
Introduction
Lynch syndrome (LS) is a prevalent cancer predisposition syndrome, originally defined by the Amsterdam criteria [1, 2] and later by pathogenic or likely pathogenic germline variants of the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, or PMS2 [3]. Compared to the general population, carriers of such variants have significantly increased risks of cancers of the colon and rectum, endometrium, ovary, kidney and urinary tract, upper gastrointestinal tract, and certain other organs [4]. In comparison with earlier retrospective and family-based studies, recent prospective studies have arrived at somewhat lower age-specific risk estimates for cancers occurring in MMR variant carriers; moreover, penetrance and expression patterns greatly depend on the MMR gene involved [5]. Among individual MMR genes, pathogenic variants in MLH1 and MSH2 have the highest and PMS2 the lowest penetrance, and MSH6 variants underlie a sex-limited trait with a high risk of gynecological cancers in females [5].
Evaluations of breast cancer risk in LS have arrived at conflicting findings. Win et al. [6] conducted a systematic review on breast cancer in LS and identified 8 studies reporting an elevated (2–18-fold) risk, whereas the remaining 13 studies found no significantly increased risk. Moreover, breast cancer risk has been reported to be specifically associated with certain MMR genes, including MLH1 (vs. MSH2 [7, 8]), MSH2 [9], and MSH6 and PMS2 (vs. MLH1 and MSH2 [10, 11]. Variability in results may reflect different methods of ascertainment, cohort sizes, ethnicity, or other factors. In the large multicenter prospective investigation by Dominguez-Valentin et al. [5], the cumulative risk of breast cancer to 75 years of age was 12–15%, similar across all four MMR genes and representing only a marginal increase vs. average population.
Instability at microsatellite sequences (MSI) and absent MMR protein expression by immunohistochemical analysis of tumor tissues are common pre-screening methods for LS. Such methods classified 51% (62/122) of breast carcinomas from predisposing MMR gene variant carriers as MMR-deficient (dMMR) in studies reviewed by Win et al. [6]. Since deficient MMR is rare (< 2%) in breast carcinomas from the average population [12], the result implied a role for MMR deficiency in LS-associated breast cancer. Furthermore, abnormal immunohistochemistry and hypermutated tumor phenotype, combined with early onset of the disease (29 years), recently led to the suggestion that breast cancer is part of the tumor spectrum of the constitutional mismatch repair deficiency (CMMRD) syndrome caused by biallelic pathogenic germline variants of MMR genes [13].
We previously showed that breast carcinoma from carriers of inherited MMR defects resembles common breast carcinoma with respect to many clinicopathological features, such as mean age at onset over 50 years; however, the fact that over half of the tumors were dMMR suggested etiologic association to LS [14]. In the present investigation, we use somatic mutation profiling of breast cancers vs. established LS-spectrum tumors as a tool to address the relationship between breast cancer and LS.
Results
Clinicopathological characteristics of patients and tumors
This study was designed to investigate if breast carcinoma, the most common form of cancer in the general female population, is in LS individuals molecularly associated with their inherited MMR defects. To this end, carcinomas of the breast (BC), ovary (OC), and colon and rectum (CRC) were ascertained through the National LS Registry of Finland. Breast carcinomas from carriers of pathogenic or likely pathogenic germline MMR variants (LS-BC, n = 20) were compared to breast carcinomas from patients shown not to carry the predisposing MMR gene variant of their families (NC-BC, n = 10) and to established LS-spectrum tumors from pathogenic MMR gene variant carriers (LS-OC, n = 16, and LS-CRC, n = 18) (Tables 1 and 2). The different predisposing MMR genes were roughly similarly distributed across all LS groups, with MLH1 associated with 67%, MSH2 with 17%, and MSH6 with 17% of the total 54 tumors (Table 2).
Table 1: Molecular characteristics of breast carcinomas case by case
| Sample ID | Predisposing germline variant | MMR protein IHC | MSI status | Second hit status* | Mutations/Mb | |
|---|---|---|---|---|---|---|
| dMMR LS-BC | BC5_31814T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | negat | MSS | L | 141 |
| BC6_31489T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | negat | MSI | L, s | 725 | |
| BC11_31501T | MLH1 ex 16, 3.5 KB genomic deletion (Mut I) | negat | MSI | L | 24 | |
| BC11_31533T | MLH1 ex 16, 3.5 Kb genomic deletion (Mut I) | negat | MSS | L | 5 | |
| BC12_31491T | MLH1 ex 16, 3,5 kb genomic deletion (Mut I) | negat | MSI | None | 22 | |
| BC14_33229T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | negat | MSS | s | 662 | |
| BC9_33225T | MLH1 c.454-1G>A (mutation II) | negat | MSI | L | 29 | |
| BC7_33223T | MSH2 c.187delG | negat | MSI | L | 15 | |
| BC37_33228T | MSH2 ex 1-7 genomic deletion/MLPA | negat | MSI | s | 12 | |
| BC1_33203T | MSH6 ex 1-2 genomic deletion/MLPA | negat | MSS | s | 35 | |
| BC1_31528T | MSH6 ex 1-2 genomic deletion/MLPA | negat | MSS | s | 246 | |
| pMMR LS-BC | BC5_31495T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | posit | MSS | None | 31 |
| BC13_31486T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | posit | MSS | s | 85 | |
| BC4_31514T | MLH1 ex 16, 3.5 kb genomic deletion (Mut I) | posit | MSS | L | 4 | |
| BC38_31510T | MLH1 c.454-1G>A (mutation II) | posit | MSS | s | 53 | |
| BC8_33226T | MSH2 ex 1-16 genomic deletion/MLPA | posit | MSS | None | 8 | |
| BC10_33221T | MSH2 c.1738insA | posit | MSS | L | 2 | |
| BC2_31518T | MSH6 ex 1-2 genomic deletion/MLPA | posit | MSS | s | 108 | |
| BC39_33217T | MSH6 ex 1-2 genomic deletion/MLPA | posit | MSS | ND | 2 | |
| BC40_31503T | MSH6 c.2983G>T (nonsense) | posit | MSS | None | 5 | |
| NC-BC | BC24_31711T | Non-carrier | posit | MSS | N/A | 1 |
| BC23_33198T | Non-carrier | posit | MSS | N/A | 1 | |
| BC21_31702T | Non-carrier | posit | MSS | N/A | 5 | |
| BC20_33207T | Non-carrier | posit | MSS | N/A (ds) | 306 | |
| BC19_31706T | Non-carrier | posit | MSS | N/A | 2 | |
| BC18_33215T | Non-carrier | posit | MSS | N/A | 3 | |
| BC16_33205T | Non-carrier | posit | MSS | N/A (ds) | 393 | |
| BC43_33196T | Non-carrier | posit | MSS | N/A | 5 | |
| BC15_31696T | Non-carrier | posit | MSS | N/A (ds) | 161 | |
| BC17_31692T | Non-carrier | posit | MSS | N/A | 2 |
Table 2: Comparison of clinicopathological characteristics of patients and tumors from different groups
| LS breast carcinomas | Non-carrier breast carcinomas | LS ovarian carcinomas | LS CRC | ||
|---|---|---|---|---|---|
| dMMR (n = 11) | pMMR (n = 9) | (All pMMR) (n = 10) | (All dMMR) (n = 16) | (All dMMR) (n = 18) | |
| Average age of onset | 53 | 63 | 59 | 46 | 44 |
| Mean no. of non-synonymous somatic mutations | 696 (174/Mb) | 131 (33/Mb) | 352 (88/Mb) | 735 (184/Mb) | 689 (172/Mb) |
| Proportion hypermutated [> 10 (ns) mutations per Mb] | 10/11 (91%) | 4/9 (44%) | 3/10 (30%) | 13/16 (81%) | 18/18 (100%) |
| Predisposing gene | |||||
| MLH1 | 7/11 (64%) | 4/9 (44%) | N/A | 13/16 (81%) | 12/18 (67%) |
| MSH2 | 2/11 (18%) | 2/9 (22%) | 3/16 (19%) | 2/18 (11%) | |
| MSH6 | 2/11 (22%) | 3/9 (33%) | 0 | 4/18 (22%) | |
| Two-hit inactivation | |||||
| Germline mutation + LOH | 6/11 (55%) | 2/9 (22%) | N/A | 7/16 (44%)XXX | 9/18 (50%)XXX |
| Germline mutation + somatic point mutation (s) | 4/11 (36%) | 3/9 (33%) | 3/16 (19%)XXX | 6/18 (33%)XXX | |
| No obvious second hit | 1/11 (9%) | 3/9 (33%) | 1/16 (6%)XXX | 2/18 (11%)XXX | |
| ND | 0 | 1/9 (11%)* | 5/16 (31%)XXX | 1/18 (6%)XXX | |
| Mutational signatures | |||||
| Average 6 | 0,072 | 0,088 | 0,017 | 0,124 | 0,115 |
| Average 20 | 0,007 | 0,138 | 0,030 | 0,027 | 0 |
| Average 26 | 0,036 | 0 | 0 | 0,006 | 0,013 |
| Average 6, 20, or 26 | 0,115 | 0,226 | 0,047 | 0,157 | 0,128 |
Baseline characterization included MMR status of the tumors, where the absence of MMR protein by IHC, presence of MSI, or both were required for MMR-deficiency (see Materials and Methods). While all LS-OCs and LS-CRCs were MMR-deficient (dMMR) and all NC-BCs MMR-proficient (pMMR), LS-BCs broke down into dMMR (n = 11) and pMMR (n = 9) subgroups (Table 1). LS-BCs that were dMMR were diagnosed at the mean age of 53 years vs. 63 years for pMMR LS-BCs (p = 0.036). NC-BCs were diagnosed at 59 years on the average (non-significant difference relative to dMMR LS-BC).
Mechanisms of second allele inactivation of MMR genes in LS-BCs
LOH, somatic mutation, and promoter methylation were considered as possible second hits (Tables 1 and 2, Supplementary Table 1). In dMMR LS-BC, the primary mechanism of the second hit was LOH (6/11, 55%) whereas only 2/9 (22%) pMMR LS-BCs showed LOH. In pMMR LS-BC, somatic point mutation (s) of the predisposing MMR genes was the predominant mechanism (3/9, 33% vs. 4/11, 36% in dMMR LS-BC). Promoter methylation analysis by MS-MLPA indicated that none of the MLH1-deficient samples exhibited promoter methylation of MLH1 as a second hit. In summary, all but one dMMR LS-BCs (10/11, 91%) had a detectable second hit, compared to 5/9 (56%) of pMMR LS-BCs (statistically non-significant). The patterns of two-hit inactivation of the predisposing MMR genes in LS-BC closely resembled those previously observed for LS-OC and LS-CRC, where a detectable second hit, mostly LOH, was present in 10/16 (63%) of LS-OCs and 15/18 (83%) of LS-CRCs (Table 2 and ref [15]).
Mutation profiles of 578 cancer-relevant genes
Numbers of mutations
Somatic mutational analysis identified an average of 174 non-synonymous somatic mutations per Mb in dMMR LS-BCs, compared with 33/Mb in pMMR LS-BCs (non-significant difference) and 88/Mb in NC-BCs (borderline significant relative to dMMR LS-BC, p = 0.053) (Table 2). The corresponding mutation counts in LS-OCs and LS-CRCs were 184/Mb and 172/Mb, respectively.
All but one dMMR LS-BCs (10/11, 91%) were hypermutated (> 10 non-synonymous mutations per Mb). The single non-hypermutated tumor (BC11_31533T) was from a carrier of the most prevalent Finnish founder variant in MLH1 and showed LOH at MLH1 and absent MLH1 protein, but stable microsatellites (Table 1). The fraction of hypermutated dMMR LS-BC tumors was comparable to that of dMMR LS tumors of other organs (13/16, 81% for LS-OC and 18/18, 100% for LS-CRC) (Table 2). Among pMMR LS-BCs, 4/9 (44%) were hypermutated; of these, an inactivating somatic “hit” to the predisposing MMR gene was detectable in three and consisted of somatic mutation in all cases (Table 1).
Interestingly, the NC-BC group, too, revealed a hypermutated subset (3/10, 30%) (Tables 1 and 2). Double somatic non-synonymous point mutations in MMR genes were identified in all three hypermutated NC-BCs, affecting MLH1 in two tumors (BC15_31696T and BC20_33207T), and MSH2 and MSH6 in one tumor (BC16_33205T) (Supplementary Table 2). All MMR gene mutations occurred with allele frequencies below 25% and no MSI-high or IHC abnormality was present in the tumors. It is possible that the results reflected clonal heterogeneity without actual two-allele inactivation in any subclone. To search for alternative explanations for the high mutational loads, the hypermutated pMMR breast carcinomas (LS and NC cases) were Sanger-sequenced for POLE exon 9 and 13 and POLD1 exon 11; no mutations were identified in any cases successful in analyses.
Top mutant genes
Mutant allele frequencies of at least 25% and involvement in approximately one-third or a higher proportion of the tumors were considered as indicators of possible cancer driver nature, as outlined in our previous study [15]. In dMMR LS-BC, 18 genes fulfilled these requirements (were affected with high-frequency mutations in a minimum of 27% of the tumors; Supplementary Table 3). The 18 “top mutant” genes included 5 known to participate in epigenetic regulation and 4 involved in DNA repair, which suggested significant enrichment when compared to the shares of epigenetic regulatory genes and DNA repair genes in the entire panel of 578 genes (5/18 vs. 47/578, p = 0.010, and 4/18 vs. 45/578, p = 0.043, respectively). Details of all mutations affecting the 18 top mutant genes are described in Supplementary Table 4. Fewer top mutant genes were identified for pMMR LS-BC and NC-BC by an analogous selection procedure (Supplementary Table 3).
Figure 1 shows the involvement of the 18 dMMR LS-BC-associated genes in the remaining carcinoma groups, with each group compared against dMMR LS-BC. Due to the modest sample sizes, no high-level significant differences were observed; however, some observations can be made. MMR-proficient breast carcinoma groups (pMMR LS-BC and NC-BC) revealed comparable patterns, which often deviated from the dMMR LS-BC mutational pattern, implying the effect of MMR proficiency/deficiency (Figure 1A and 1B). The three MMR-deficient carcinoma groups (dMMR LS-BC, LS-OC, and LS-CRC) showed similarities and differences likely to at least in part reflect the tissues of origin (Figure 1C and 1D).
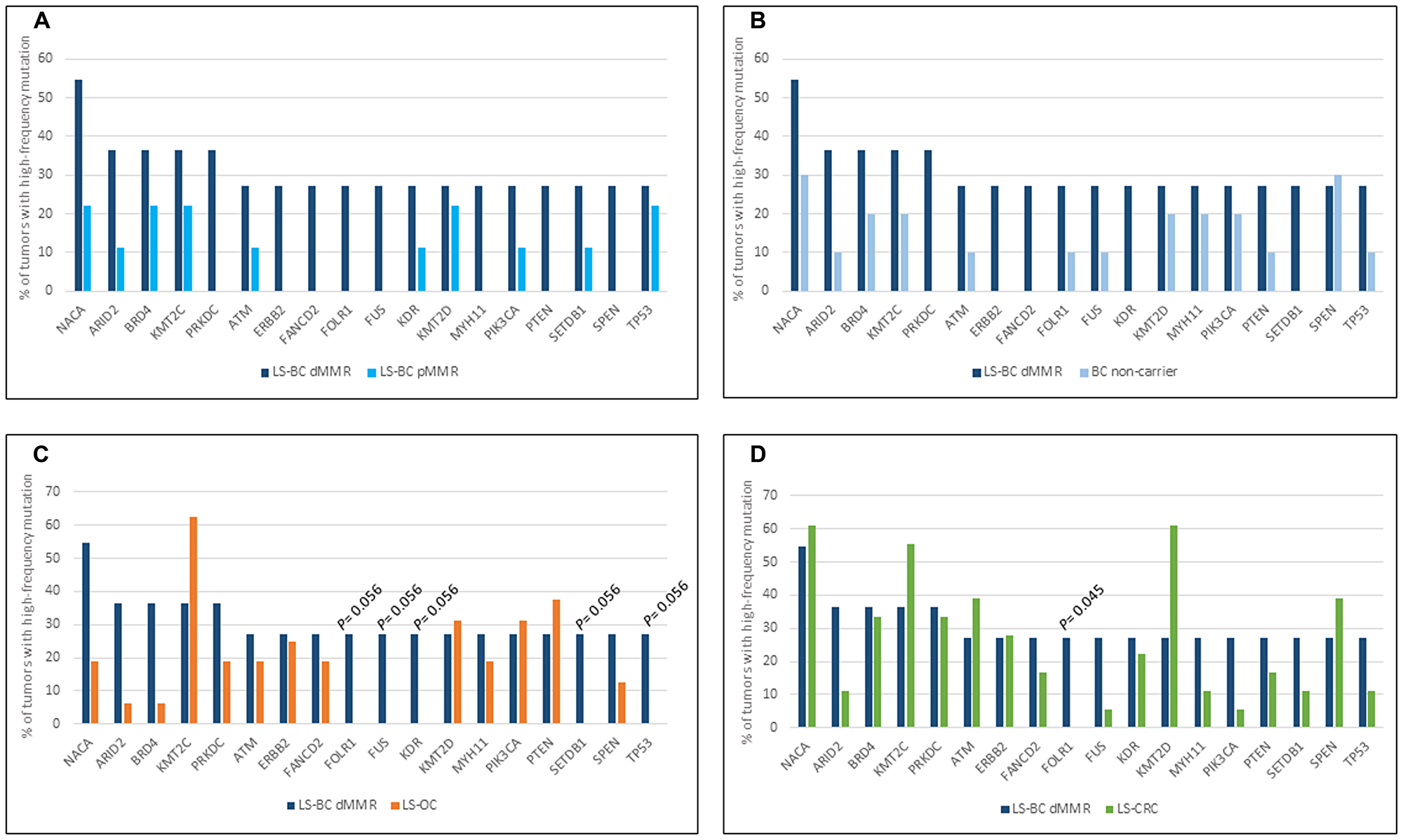
Figure 1: Top mutant genes across tumor types. Involvement of 18 LS-BC-associated top mutant genes (x-axis) in the dMMR subset of LS-BC (dark blue bars in (A–D), compared to the pMMR subset of LS-BC (turquoise bars in A), non-carrier BC (light blue bars in B), LS-OC (orange bars in C), and LS-CRC (green bars in D).
According to the predicted consequences, non-synonymous somatic mutations were classified into non-truncating (missense or in frame insertion/deletion) and truncating (frameshift or nonsense). As expected, the relative proportion of truncating mutations affecting our top mutant genes was higher in dMMR LS-BC compared to pMMR LS-BC (Supplementary Figure 1).
Mutational signature analysis
Somatic non-synonymous (high- and low-frequency) mutations detected in the panel of 578 genes were used to determine mutational signatures for each group of tumors (Figure 2) As the total number of mutations was low in a proportion of tumors, we preferred not to compare individual tumors, but instead determined group-specific averages for mutational signatures and used these for group-wise comparisons. Figure 2A shows the distribution of the 96 possible trinucleotide mutations [16] in breast carcinomas. Previous studies have linked C > T transitions at NpCpG sites to MMR deficiency [16]. Such mutations represented the most frequent type of substitutions in both the dMMR and pMMR subsets of LS-BC and were less common in NC-BC. Thirty reference mutational signatures are indexed in COSMIC, and Figure 2B shows the relative percentages of these signatures in BC, OC, and CRC. Among the main MMR-deficiency associated signatures 6, 20, and 26 [16], signature 6 was well represented in all tumor groups from originating from carriers of inherited MMR gene defects, including LS-BC (both dMMR and pMMR), LS-OC, and LS-CRC. Signature 20 was prominent in pMMR LS-BC. When the average frequencies of signatures 6, 20, and 26 were combined, dMMR LS-BC (0.115) and pMMR LS-BC (0.226) resembled LS-OC (0.157) and LS-CRC (0.128), whereas the share of MMR-deficiency associated signatures in NC-BC was clearly lower (0.047) (Table 2). The POLE-associated signature 10 [16] was absent in our entire breast cancer series. Of curiosity, all breast carcinoma subgroups (LS and NC) shared a notable signature 30 (Figure 2B), recently linked to defective base excision repair characteristic of breast and other carcinomas from biallelic NTHL1-mutation carriers [17]. NTHL1 was not included in our CCP panel, and the origin of signature 30 in our series remains unknown.
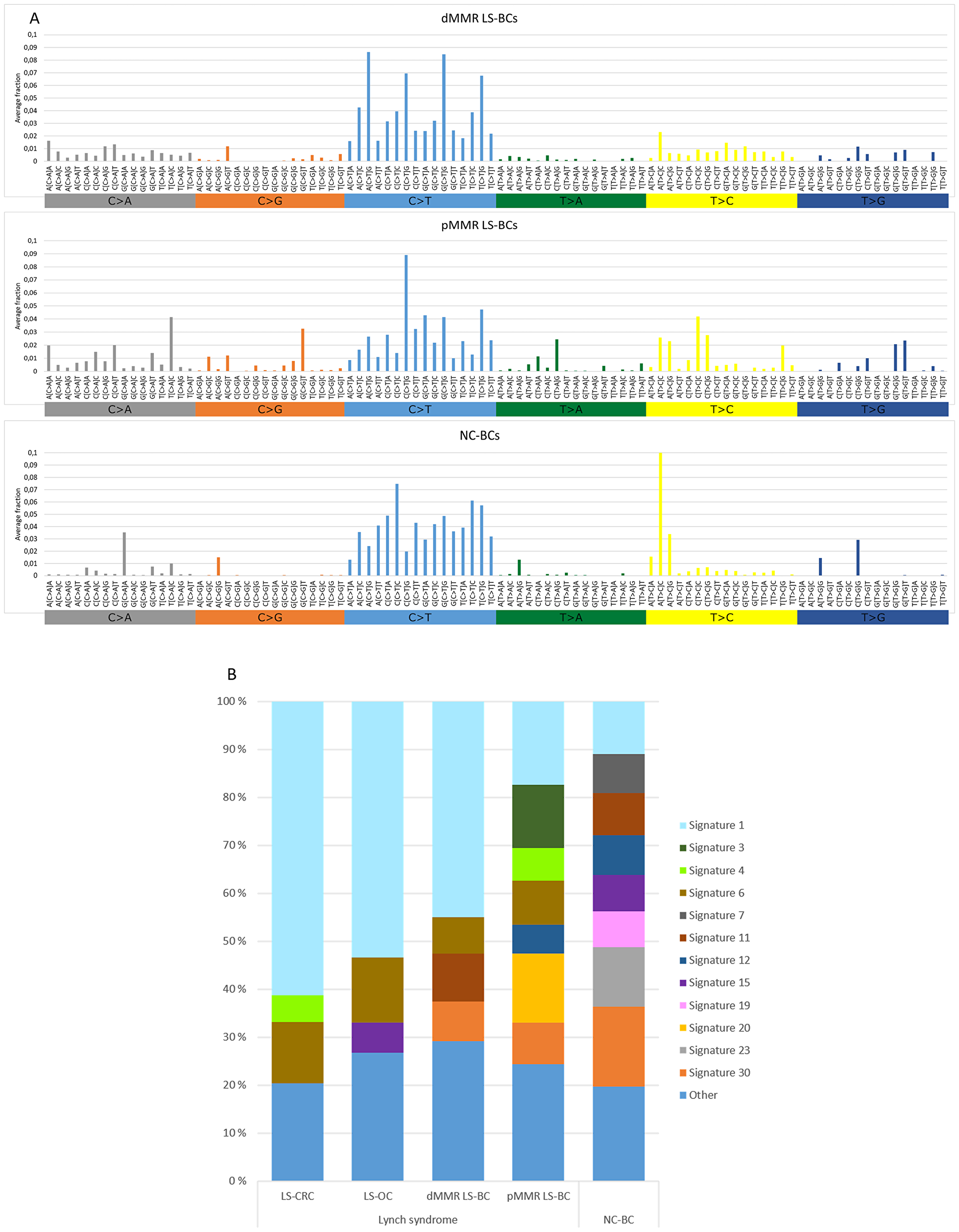
Figure 2: Mutational signatures of tumor groups. (A) Distributions of the average fractions of each of the 96 possible trinucleotide substitutions across dMMR LS-BCs, pMMR LS-BCs, and NC-BCs. (B) Proportions of COSMIC signatures 1–30 in LS-CRC, LS-OC, dMMR LS-BC, pMMR LS-BC, and NC-BC, based on average frequencies across each group. Signatures with average frequencies below 0.05 are combined into the ‘Other’ group.
DISCUSSION
The standardized incidence ratio of breast cancer in the Finnish LS families is not elevated [18]. Moreover, breast carcinoma from LS patients is not associated with any specific histological phenotype or clinical features that would distinguish it from breast carcinomas that occur sporadically. LS-associated breast carcinoma is typically ductal and diagnosed above 50 years of age on the average [19–22]. Hormone receptor status may vary: Walsh et al. [22] observed that most dMMR breast carcinomas from LS patients were hormone receptor negative compared to the pMMR subgroup, whereas the majority of LS breast carcinomas from our series were estrogen receptor-positive (15/18, 83%) and no difference between the dMMR and pMMR subgroups was observed. Like the series of Walsh et al. [22], our LS breast cancers were predominantly HER2-negative (15/18, 83%).
Microsatellite and immunohistochemical analysis divided our LS breast carcinomas into dMMR (55%) and pMMR (45%) subgroups in agreement with published studies (average 51% dMMR [6];). We have previously noted that immunohistochemical analysis is more sensitive than MSI to detect MMR deficiency in breast and other cancers from LS patients [14]. This likely reflects clonal heterogeneity characteristic of LS and sporadic MMR-deficient tumors [23, 24]. Consequently, all (100%) dMMR LS-BCs revealed immunohistochemical abnormality, but only 6 (55%) showed MSI-high. Neither MSI nor immunohistochemical change was present in the pMMR subgroup of LS-BCs.
Incorporation of mutational signatures which reflect the underlying pathophysiologic processes [16] may increase sensitivity even further. By studying large series of unselected breast carcinomas by genome-wide sequencing, Davies et al. [25] found that mutational signatures 6, 20, and 26 recognized MMR deficiency more faithfully than sequencing of MMR genes for mutations or analyzing exome data for MSI. We used non-synonymous mutations of 578 genes as the basis of mutation signature analysis. This limitation must be kept in mind when interpreting differences in mutation spectra, although targeted capture by panel sequencing and/or restriction to non-synonymous mutations have turned out informative in previous studies [26, 27]. The combined average proportion of MMR-deficiency-associated signatures, while low in NC-BC, was in our dMMR subset of LS-BC comparable to the averages observed in the established LS-spectrum tumors LS-OC and LS-CRC, and in the pMMR subset of LS-BC even higher (Table 2), suggesting that inherited MMR deficiency was likely to play an important role in the etiology of LS-BC irrespective of IHC/MSI status.
The total mutational burden offers another opportunity to reveal MMR deficiency. Nowak et al. [27] compared panel sequencing results from unselected colorectal carcinomas with immunohistochemical and MSI data and found that false negative results relative to MSI were attributable to tumor heterogeneity, whereas false positive results were explained by POLE mutations. Using the commonly accepted threshold of over 10 somatic mutations/Mb [28], 100% LS-CRCs and 91% dMMR LS-BCs were hypermutated, compared to 81% of LS-OCs and 44% of pMMR LS-BCs (Table 2). Importantly, the NC-BC group also had a notable hypermutated subset (3/10, 30%) in the absence of MSI or extinct MMR protein expression or evidence of DNA polymerase proofreading defects. While somatic low-frequency mutations were detected in MMR genes and might play a role, the ultimate mechanism of hypermutated phenotype in NC-BC breast carcinomas remained unsettled.
Even for such cancers whose risks have consistently been shown to be elevated in LS compared to the average population, considerable MSS fractions exist. For example, brain tumors we previously examined from LS patients all lacked MSI-high [29]. Additionally, one-fourth of colorectal adenomas developing in LS patients are pMMR by immunohistochemical and MSI analysis, suggesting that MMR deficiency is not a prerequisite for tumor formation [30, 31]. In LS, even immunoactivation may take place in premalignant lesions that have neither dMMR nor elevated somatic mutational loads [32]. Significant heterogeneity has been demonstrated on genome-wide level: genomic and transcriptomic analyses conducted by Binder et al. [33] divided LS-CRCs into two subgroups, one with high numbers of somatic mutations reminiscent of sporadic MSI CRC and another one with lower mutational loads resembling sporadic MSS CRC. As for mechanisms that might mediate predisposition to pMMR cancers in carriers of inherited MMR defects, several possibilities exist. Apart from the repair of replication errors, the MMR system has many other anti-carcinogenic functions, such as cell cycle checkpoint control in response to DNA damage [34]. For MLH1, the predominant predisposing gene among our LS-BC cases, it was demonstrated that even low reductions of the protein product can impair cell cycle checkpoint activation while the cells remain MMR-proficient [35]. Chromosomal segregation represents another function sensitive to the dosage of the MLH1 gene product in an analogous manner [36]. Finally, MMR proteins participate in additional repair mechanisms, whose failure may contribute to hypermutability despite MMR proficiency; for example, MSH2 is part of BRCA1-associated genome surveillance complex that protects against DNA double-strand breaks [37]. In this context, it is of interest that the pMMR subgroup of LS-BC had a notable signature 3 (Figure 2B) which is known to be associated with defects in homologous recombination [16].
A whole-genome investigation of 560 breast carcinomas [38] identified TP53, PIK3CA, MYC, CCND1, PTEN, ERBB2, FGFR1, GATA3, RB1, and MAP3K1 as the most frequently mutant genes. Of these, TP53, PIK3CA, PTEN, and ERBB2 were among the top mutated genes selected by the criteria we used (at least 27% of tumors affected by mutations with allele frequency 25% or higher) (Figure 1). While seldom affected in LS-CRC and LS-OC, TP53 was mutant in 5/20 (25%) of our LS-BCs. Interestingly, somatic TP53 mutations in breast cancer were recently associated with immune-rich status [39]. In a function-based classification, epigenetic regulatory genes and DNA repair genes were significantly enriched as mutational targets among our LS-BC-associated genes. The histone lysine methyltransferases KMT2C (MLL3) and KMT2D (MLL2) (Figure 1) also belong to the driver genes detected by Nik-Zainal et al. [38]. Mutations in these genes may alter the expression of other genes (e. g., inactivating KMT2C mutations were shown to downregulate genes involved in homologous recombination-mediated DNA repair, making the tumor cells chromosomally unstable [40];) or be harmful by other mechanisms (e. g., KMT2D mutations were found to increase mutational burden and genome instability in cancer through transcription stress [41],). Combined with our previous findings [15], frequent mutations in epigenetic regulatory genes appear to be a common feature of LS tumors, applicable to LS-OC and LS-CRC as well.
Mismatch repair deficiency and the associated hypermutability may indicate responsiveness to PD-1 blockade, as recently reported for metastatic dMMR breast cancer [42]. Rampias T et al. [40] showed that inactivating KMT2C mutations (see above) caused sensitivity to PARP1/2 inhibition through synthetic lethality. Several other genes involved in LS-associated breast cancer may also be clinically actionable [43].
In conclusion, we demonstrate that LS-BCs which fell into dMMR and pMMR subsets by conventional methods shared MMR-deficiency-associated consensus signatures with the established LS spectrum tumors LS-OC and LS-CRC. Our results suggest that inherited MMR deficiency likely contributed to the development of LS-BC through disruption of MMR-related and non-MMR-related functions, thereby facilitating tumor initiation or progression. As this study was based on a modest number of cases retrieved from a national LS registry and the predisposing genes (MLH1, MSH2, and MSH6) were unevenly distributed, our tumors may not be considered representative of all tumors of the respective organs occurring in LS. Therefore, our results need to be confirmed in larger sample sets preferably representing multiple populations.
Materials and Methods
Patients and samples
All available breast carcinomas (LS-BC) (n = 20) and corresponding normal DNA samples from 17 females were collected from the National LS registry of Finland (LSRFi) that includes information of approximately 300 LS families and over 1600 tested carriers of inherited MMR defects. All predisposing variants were pathogenic or likely pathogenic, representing pathogenicity classes 5 and 4, respectively. MLH1 was affected in 9 patients (with 11 tumors) of which 7 patients had the prevalent Finnish founder variant (“mutation I”), which is a 3.5-kb genomic deletion of exon 16 and its flanking introns [44]. Four individuals (with one tumor each) had a pathogenic or likely pathogenic germline variant in MSH2, and one individual with two tumors had a predisposing variant in MSH6 (Table 1). Estrogen receptor (ER) status was positive in 15 LS-BC samples, 3 tested negative and the status could not be determined for 2. Human epidermal growth factor receptor 2 (HER2) status was positive for 3, negative for 15, and could not be determined for 2 LS-BC tumors.
For comparison, 10 breast carcinomas from non-carrier members of families registered in LSRFi were included (the NC-BC group in Table 1). The ER status and HER2 status (mainly positive and negative, respectively) of NC-BCs matched with those of LS-BCs. The average age at breast cancer diagnosis was similar in LS and NC-BC groups (57 and 59 years, respectively). In addition, we analyzed 16 LS ovarian and 18 LS colorectal carcinomas (LS-OCs and LS-CRCs, respectively) from the same registry [15].
All tumor samples and the majority of normal DNA samples (19/27) were formalin-fixed paraffin embedded (FFPE) samples. DNA was extracted from selected high tumor percentage areas according to the modified protocol described by Isola et al. [45]. The remaining eight normal DNA samples were extracted from blood according to the non-enzymatic protocol described by Lahiri and Nurnberger [46].
This study was approved by the Institutional Review Board of the Helsinki University Central Hospital (466/E6/01). The National Supervisory Authority for Welfare and Health (Dnro 1272/04/044/07 and Dnro 10741/06.01.03.01/2015) approved the collection of archival specimens. Informed consent procedures defined by the Ethics approvals were followed in sample collection.
MMR status of breast carcinomas
Results from immunohistochemical (IHC) analyses for MMR protein expression and microsatellite instability (MSI) analyses were available from our previous investigation [14]. Breast carcinomas were considered MMR-deficient (dMMR) when MMR protein was absent by IHC and/or the tumors showed MSI (at least one of the two mononucleotide repeat markers BAT25 and BAT26 was unstable).
MLH1 promoter methylation analysis
Promoter methylation status of MLH1 in breast cancer samples with deficient MLH1 expression was determined by methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) using SALSA MS-MLPA probemix ME001-C1 (MRC Holland, Amsterdam, Netherlands), as described in Lotsari et al. [14]. Of the two MLH1-associated probe pairs, the one closest to the transcription start site was considered. Methylation dosage ratio of 0.25 or higher (corresponding to at least 25% of methylated DNA) provided the best discrimination between tumor and paired normal DNA and was used as the cut-off for hypermethylation [14].
Comprehensive cancer panel (CCP) sequencing
Tumor and matching normal DNA samples from LS-BC and NC-BC cases were sequenced in the Institute for Molecular Medicine Finland (FIMM) on Illumina HiSeq 2500 platform (San Diego, CA, USA) using Nimblegen Comprehensive cancer panel (Roche Diagnostics). The panel is a 4 Mb design covering 578 known cancer-related genes and their intronic regions compiled from the Sanger Institute Cancer Gene Census database (https://www.ncbi.nlm.nih.gov/pubmed) and the NCBI Gene tests database (https://www.ncbi.nlm.nih.gov/). ThruPLEX® DNA-seq Kit was used for library preparation, and exon capture was conducted according to the manufacturer’s protocol (Rubicon Genomics). The variant calling pipeline is described in detail by Sulonen et al. [47]; in the present study, we used version 3.6. To enable comparison with breast cancer data, LS-OC and LS-CRC data generated previously [15] were re-analyzed with the VCP 3.6 pipeline. Supplementary Table 5 shows performance characteristics of breast carcinomas. The mean target coverage was 160-fold for dMMR LS-BC, 114-fold for pMMR LS-BC, and 78-fold for NC-BC. Please see ref [15]. for the performance characteristics of LS-OC and LS-CRC.
Somatic mutation analysis of CCP data
Paired tumor and normal sample sequencing data were analyzed by the VarScan 2 mutation detection algorithm version 2.3.2 [48] to identify non-synonymous (missense, nonsense, frameshift, in-frame coding deletion/insertion and splice site) changes of somatic origin. Variants with VarScan somatic p-value below 0.01 were considered significant and are referred to as `somatic mutations` throughout this paper. Such variants had the possibility of being pathogenic according to traditional pathogenicity classes 3–5 and were selected for subsequent analyses. The variants were categorized based on variant allele frequency (low frequency < 0.25 vs. high frequency ≥ 0.25) and effect (truncating vs. non-truncating) as described previously [15, 49].
POLD1 and POLE sequencing
Hypermutated pMMR breast carcinomas were screened for proofreading mutations in POLE exons 9 and 13 by Sanger sequencing with primers described by Church et al. [50]. POLD1 exon 11 was examined by Sanger sequencing with primers described in Valle et al. 2014 [51].
Two-hit inactivation of MMR genes
Somatic point mutations in MMR genes that could serve as second hits were identified from the CCP sequencing data. Loss of heterozygosity (LOH) was also evaluated as a possible second hit and the method depended on the type of the predisposing MMR gene alteration. When the predisposing MMR gene change was a point mutation, VarSeq software (GoldenHelix®) with VCP filtered CCP sequencing data (. vcf-files) was used to compare sequence data from tumor and their corresponding normal samples. The variant allele reads (Alt) to reference allele reads (Ref) ratio was determined in tumor (T) and normal (N) DNA, and LOH ratio (R) calculated as follows: R = (Alt: Ref)T/(Alt: Ref)N. The thresholds for “strict” LOH and “putative” LOH were as specified by Ollikainen et al. [52]. When the predisposing change was a large deletion, MLPA-based data (SALSA P003-C1 for MLH1 and MSH2 and SALSA 072-C1 for MSH6; MRC Holland, Amsterdam, The Netherlands) were utilized for LOH analysis and the results interpreted according to Zhang et al. [53]. “Putative” and “strict” LOH are called LOH throughout this paper. In MLH1-associated cases, promoter methylation of MLH1 was tested as a second hit by MS-MLPA as described above under “MLH1 promoter methylation analysis”.
Definition of top-mutated genes
We used a procedure developed in our previous study [15]. In brief, for each of the 578 genes of the CCP panel, we determined the proportion of tumors having that gene in a mutant form. We focused on mutations with high (≥ 25%) variant allele frequency to increase the likelihood of clonal (driver) as opposed to subclonal (passenger) mutations [54]. Based on the distribution of the proportions of tumors with individual genes mutant, a cut-off of one-third was established to divide the genes into top-mutated and less commonly mutated categories. Finally, a pathway annotation was performed on each top gene according to GeneCards (http://www.genecards.org) and relevant publications from PubMed (https://www.ncbi.nlm.nih.gov/pubmed).
In silico predictions of somatic mutations
We utilized in silico predictions from Varsome-database [55] to assign a pathogenic significance category for somatic variants identified for MMR genes in the second hit analysis.
Mutational signature analysis
Mutational signatures by Alexandrov et al. [16] were determined by applying the R package deconstructSigs [56] to significant (p < 0.01) non-synonymous somatic mutations from VarScan2 analysis (see “Somatic mutation analysis of CCP data” above) using default parameters against signatures recognized by the COSMIC database [57]. In the analysis, deconstructSigs determines the mutational profile of tumor samples by applying a multiple linear regression model to the input data. Mutational signatures were called for each sample individually and collectively for groups of samples (BC vs. OC vs. CRC; dMMR vs. pMMR).
Statistical analyses
Statistical analyses were performed using IBM SPSS Statistical software version 25.0 (IBM SPSS Inc., Chicago, IL, USA). The applicability of the data for parametric vs. non-parametric tests was tested first. Statistical significance of distribution of mutated genes in independent groups was evaluated using the Mann-Whitney U test. Pairwise comparisons of frequency data were conducted by the Fisher’s exact test. Differences with p-value < 0.05 (two-tailed) were considered significant.
Abbreviations
CMMRD: constitutional mismatch repair deficiency; COSMIC: Catalogue of Somatic Mutations in Cancer; dMMR: mismatch repair deficient; ER: Estrogen receptor; FFPE: formalin-fixed paraffin embedded; HER2: Human epidermal growth factor receptor 2; IHC: immunohistochemistry; LOH: loss of heterozygosity; LS: Lynch syndrome; LS-BC: Lynch syndrome breast carcinoma; LS-CRC: Lynch syndrome colorectal carcinoma; LS-OC: Lynch syndrome ovarian carcinoma; LSRFi: the National LS registry of Finland; MMR: DNA mismatch repair; MSI: microsatellite instability; MS-MLPA: methylation-specific multiplex ligation-dependent probe amplification; NC-BC: non-carrier breast carcinoma; pMMR: mismatch repair proficient.
Data availability
All relevant data generated during this study are included in this published article and its additional materials.
ACKNOWLEDGMENTS
We thank the clinical staff and patients for participating in the study. Saila Saarinen is sincerely thanked for expert technical assistance.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interests.
FUNDING
This work was supported by Jane and Aatos Erkko Foundation (to P.P. and J.-P.M); the Academy of Finland (grant number 294643 to P.P); the Finnish Cancer Foundation (to N.P, P.P., and J.-P.M); the Sigrid Juselius Foundation (to P.P); the HiLIFE Fellows 2017–2020 (to P.P); K. Albin Johansson Stiftelse (to N.P). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991; 34:424–425. https://doi.org/10.1007/BF02053699. [PubMed].
2. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999; 116:1453–1456. https://doi.org/10.1016/S0016-5085(99)70510-X. [PubMed].
3. Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, Farrell MP, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014; 46:107–115. https://doi.org/10.1038/ng.2854. [PubMed].
4. Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, Macrae FA, Goldblatt J, Southey MC, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012; 30:958–964. https://doi.org/10.1200/JCO.2011.39.5590. [PubMed].
5. Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, Engel C, Aretz S, Jenkins MA, Sunde L, Bernstein I, Capella G, Balaguer F, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020; 22:15–25. https://doi.org/10.1038/s41436-019-0596-9. [PubMed].
6. Win AK, Lindor NM, Jenkins MA. Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res. 2013; 15:R27. https://doi.org/10.1186/bcr3405. [PubMed].
7. Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, du Sart D, Tucker K, Kirk J. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet. 2001; 68:118–127. https://doi.org/10.1086/316942. [PubMed].
8. Harkness EF, Barrow E, Newton K, Green K, Clancy T, Lalloo F, Hill J, Evans DG. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet. 2015; 52:553–556. https://doi.org/10.1136/jmedgenet-2015-103216. [PubMed].
9. Goldberg M, Bell K, Aronson M, Semotiuk K, Pond G, Gallinger S, Zbuk K. Association between the Lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. J Med Genet. 2017; 54:742–746. https://doi.org/10.1136/jmedgenet-2017-104542. [PubMed].
10. Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J Clin Oncol. 2017; 35:2568–2575. https://doi.org/10.1200/JCO.2016.71.9260. [PubMed].
11. Roberts ME, Jackson SA, Susswein LR, Zeinomar N, Ma X, Marshall ML, Stettner AR, Milewski B, Xu Z, Solomon BD, Terry MB, Hruska KS, Klein RT, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med. 2018; 20:1167–1174. https://doi.org/10.1038/gim.2017.254. [PubMed].
12. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016; 22:1342–1350. https://doi.org/10.1038/nm.4191. [PubMed].
13. Bush L, Aronson M, Tabori U, Campbell BB, Bedgood RB, Jasperson K. Delineating a new feature of constitutional mismatch repair deficiency (CMMRD) syndrome: breast cancer. Fam Cancer. 2019; 18:105–108. https://doi.org/10.1007/s10689-018-0088-0. [PubMed].
14. Lotsari JE, Gylling A, Abdel-Rahman WM, Nieminen TT, Aittomaki K, Friman M, Pitkanen R, Aarnio M, Jarvinen HJ, Mecklin JP, Kuopio T, Peltomaki P. Breast carcinoma and Lynch syndrome: molecular analysis of tumors arising in mutation carriers, non-carriers, and sporadic cases. Breast Cancer Res. 2012; 14:R90. https://doi.org/10.1186/bcr3205. [PubMed].
15. Porkka N, Valo S, Nieminen TT, Olkinuora A, Maki-Nevala S, Eldfors S, Peltomaki P. Sequencing of Lynch syndrome tumors reveals the importance of epigenetic alterations. Oncotarget. 2017; 8:108020–108030. https://doi.org/10.18632/oncotarget.22445. [PubMed].
16. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. https://doi.org/10.1038/nature12477. [PubMed].
17. Grolleman JE, de Voer RM, Elsayed FA, Nielsen M, Weren RDA, Palles C, Ligtenberg MJL, Vos JR, Ten Broeke SW, de Miranda N, Kuiper RA, Kamping EJ, Jansen EAM, et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell. 2019; 35:256–66.e5. https://doi.org/10.1016/j.ccell.2018.12.011. [PubMed].
18. Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999; 81:214–218. https://doi.org/10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L. [PubMed].
19. Buerki N, Gautier L, Kovac M, Marra G, Buser M, Mueller H, Heinimann K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer. 2012; 51:83–91. https://doi.org/10.1002/gcc.20935. [PubMed].
20. Jensen UB, Sunde L, Timshel S, Halvarsson B, Nissen A, Bernstein I, Nilbert M. Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat. 2010; 120:777–782. https://doi.org/10.1007/s10549-009-0449-3. [PubMed].
21. Shanley S, Fung C, Milliken J, Leary J, Barnetson R, Schnitzler M, Kirk J. Breast cancer immunohistochemistry can be useful in triage of some HNPCC families. Fam Cancer. 2009; 8:251–255. https://doi.org/10.1007/s10689-008-9226-4. [PubMed].
22. Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, Walters R, McKeone DM, Spurdle AB, Hopper JL, Jenkins MA, Phillips KD, Suthers GK, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010; 16:2214–2224. https://doi.org/10.1158/1078-0432.CCR-09-3058. [PubMed].
23. Beggs AD, Domingo E, Abulafi M, Hodgson SV, Tomlinson IP. A study of genomic instability in early preneoplastic colonic lesions. Oncogene. 2013; 32:5333–5337. https://doi.org/10.1038/onc.2012.584. [PubMed].
24. Giuffrè G, Müller A, Brodegger T, Bocker-Edmonston T, Gebert J, Kloor M, Dietmaier W, Kullmann F, Büttner R, Tuccari G, Rüschoff J; German HNPCC Consortium, German Cancer Aid, (Deutsche Krebshilfe). Microsatellite analysis of hereditary nonpolyposis colorectal cancer-associated colorectal adenomas by laser-assisted microdissection: correlation with mismatch repair protein expression provides new insights in early steps of tumorigenesis. J Mol Diagn. 2005; 7:160–170. https://doi.org/10.1016/S1525-1578(10)60542-9. [PubMed].
25. Davies H, Morganella S, Purdie CA, Jang SJ, Borgen E, Russnes H, Glodzik D, Zou X, Viari A, Richardson AL, Borresen-Dale AL, Thompson A, Eyfjord JE, et al. Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017; 77:4755–4762. https://doi.org/10.1158/0008-5472.CAN-17-1083. [PubMed].
26. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017; 23:703–713. https://doi.org/10.1038/nm.4333. [PubMed].
27. Nowak JA, Yurgelun MB, Bruce JL, Rojas-Rudilla V, Hall DL, Shivdasani P, Garcia EP, Agoston AT, Srivastava A, Ogino S, Kuo FC, Lindeman NI, Dong F. Detection of Mismatch Repair Deficiency and Microsatellite Instability in Colorectal Adenocarcinoma by Targeted Next-Generation Sequencing. J Mol Diagn. 2017; 19:84–91. https://doi.org/10.1016/j.jmoldx.2016.07.010. [PubMed].
28. Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, Sander C, Pursell ZF, Wheeler DA. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014; 24:1740–1750. https://doi.org/10.1101/gr.174789.114. [PubMed].
29. Gylling AH, Nieminen TT, Abdel-Rahman WM, Nuorva K, Juhola M, Joensuu EI, Jarvinen HJ, Mecklin JP, Aarnio M, Peltomaki PT. Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis. 2008; 29:1351–1359. https://doi.org/10.1093/carcin/bgn133. [PubMed].
30. Ahadova A, Gallon R, Gebert J, Ballhausen A, Endris V, Kirchner M, Stenzinger A, Burn J, von Knebel Doeberitz M, Blaker H, Kloor M. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int J Cancer. 2018; 143:139–150. https://doi.org/10.1002/ijc.31300. [PubMed].
31. Maki-Nevala S, Valo S, Ristimaki A, Sarhadi V, Knuutila S, Nystrom M, Renkonen-Sinisalo L, Lepisto A, Mecklin JP, Peltomaki P. DNA methylation changes and somatic mutations as tumorigenic events in Lynch syndrome-associated adenomas retaining mismatch repair protein expression. EBioMedicine. 2019; 39:280–291. https://doi.org/10.1016/j.ebiom.2018.12.018. [PubMed].
32. Chang K, Taggart MW, Reyes-Uribe L, Borras E, Riquelme E, Barnett RM, Leoni G, San Lucas FA, Catanese MT, Mori F, Diodoro MG, You YN, Hawk ET, et al. Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol. 2018; 4:1085–1092. https://doi.org/10.1001/jamaoncol.2018.1482. [PubMed].
33. Binder H, Hopp L, Schweiger MR, Hoffmann S, Juhling F, Kerick M, Timmermann B, Siebert S, Grimm C, Nersisyan L, Arakelyan A, Herberg M, Buske P, et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J Pathol. 2017; 243:242–254. https://doi.org/10.1002/path.4948. [PubMed].
34. Gupta D, Heinen CD. The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair (Amst). 2019; 78:60–69. https://doi.org/10.1016/j.dnarep.2019.03.009. [PubMed].
35. Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavo E, di Pietro M, Marra G, Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003; 22:2245–2254. https://doi.org/10.1093/emboj/cdg216. [PubMed].
36. Pussila M, Toronen P, Einarsdottir E, Katayama S, Krjutskov K, Holm L, Kere J, Peltomaki P, Makinen MJ, Linden J, Nystrom M. Mlh1 deficiency in normal mouse colon mucosa associates with chromosomally unstable colon cancer. Carcinogenesis. 2018; 39:788–797. https://doi.org/10.1093/carcin/bgy056. [PubMed].
37. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000; 14:927–939. [PubMed].
38. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. https://doi.org/10.1038/nature17676. [PubMed].
39. Behring M, Vazquez AI, Cui X, Irvin MR, Ojesina AI, Agarwal S, Manne U, Shrestha S. Gain of function in somatic TP53 mutations is associated with immune-rich breast tumors and changes in tumor-associated macrophages. Mol Genet Genomic Med. 2019; 7:e1001. https://doi.org/10.1002/mgg3.1001. [PubMed].
40. Rampias T, Karagiannis D, Avgeris M, Polyzos A, Kokkalis A, Kanaki Z, Kousidou E, Tzetis M, Kanavakis E, Stravodimos K, Manola KN, Pantelias GE, Scorilas A, et al. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. 2019; 20. https://doi.org/10.15252/embr.201846821. [PubMed].
41. Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, Aygun O, Kelly GP, Matthews N, Stewart A, Stewart AF, Svejstrup JQ. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016; 30:408–420. https://doi.org/10.1101/gad.275453.115. [PubMed].
42. Kok M, Horlings HM, Snaebjornsson P, Chalabi M, Schumacher TN, Blank CU, Linn SC, van Dieren J. Case report: Profound Immunotherapy Response in Mismatch Repair-Deficient Breast Cancer. JCO Precis Oncol. 2017; 10:1–3.
43. Chamberlin MD, Bernhardt EB, Miller TW. Clinical Implementation of Novel Targeted Therapeutics in Advanced Breast Cancer. J Cell Biochem. 2016; 117:2454–2463. https://doi.org/10.1002/jcb.25590. [PubMed].
44. Nystrom-Lahti M, Kristo P, Nicolaides NC, Chang SY, Aaltonen LA, Moisio AL, Jarvinen HJ, Mecklin JP, Kinzler KW, Vogelstein B, De La Chapelle A, Peltomäki P. Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med. 1995; 1:1203–1206. https://doi.org/10.1038/nm1195-1203. [PubMed].
45. Isola J, DeVries S, Chu L, Ghazvini S, Waldman F. Analysis of changes in DNA sequence copy number by comparative genomic hybridization in archival paraffin-embedded tumor samples. Am J Pathol. 1994; 145:1301–1308. [PubMed].
46. Lahiri DK, Nurnberger JI Jr. A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991; 19:5444. https://doi.org/10.1093/nar/19.19.5444. [PubMed].
47. Sulonen AM, Ellonen P, Almusa H, Lepisto M, Eldfors S, Hannula S, Miettinen T, Tyynismaa H, Salo P, Heckman C, Joensuu H, Raivio T, Suomalainen A, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011; 12:R94. https://doi.org/10.1186/gb-2011-12-9-r94. [PubMed].
48. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–576. https://doi.org/10.1101/gr.129684.111. [PubMed].
49. Porkka N, Lahtinen L, Ahtiainen M, Bohm JP, Kuopio T, Eldfors S, Mecklin JP, Seppala TT, Peltomaki P. Epidemiological, clinical and molecular characterization of Lynch-like syndrome: A population-based study. Int J Cancer. 2019; 145:87–98. https://doi.org/10.1002/ijc.32085. [PubMed].
50. Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, ter Haar N, Noske A, Amant F, Tomlinson IP, Wild PJ, Lambrechts D, Jurgenliemk-Schulz IM, Jobsen JJ, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst. 2015; 107:402. https://doi.org/10.1093/jnci/dju402. [PubMed].
51. Valle L, Hernandez-Illan E, Bellido F, Aiza G, Castillejo A, Castillejo MI, Navarro M, Segui N, Vargas G, Guarinos C, Juarez M, Sanjuan X, Iglesias S, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014; 23:3506–3512. https://doi.org/10.1093/hmg/ddu058. [PubMed].
52. Ollikainen M, Abdel-Rahman WM, Moisio AL, Lindroos A, Kariola R, Jarvela I, Poyhonen M, Butzow R, Peltomaki P. Molecular analysis of familial endometrial carcinoma: a manifestation of hereditary nonpolyposis colorectal cancer or a separate syndrome? J Clin Oncol. 2005; 23:4609–4616. https://doi.org/10.1200/JCO.2005.06.055. [PubMed].
53. Zhang J, Lindroos A, Ollila S, Russell A, Marra G, Mueller H, Peltomaki P, Plasilova M, Heinimann K. Gene conversion is a frequent mechanism of inactivation of the wild-type allele in cancers from MLH1/MSH2 deletion carriers. Cancer Res. 2006; 66:659–664. https://doi.org/10.1158/0008-5472.CAN-05-4043. [PubMed].
54. Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016; 48:238–244. https://doi.org/10.1038/ng.3489. [PubMed].
55. Varsome - The Human Genomics Community. https://varsome.com/about/about-varsome/.
56. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 17:31. https://doi.org/10.1186/s13059-016-0893-4. [PubMed].
57. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. Clock-like mutational processes in human somatic cells. Nat Genet. 2015; 47:1402–1407. https://doi.org/10.1038/ng.3441. [PubMed].