
Introduction
The American Cancer Society estimates that > 190,000 new cases of prostate cancer (PCa) will be diagnosed in the United States in 2020 along with about 33,330 deaths [1]. The first-line therapy for PCa includes hormonal ablation or androgen deprivation therapy (ADT). Although ADT is effective in debulking tumors in most cases, the majority of treated PCa patients develop ADT resistance leading to emergence of more aggressive castration-resistant PCa (CRPC), which is the primary cause of PCa related deaths [2]. Development of acquired resistance to therapeutic regimens is common in virtually all targeted therapies and recent research implicates intratumor heterogeneity [3, 4]. Intratumor heterogeneity is maintained partly through the expression of distinct sets of genes among tumor cells, a phenomenon called cellular transcriptomic heterogeneity (CTH) [4], which is controlled by transcription factors and histone-modifying enzymes [3–6]. These enzymes work through regulating chromatin structure, which is an important determinant of gene activity. Chromatin, consisting of histones wrapped by DNA, is regulated by histone-modifying enzymes via methylation, phosphorylation, acetylation, ubiquitination, sumoylation, and ribosylation at lysine, arginine, serine, threonine, tyrosine, and other residues of histone tails [7, 8]. These histone modifications not only impact gene expression but also influence how the 3D structure of the chromatin is organized within the nucleus [9–12]. Depending on the type of modifications and the residues modified on the histone, the target gene can be transcriptionally activated or repressed. Genome-wide studies have indicated that histone acetylation is associated with higher transcriptional activity [5, 13, 14] whereas DNA methylation in the CpG islands is correlated with transcriptional repression. The methylation of histones impacts gene expression in a context-dependent manner. For example, trimethylation of histone 3 at lysine 4 and lysine 36 (H3K4me3 and H3K36me3) is generally associated with gene activation [15–18] whereas methylation at lysine 9 and 27 correlates with gene repression [16, 19–21].
KDM5B, also called JARID1B or PLU1, is a Jumonji C-containing (jmjC) histone lysine demethylase that plays important roles in organogenesis, stem cell functions and cancer development [6, 22]. KDM5B was initially identified as a critical regulator of embryonic cell differentiation (via decreasing H3K4 methylation in D3-D5 embryos) and as a determinant of zygotic genome activation and cellular fate changes during development [23]. In mouse embryonic stem cells, KDM5B binds various developmental genes to ensure proper neuronal differentiation [24]. Given its crucial role, KDM5B knockout in mice is embryonically lethal and the embryos exhibit extensive developmental defects [25]. These studies highlight an important role for KDM5B during normal development.
Genes involved in organogenesis and development are often dysregulated in tumorigenesis. Not surprisingly, KDM5B is overexpressed, and has been reported to play an oncogenic function, in a variety of cancers including breast cancer [4, 22], melanoma [26], PCa [27], lung cancer [28], hepatocellular carcinoma [29], gastric cancer [30], neuroblastoma [31] and leukemia [32]. On the other hand, potential tumor-suppressive functions of KDM5B have also been documented in some cases of melanoma and subtypes of breast cancer [33, 34]. Mechanistically, KDM5B has been reported to interact with transcription factors such as estrogen receptor α (ERα), androgen receptor (AR), progesterone receptor, PAX9, FOXG1, etc, and such interactions direct its localization to a diverse repertoire of genes in different cell types that further leads to distinct gene expression and contributes to CTH and intratumor heterogeneity [6].
Interestingly, KDM5B is specifically expressed in breast luminal cells and its loss induces basal-type gene expression, suggesting that KDM5B is a luminal lineage-driving oncogene in breast cancer [22]. Of note, KDM5B induces CTH and mediates therapeutic resistance in breast cancer [22]. As PCa is also a hormone-driven, luminal-type cancer with significant cellular heterogeneity and CTH [35, 36], KDM5B might play similar functions in PCa. Indeed, epigenetic events, mediated by histone-modifying enzymes, may be intimately involved in regulating AR signaling and PCa heterogeneity [27, 37, 38]. Our recent studies [35, 36] indicate that not only the expression of AR, but its transcriptional activity (as judged by the expression levels of AR transcriptional targets such as LRIG1) is also highly heterogeneous contributing to intratumor cellular heterogeneity in PCa.
Here, we first present bioinformatics data on KDM5B that supports an oncogenic function of KDM5B in PCa as previously reported [27]. We then present the surprising findings that Kdm5b deletion in the mouse prostate results in mild hyperplasia. Altogether, our study provides evidence for context-dependent roles of KDM5B in prostate organogenesis and tumorigenesis.
Results
KDM5B mRNA is upregulated in PCa and correlates with genomic amplifications, tumor grade and poor survival
We first analyzed the KDM5B mRNA levels in GTEx (Genotype-Tissue Expression; https://gtexportal.org/) RNA-seq database. The results revealed wide expression of KDM5B mRNA across many human tissues including the prostate with the highest expression in the testis (Supplementary Figure 1A). Next we analyzed the KDM5B mRNA levels in TCGA PRAD dataset comparing normal and prostate tumor tissues. The KDM5B mRNA levels were significantly higher in PCa than the normal/benign tissue in both matched pair (52N/T; Figure 1A) and overall (52N/498T; Figure 1B) comparisons. Notably, KDM5B mRNA levels were elevated in PCa patient tumors with high tumor grade, i.e., combined Gleason Scores (GS) of 7–9 (Figure 1C). Furthermore, analysis of KDM5B mRNA levels in several Oncomine PCa datasets, including the Glinsky [39], Grasso (GSE35988; [40]) and Setlur (GSE8402; [41]) datasets revealed that high KDM5B mRNA levels correlated with poor PCa patients’ overall survival (Figure 1D–1F). These patients’ data, collectively, suggest an AR-regulated oncogenic role of KDM5B in PCa. Interestingly, when we interrogated the KDM5B mRNA levels in two RNA-seq datasets (GSE48403 and GSE111177) of matched patient tumors before ADT (pre-ADT) and post ADT failure (post-ADT), we observed reduced KDM5B mRNA levels in post-ADT tumors (Figure 1G and 1H). These results suggest that, consistent with an early study [27], KDM5B is regulated by AR.
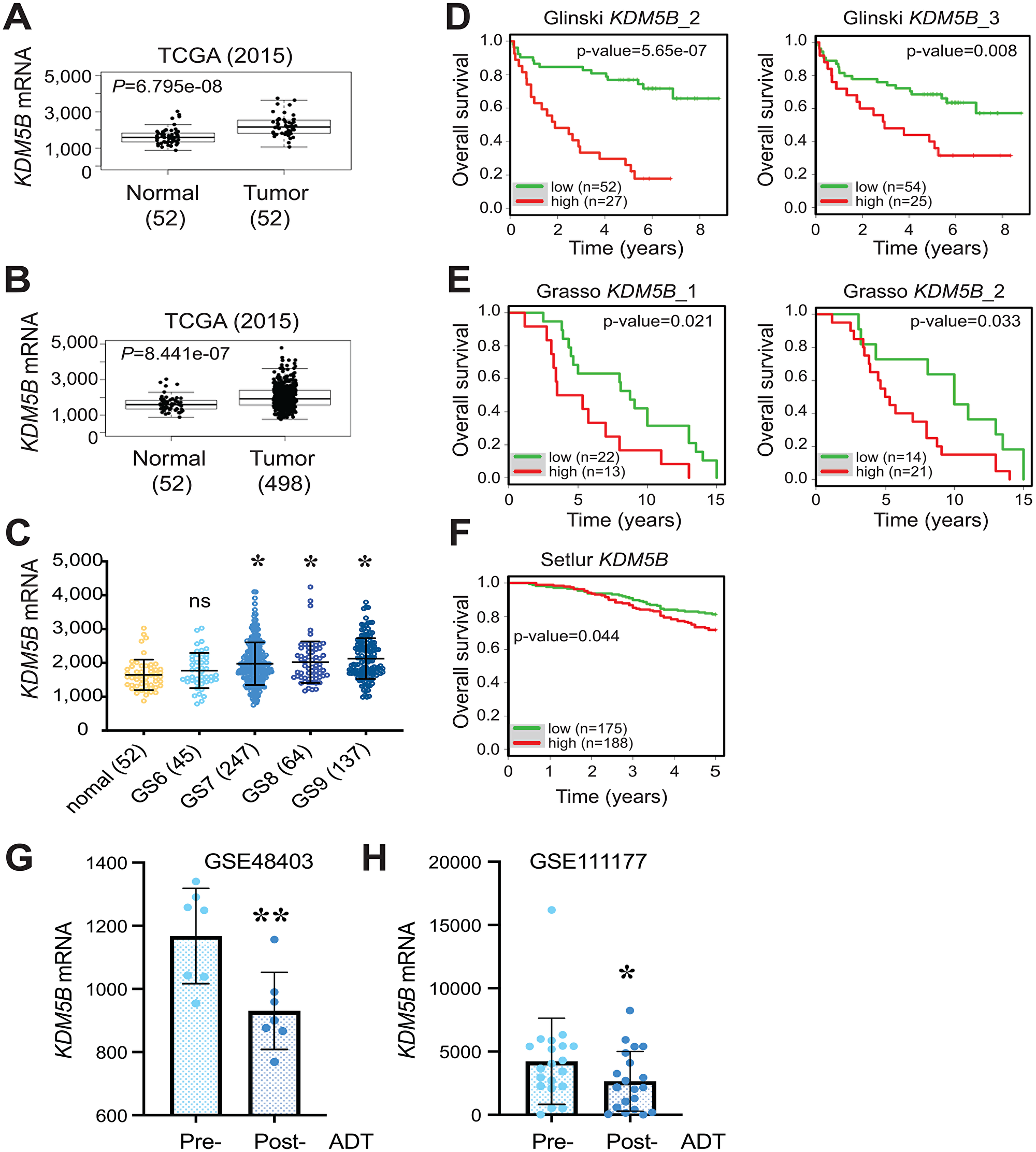
Figure 1: Overexpression of KDM5B mRNA in PCa and correlation with poor patient survival. (A and B) Elevated KDM5B mRNA levels in the TCGA PRAD dataset comparing 52 normal and 52 paired cancer tissues (A) or 52 normal and 498 cancer tissues (B). P values (two-tailed unpaired Student’s t-test) were indicated. (C) Elevated KDM5B mRNA levels in high-grade prostate tumors. Shown are KDM5B mRNA levels in the TCGA PRAD dataset PCa of increasing tumor grade (i.e., Gleason Score, GS6-10) and the 52 normal prostate tissues. *P < 0.05 compared with the normal (One-way ANOVA). ns, not significant. (D–F) High KDM5B mRNA levels correlate with poor PCa patient overall survival. Shown are Kaplan-Meier survival plots in the 3 indicated Oncomine datasets (1, 2, and 3 refer to different cDNA microarray probes used). p-values were determined using the Log-Rank test. (G and H) Reduced KDM5B mRNA levels in post-ADT patient tumors. KDM5B mRNA levels (read counts) were extracted from two RNA-seq datasets (GSE48403, n = 7; GSE111177, n = 20) and presented as the mean +/– S. D. *P = 0.032 and **P = 0.007 (two-tailed, paired Student’s t-test).
An analysis of KDM5B mRNA levels in 31 human cancers in TCGA with the corresponding normal tissues pooled from TCGA and GTEx revealed increased or a trend of increased KDM5B mRNA in multiple cancers including bladder, breast, esophageal, pancreatic and prostate cancers as well as thymoma and acute myeloid lymphoma (Supplementary Figure 1B). Interestingly, KDM5B, most abundantly expressed in the normal testis (Supplementary Figure 1A), is reduced in testicular germ cell tumors (Supplementary Figure 1B). We further examined the mutational landscape of the KDM5B gene across a spectrum of cancers (Supplementary Figure 2). Multiple mutations representing amplification, missense mutations, and truncating mutations as well as copy number alterations (CNA) were observed in many cancers (Supplementary Figure 2A). Strikingly, in two hormone-driven cancers, breast (Supplementary Figure 2A) and prostate (Supplementary Figure 2B) cancers, we observed prevalent KDM5B genomic amplifications, which were accompanied by increased KDM5B mRNA expression in invasive breast cancer (BRCA; Supplementary Figure 1B), or increased (Figure 1A–1C) or an increased trend of (Supplementary Figure 1B) KDM5B mRNA levels in PCa. Several other cancers that showed genomic amplifications of KDM5B gene (Supplementary Figure 2A), e.g., thymic tumors (THYM), cholangiocarcinoma (CHOL), esophageal cancers (ESCA), GBM and bladder cancer (BLCA), also exhibited increased or trend of increased KDM5B expression (Supplementary Figure 1B). Overall, the analysis of genomic amplifications in KDM5B gene supports an oncogenic role of KDM5B in many human cancers including PCa. On the other hand, several cancers that manifested KDM5B genomic amplifications, e.g., hepatocellular carcinoma (LIHC) and ovarian cancer (OV), were not accompanied with (trend of) increased KDM5B mRNA expression (Supplementary Figure 1B; Supplementary Figure 2A).
Kdm5b knockout induces mild hyperplasia in the mouse prostate
Given the role of KDM5B in normal development and organogenesis (see Introduction), we intend to test its role in normal mouse prostate development in order to better understand its potential involvement in prostate tumorigenesis. To specifically knock out Kdm5b in the mouse prostatic epithelium, we crossed Kdm5b-floxed (Kdm5bf/f) mice [24] with the Pb-Cre4 line, which has Cre recombinase expression under the control of the ARR2PB promoter comprising a proximal element of the rat Probasin (Pb) promoter and two androgen responsive regions (Figure 2A and 2B). The ARR2PB promoter confines transgene expression predominantly in prostate luminal epithelial cells [42]. The mice were genotyped as described in the Methods with a representative gel image showing detection of the Kdm5b floxed and wilt-type (wt) alleles along with Pb-Cre4 (Figure 2C).
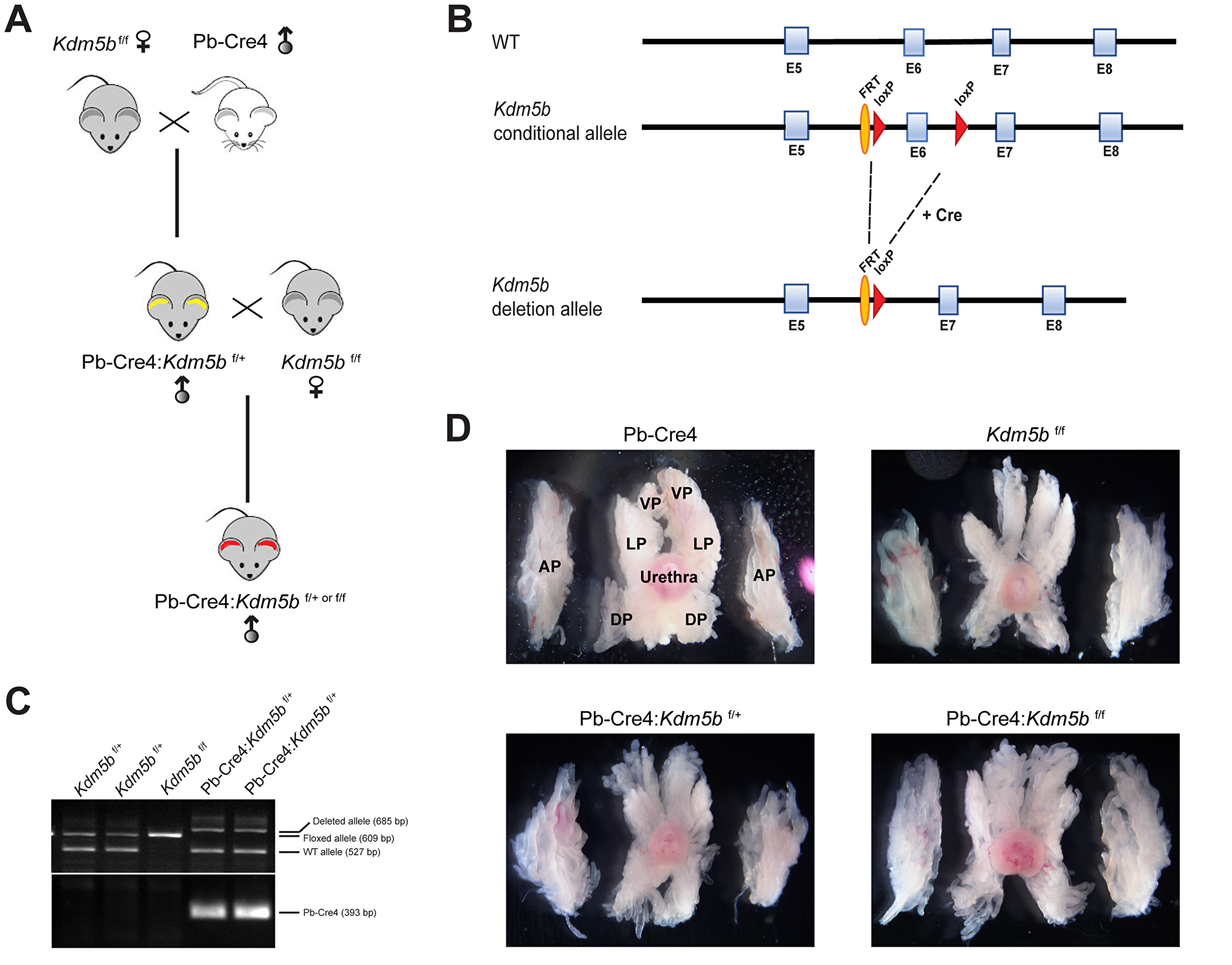
Figure 2: Generation of conditional Kdm5b knockout mice. (A) Breeding strategy for generating Pb-Cre4:Kdm5bf/+ or Pb-Cre4:Kdm5bf/f mice. (B) Schematic of the KDM5B construct showing the loxP sites flanking the Exon (E) 6 of the KDM5B allele. (C) Representative genotyping gel image showing different Kdm5b alleles (Floxed, WT and deleted) and Pb-Cre4 PCR product bands. (D) Representative images of microdissected whole-mount prostates in WT and Kdm5b-deleted mice of different genotypes at 3 months of age (n = 3). AP, DP, LP and VP indicate anterior, dorsal, lateral, and ventral prostate lobes, respectively. The orientations of prostatic lobes are the same for all images shown.
Kdm5b knockout did not affect mouse prostate development (data not shown). Microdissected prostatic lobes (i.e., the anterior, dorsal, lateral and ventral prostates; AP, DP, LP and VP, respectively) from the 3-month-old mice of various genotypes showed overall similar gross morphologies and structures (Figure 2D). Immunofluorescence staining revealed beautiful and specific Kdm5b protein in the nucleus of luminal cells (Figure 3A). As expected, Kdm5b protein expression was significantly reduced in the heterozygous Pb-Cre4:Kdm5bf/+ mouse prostate and completely lost in the Pb-Cre4;Kdm5bf/f prostate (Figure 3A). Interestingly, H&E analysis revealed mild hyperplasia in both heterozygous and homozygous Kdm5b-knockout mouse prostate, especially in the LP and VP (Figure 3B). In the control (i.e., Pb-Cre4 and Kdm5bf/+) mice, the LP and VP consisted of uniform glands with single luminal epithelial cell layer (Figure 3B). In contrast, the Kdm5b-deleted hyperplastic glands displayed thickened luminal cell compartment with more than one cell layer and, frequently, papillary structures protruded into the lumen (Figure 3B). In support of the hyperplasia phenotype, quantitative analysis revealed significantly increased cellularity in both the LP and VP of Pb-Cre4:Kdm5bf/+ and Pb-Cre4:Kdm5bf/f prostates (Figure 4A and 4B). Hyperplasia persisted, but no PIN (Prostate Intraepithelial Neoplasia) or apparent tumors developed, in 1–1.5 years old Pb-Cre4:Kdm5bf/+ and Pb-Cre4:Kdm5bf/f prostates (data not shown).
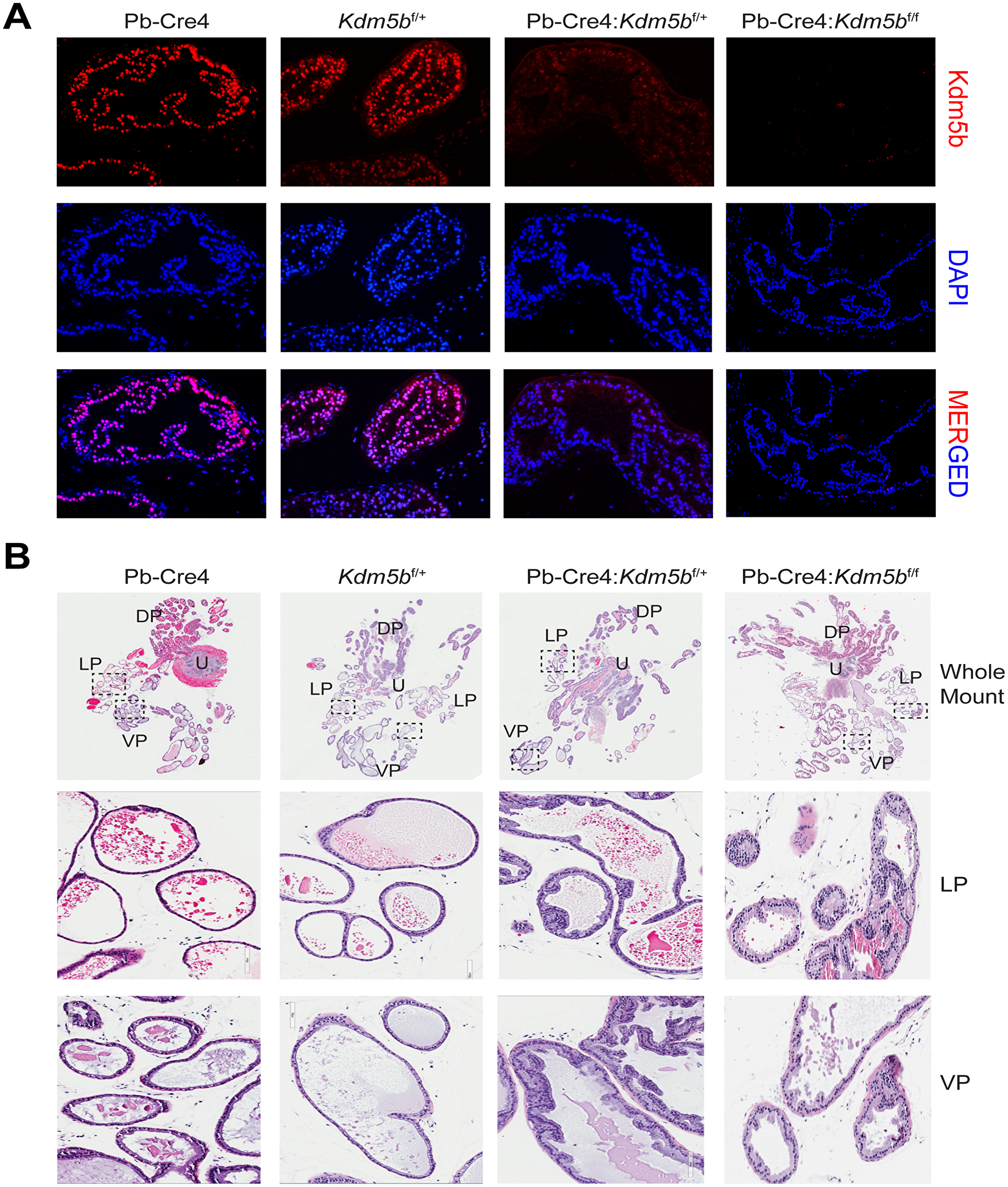
Figure 3: Kdm5b deficient mouse prostates manifest low-grade hyperplasia. (A) Immunofluorescence using anti-KDM5B antibody on formalin-fixed paraffin-embedded prostatic tissues obtained from the indicated genotypes. DAPI is used to stain the nuclei. (B) Representative HE images of whole-mount prostate sections (upper panels) from 3 months old mice of indicated genotypes. The middle and lower panel are representative blow-up images of LP and VP of the boxed areas in the whole mounts.
DISCUSSION
Epigenetic modifications lie at the heart of normal development and organogenesis, and analysis of epigenetic landscape comparing various epigenetic modifications between normal and cancer tissues has revealed massive epigenetic dysregulation during cancer development. Molecular dissection of the epigenetic modifiers may shed light on their roles in normal vs. cancer development. In the present study, we provide evidence that KDM5B, a H3K4 demethylase, may exhibit two contrasting functions: in human PCa, it is significantly upregulated (consistent with an earlier report; 27) and correlates with poor patient survival thus pointing to an oncogenic role; in contrast, genetic deletion of Kdm5b leads to mild hyperplasia in the mouse prostate, pointing to a potentially tumor-suppressive function.
KDM5B has been traditionally thought to repress transcription since it catalyzes the demethylation of H3K4me1/me2/me3: H3K4me2/me3 are enriched at the promoter region of actively transcribed genes [16] while H3K4me1 marks the enhancer regions [43–45]. Surprisingly, KDM5B is overexpressed (or shows the trend of overexpression) in many human cancers in addition to PCa (Supplementary Figure 1B). Though the role of KDM5B in tumorigenesis is not well understood, its higher expression in cancer cells may regulate the distribution of H3K4me3 near promoter regions of tumor suppressors and modulate their expression, thus affecting cancer cell proliferation [6]. Recently, several KDM5B inhibitors have been identified and reported in different cancers such as the breast and prostate cancers [46, 47]. In all these studies, genetic or pharmacologic inhibition of KDM5B upregulates the expression of tumor suppressor genes and caused growth arrest and apoptotic cell death in cancer cells suggesting that KDM5B may mostly act as a repressor of tumor suppressor genes. KDM5B inhibitors have also been reported to overcome radioresistance in cancer cells by preventing the demethylation of H3K4 at the sites of double-strand breaks induced by radiation [48]. Therefore, DNA damage repair machinery failed to resolve the damage leading to increased radiosensitivity of KDM5B-overexpressing cancer cells. Overall these studies suggest a tumor promoting potential of KDM5B, which is consistent with our observations that it is highly expressed in human PCa and its expression correlates with poor patient survival.
Interestingly, Kdm5b deletion in the mouse prostate leads to mild hyperplasia, which is surprising given its reported pro-oncogenic role in prostate and other cancers. Earlier studies demonstrate that KDM5B is not only overexpressed in hormone-driven cancers such as the breast and prostate cancers, but also interacts with hormone receptors, ER and AR, to positively regulate their transcriptional activities [25, 27]. Therefore, it is plausible to speculate that KDM5B might function in the prostate and PCa through modulating AR signaling. In partial support of this connection, KDM5B mRNA levels were significantly reduced in post-ADT (castration-resistant) patient prostate tumors (Figure 1G and 1H). Whether the hyperplasia in Kdm5b-deficient prostate could be related to anomalies in the AR signaling axis is currently under investigation. On the other hand, increased cellularity in Kdm5b-deleted mouse prostate (Figure 3B and Figure 4) implies that Kdm5b might demethylate the H3K4 in the promoter/enhancer regions of tumor-promoting genes and keep them in a repressed state during normal prostate development. Kdm5b deletion in the prostate leads to upregulation of these genes and subsequent luminal hyperplasia. Further experiments such as RNA-seq analysis of the Kdm5b-depleted mouse prostate need to be performed to reveal the full spectrum of its functions in prostate development.
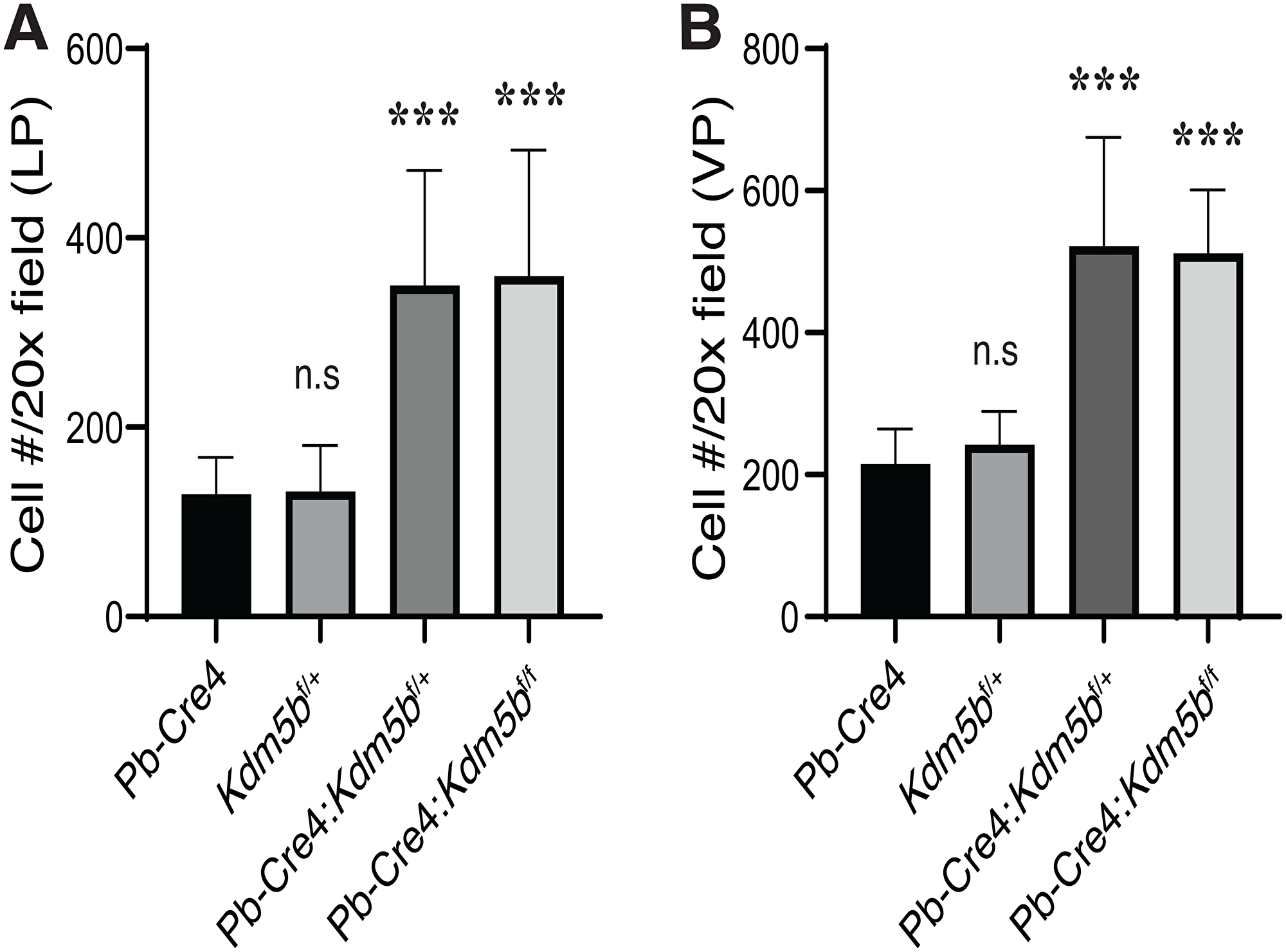
Figure 4: Kdm5b deficiency causes increased cellularity in the mouse prostates. The mouse LP (A) and VP (B) lobes were micro-dissected out from animals of the indicated genotypes (n = 8/genotype) as shown in Figure 3B. The number of cells, as identified by DAPI staining, were counted in 10 random microscopic fields under a 20× objective lens. Presented are bar graphs (mean +/– S. D) of the cell numbers per 20× field. ***P < 0.0001 (Student’s t-test); n.s, not (statistically) significant.
Collectively, our data suggest that KDM5B may possess context-dependent functions in prostate organ development and prostate tumorigenesis. As it has been previously reported that KDM5B interacts with a variety of transcription factors and such interactions determine its localization on the specific set of genes, it will be very interesting to investigate its interacting partners during different stages of prostate and PCa development.
Materials and Methods
KDM5B bioinformatics, survival and mutational landscape analyses
General strategies in bioinformatically analyzing KDM5B mRNA expression and correlation with patient survival have been described in our recent publications [35, 36]. Briefly, KDM5B mRNA levels in normal human tissues (Supplementary Figure 1A) were extracted from the GTEx (Genotype-Tissue Expression) data portal (https://gtexportal.org/). For TCGA data analysis (Figure 1A–1C), we obtained TCGA level-3 data for KDM5B mRNA from TCGA data portal (https://tcga-data.nci.nih.gov). We performed the Student’s t-test for normal and tumor tissue comparisons and one-way ANOVA for determining expression levels among different Gleason scores, respectively (Figure 1A–1C).
We performed survival analysis and generated Kaplan-Meier survival plots using the survival package in R (Figure 1D–1F). In brief, we obtained the individual normalized gene expression data from patients with both survival time and survival status from Oncomine (https://www.oncomine.com; Compendia Bioscience) datasets and ranked the data according to KDM5B mRNA expression. Then, we assigned the sample with rank from the first quartile to the third quartile into two groups and compared the p-value between these two groups along with different cutoffs. Finally, we set the ultimate cutoff with the smallest p-value and plotted a Kaplan-Meier survival curve.
We also compared KDM5B mRNA expression levels of 31 human tumors in TCGA with the corresponding matched normal tissues pooled from TCGA and GTEx (Supplementary Figure 1B). Briefly, KDM5B gene expression across the 31 cancer types and paired normal samples was generated from GEPIA (http://gepia.cancer-pku.cn), with each dot representing a distinct tumor (from TCGA) or normal sample (from TCGA and GTEx). Four-way analysis of variance (ANOVA) was employed, using sex, age, ethnicity and disease state (Tumor or Normal) as variables, to determine differential expression. The expression data are first log2(TPM+1) transformed and the log2FC is defined as median (Tumor) - median (Normal). The Benjamini and Hochberg false discovery rate (FDR) method was used to adjust.
Finally, we analyzed the KDM5B mutations in the prostate and multiple cancer datasets using cBioportal (http://www.cbioportal.org) (Supplementary Figure 2).
Generation of conditional KDM5B knockout mice
Generation of conditional knockout mice with a floxed Kdm5b allele has been described previously [24]. The overall breeding strategy is illustrated in Figure 2A. Male PB-Cre4 mice were crossed with female Kdm5bf/f to obtain PB-Cre4:Kdm5bf/+ or PB-Cre4:Kdm5bf/f mice. For genotyping, genomic DNA was isolated from tail snips using EZNA tissue DNA kit (Omega Bio-Tek, GA, USA) as per manufacturer’s instructions. Briefly, tail snips were minced in supplied TL buffer and Proteinase K solution followed by incubation at 55°C overnight. Genomic DNA were purified using supplied HiBind DNA mini columns and 2 μl of genomic DNA extracts were subjected to PCR reaction for genotyping of different alleles (WT and floxed; PB-Cre4) using the following primers: Kdm5b floxed allele (Fwd: CCCTGG-GATTGCAGTTAAAG; Rev (floxed allele): TGGCTTCCACAATCTTCAATG; Rev (deleted allele): GTCAACTGCAAACTGACCTCTG; PCR product size: WT = 527 bp; floxed allele = 609 bp; and deleted allele = 685 bp), and Pb-Cre4 transgene (Fwd: CTGAAGAATGGGACAGGCATTG; Rev: CATCACTC-GTTGCATCGACC; PCR product size: 393 bp).
Prostate isolation, microdissection, Aperio ScanScope analysis, and immunofluorescence
Basic procedures for these experiments have been previously described [35, 36, 49–52]. After sacrificing mice at the age of 3 months, the prostates were surgically removed along with the urogenital tract. The prostates were placed immediately in ice-cold phosphate-buffered saline (PBS) and microdissected under a dissection microscope to remove fat and connective tissues. The isolated whole-mount prostates were photographed with a Nikon digital camera and then fixed in 10% formalin for further histological analysis. For Aperio Scanscope analysis, H&E-stained glass slides containing sections of WM mouse prostates were scanned via an Aperio ScanScope imaging platform (Aperio Technologies, Vista, CA, USA) and images analyzed on Imagescope analysis software.
For immunofluorescence staining, formalin fixed mouse prostate sections were deparaffinized using xylene and rehydrated through incubating sections in serial decreasing concentrations of ethanol (95–50%). After washing with distilled water, antigen was retrieved in antigen-retrieval solution (Sodium citrate buffer, pH6.0). After washing with wash buffer (Tris Buffered Saline-Tween 20), sections were blocked with Background sniper (Biocare Medical, CA, USA) for 1 hr at room temperature (RT) followed by anti-KDM5B antibody (Bethyl lab, Cat # A301-813A) incubation at 4°C for overnight. After washing with wash buffer, sections were incubated with anti-rabbit IgG-PE secondary antibody (Santa Cruz, Cat # sc-3753) for 1 hr at RT followed by 10 minutes incubation in DAPI (0.1 μg/ml). Sections were washed extensively with wash buffer and mounted with Prolong Gold mounting medium (Thermo Fisher Scientific, MA, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software or using R. In general, paired or unpaired two-tailed Student’s t-test was used to calculate the statistical significance between comparisons. Differences in patients’ overall survival were determined by the Log-Rank Test. P < 0.05 is considered statistically significant.
Ethics statement
All animal work was approved by our institutional animal care and use committee at the University of Texas M.D Anderson Cancer Center or the Roswell Park Comprehensive Cancer Center. All animals were maintained in standard conditions according to the institutional guidelines. Animal housing rooms were under temperature and humidity control and mice were not subjected to water or food restrictions.
ACKNOWLEDGMENTS
We acknowledge the support of the Animal Facility at the M.D Anderson Cancer Center (Smithville) as well as several Shared Resources (SR) at the Roswell Park Comprehensive Cancer Center including BSGSR, ETM, FICSR, GSR, LASR, and PNSR. We also thank Dr. K. Helin for providing Kdm5b-floxed mice.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This project was supported, in part, by grants from the U.S NIH (R01CA237027, R01CA240290, R21CA237939, and R21CA218635) and Department of Defense (W81XWH-16-1-0575), and by RPCCC and NCI center grant P30CA016056.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70:7–30. https://doi.org/10.3322/caac.21590. [PubMed].
2. Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, Kelly K, Lin D, Dicker A, Arnold J, Hecht T, Wicha M, Sears R, et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res. 2019; 25:6916–6924. https://doi.org/10.1158/1078-0432.CCR-19-1423. [PubMed].
3. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018; 15:81–94. https://doi.org/10.1038/nrclinonc.2017.166. [PubMed].
4. Hinohara K, Wu HJ, Sebastien V, McDonald TO, Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou M, Ding L, Peluffo G, Cohen O, et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell. 2019; 35:330–332. https://doi.org/10.1016/j.ccell.2019.01.012. [PubMed].
5. Schübeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill LP, Turner BM, Delrow J, Bell SP, Groudine M. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004; 18:1263–1271. https://doi.org/10.1101/gad.1198204. [PubMed].
6. Xhabija B, Kidder BL. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin Cancer Biol. 2019; 57:79–85. https://doi.org/10.1016/j.semcancer.2018.11.001. [PubMed].
7. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128:693–705. https://doi.org/10.1016/j.cell.2007.02.005. [PubMed].
8. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007; 447:407–412. https://doi.org/10.1038/nature05915. [PubMed].
9. Zinner R, Albiez H, Walter J, Peters AH, Cremer T, Cremer M. Histone lysine methylation patterns in human cell types are arranged in distinct three-dimensional nuclear zones. Histochem Cell Biol. 2006; 125:3–19. https://doi.org/10.1007/s00418-005-0049-1. [PubMed].
10. Bartova E, Galiova G, Krejci J, Harnicarova A, Strasak L, Kozubek S. Epigenome and chromatin structure in human embryonic stem cells undergoing differentiation. Dev Dyn. 2008; 237:3690–3702. https://doi.org/10.1002/dvdy.21773. [PubMed].
11. Zhu Y, Chen Z, Zhang K, Wang M, Medovoy D, Whitaker JW, Ding B, Li N, Zheng L, Wang W. Constructing 3D interaction maps from 1D epigenomes. Nat Commun. 2016; 7:10812. https://doi.org/10.1038/ncomms10812. [PubMed].
12. Sequeira-Mendes J, Gutierrez C. Genome architecture: from linear organisation of chromatin to the 3D assembly in the nucleus. Chromosoma. 2016; 125:455–469. https://doi.org/10.1007/s00412-015-0538-5. [PubMed].
13. Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005; 122:517–527. https://doi.org/10.1016/j.cell.2005.06.026. [PubMed].
14. Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005; 19:542–552. https://doi.org/10.1101/gad.1272505. [PubMed].
15. Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005; 280:17732–17736. https://doi.org/10.1074/jbc.M500796200. [PubMed].
16. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. https://doi.org/10.1016/j.cell.2007.05.009. [PubMed].
17. Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002; 99:8695–8700. https://doi.org/10.1073/pnas.082249499. [PubMed].
18. Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ 3rd, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005; 120:169–181. https://doi.org/10.1016/j.cell.2005.01.001. [PubMed].
19. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039–1043. https://doi.org/10.1126/science.1076997. [PubMed].
20. Hublitz P, Albert M, Peters AH. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol. 2009; 53:335–354. https://doi.org/10.1387/ijdb.082717ph. [PubMed].
21. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001; 410:120–124. https://doi.org/10.1038/35065138. [PubMed].
22. Yamamoto S, Wu Z, Russnes HG, Takagi S, Peluffo G, Vaske C, Zhao X, Moen Vollan HK, Maruyama R, Ekram MB, Sun H, Kim JH, Carver K, et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell. 2014; 25:762–777. https://doi.org/10.1016/j.ccr.2014.04.024.
23. Glanzner WG, Gutierrez K, Rissi VB, de Macedo MP, Lopez R, Currin L, Dicks N, Baldassarre H, Agellon LB, Bordignon V. Histone Lysine Demethylases KDM5B and KDM5C Modulate Genome Activation and Stability in Porcine Embryos. Front Cell Dev Biol. 2020; 8:151. https://doi.org/10.3389/fcell.2020.00151. [PubMed].
24. Schmitz SU, Albert M, Malatesta M, Morey L, Johansen JV, Bak M, Tommerup N, Abarrategui I, Helin K. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 2011; 30:4586–4600. https://doi.org/10.1038/emboj.2011.383. [PubMed].
25. Catchpole S, Spencer-Dene B, Hall D, Santangelo S, Rosewell I, Guenatri M, Beatson R, Scibetta AG, Burchell JM, Taylor-Papadimitriou J. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol. 2011; 38:1267–1277. [PubMed].
26. Liu X, Zhang SM, McGeary MK, Krykbaeva I, Lai L, Jansen DJ, Kales SC, Simeonov A, Hall MD, Kelly DP, Bosenberg MW, Yan Q. KDM5B Promotes Drug Resistance by Regulating Melanoma-Propagating Cell Subpopulations. Mol Cancer Ther. 2019; 18:706–717. https://doi.org/10.1158/1535-7163.MCT-18-0395. [PubMed].
27. Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007; 104:19226–19231. https://doi.org/10.1073/pnas.0700735104. [PubMed].
28. Kuo KT, Huang WC, Bamodu OA, Lee WH, Wang CH, Hsiao M, Wang LS, Yeh CT. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin Epigenetics. 2018; 10:107. https://doi.org/10.1186/s13148-018-0533-9. [PubMed].
29. Shigekawa Y, Hayami S, Ueno M, Miyamoto A, Suzaki N, Kawai M, Hirono S, Okada KI, Hamamoto R, Yamaue H. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget. 2018; 9:34320–34335. https://doi.org/10.18632/oncotarget.26144. [PubMed].
30. Wang Z, Tang F, Qi G, Yuan S, Zhang G, Tang B, He S. KDM5B is overexpressed in gastric cancer and is required for gastric cancer cell proliferation and metastasis. Am J Cancer Res. 2014; 5:87–100. [PubMed].
31. Kuo YT, Liu YL, Adebayo BO, Shih PH, Lee WH, Wang LS, Liao YF, Hsu WM, Yeh CT, Lin CM. JARID1B Expression Plays a Critical Role in Chemoresistance and Stem Cell-Like Phenotype of Neuroblastoma Cells. PLoS One. 2015; 10:e0125343. https://doi.org/10.1371/journal.pone.0125343. [PubMed].
32. Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, Schneidawind D, Duque-Afonso J, Weng Z, Cleary ML. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell. 2015; 28:198–209. https://doi.org/10.1016/j.ccell.2015.06.003. [PubMed].
33. Roesch A, Becker B, Schneider-Brachert W, Hagen I, Landthaler M, Vogt T. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J Invest Dermatol. 2006; 126:1850–1859. https://doi.org/10.1038/sj.jid.5700324. [PubMed].
34. Yeh IJ, Esakov E, Lathia JD, Miyagi M, Reizes O, Montano MM. Phosphorylation of the histone demethylase KDM5B and regulation of the phenotype of triple negative breast cancer. Sci Rep. 2019; 9:17663. https://doi.org/10.1038/s41598-019-54184-0. [PubMed].
35. Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, Liu B, Tang GW, Zhang D, Tracz A, Jeter C, Rycaj K, Calhoun-Davis T, et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun. 2018; 9:3600. https://doi.org/10.1038/s41467-018-06067-7. [PubMed].
36. Li Q, Liu B, Chao HP, Ji Y, Lu Y, Mehmood R, Jeter C, Chen T, Moore JR, Li W, Liu C, Rycaj K, Tracz A, et al. LRIG1 is a pleiotropic androgen receptor-regulated feedback tumor suppressor in prostate cancer. Nat Commun. 2019; 10:5494. https://doi.org/10.1038/s41467-019-13532-4. [PubMed].
37. Cucchiara V, Yang JC, Mirone V, Gao AC, Rosenfeld MG, Evans CP. Epigenomic Regulation of Androgen Receptor Signaling: Potential Role in Prostate Cancer Therapy. Cancers (Basel). 2017; 9:9. https://doi.org/10.3390/cancers9010009. [PubMed].
38. Gao L, Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics. 2010; 5:100–104. https://doi.org/10.4161/epi.5.2.10778. [PubMed].
39. Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WL. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest. 2004; 113:913–923. https://doi.org/10.1172/JCI20032. [PubMed].
40. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243. https://doi.org/10.1038/nature11125. [PubMed].
41. Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, Perner S, Sboner A, Pawitan Y, Andrén O, Johnson LA, Tang J, Adami HO, Calza S, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst. 2008; 100:815–825. https://doi.org/10.1093/jnci/djn150. [PubMed].
42. Zhang J, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000; 141:4698–4710. https://doi.org/10.1210/endo.141.12.7837. [PubMed].
43. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470:279–283. https://doi.org/10.1038/nature09692. [PubMed].
44. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010; 107:21931–21936. https://doi.org/10.1073/pnas.1016071107. [PubMed].
45. Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, Ragoussis J, Natoli G. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010; 32:317–328. https://doi.org/10.1016/j.immuni.2010.02.008. [PubMed].
46. Leadem BR, Kagiampakis I, Wilson C, Cheung TK, Arnott D, Trojer P, Classon M, Easwaran H, Baylin SB. A KDM5 Inhibitor Increases Global H3K4 Trimethylation Occupancy and Enhances the Biological Efficacy of 5-Aza-2′-Deoxycytidine. Cancer Res. 2018; 78:1127–1139. https://doi.org/10.1158/0008-5472.CAN-17-1453. [PubMed].
47. Sayegh J, Cao J, Zou MR, Morales A, Blair LP, Norcia M, Hoyer D, Tackett AJ, Merkel JS, Yan Q. Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J Biol Chem. 2013; 288:9408–9417. https://doi.org/10.1074/jbc.M112.419861. [PubMed].
48. Bayo J, Tran TA, Wang L, Pena-Llopis S, Das AK, Martinez ED. Jumonji Inhibitors Overcome Radioresistance in Cancer through Changes in H3K4 Methylation at Double-Strand Breaks. Cell Rep. 2018; 25:1040–1050.e1045. https://doi.org/10.1016/j.celrep.2018.09.081. [PubMed].
49. Suraneni MV, Schneider-Broussard R, Moore JR, Davis TC, Maldonado CJ, Li H, Newman RA, Kusewitt D, Hu J, Yang P, Tang DG. Transgenic expression of 15-lipoxygenase 2 (15-LOX2) in mouse prostate leads to hyperplasia and cell senescence. Oncogene. 2010; 29:4261–4275. https://doi.org/10.1038/onc.2010.197. [PubMed].
50. Badeaux MA, Jeter CR, Gong S, Liu B, Suraneni MV, Rundhaug J, Fischer SM, Yang T, Kusewitt D, Tang DG. In vivo functional studies of tumor-specific retrogene NanogP8 in transgenic animals. Cell Cycle. 2013; 12:2395–2408. https://doi.org/10.4161/cc.25402. [PubMed].
51. Liu B, Gong S, Li Q, Chen X, Moore J, Suraneni MV, Badeaux MD, Jeter CR, Shen J, Mehmood R, Fan Q, Tang DG. Transgenic overexpression of NanogP8 in the mouse prostate is insufficient to initiate tumorigenesis but weakly promotes tumor development in the Hi-Myc mouse model. Oncotarget. 2017; 8:52746–52760. https://doi.org/10.18632/oncotarget.17186. [PubMed].
52. Zhang D, Jeter C, Gong S, Tracz A, Lu Y, Shen J, Tang DG. Histone 2B-GFP Label-Retaining Prostate Luminal Cells Possess Progenitor Cell Properties and Are Intrinsically Resistant to Castration. Stem Cell Reports. 2018; 10:228–242. https://doi.org/10.1016/j.stemcr.2017.11.016. [PubMed].