
Introduction
Fms-like tyrosine kinase 3 (FLT3) is a member of the class III family of receptor tyrosine kinases expressed on early hematopoietic stem and progenitor cells, and plays an important role in hematopoiesis [1]. Wild-type FLT3 (FLT3wt) is activated by FLT3 ligand (FL), which promotes receptor dimerization, auto-phosphorylation, and cell proliferation via activation of its downstream signaling molecules, including STAT5, ERK1/2, and AKT [2]. FLT3 is made up of 5 functional domains: an immunoglobulin-like extracellular domain, a transmembrane domain, a juxtamembrane domain (JMD), a tyrosine kinase domain (TKD), and a small C-terminal domain [3]. Mutations at the JMD or TKD of FLT3 induce constitutive kinase activation that is independent of FL, and occurs in approximately one-third of acute myeloid leukemia (AML) patients [4, 5]. In particular, in-frame duplications of 3 to >400 base pairs in the JMD, known as internal tandem duplications (ITDs), are the most common mutations, occurring in up to 30% of patients with AML, and are associated with poor prognosis [4–7]. Activating point mutations in the TKD are also observed in patients with AML, but at a lower frequency than ITD mutations [5, 8]. These activating mutations are oncogenic and render a state of “oncogene addiction” in this disease [5, 9–11]. Therefore, FLT3 is considered a promising drug target in AML patients with FLT3 mutations.
A number of FLT3 inhibitors, including gilteritinib, midostaurin, quizartinib, and sorafenib, have been evaluated in clinical trials [12–15]. In 2017, the US Food and Drug Administration (FDA) and European Medicines Agency approved midostaurin for the treatment of adult patients with newly diagnosed AML with FLT3 mutation in combination with standard chemotherapy [16]. Gilteritinib is a selective FLT3 inhibitor that inhibits both FLT3-ITD and FLT3-TKD mutations, and is classified as an ATP-competitive type I inhibitor [17]. Based on a phase 3 clinical trial, gilteritinib was recently approved by the Pharmaceuticals and Medical Devices Agency and FDA as monotherapy for patients with relapsed/refractory FLT3-mutated AML [18].
A number of resistance mechanisms to FLT3 inhibitors have been identified in preclinical and clinical studies, including acquired FLT3 resistance mutations [19], other gene mutations such as NRAS [20], and altered protein expression such as that of FL [21], AXL kinase [22, 23], Pim kinase [24], or FGF2 [25]. In particular, one study reported that increased plasma concentrations of FL after chemotherapy induces resistance to FLT3 inhibitors—including midostaurin, quizartinib, sorafenib, and lestaurtinib—in AML cells with FLT3wt or mutant FLT3 (FLT3mut) [21]. The mechanism by which FL impedes the efficacy of FLT3 inhibitors is reportedly based on FL-dependent activation of FLT3wt [26]. One study showed that the efficacy of quizartinib, a selective FLT3 inhibitor that binds to inactive forms of FLT3, was attenuated when cells co-expressing FLT3wt/mut were stimulated with FL, resulting in activation of survival/growth signals via FLT3wt. Given that AML cells that harbor FLT3 mutations co-express FLT3wt [27], FL-dependent resistance to FLT3 inhibitors is considered one of the obstacles to treatment of AML patients with FLT3wt/mut. However, it remains unclear whether this effect is the same for all FLT3 inhibitors.
Here, we investigated whether the inhibitory effects of gilteritinib on tumor growth and FLT3 signal transduction are affected by FL in the human AML cell line MOLM-13, which harbors both FLT3wt and FLT3-ITD. We subsequently generated xenograft mouse models using FL-expressing MOLM-13 cells to mimic the upregulated condition of soluble FL observed in AML patients treated with chemotherapy and FLT3 inhibitor, and evaluated the antitumor activity of gilteritinib. In addition, we investigated the inhibitory effects of gilteritinib on FLT3wt and FLT3-ITD in HEK293 and Ba/F3 cells. Our results demonstrate that gilteritinib potently inhibits tumor growth even in the presence of FL due to comparable inhibitory efficacy against FLT3wt and FLT3-ITD. These findings indicate that FL has no effect on the inhibitory effects of gilteritinib and suggest that gilteritinib has therapeutic potential in FLT3-mutated AML patients with FL overexpression.
Results
Growth inhibition and apoptosis induction by gilteritinib with or without FL
To investigate whether the growth inhibitory effect of gilteritinib, a FLT3 inhibitor, is affected by FL stimulation, we treated MOLM-13 cells harboring both FLT3wt and FLT3-ITD with gilteritinib in the presence or absence of FL. Given that FL stimulation reportedly impedes the effects of other FLT3 inhibitors [21, 26], we used quizartinib and midostaurin as reference compounds. Quizartinib inhibited the growth of MOLM-13 cells with IC50 values of 1.6 nM (95% CI: 1.2–2.0) and 5.6 nM (95% CI: 3.8–8.1) in the absence and presence of FL, respectively (Figure 1A and 1D). Midostaurin inhibited the growth of MOLM-13 cells with IC50 values of 20.7 nM (95% CI: 13.8–31.2) and 63.7 nM (95% CI: 53.9–75.2) in the absence and presence of FL, respectively (Figure 1A and 1D). Consistent with previous reports, our findings demonstrate that the growth inhibitory effects of quizartinib and midostaurin on MOLM-13 cells were attenuated in the presence compared with the absence of FL. In contrast, gilteritinib inhibited the growth of MOLM-13 cells with IC50 values of 15.5 nM (95% CI: 11.7–20.5) and 20.3 nM (95% CI: 14.7–28.0) in the absence and presence of FL, respectively, indicating that FL stimulation had little effect on the growth inhibitory effect of gilteritinib in MOLM-13 cells (Figure 1A and 1D).
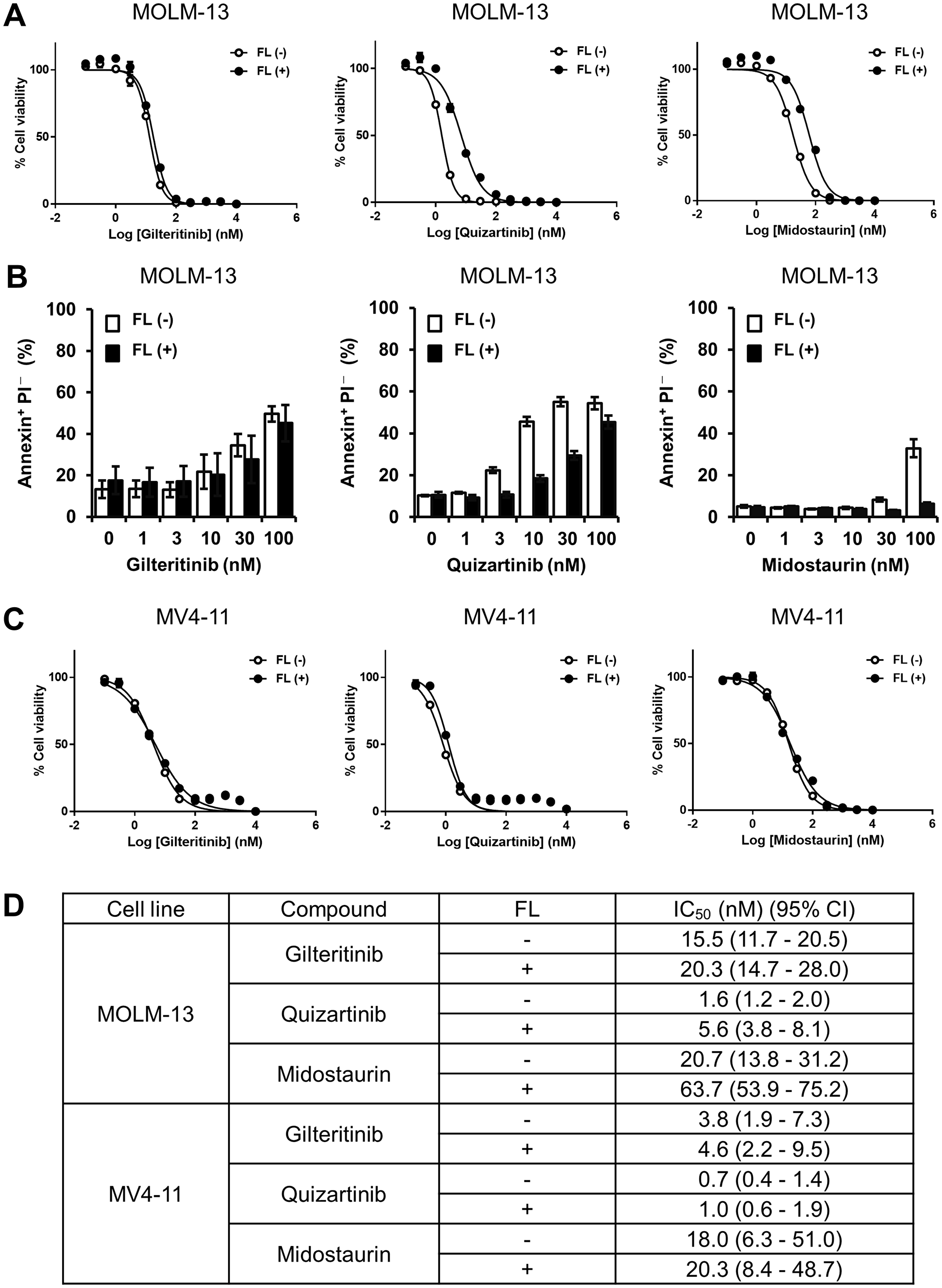
Figure 1: Growth inhibition and apoptosis induction by gilteritinib and quizartinib. (A and C) MOLM-13 and MV4-11 cells were treated with gilteritinib, quizartinib or midostaurin in the presence or absence of FL at 25 ng/mL for 3 days. Cell viability was measured using the CellTiter-Glo 2.0 Assay. At least 3 experiments were performed in duplicate or triplicate, and representative data are shown as mean ± SD. (B) MOLM-13 cells were treated with gilteritinib, quizartinib or midostaurin in the presence or absence of FL at 25 ng/mL for 2 days. Cells were harvested and stained with Annexin V and PI. Apoptotic cells were measured using flow cytometry. The experiment was performed in triplicate, and data are shown as mean ± SEM. (D) Cell viability analysis of MOLM-13 and MV4-11 cells treated with gilteritinib, quizartinib or midostaurin as in Figure 1A and 1C. Geometric mean IC50 values and 95% CIs were determined from at least 3 independent experiments. Abbreviations: CI, confidence interval; FL, FLT3 ligand.
In addition, we analyzed the ability of gilteritinib, quizartinib and midostaurin to induce apoptosis of MOLM-13 cells. We found that while FL stimulation had no effect on apoptosis induction by gilteritinib, the presence of FL attenuated apoptosis induction by quizartinib and midostaurin compared with the absence of FL (Figure 1B). Given that a previous study showed that FLT3wt was responsible for inducing the FL-dependent attenuation of the growth inhibitory effect of quizartinib [26], we investigated the growth inhibitory effects of quizartinib and midostaurin using the human AML cell line MV4-11, which harbors a homozygous FLT3-ITD mutation. The inhibitory effects of quizartinib and midostaurin on the growth of MV4-11 cells were comparable between the presence and absence of FL (Figure 1C and 1D). These results indicate that unlike quizartinib and midostaurin, FL had no effect on the growth inhibitory effect of gilteritinib in AML cells with FLT3wt/FLT3-ITD.
Antitumor activity of gilteritinib in xenograft mouse models
Given that FL had no effect on the growth inhibitory effect of gilteritinib in vitro, we next investigated the potential effect of FL on the antitumor activity of gilteritinib in xenograft mouse models. To generate a xenograft mouse model that mimics the upregulated condition of soluble FL observed in AML patients treated with chemotherapy or FLT3 inhibitor [21], we infected MOLM-13 cells with a retrovirus encoding a soluble FL or mock gene to generate FL-expressing or mock MOLM-13 cells, respectively. Analysis of the expression of FL and activities of FLT3 downstream molecules in FL-expressing and mock MOLM-13 cells demonstrated FL expression in the supernatant of FL-expressing cells but not mock cells. Further, the activities of the FLT3 downstream molecules were not significantly altered in FL-expressing cells compared to mock cells under the normal condition (Figure 2A). These cells were subcutaneously inoculated into the flank of mice and allowed to grow. Tumor volume and plasma FL concentrations were measured 18 and 22 days after inoculation, respectively. The growth rate of FL-expressing MOLM-13 tumors was not significantly different from that of mock MOLM-13 tumors. Further, plasma FL concentrations were detected in nude mice engrafted with FL-expressing cells but not mock cells (Figure 2B). Interestingly, plasma FL concentrations in FL-expressing tumor xenograft models on day 22 were similar to those observed in AML patients treated with chemotherapy and FLT3 inhibitor [21].
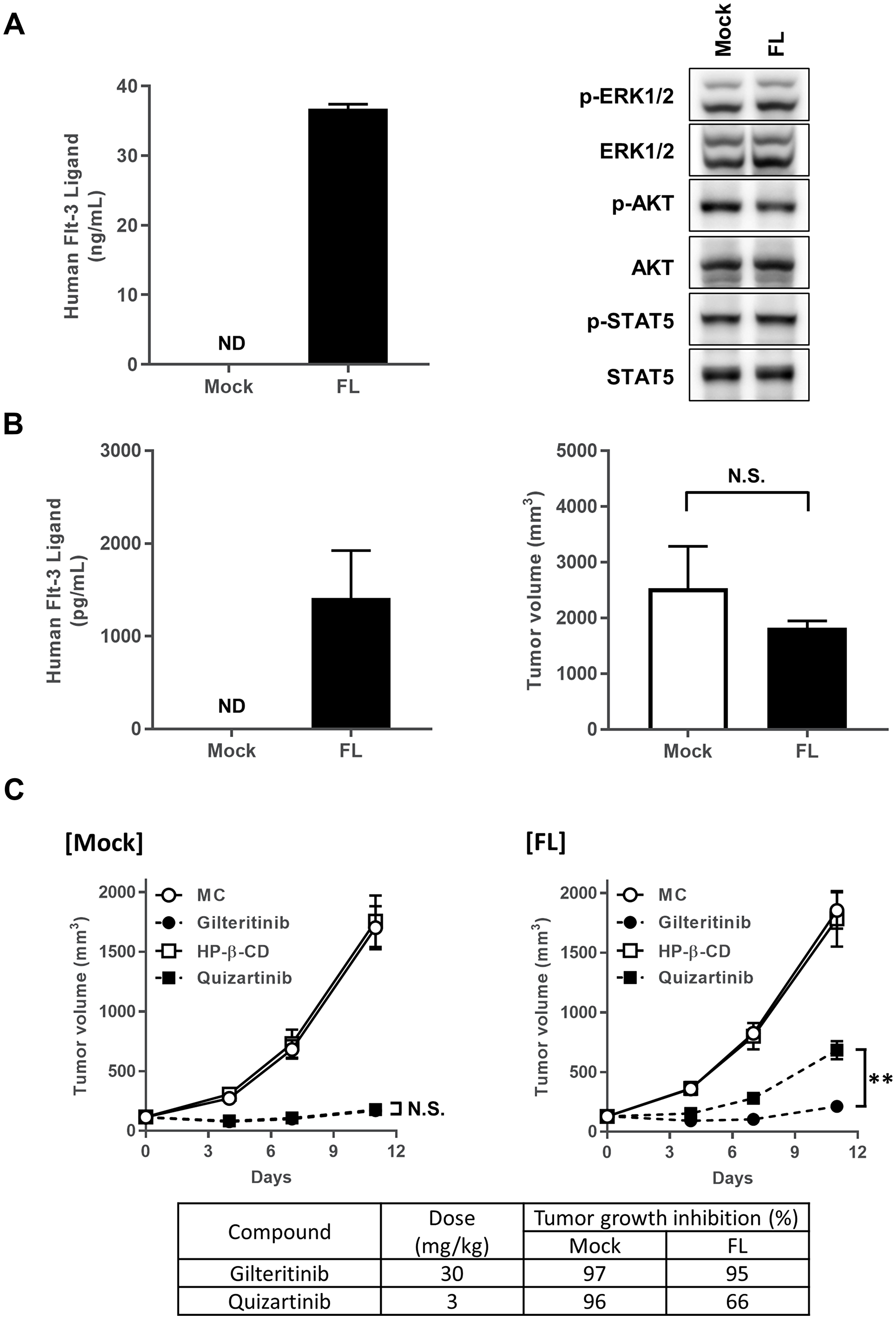
Figure 2: Antitumor efficacy of gilteritinib and quizartinib. (A) FL expression was detected in FL-expressing or mock MOLM-13 cells. FL-expressing or mock MOLM-13 cells were subjected to western blotting analysis using the indicated antibodies. (B) Plasma FL concentrations were determined on day 22, and data are shown as mean ± SEM (n = 5). Tumor volume was measured on day 18, and data are shown as mean ± SEM (n = 6). (C) Mice engrafted with FL-expressing or mock MOLM-13 cells were orally administered gilteritinib or quizartinib at 30 mg/kg or 3 mg/kg, respectively. Tumor volume was measured, and data are shown as mean ± SEM (n = 10). Tumor volume on day 11 was compared between the gilteritinib-treated group and quizartinib-treated group using Student’s t-test. **P < 0.01. Abbreviations: FL, FLT3 ligand; ND, not detected; N. S., not significantly different.
Next, we evaluated the antitumor activities of gilteritinib and quizartinib in these xenograft mouse models. Once-daily administration of gilteritinib at 30 mg/kg or quizartinib at 3 mg/kg per day for 11 days inhibited the growth of mock MOLM-13 tumors by 97% or 96%, respectively, indicating that the antitumor efficacies of gilteritinib (30 mg/kg) and quizartinib (3 mg/kg) in the mock-cell xenograft model were comparable (Figure 2C). When quizartinib (3 mg/kg) was administered to mice with FL-expressing MOLM-13 tumors, tumor growth was inhibited by 66%, indicating that the presence of FL attenuated the antitumor activity of quizartinib compared with that of gilteritinib (Figure 2C). As expected from our in vitro results, gilteritinib (30 mg/kg) showed similar efficacy to that for mock MOLM-13 tumors, inhibiting FL-expressing MOLM-13 tumor growth by 95% (Figure 2C). These results indicate that, unlike quizartinib, FL had no effect on the antitumor efficacy of gilteritinib in vivo.
Inhibitory effects of gilteritinib on FLT3wt and FLT3-ITD
We next investigated the mechanism of the lack of effect of FL on the antitumor activity of gilteritinib. Studies have reported that FL-induced activation of FLT3wt is a key mechanism in FL-dependent resistance of FLT3 inhibitors: quizartinib has higher potency against FLT3-ITD than FLT3wt, which leads to FL-mediated impairment of antitumor activity by quizartinib [26, 28]. Therefore, we hypothesized that gilteritinib inhibits FLT3wt with the same or greater efficacy than it inhibits FLT3-ITD.
Because it is difficult to investigate the inhibitory effects of gilteritinib against FLT3wt and FLT3-ITD in MOLM-13 cells, we instead examined the inhibitory effects of gilteritinib and quizartinib on auto-phosphorylation of the 2 types of FLT3 and on the cell proliferation of HEK293 and Ba/F3 cells. HEK293 cells were transfected with FLT3wt- or FLT3-ITD-expressing vectors and auto-phosphorylation levels of FLT3 were determined using western blotting. We found that while the inhibitory effects of gilteritinib on the auto-phosphorylation levels of FLT3wt and FLT3-ITD were comparable, those of quizartinib were weaker on FLT3wt compared with FLT3-ITD (Figure 3A and 3B). We subsequently generated FLT3wt- and FLT3-ITD-expressing Ba/F3 cells (Figure 3C) and investigated the inhibitory effects of gilteritinib and quizartinib on their growth. Gilteritinib inhibited the growth of FLT3wt- and FLT3-ITD-expressing Ba/F3 cells with IC50 values of 19.7 nM (95% CI: 13.5–28.7) and 9.2 nM (95% CI: 5.5–15.3), respectively (Figure 3D). In contrast, quizartinib inhibited the growth of FLT3wt- and FLT3-ITD-expressing Ba/F3 cells with IC50 values of 6.3 nM (95% CI: 5.7–7.0) and 0.4 nM (95% CI: 0.4–0.5), respectively (Figure 3D). In addition, midostaurin inhibited the growth of FLT3wt- and FLT3-ITD-expressing Ba/F3 cells with IC50 values of 28.5 nM (95% CI: 23.5–34.5) and 4.2 nM (95% CI: 3.7–4.7), respectively (Figure 3D). These results indicate that gilteritinib had similar inhibitory action on FLT3wt and FLT3-ITD, while quizartinib and midostaurin had more potent inhibitory action on FLT3-ITD than on FLT3wt. The similar inhibitory potency against FLT3wt and FLT3-ITD suggests that FL has no effect on the efficacy of gilteritinib.
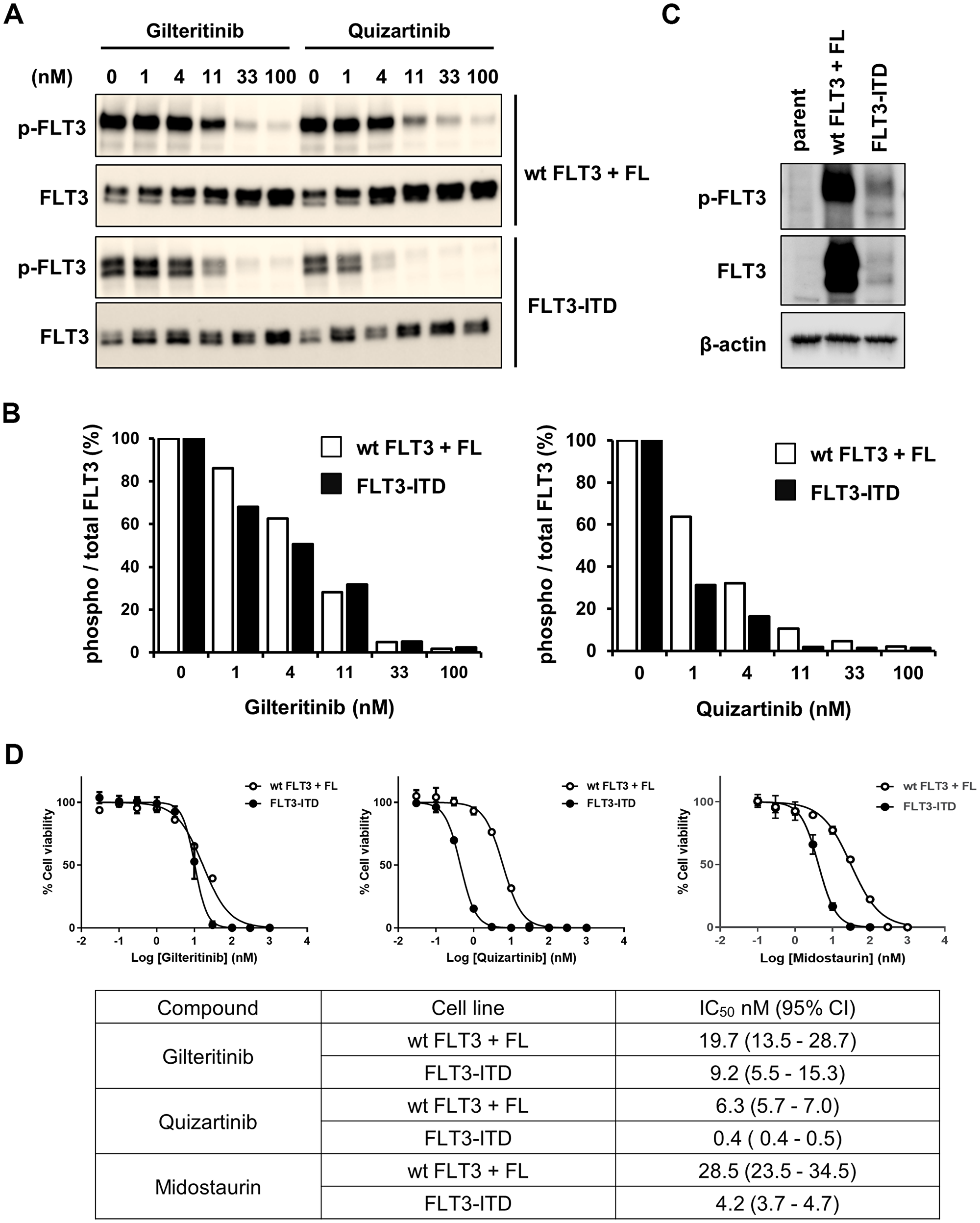
Figure 3: Inhibitory effects of gilteritinib and quizartinib on FLT3wt and FLT3-ITD. (A and B) HEK293 cells were transfected with an FLT3wt- or FLT3-ITD-expressing vector. After culturing overnight, cells were treated with gilteritinib or quizartinib for 1 hour. FL (25 ng/mL) was added to FLT3wt-expressing cells simultaneously with the compounds. Cells were lysed and subjected to western blotting analysis using antibodies against total or phospho-FLT3 (Y591). The ratio of phospho/total FLT3 was calculated and is shown in the graph. (C) FLT3wt- or FLT3-ITD-expressing Ba/F3 cells and parental cells were subjected to western blotting analysis using antibodies against FLT3, phospho-FLT3 and β-actin. (D) FLT3wt- or FLT3-ITD-expressing Ba/F3 cells were treated with gilteritinib, quizartinib or midostaurin for 3 days. FL (25 ng/mL) was added to FLT3wt-expressing cells simultaneously with the compounds. Cell viability was measured using the CellTiter-Glo 2.0 Assay. Three experiments were performed in triplicate, and representative data are shown as the mean ± SD. Geometric mean IC50 values and 95% CIs were determined from 3 independent experiments. Abbreviations: CI, confidence interval; FL, FLT3 ligand; ITD, internal tandem duplication; wt, wild-type.
Inhibitory effects of gilteritinib on FLT3 downstream signaling molecules
To investigate whether the presence or absence of FL affects the gilteritinib-mediated suppression of FLT3 downstream signaling molecules, we next evaluated the phosphorylation statuses of ERK1/2, STAT5, and AKT in MOLM-13 cells treated with gilteritinib or quizartinib. Treatment with gilteritinib and quizartinib without FL stimulation suppressed phosphorylation of all FLT3 downstream signaling molecules tested. In the presence of FL, quizartinib showed similar suppression of the phosphorylation of STAT5, but attenuated inhibitory efficacy for the phosphorylation of ERK1/2 and AKT (Figure 4A and Supplementary Figure 1). In contrast, in the presence of FL, gilteritinib showed similar suppression of the phosphorylation of ERK1/2 and STAT5, but not AKT (Figure 4A and Supplementary Figure 1).
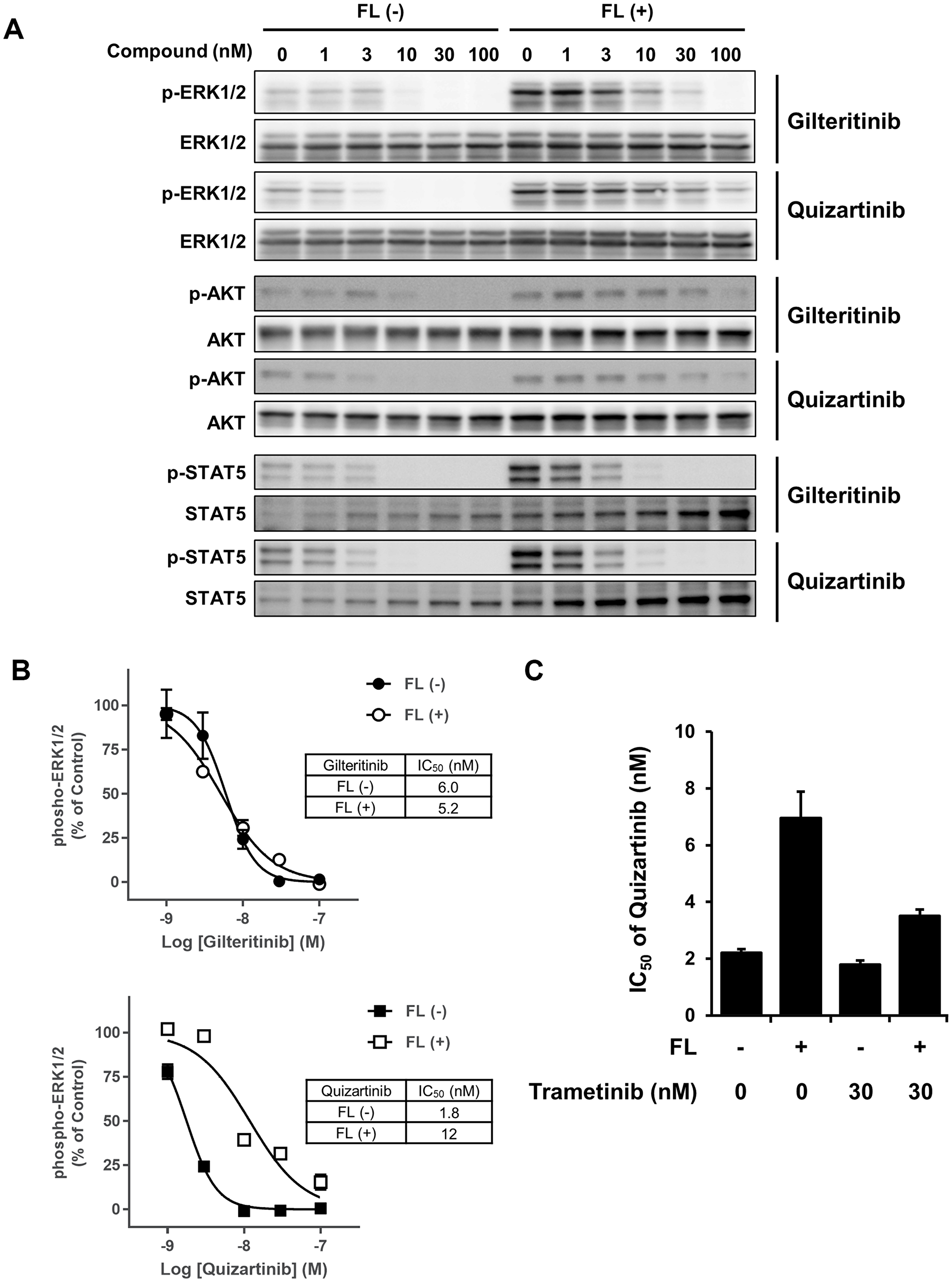
Figure 4: FLT3 downstream signaling analysis. (A) MOLM-13 cells were treated with gilteritinib or quizartinib in the presence or absence of FL at 25 ng/mL for 2 hours. Cells were lysed and subjected to western blotting analysis using the indicated antibodies. (B) Phospho-ERK1/2 levels were determined using the phospho-ERK assay kit. The experiment was performed in triplicate, and data are shown as mean ± SD. (C) MOLM-13 cells were treated with quizartinib in combination with or without trametinib in the presence or absence of FL at 25 ng/mL for 2 days. Cell viability was measured using the CellTiter-Glo 2.0 Assay. Cell viability was calculated by defining the survival of quizartinib-untreated cells with or without trametinib treatment and medium control wells as 100% and 0%, respectively. Three experiments were performed in triplicate, and data are shown as mean ± SEM. Abbreviation: FL, FLT3 ligand.
Given that gilteritinib and quizartinib differed in their suppression of the phosphorylation of ERK1/2 in the presence of FL, we further investigated the effect of gilteritinib and quizartinib on the phosphorylation status of ERK1/2 in MOLM-13 cells in the presence or absence of FL using the advanced ERK phospho-T202/Y204 detection system. Similar to our initial findings, this assay showed that while the presence of FL attenuated the effects of quizartinib, it did not affect those of gilteritinib (Figure 4B). Given that Fangli et al reported that activation of the MAPK pathway plays a crucial role in FL-dependent resistance to FLT3 inhibitors in FLT3wt and FLT3-ITD co-expressing 32D cells [26], we investigated whether trametinib, a potent and selective MEK inhibitor, recovers the efficacy of quizartinib in the presence of FL. We confirmed that treatment with trametinib to inhibit the MAPK pathway abolished the FL-dependent resistance to quizartinib (Figure 4C and Supplementary Figure 2). Therefore, these results indicate that the inhibitory effect of gilteritinib on the FLT3wt-MAPK pathway is crucial for overcoming FL-dependent resistance.
Crystal structure of FLT3 in complex with gilteritinib
To reveal the binding mode, we determined the crystal structure of the FLT3 kinase domain in complex with gilteritinib. As expected from the computational modeling performed in our previous report [17], gilteritinib bound to the ATP pocket of FLT3 (Figure 5A). While the JMD was clearly observed in the overall structure, a portion of the activation loop including Gly831 in the DFG motif was disordered. Asp829 and Phe830 in the DFG motif adopted the “DFG-out” conformation. In the binding mode of gilteritinib, two hydrogen bonds were formed between the carbamoyl group and the main chain atoms of Glu692 and Cys694 (Figure 5B). There was no distinct interaction between gilteritinib and the activation loop including Asp829 and Phe830.
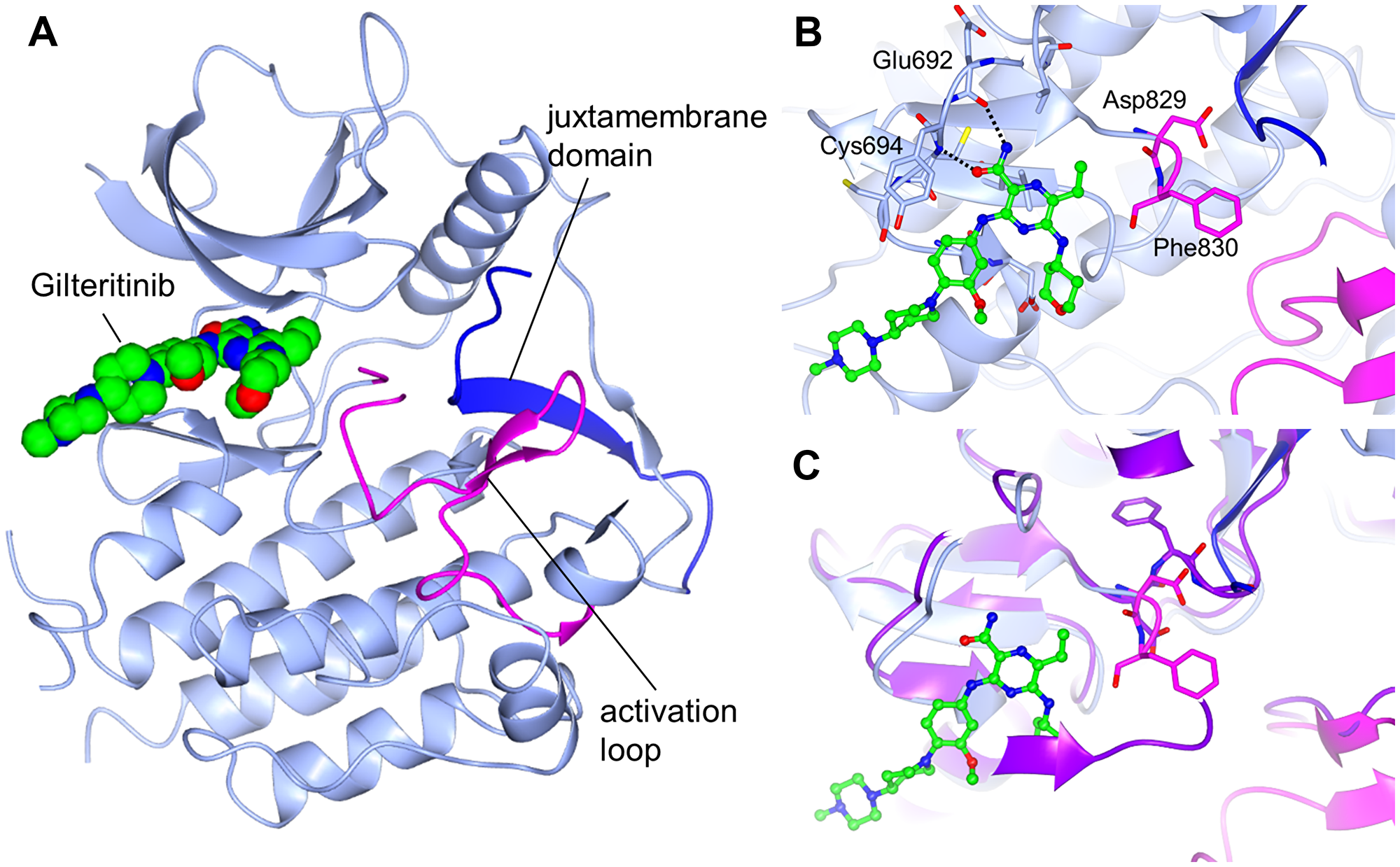
Figure 5: Crystal structure of FLT3 bound to gilteritinib. FLT3 is shown in light blue, the activation loop is shown in magenta, and the juxtamembrane domain is shown in blue. (A) Overall structure. (B) Binding mode of gilteritinib. Hydrogen bonds are shown as dashed lines. (C) Overlay with the c-Kit active structure (PDB 1PKG). c-Kit is shown in purple. DFG motifs are shown as a stick model.
DISCUSSION
FLT3 is a promising molecular target in a subset of AML cases, and the therapeutic effects of FLT3 inhibitors have been reported in AML patients with FLT3 mutations. One of the critical issues of treatment with FLT3 inhibitors in FLT3-mutated AML is drug-resistance. A number of resistance mechanisms against FLT3 inhibitors have been identified, including acquired resistance mutations of FLT3, other somatic mutations of cancer-related genes, and activation of alternative signaling pathways. Our previous report showed that gilteritinib has inhibitory activity against the TKD mutation and gate keeper mutation of FLT3, which confer resistance to quizartinib [17]. Here, we investigated the effect of increasing FL, another potential mechanism of resistance to FLT3 inhibitors, on the efficacy of gilteritinib.
First, we confirmed previous findings that FL attenuates the inhibitory effects of FLT3 inhibitors including midostaurin and quizartinib in FLT3-mutated AML cells (Figure 1). Surprisingly, and in contrast to the profile of other FLT3 inhibitors, our results demonstrated that FL had no effect on the growth inhibitory effect of gilteritinib. These findings were also confirmed in xenograft mouse models injected with FL-overexpressing AML cells.
Next, we investigated the underlying mechanism of the unique profile of gilteritinib. Previous report showed that the FL-mediated attenuation in the efficacy of quizartinib is a result of its weaker inhibitory effect on FLT3wt compared with FLT3-ITD [26]. We therefore hypothesized that gilteritinib may show similar inhibitory potency against FLT3wt and FLT3-ITD. Indeed, in the analyses of FLT3 signaling, gilteritinib inhibited FLT3wt and FLT3-ITD to a similar extent in HEK293 and Ba/F3 cells, and also similarly suppressed FLT3 downstream signaling molecules, including ERK1/2 and STAT5, even in the presence of FL in MOLM-13 cells.
These findings may be clinically meaningful for several reasons. First, an FL increase has been observed in AML patients treated with a FLT3 inhibitor or chemotherapy [21]. The plasma concentrations of FL in the xenograft model established by inoculating FL-expressing MOLM-13 cells in our study were similar to those in AML patients [21]. In addition, detectable phosphorylation of FLT3wt, but not FLT3-ITD, has been observed using the plasma inhibitory activity assay with human plasma samples from patients administered quizartinib [28], suggesting that the efficacy of quizartinib may also be attenuated in the presence of FL in AML patients. Furthermore, single cell analysis of AML blasts in patients has revealed the presence of AML cells with FLT3-ITD co-expressing FLT3wt [27]. Therefore, the FL increase may be correlated with resistance to treatment with FLT3 inhibitors. Our results suggest that gilteritinib may overcome the FL increase and lengthen the duration of efficacy, although further research is needed to confirm this in patients. In this study, we investigated the inhibitory activity of gilteritinib using the ITD sequence derived from MOLM-13 cells. A number of ITD mutations have been observed in AML patients with FLT3-ITD due to the presence of different ITD insertion sequences. Therefore, future studies should examine the efficacy of gilteritinib in the presence of FL using primary AML cells with various ITD mutations.
Tyrosine kinase inhibitors are classified into 2 types based on their affinity to receptors. Type I inhibitors such as gilteritinib, midostaurin, and lestaurtinib bind to both the active and inactive forms of FLT3, while type II inhibitors such as quizartinib bind only to inactive FLT3. However, given that both type I and II inhibitors are affected by FL—with the exception of gilteritinib—we propose that selectivity against FLT3wt and FLT3-ITD may be more important than the inhibitors’ classification type.
The crystal structure of the complex formed between FLT3 and gilteritinib was obtained using the kinase domain of FLT3wt, including the JMD, which has been reported to bind to the active site of FLT3 and to stabilize the autoinhibited form [29]. In the complex structure with gilteritinib, the JMD bound to the active site, indicating that FLT3 adopted the autoinhibited form. Given that gilteritinib did not interact with the activation loop or the JMD, gilteritinib may also bind to the active form of FLT3, including the constitutively active form of FLT3-ITD. To elucidate whether gilteritinib binds to the active form, the c-Kit active structure [30] was superimposed onto the FLT3 complex structure with gilteritinib (Figure 5C). The activation loop of FLT3 was predicted to adopt a similar conformation to the c-Kit activation loop because the amino acid residues are well conserved. Gilteritinib was far from the active conformation of the activation loop, suggesting that it likely binds to the active form of FLT3 as well as the autoinhibited form. These findings correspond to the similar inhibitory activity of gilteritinib against FLT3wt and FLT3-ITD.
FL is reportedly involved in promoting the differentiation of hematopoietic progenitor cells into dendritic cells (DC) [31]. A potential downside of the inhibitory effect of gilteritinib on FLT3wt is that it may affect DC differentiation and immune function. However, gilteritinib has inhibitory activity against AXL, which has cancer immunosuppressive activity [17, 32]. The mechanism by which gilteritinib affects cancer immunity is currently unknown.
Our results suggest that gilteritinib has therapeutic potential in FLT3-mutated AML patients with FL overexpression. In addition, we propose that gilteritinib may be a good combination drug with chemotherapies that increase plasma concentrations of FL in FLT3-mutated AML patients and/or an effective monotherapy for AML patients with FL upregulation after chemotherapy.
Materials and Methods
Regents and antibodies
Gilteritinib hemifumarate (gilteritinib) and quizartinib dihydrochloride were synthesized by Astellas Pharma, Inc. and Sumika Technoservice Co., respectively. Quizartinib and midostaurin were purchased from Sellek Chemicals and LC Laboratories, respectively. Trametinib was synthesized by OSI Pharma, Inc. Gilteritinib hemifumarate was dissolved in dimethyl sulfoxide (DMSO) or suspended in 0.5% methylcellulose (MC) for in vitro or in vivo experiments, respectively. Quizartinib, midostaurin, and trametinib were dissolved in DMSO for in vitro experiments. Quizartinib dihydrochloride was dissolved in 22% 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) for in vivo experiments. Recombinant human FLT3 ligand protein was purchased from R&D Systems, Inc. Matrigel was purchased from BD Biosciences. The following antibodies were used for immunoblotting: anti-phospho-Stat5 (Y694) (BD biosciences), anti-FLT3, anti-β-actin, anti-p44/42 MAPK (Erk1/2), anti-Akt, anti-Stat5, anti-phospho-FLT3 (Y591), anti-phospho-p44/43 MAPK (Erk1/2) (T202/Y204), and anti-phospho-Akt (S473) (Cell Signaling Technology).
Plasmids
Human FLT3wt (NM_004119) and FLT3-ITD were N-terminally tagged with flag and cloned into the pMXs-Puro retroviral vector (Cell Biolabs). The ITD sequence of FLT3-ITD was derived from MOLM-13 cells [33]. FLT3LG (a. a. 1-181, NM_001204502) encoding the soluble form of FL was cloned into the pMXs-Puro retroviral vector.
Cell lines, cell culture, and stable cell lines
MOLM-13 cells were purchased from the German Collection of Microorganisms and Cell Cultures; MV4-11 and HEK293 cells from the American Type Culture Collection; retroviral packaging cell line GP2-293 from Clontech; and Ba/F3 cells from RIKEN Cell bank. MOLM-13 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Sigma-Aldrich) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS); MV4-11 in Iscove’s Modified Dulbecco’s Medium (IMDM; Thermo Fisher Scientific) supplemented with 10% FBS and 1% PS; HEK293 and GP2-293 cells in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich) supplemented with 10% FBS and 1% PS; and Ba/F3 cells in RPMI-1640 supplemented with 10% FBS, 1% PS, and 10 ng/mL mouse interleukin (mIL)-3 (R&D systems).
FL-expressing or mock MOLM-13 cells were generated by Astellas Pharma, Inc. The pMXs-Puro retroviral vector containing the human soluble FLT3LG gene or empty vector was co-transfected with pVSV-G (Clontech) into GP2-293 cells using lipofectamine 2000 (Invitrogen) to produce the virus. MOLM-13 cells were then infected with the viral supernatant containing the FLT3LG gene or empty vector. Stable transfectants were obtained and maintained under selection pressure using puromycin (Nacalai Tesque) at 1.0 μg/mL. FLT3wt- or FLT3-ITD-expressing Ba/F3 cells were also generated by Astellas Pharma, Inc. The pMXs-Puro retroviral vector containing the FLT3wt- or FLT3-ITD gene was transfected into Platinum-E cells using lipofectamine 2000 to produce the virus. Ba/F3 cells were then infected with viral supernatant containing each FLT3 gene. FLT3wt- or FLT3-ITD-expressing Ba/F3 cells were selected using puromycin at 1.0 μg/mL and subsequently cultured in mIL-3-deprived medium with or without 10 ng/mL FL, respectively.
Cell viability assay
MOLM-13 and MV4-11 cells were seeded at 10,000 cells/well in 96-well plates. After culturing overnight, cells were treated with gilteritinib, quizartinib, or midostaurin in the presence or absence of FL at 25 ng/mL for 3 days [21, 26]. FLT3wt- or FLT3-ITD-expressing Ba/F3 cells were seeded at 1,000 cells/well in 96-well plates and treated with gilteritinib or quizartinib for 3 days in the presence or absence of FL at 25 ng/mL for 3 days. Cell viability was measured using the CellTiter-Glo 2.0 Assay (Promega). Measurement of luciferase activity was performed using the ARVO X3 (PerkinElmer) or SpectraMax Paradigm (Molecular Devices). Data were analyzed using GraphPad PRISM 7 software (GraphPad). Cell viability was calculated by defining the survival of untreated cells and medium control wells as 100% and 0%, respectively.
Apoptosis assay
MOLM-13 cells were seeded at 2 × 105 cells/well in 12-well plates and treated with gilteritinib, quizartinib or midostaurin in the presence or absence of FL at 25 ng/mL for 2 days. Cells were harvested and stained with Annexin V and PI using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences). Apoptotic cells were analyzed using MACS Quant Analyzer10 (Miltenyi Biotec). Flow cytometric data were analyzed using FlowJo software (FlowJo).
Animal models for in vivo studies
All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of Astellas Pharma, Inc. The Tsukuba Research Center of Astellas Pharma, Inc. has been awarded accreditation status by AAALAC International. Mice were maintained on water and a standard diet throughout the experimental procedures. Four-week-old male nude mice (BALB/c nu/nu) were purchased from Charles River Laboratories Japan, Inc.
In vitro FL detection
FL-expressing or mock MOLM-13 cells were seeded at 3 × 105 cells/well in 6-well plates. After culturing for 4 days, the supernatant of both cell types was collected and the amount of FL was quantified using the Human Flt-3 Ligand Quantikine ELISA Kit (R&D Systems).
In vivo xenograft studies for FL detection
FL-expressing or mock MOLM-13 cells were subcutaneously inoculated into the flank of mice at 5 × 106 cells/0.1 mL (matrigel: PBS = 1:1)/mouse and allowed to grow. Blood samples were collected 22 days after inoculation and the amount of FL in mouse plasma samples was quantified using the Human Flt-3 Ligand Quantikine ELISA Kit.
In vivo xenograft studies on the antitumor activity of gilteritinib and quizartinib
FL-expressing or mock MOLM-13 cells were subcutaneously inoculated into the flank of mice at 5 × 106 cells/0.1 mL (matrigel: PBS = 1:1)/mouse and allowed to grow. Seven days after inoculation, the mice were divided into 4 groups (gilteritinib, quizartinib, vehicle for gilteritinib, and vehicle for quizartinib; n = 10 each) such that the mean tumor volume was similar among the groups on the first day of administration (day 0). Gilteritinib (30 mg/kg) or quizartinib (3 mg/kg) was orally administered once daily to these mice for 11 days. The dose of each compound was expressed as the salt form. Tumor diameter was measured using a caliper on days 0, 4, 7, and 11, and tumor volume was determined by calculating the volume of an ellipsoid using the formula: length × width2 × 0.5. Body weight was measured using a standard balance.
Statistical analysis
For the in vivo subcutaneous xenograft mouse model, values are expressed as mean ± SEM. Tumor volume in the gilteritinib-treated group was compared with that in the quizartinib-treated group using Student’s t test. P < 0.05 was considered statistically significant. Microsoft Excel (Microsoft) and GraphPad Prism (GraphPad Software) were used for data processing.
Western blotting analysis
Cells were lysed with cell lysis buffer (Cell Signaling Technology) or RIPA buffer (Thermo Fisher Scientific) supplemented with Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of total protein were resolved by SDS-PAGE and transferred to PVDF membranes. After blocking at room temperature with Blocking One (Nacalai Tesque) or PVDF Blocking Reagent for Can Get Signal (TOYOBO Life Science), the membranes were incubated overnight with primary antibodies. After washing with TBS-T, membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. Proteins of interest were visualized by enhanced chemiluminescence using the ECL Prime Western Blotting Detection System (GE Healthcare), Western Lightning Plus-ECL (PerkinElmer), or Western Lightning Ultra (PerkinElmer), and detected with LAS-4000 (GE Healthcare) or LAS-4000EPUVmini (Fujifilm) and quantified using ImageQuant software (GE Healthcare) or Multi Gauge software (Fujifilm).
Quantification of phospho-ERK1/2
MOLM-13 cells were seeded at 3 ×106 cells/well in 6-well plates and treated with gilteritinib or quizartinib in the presence or absence of FL at 25 ng/mL for 2 hours. Cells were harvested and phospho-ERK levels were evaluated using the Advanced ERK phospho-T202 /Y204 kit (Cisbio) and EnVision (Perkin Elmer).
X-ray structural analysis
X-ray diffraction data were collected from crystals of the complex formed between human FLT3 and gilteritinib using the PXII/X10SA beamline at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland) under cryogenic conditions. Data were processed using XDS and XSCALE software [34]. A previously solved structure of hFLT3 was used as the search model. Subsequent model building and refinement was performed according to standard protocols using CCP4 [35] and COOT [36] software packages. Refinement using the translation/libration/screw-rotation (TLS) model (using REFMAC5 [37], CCP4) was performed. Ligand parameters and corresponding library files were generated using CORINA (Molecular Networks GmbH, Nürnberg, Germany). The water model was built using the COOT “Find waters” algorithm by placing water molecules at peaks of the Fo-Fc map contoured at 3.0 with subsequent refinement using REFMAC5, and checking all waters using the COOT validation tool. The coordinates were deposited into the Protein Data Bank (accession code 6JQR). All crystallographic work was performed by Proteros Biostructures GmbH (Martinsried, Germany).
ACKNOWLEDGMENTS
The authors thank T. Inoue (World Intec Co., Ltd.) for help with in vitro and in vivo experiments.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research was supported by funding from Astellas Pharma, Inc.
References
1. Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood. 1996; 88:3383–3390. [PubMed].
2. Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res. 2009; 15:4263–4269. https://doi.org/10.1158/1078-0432.CCR-08-1123. [PubMed].
3. Patnaik MM. The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma. 2018; 59:2273–2286. https://doi.org/10.1080/10428194.2017.1399312. [PubMed].
4. Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996; 10:1911–1918. [PubMed].
5. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001; 97:2434–2439. [PubMed].
6. Pratz KW, Luger SM. Will FLT3 inhibitors fulfill their promise in acute meyloid leukemia? Curr Opin Hematol. 2014; 21:72–78. https://doi.org/10.1097/MOH.0000000000000022. [PubMed].
7. Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, Döhner H, Döhner K; AML Study Group Ulm. Acute myeloid leukemia. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002; 100:4372–4380. https://doi.org/10.1182/blood-2002-05-1440. [PubMed].
8. Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001; 113:983–988. https://doi.org/10.1046/j.1365-2141.2001.02850.x. [PubMed].
9. Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, Naoe T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000; 19:624–631. https://doi.org/10.1038/sj.onc.1203354. [PubMed].
10. Tse KF, Allebach J, Levis M, Smith BD, Bohmer FD, Small D. Inhibition of the transforming activity of FLT3 internal tandem duplication mutants from AML patients by a tyrosine kinase inhibitor. Leukemia. 2002; 16:2027–2036. https://doi.org/10.1038/sj.leu.2402674. [PubMed].
11. Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003; 102:646–651. https://doi.org/10.1182/blood-2002-11-3441. [PubMed].
12. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Dohner K, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017; 377:454–464. https://doi.org/10.1056/NEJMoa1614359. [PubMed].
13. Cortes J, Perl AE, Dohner H, Kantarjian H, Martinelli G, Kovacsovics T, Rousselot P, Steffen B, Dombret H, Estey E, Strickland S, Altman JK, Baldus CD, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018; 19:889–903. https://doi.org/10.1016/S1470-2045(18)30240-7. [PubMed].
14. Ohanian M, Garcia-Manero G, Levis M, Jabbour E, Daver N, Borthakur G, Kadia T, Pierce S, Burger J, Richie MA, Patel K, Andreeff M, Estrov Z, et al. Sorafenib Combined with 5-azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am J Hematol. 2018; 93:1136–1141. https://doi.org/10.1002/ajh.25198. [PubMed].
15. Sutamtewagul G, Vigil CE. Clinical use of FLT3 inhibitors in acute myeloid leukemia. Onco Targets Ther. 2018; 11:7041–7052. https://doi.org/10.2147/OTT.S171640. [PubMed].
16. Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017; 129:3403–3406. https://doi.org/10.1182/blood-2017-05-782292. [PubMed].
17. Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K, Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs. 2017; 35:556–565. https://doi.org/10.1007/s10637-017-0470-z. [PubMed].
18. Gorcea CM, Burthem J, Tholouli E. ASP2215 in the treatment of relapsed/refractory acute myeloid leukemia with FLT3 mutation: background and design of the ADMIRAL trial. Future Oncol. 2018; 14:1995–2004. https://doi.org/10.2217/fon-2017-0582. [PubMed].
19. Staudt D, Murray HC, McLachlan T, Alvaro F, Enjeti AK, Verrills NM, Dun MD. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int J Mol Sci. 2018; 19:3198. https://doi.org/10.3390/ijms19103198. [PubMed].
20. McMahon CM, Canaani J, Rea B, Sargent RL, Morrissette JJD, Lieberman DB, Watt C, Schwartz GW, Faryabi RB, Ferng TT, Shah NP, Smith CC, Carroll M, et al. Mechanisms of Acquired Resistance to Gilteritinib Therapy in Relapsed and Refractory FLT3 -Mutated Acute Myeloid Leukemia. Blood. 2017; 130:295.
21. Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, Small D, Burnett A, Levis M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011; 117:3286–3293. https://doi.org/10.1182/blood-2010-01-266742. [PubMed].
22. Park IK, Mishra A, Chandler J, Whitman SP, Marcucci G, Caligiuri MA. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: implications for Axl as a potential therapeutic target. Blood. 2013; 121:2064–2073. https://doi.org/10.1182/blood-2012-07-444018. [PubMed].
23. Weisberg E, Ray A, Nelson E, Adamia S, Barrett R, Sattler M, Zhang C, Daley JF, Frank D, Fox E, Griffin JD. Reversible resistance induced by FLT3 inhibition: a novel resistance mechanism in mutant FLT3-expressing cells. PLoS One. 2011; 6:e25351. https://doi.org/10.1371/journal.pone.0025351. [PubMed].
24. Green AS, Maciel TT, Hospital MA, Yin C, Mazed F, Townsend EC, Pilorge S, Lambert M, Paubelle E, Jacquel A, Zylbersztejn F, Decroocq J, Poulain L, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv. 2015; 1:e1500221. https://doi.org/10.1126/sciadv.1500221. [PubMed].
25. Traer E, Martinez J, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, Kovacsovics T, Tyner JW, Wong M, Druker BJ. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016; 76:6471–6482. https://doi.org/10.1158/0008-5472.CAN-15-3569. [PubMed].
26. Chen F, Ishikawa Y, Akashi A, Naoe T, Kiyoi H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. 2016; 7:47018–47032. https://doi.org/10.18632/oncotarget.10147. [PubMed].
27. Stirewalt DL, Pogosova-Agadjanyan EL, Tsuchiya K, Joaquin J, Meshinchi S. Copy-neutral loss of heterozygosity is prevalent and a late event in the pathogenesis of FLT3/ITD AML. Blood Cancer J. 2014; 4:e208. https://doi.org/10.1038/bcj.2014.27. [PubMed].
28. Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, Gammon G, Trone D, Armstrong RC, James J, Levis M. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013; 31:3681–3687. https://doi.org/10.1200/JCO.2013.48.8783. [PubMed].
29. Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004; 13:169–178. https://doi.org/10.1016/S1097-2765(03)00505-7. [PubMed].
30. Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003; 278:31461–31464. https://doi.org/10.1074/jbc.C300186200. [PubMed].
31. Harada S, Kimura T, Fujiki H, Nakagawa H, Ueda Y, Itoh T, Yamagishi H, Sonoda Y. Flt3 ligand promotes myeloid dendritic cell differentiation of human hematopoietic progenitor cells: possible application for cancer immunotherapy. Int J Oncol. 2007; 30:1461–1468. https://doi.org/10.3892/ijo.30.6.1461. [PubMed].
32. Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014; 14:769–785. https://doi.org/10.1038/nrc3847. [PubMed].
33. Komeno Y, Kurokawa M, Imai Y, Takeshita M, Matsumura T, Kubo K, Yoshino T, Nishiyama U, Kuwaki T, Kubo K, Osawa T, Ogawa S, Chiba S, et al. Identification of Ki23819, a highly potent inhibitor of kinase activity of mutant FLT3 receptor tyrosine kinase. Leukemia. 2005; 19:930–935. https://doi.org/10.1038/sj.leu.2403736. [PubMed].
34. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010; 66:125–32. https://doi.org/10.1107/S0907444909047337. [PubMed].
35. Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994; 50:760–763. https://doi.org/10.1107/S0907444994003112. [PubMed].
36. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004; 60:2126–2132. https://doi.org/10.1107/S0907444904019158. [PubMed].
37. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011; 67:355–367. https://doi.org/10.1107/S0907444911001314. [PubMed].