]
133. Pandya PH, Murray ME, Pollok KE, Renbarger JL. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J Immunol Res. 2016; 2016:4273943. https://doi.org/10.1155/2016/4273943. [PubMed].
134. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 121:1–14. https://doi.org/10.1111/j.1365-2567.2007.02587.x. [PubMed].
135. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol. 2014; 27:16–25. https://doi.org/10.1016/j.coi.2014.01.004. [PubMed].
136. Castagnoli L, De Santis F, Volpari T, Vernieri C, Tagliabue E, Di Nicola M, Pupa SM. Cancer Stem Cells: Devil or Savior-Looking behind the Scenes of Immunotherapy Failure. Cells. 2020; 9:555. https://doi.org/10.3390/cells9030555. [PubMed].
137. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, Galli R, Parmiani G, Maccalli C. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010; 16:800–13. https://doi.org/10.1158/1078-0432.CCR-09-2730. [PubMed].
138. Hirohashi Y, Torigoe T, Tsukahara T, Kanaseki T, Kochin V, Sato N. Immune responses to human cancer stem-like cells/cancer-initiating cells. Cancer Sci. 2016; 107:12–17. https://doi.org/10.1111/cas.12830. [PubMed].
139. Ma J, Frank MH. Tumor initiation in human malignant melanoma and potential cancer therapies. Anticancer Agents Med Chem. 2010; 10:131–36. https://doi.org/10.2174/187152010790909254. [PubMed].
140. Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, Zhou J, Hodi FS, Spagnoli GC, Murphy GF, Frank MH. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010; 70:697–708. https://doi.org/10.1158/0008-5472.CAN-09-1592. [PubMed].
141. Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci. 2009; 1176:154–69. https://doi.org/10.1111/j.1749-6632.2009.04568.x. [PubMed].
142. Chartrain M, Riond J, Stennevin A, Vandenberghe I, Gomes B, Lamant L, Meyer N, Gairin JE, Guilbaud N, Annereau JP. Melanoma chemotherapy leads to the selection of ABCB5-expressing cells. PLoS One. 2012; 7:e36762. https://doi.org/10.1371/journal.pone.0036762. [PubMed].
143. Frank NY, Frank MH. ABCB5 gene amplification in human leukemia cells. Leuk Res. 2009; 33:1303–05. https://doi.org/10.1016/j.leukres.2009.04.035. [PubMed].
144. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016; 37:208–20. https://doi.org/10.1016/j.it.2016.01.004. [PubMed].
145. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010; 70:99–108. https://doi.org/10.1158/0008-5472.CAN-09-1882. [PubMed].
146. Argyle D, Kitamura T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front Immunol. 2018; 9:2629. https://doi.org/10.3389/fimmu.2018.02629. [PubMed].
147. Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1-FOXK1 axis promotes recruitment of tumor-associated macrophages. Cell Rep. 2017; 21:2471–86. https://doi.org/10.1016/j.celrep.2017.11.014. [PubMed].
148. Wu Y, Chen M, Wu P, Chen C, Xu ZP, Gu W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol. 2017; 44:602–04. https://doi.org/10.1111/1440-1681.12732. [PubMed].
149. Wu T, Dai Y, Wang W, Teng G, Jiao H, Shuai X, Zhang R, Zhao P, Qiao L. Macrophage targeting contributes to the inhibitory effects of embelin on colitis-associated cancer. Oncotarget. 2016; 7:19548–58. https://doi.org/10.18632/oncotarget.6969. [PubMed].
150. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013; 18:43–73. https://doi.org/10.1615/CritRevOncog.v18.i1-2.40. [PubMed].
151. Zarif JC, Taichman RS, Pienta KJ. TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology. 2014; 3:e941734. https://doi.org/10.4161/21624011.2014.941734. [PubMed].
152. Wang H, Wang X, Li X, Fan Y, Li G, Guo C, Zhu F, Zhang L, Shi Y. CD68(+)HLA-DR(+) M1-like macrophages promote motility of HCC cells via NF-κB/FAK pathway. Cancer Lett. 2014; 345:91–99. https://doi.org/10.1016/j.canlet.2013.11.013. [PubMed].
153. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014; 5:75. https://doi.org/10.3389/fphys.2014.00075. [PubMed].
154. Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, Lopez-Camarillo C, Hernadez de la Cruz ON, Lopez-Gonzalez JS. Contribution of Angiogenesis to Inflammation and Cancer. Front Oncol. 2019; 9:1399. https://doi.org/10.3389/fonc.2019.01399. [PubMed].
155. Pelekanou V, Villarroel-Espindola F, Schalper KA, Pusztai L, Rimm DL. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in ER-positive and -negative cancers. Breast Cancer Res. 2018; 20:154. https://doi.org/10.1186/s13058-018-1076-x. [PubMed].
156. Vinogradov S, Warren G, Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine (Lond). 2014; 9:695–707. https://doi.org/10.2217/nnm.14.13. [PubMed].
157. Hu G, Guo M, Xu J, Wu F, Fan J, Huang Q, Yang G, Lv Z, Wang X, Jin Y. Nanoparticles Targeting Macrophages as Potential Clinical Therapeutic Agents Against Cancer and Inflammation. Front Immunol. 2019; 10:1998. https://doi.org/10.3389/fimmu.2019.01998. [PubMed].
158. Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017; 9:289–302. https://doi.org/10.2217/imt-2016-0135. [PubMed].
159. Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017; 316:1–10. https://doi.org/10.1016/j.cellimm.2017.04.005. [PubMed].
160. Zhu C, Kros JM, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol. 2017; 19:1435–46. https://doi.org/10.1093/neuonc/nox081. [PubMed].
161. Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P. Cancer stem cells and tumor-associated macrophages: a roadmap for multitargeting strategies. Oncogene. 2016; 35:671–82. https://doi.org/10.1038/onc.2015.132. [PubMed].
162. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011; 108:1397–402. https://doi.org/10.1073/pnas.1018898108. [PubMed].
163. Werner-Klein M, Grujovic A, Irlbeck C, Obradović M, Hoffmann M, Koerkel-Qu H, Lu X, Treitschke S, Köstler C, Botteron C, Weidele K, Werno C, Polzer B, et al. Interleukin-6 trans-signaling is a candidate mechanism to drive progression of human DCCs during clinical latency. Nat Commun. 2020; 11:4977. https://doi.org/10.1038/s41467-020-18701-4. [PubMed].
164. Sternberg C, Gruber W, Eberl M, Tesanovic S, Stadler M, Elmer DP, Schlederer M, Grund S, Roos S, Wolff F, Kaur S, Mangelberger D, Lehrach H, et al. Synergistic cross-talk of hedgehog and interleukin-6 signaling drives growth of basal cell carcinoma. Int J Cancer. 2018; 143:2943–54. https://doi.org/10.1002/ijc.31724. [PubMed].
165. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, Zou W, Welling TH. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014; 147:1393–404. https://doi.org/10.1053/j.gastro.2014.08.039. [PubMed].
166. Wang T, Song P, Zhong T, Wang X, Xiang X, Liu Q, Chen H, Xia T, Liu H, Niu Y, Hu Y, Xu L, Shao Y, et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene. 2019; 38:4932–47. https://doi.org/10.1038/s41388-019-0763-0. [PubMed].
167. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH, Wei LX. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014; 352:160–68. https://doi.org/10.1016/j.canlet.2014.05.008. [PubMed].
168. Velasco-Velázquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011; 179:2–11. https://doi.org/10.1016/j.ajpath.2011.03.005. [PubMed].
169. Zhang C, Ma K, Li WY. IL-6 Promotes Cancer Stemness and Oncogenicity in U2OS and MG-63 Osteosarcoma Cells by Upregulating the OPN-STAT3 Pathway. J Cancer. 2019; 10:6511–25. https://doi.org/10.7150/jca.29931. [PubMed].
170. Chen Y, Zhang F, Tsai Y, Yang X, Yang L, Duan S, Wang X, Keng P, Lee SO. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat Oncol. 2015; 10:227. https://doi.org/10.1186/s13014-015-0534-1. [PubMed].
171. Zhang F, Duan S, Tsai Y, Keng PC, Chen Y, Lee SO, Chen Y. Cisplatin treatment increases stemness through upregulation of hypoxia-inducible factors by interleukin-6 in non-small cell lung cancer. Cancer Sci. 2016; 107:746–54. https://doi.org/10.1111/cas.12937. [PubMed].
172. Lee SO, Yang X, Duan S, Tsai Y, Strojny LR, Keng P, Chen Y. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget. 2016; 7:6626–38. https://doi.org/10.18632/oncotarget.6570. [PubMed].
173. Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, Wang X, Zhang J, Guo L, Wang S, Chen Z, Wang Z, Xiang S. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis. 2020; 11:234. https://doi.org/10.1038/s41419-020-2435-y. [PubMed].
174. Patel S, Alam A, Pant R, Chattopadhyay S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front Immunol. 2019; 10:2872. https://doi.org/10.3389/fimmu.2019.02872. [PubMed].
175. López de Andrés J, Griñán-Lisón C, Jiménez G, Marchal JA. Cancer stem cell secretome in the tumor microenvironment: a key point for an effective personalized cancer treatment. J Hematol Oncol. 2020; 13:136. https://doi.org/10.1186/s13045-020-00966-3. [PubMed].
176. Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009; 5:1145–68. https://doi.org/10.2217/fon.09.90. [PubMed].
177. Hou YC, Chao YJ, Tung HL, Wang HC, Shan YS. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer. 2014; 120:2766–77. https://doi.org/10.1002/cncr.28774. [PubMed].
178. Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, Reinhardt F, Donnenberg VS, Bhargava R, Carr SA, Weinberg RA. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014; 16:1105–17. https://doi.org/10.1038/ncb3041. [PubMed].
179. Ma Z, Zhao X, Deng M, Huang Z, Wang J, Wu Y, Cui D, Liu Y, Liu R, Ouyang G. Bone Marrow Mesenchymal Stromal Cell-Derived Periostin Promotes B-ALL Progression by Modulating CCL2 in Leukemia Cells. Cell Rep. 2019; 26:1533–1543.e4. https://doi.org/10.1016/j.celrep.2019.01.034. [PubMed].
180. Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012; 2:840–55. https://doi.org/10.1158/2159-8290.CD-12-0101. [PubMed].
181. Park TS, Donnenberg VS, Donnenberg AD, Zambidis ET, Zimmerlin L. Dynamic Interactions Between Cancer Stem Cells And Their Stromal Partners. Curr Pathobiol Rep. 2014; 2:41–52. https://doi.org/10.1007/s40139-013-0036-5. [PubMed].
182. Tao W, Chu C, Zhou W, Huang Z, Zhai K, Fang X, Huang Q, Zhang A, Wang X, Yu X, Huang H, Wu Q, Sloan AE, et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat Commun. 2020; 11:3015. https://doi.org/10.1038/s41467-020-16827-z. [PubMed].
183. Gong M, Wang X, Mu L, Wang Y, Pan J, Yuan X, Zhou H, Xing J, Wang R, Sun J, Liu Q, Zhang X, Wang L, et al. SRC-1 enhances the stemness of glioblastoma by activating lncRNA XIST/ miR-152/ KLF4 pathway. Cancer Sci. 2020 Oct 14. https://doi.org/10.1111/cas.14685. [Epub ahead of print]. [PubMed]
184. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, Xing F, Liu W, Pandey PR, Fukuda K, Modur V, Ghosh A, Wilber A, Watabe K. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012; 72:537–47. https://doi.org/10.1158/0008-5472.CAN-11-1678. [PubMed].
185. Tseng HC, Arasteh A, Kaur K, Kozlowska A, Topchyan P, Jewett A. Differential Cytotoxicity but Augmented IFN-γ Secretion by NK Cells after Interaction with Monocytes from Humans, and Those from Wild Type and Myeloid-Specific COX-2 Knockout Mice. Front Immunol. 2015; 6:259. https://doi.org/10.3389/fimmu.2015.00259. [PubMed].
186. Tseng HC, Bui V, Man YG, Cacalano N, Jewett A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front Immunol. 2014; 5:269. https://doi.org/10.3389/fimmu.2014.00269. [PubMed].
187. Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. 2014; 5:127. https://doi.org/10.3389/fimmu.2014.00127. [PubMed].
188. Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006; 42:717–27. https://doi.org/10.1016/j.ejca.2006.01.003. [PubMed].
189. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014; 35:3945–51. https://doi.org/10.1007/s13277-013-1561-x. [PubMed].
190. Rinkenbaugh AL, Baldwin AS. The NF-κB Pathway and Cancer Stem Cells. Cells. 2016; 5:16. https://doi.org/10.3390/cells5020016. [PubMed].
191. Jinushi M, Chiba S, Yoshiyama H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011; 108:12425–12430. https://doi.org/10.1073/pnas.1106645108. [PubMed].
192. Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, Markowitz D, Reisfeld RA, Luo Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. 2013; 31:248–258. https://doi.org/10.1002/stem.1281. [PubMed].
193. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, Jiang XF, Yang L, Zhang P, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012; 189:444–453. https://doi.org/10.4049/jimmunol.1103248. [PubMed].
194. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010; 89:219–29. https://doi.org/10.1177/0022034509359125. [PubMed].
195. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-Stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J Exp Med. 2001; 193:727–740. https://doi.org/10.1084/jem.193.6.727. [PubMed].
196. Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013; 5:1075–87. https://doi.org/10.2217/imt.13.102. [PubMed].
197. Ding J, Jin W, Chen C, Shao Z, Wu J. Tumor associated macrophage × cancer cell hybrids may acquire cancer stem cell properties in breast cancer. PLoS One. 2012; 7:e41942. https://doi.org/10.1371/journal.pone.0041942. [PubMed].
198. Pawelek J, Chakraborty A, Lazova R, Yilmaz Y, Cooper D, Brash D, Handerson T. Co-opting macrophage traits in cancer progression: a consequence of tumor cell fusion? Contrib Microbiol. 2006; 13:138–55. https://doi.org/10.1159/000092970. [PubMed].
199. Dittmar T, Nagler C, Niggemann B, Zänker KS. The dark side of stem cells: triggering cancer progression by cell fusion. Curr Mol Med. 2013; 13:735–50. https://doi.org/10.2174/1566524011313050005. [PubMed].
200. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007; 25:267–96. https://doi.org/10.1146/annurev.immunol.25.022106.141609. [PubMed].
201. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: t-cell immunotherapy revives an old story. Mol Med Today. 1999; 5:178–86. https://doi.org/10.1016/s1357-4310(99)01451-3. [PubMed].
202. Bicknell DC, Rowan A, Bodmer WF. Beta 2-microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proc Natl Acad Sci U S A. 1994; 91:4751–55. https://doi.org/10.1073/pnas.91.11.4751. [PubMed].
203. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016; 17:e542–e551. https://doi.org/10.1016/S1470-2045(16)30406-5. [PubMed].
204. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019; 19:133–50. https://doi.org/10.1038/s41568-019-0116-x. [PubMed].
205. Stankovic B, Bjørhovde HA, Skarshaug R, Aamodt H, Frafjord A, Müller E, Hammarström C, Beraki K, Bækkevold ES, Woldbæk PR, Helland Å, Brustugun OT, Øynebråten I. Immune Cell Composition in Human Non-small Cell Lung Cancer. Front Immunol. 2019; 9:3101. https://doi.org/10.3389/fimmu.2018.03101. [PubMed].
206. Wang Z, Cao Y, Albino AP, Zeff RA, Houghton A, Ferrone S. Lack of HLA class I antigen expression by melanoma cells SK-MEL-33 caused by a reading frameshift in beta 2-microglobulin messenger RNA. J Clin Invest. 1993; 91:684–92. https://doi.org/10.1172/JCI116249. [PubMed].
207. Natali PG, Nicotra MR, Bigotti A, Venturo I, Marcenaro L, Giacomini P, Russo C. Selective changes in expression of HLA class I polymorphic determinants in human solid tumors. Proc Natl Acad Sci U S A. 1989; 86:6719–23. https://doi.org/10.1073/pnas.86.17.6719. [PubMed].
208. Lanier LL. NK cell recognition. Annu Rev Immunol. 2005; 23:225–74. https://doi.org/10.1146/annurev.immunol.23.021704.115526. [PubMed].
209. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002; 419:734–38. https://doi.org/10.1038/nature01112. [PubMed].
210. Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2006; 103:9190–95. https://doi.org/10.1073/pnas.0603503103. [PubMed].
211. Mizoguchi H, O’Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992; 258:1795–98. https://doi.org/10.1126/science.1465616. [PubMed].
212. Koneru M, Schaer D, Monu N, Ayala A, Frey AB. Defective proximal TCR signaling inhibits CD8+ tumor-infiltrating lymphocyte lytic function. J Immunol. 2005; 174:1830–40. https://doi.org/10.4049/jimmunol.174.4.1830. [PubMed].
213. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, Wang X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front Immunol. 2019; 10:2298. https://doi.org/10.3389/fimmu.2019.02298. [PubMed].
214. Kim HR, Ha SJ, Hong MH, Heo SJ, Koh YW, Choi EC, Kim EK, Pyo KH, Jung I, Seo D, Choi J, Cho BC, Yoon SO. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci Rep. 2016; 6:36956. https://doi.org/10.1038/srep36956. [PubMed].
215. Codony-Servat J, Rosell R. Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res. 2015; 4:689–703. https://doi.org/10.3978/j.issn.2218-6751.2015.12.11. [PubMed].
216. Marshall HT, Djamgoz MB. Immuno-Oncology: Emerging Targets and Combination Therapies. Front Oncol. 2018; 8:315. https://doi.org/10.3389/fonc.2018.00315. [PubMed].
217. Lackey A, Donington JS. Surgical management of lung cancer. Semin Intervent Radiol. 2013; 30:133–40. https://doi.org/10.1055/s-0033-1342954. [PubMed].
218. Howington JA, Blum MG, Chang AC, Balekian AA, Murthy SC. Treatment of stage I and II non-small cell lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013; 143:e278S–e313S. https://doi.org/10.1378/chest.12-2359. [PubMed].
219. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, Lin SH. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front Pharmacol. 2018; 9:185. https://doi.org/10.3389/fphar.2018.00185. [PubMed].
220. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancers (Basel). 2014; 6:1769–92. https://doi.org/10.3390/cancers6031769. [PubMed].
221. Tu LC, Foltz G, Lin E, Hood L, Tian Q. Targeting stem cells-clinical implications for cancer therapy. Curr Stem Cell Res Ther. 2009; 4:147–53. https://doi.org/10.2174/157488809788167373. [PubMed].
222. Hussain F, Freissmuth M, Völkel D, Thiele M, Douillard P, Antoine G, Thurner P, Ehrlich H, Schwarz HP, Scheiflinger F, Kerschbaumer RJ. Human anti-macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo. Mol Cancer Ther. 2013; 12:1223–34. https://doi.org/10.1158/1535-7163.MCT-12-0988. [PubMed].
223. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014; 25:846–59. https://doi.org/10.1016/j.ccr.2014.05.016. [PubMed].
224. Guda MR, Rashid MA, Asuthkar S, Jalasutram A, Caniglia JL, Tsung AJ, Velpula KK. Pleiotropic role of macrophage migration inhibitory factor in cancer. Am J Cancer Res. 2019; 9:2760–73. [PubMed].
225. Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: the root of tumor recurrence and metastases. Semin Cancer Biol. 2017; 44:10–24. https://doi.org/10.1016/j.semcancer.2017.02.011. [PubMed].
226. Iqbal W, Alkarim S, AlHejin A, Mukhtar H, Saini KS. Targeting signal transduction pathways of cancer stem cells for therapeutic opportunities of metastasis. Oncotarget. 2016; 7:76337–53. https://doi.org/10.18632/oncotarget.10942. [PubMed].
227. Yamashina T, Baghdadi M, Yoneda A, Kinoshita I, Suzu S, Dosaka-Akita H, Jinushi M. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 2014; 74:2698–709. https://doi.org/10.1158/0008-5472.can-13-2169. [PubMed].
228. Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, Kleeff J, Sainz B Jr, Heeschen C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin Cancer Res. 2015; 21:2325–37. https://doi.org/10.1158/1078-0432.CCR-14-1399. [PubMed].
229. Lee TK, Cheung VC, Lu P, Lau EY, Ma S, Tang KH, Tong M, Lo J, Ng IO. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology. 2014; 60:179–91. https://doi.org/10.1002/hep.27070. [PubMed].
230. Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009; 19:72–80. https://doi.org/10.1016/j.tcb.2008.12.001. [PubMed].
231. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia. 2012; 26:2538–45. https://doi.org/10.1038/leu.2012.141. [PubMed].
232. Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J Jr, Chang HY, van de Rijn M, Shortliffe L, Weissman IL. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. 2009; 106:14016–21. https://doi.org/10.1073/pnas.0906549106. [PubMed].
233. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, van Rooijen N, Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009; 138:286–99. https://doi.org/10.1016/j.cell.2009.05.045. [PubMed].
234. Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009; 138:722–37. https://doi.org/10.1016/j.cell.2009.07.039. [PubMed].
235. Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine (Lond). 2013; 9:1–14. https://doi.org/10.1016/j.nano.2012.05.013. [PubMed].
236. Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006; 24:1211–17. https://doi.org/10.1038/nbt1006-1211. [PubMed].
237. Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015; 200:138–57. https://doi.org/10.1016/j.jconrel.2014.12.030. [PubMed].
238. Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012; 338:903–10. https://doi.org/10.1126/science.1226338. [PubMed].
239. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010; 7:653–64. https://doi.org/10.1038/nrclinonc.2010.139. [PubMed].
240. Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003; 20:1337–50. https://doi.org/10.1023/a:1025785505977. [PubMed].
241. Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011; 153:198–205. https://doi.org/10.1016/j.jconrel.2011.06.001. [PubMed].
242. Fojo T, Parkinson DR. Biologically targeted cancer therapy and marginal benefits: are we making too much of too little or are we achieving too little by giving too much? Clin Cancer Res. 2010; 16:5972–80. https://doi.org/10.1158/1078-0432.CCR-10-1277. [PubMed].
243. Kim SS, Rait A, Rubab F, Rao AK, Kiritsy MC, Pirollo KF, Wang S, Weiner LM, Chang EH. The clinical potential of targeted nanomedicine: delivering to cancer stem-like cells. Mol Ther. 2014; 22:278–91. https://doi.org/10.1038/mt.2013.231. [PubMed].
244. Kruyt FA, Schuringa JJ. Apoptosis and cancer stem cells: implications for apoptosis targeted therapy. Biochem Pharmacol. 2010; 80:423–30. https://doi.org/10.1016/j.bcp.2010.04.010. [PubMed].
245. Massard C, Deutsch E, Soria JC. Tumour stem cell-targeted treatment: elimination or differentiation. Ann Oncol. 2006; 17:1620–24. https://doi.org/10.1093/annonc/mdl074. [PubMed].
246. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007; 2:751–60. https://doi.org/10.1038/nnano.2007.387. [PubMed].
247. Gao W, Xiao Z, Radovic-Moreno A, Shi J, Langer R, Farokhzad OC. Progress in siRNA delivery using multifunctional nanoparticles. Methods Mol Biol. 2010; 629:53–67. https://doi.org/10.1007/978-1-60761-657-3_4. [PubMed].
248. Yap TA, Sandhu SK, Workman P, de Bono JS. Envisioning the future of early anticancer drug development. Nat Rev Cancer. 2010; 10:514–23. https://doi.org/10.1038/nrc2870. [PubMed].
249. Biddlestone-Thorpe L, Marchi N, Guo K, Ghosh C, Janigro D, Valerie K, Yang H. Nanomaterial-mediated CNS delivery of diagnostic and therapeutic agents. Adv Drug Deliv Rev. 2012; 64:605–13. https://doi.org/10.1016/j.addr.2011.11.014. [PubMed].
250. Castro MG, Candolfi M, Kroeger K, King GD, Curtin JF, Yagiz K, Mineharu Y, Assi H, Wibowo M, Ghulam Muhammad AK, Foulad D, Puntel M, Lowenstein PR. Gene therapy and targeted toxins for glioma. Curr Gene Ther. 2011; 11:155–80. https://doi.org/10.2174/156652311795684722. [PubMed].
251. Kievit FM, Zhang M. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater. 2011; 23:H217–47. https://doi.org/10.1002/adma.201102313. [PubMed].
252. McCubrey JA, Steelman LS, Abrams SL, Misaghian N, Chappell WH, Basecke J, Nicoletti F, Libra M, Ligresti G, Stivala F, Maksimovic-Ivanic D, Mijatovic S, Montalto G, et al. Targeting the cancer initiating cell: the ultimate target for cancer therapy. Curr Pharm Des. 2012; 18:1784–95. https://doi.org/10.2174/138161212799859701. [PubMed].
253. Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials. 2011; 32:5417–26. https://doi.org/10.1016/j.biomaterials.2011.04.006. [PubMed].
254. Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010; 188:759–68. https://doi.org/10.1083/jcb.200910104. [PubMed].
255. Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012; 63:185–98. https://doi.org/10.1146/annurev-med-040210-162544. [PubMed].
256. Vlashi E, Kelderhouse LE, Sturgis JE, Low PS. Effect of folate-targeted nanoparticle size on their rates of penetration into solid tumors. ACS Nano. 2013; 7:8573–82. https://doi.org/10.1021/nn402644g. [PubMed].
257. Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, Wright WE, Shay JW, Go NF. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010; 70:9494–504. https://doi.org/10.1158/0008-5472.CAN-10-0233. [PubMed].
258. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014; 66:2–25. https://doi.org/10.1016/j.addr.2013.11.009. [PubMed].
259. Mirny L. Cell commuters avoid delays. Nat Phys. 2008; 4:93–95. https://doi.org/10.1038/nphys848.
260. Huai Y, Hossen MN, Wilhelm S, Bhattacharya R, Mukherjee P. Nanoparticle Interactions with the Tumor Microenvironment. Bioconjug Chem. 2019; 30:2247–63. https://doi.org/10.1021/acs.bioconjchem.9b00448. [PubMed].
261. Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011; 8:2101–41. https://doi.org/10.1021/mp200394t. [PubMed].
262. Din FU, Aman W, Ullah I, Qureshi OS, Mustapha O, Shafique S, Zeb A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int J Nanomedicine. 2017; 12:7291–309. https://doi.org/10.2147/IJN.S146315. [PubMed].
263. Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015; 93:52–79. https://doi.org/10.1016/j.ejpb.2015.03.018. [PubMed].
264. Boado RJ, Hui EK, Lu JZ, Sumbria RK, Pardridge WM. Blood-brain barrier molecular trojan horse enables imaging of brain uptake of radioiodinated recombinant protein in the rhesus monkey. Bioconjug Chem. 2013; 24:1741–49. https://doi.org/10.1021/bc400319d. [PubMed].
265. Gutman M, Laufer R, Eisenthal A, Goldman G, Ravid A, Inbar M, Klausner JM. Increased microvascular permeability induced by prolonged interleukin-2 administration is attenuated by the oxygen-free-radical scavenger dimethylthiourea. Cancer Immunol Immunother. 1996; 43:240–44. https://doi.org/10.1007/s002620050328. [PubMed].
266. Black KL, Chio CC. Increased opening of blood-tumour barrier by leukotriene C4 is dependent on size of molecules. Neurol Res. 1992; 14:402–04. https://doi.org/10.1080/01616412.1992.11740093. [PubMed].
267. Nowacek AS, Miller RL, McMillan J, Kanmogne G, Kanmogne M, Mosley RL, Ma Z, Graham S, Chaubal M, Werling J, Rabinow B, Dou H, Gendelman HE. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine (Lond). 2009; 4:903–17. https://doi.org/10.2217/nnm.09.71. [PubMed].
268. Dash PK, Gendelman HE, Roy U, Balkundi S, Alnouti Y, Mosley RL, Gelbard HA, McMillan J, Gorantla S, Poluektova LY. Long-acting nanoformulated antiretroviral therapy elicits potent antiretroviral and neuroprotective responses in HIV-1- infected humanized mice. AIDS. 2012; 26:2135–44. https://doi.org/10.1097/QAD.0b013e328357f5a. [PubMed].
269. Wan L, Zhang X, Pooyan S, Palombo MS, Leibowitz MJ, Stein S, Sinko PJ. Optimizing size and copy number for PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarrier uptake by macrophages. Bioconjug Chem. 2008; 19:28–38. https://doi.org/10.1021/bc070066k. [PubMed].
270. Chao Y, Karmali PP, Simberg D. Role of carbohydrate receptors in the macrophage uptake of dextran-coated iron oxide nanoparticles. Adv Exp Med Biol. 2012; 733:115–23. https://doi.org/10.1007/978-94-007-2555-3_11. [PubMed].
271. Li X, Liu R, Su X, Pan Y, Han X, Shao C, Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019; 18:177. https://doi.org/10.1186/s12943-019-1102-3. [PubMed].
272. Guerriero JL. Macrophages: the road less traveled, changing anti-cancer therapy. Trends Mol Med. 2018; 24:472–89. https://doi.org/10.1016/j.molmed.2018.03.006. [PubMed].
273. Cassetta L, Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology. 2018; 155:285–93. https://doi.org/10.1111/imm.12976. [PubMed].