
Introduction
End-stage kidney disease (ESKD) represents irreversible kidney failure through a variety of causes. Ageing of the population, with frequently occurring diabetes, atherosclerotic vascular disease, and hypertension, is predominantly responsible for an increasing prevalence of ESKD [1–3]. Despite the large socioeconomic impact of ESKD [4], innovative novel therapies have thus far failed to reach the clinic. Recognition of the problems related to currently available treatments, has spurred the development of novel approaches of which cell-based systems, also known as bioartificial kidney (BAK), that seek to replicate the kidney’s function through the integration of proximal tubule cells are of promise [5].
One of the crucial issues to take into consideration when developing a BAK is the sufficient availability of suitable cells. Human primary proximal tubule epithelial cells (PTEC) have a limited life span in vitro and presenting risks, such as functional changes occurring upon culturing as well as dedifferentiation and senescence of cells [6, 7]. Several studies have therefore focused on animal cells [8–10], or cell lines [11–15]. Issues related to the use of animal-derived cells in BAK are safety concerns compromising approval for clinical application and species differences in cell behaviour.
To overcome these limitations, we employed human urine-derived PTEC that were conditionally immortalized using the essential catalytic subunit of human telomerase (hTERT) and a temperature-sensitive mutant U19tsA58 of SV40 large T antigen (SV40T), creating conditionally immortalized PTEC (ciPTEC) [16]. Due to the expression of temperature-sensitive SV40T, cells can be expanded at permissive temperature of 33° C and differentiated into mature cells at non-permissive temperature of 37° C [16–18]. While hTERT acts by stabilizing telomeres, thus preventing the occurrence of replicative senescence [19], SV40T involves the activation of E2F-mediated transcription through binding with Rb-E2F complex, as well as the inhibition of p53 [20, 21]. The cell line has been thoroughly characterized over the years [16, 22–25], but the absence of the physiologically important organic anion transporter 1 (OAT1) protein led us to modify ciPTEC further by an overexpression of the transporter [26]. With this cell line we demonstrated the capacity of an efficient removal of uremic toxins when cells are cultured on hollow fiber membranes (HFM), thereby creating fully functional kidney tubules [27]. In addition to the proven lack of ciPTEC allostimulatory potential in vitro [28], a successful upscaling of the biofunctionalized HFM with tight epithelial monolayers and cell function has been achieved, encouraging further efforts towards the BAK development [29].
Eyeing possible clinical applications of ciPTEC, a thorough safety evaluation is warranted to exclude any risks related to oncogenesis and tumorigenesis [30]. Even though SV40T mediated inhibition of p53 and Rb pathways or telomere length maintenance by hTERT are not sufficient to induce oncogenic transformation, various parameters related to cell growth and proliferation, apoptosis and migration have to be examined [31–35]. Given that the transgenes were introduced by retroviral and lentiviral transductions, which can be oncogenic through insertional mutagenesis [36–38], we evaluated if the transgenes had disrupted proto-oncogenes or important genes required for PTEC function. Finally, we evaluated cell transforming properties and tumorigenic potential in vivo to gain more insight into safety and suitability of these cells for applications in renal replacement therapies.
Results
The proliferative capacity and apoptosis resistance of ciPTEC in relation to SV40T expression
As expected [16], the expression of SV40T was abundant at permissive temperature (33° C) but went down (90% reduction) within one day of culturing at a non-permissive temperature (37° C) and remained low for up to 7 days (Figure 1A). Clinical studies indicated that cooling the dialysate down to 35° C can be beneficial as it may enhance the patient’s hemodynamic stability by preventing intradialytic hypotension [39, 40]. A transient drop in temperature (4 h, 33° C), however, did not result in an increase in SV40T expression compared to cells maintained at 37° C for 7 days (Figure 1A).
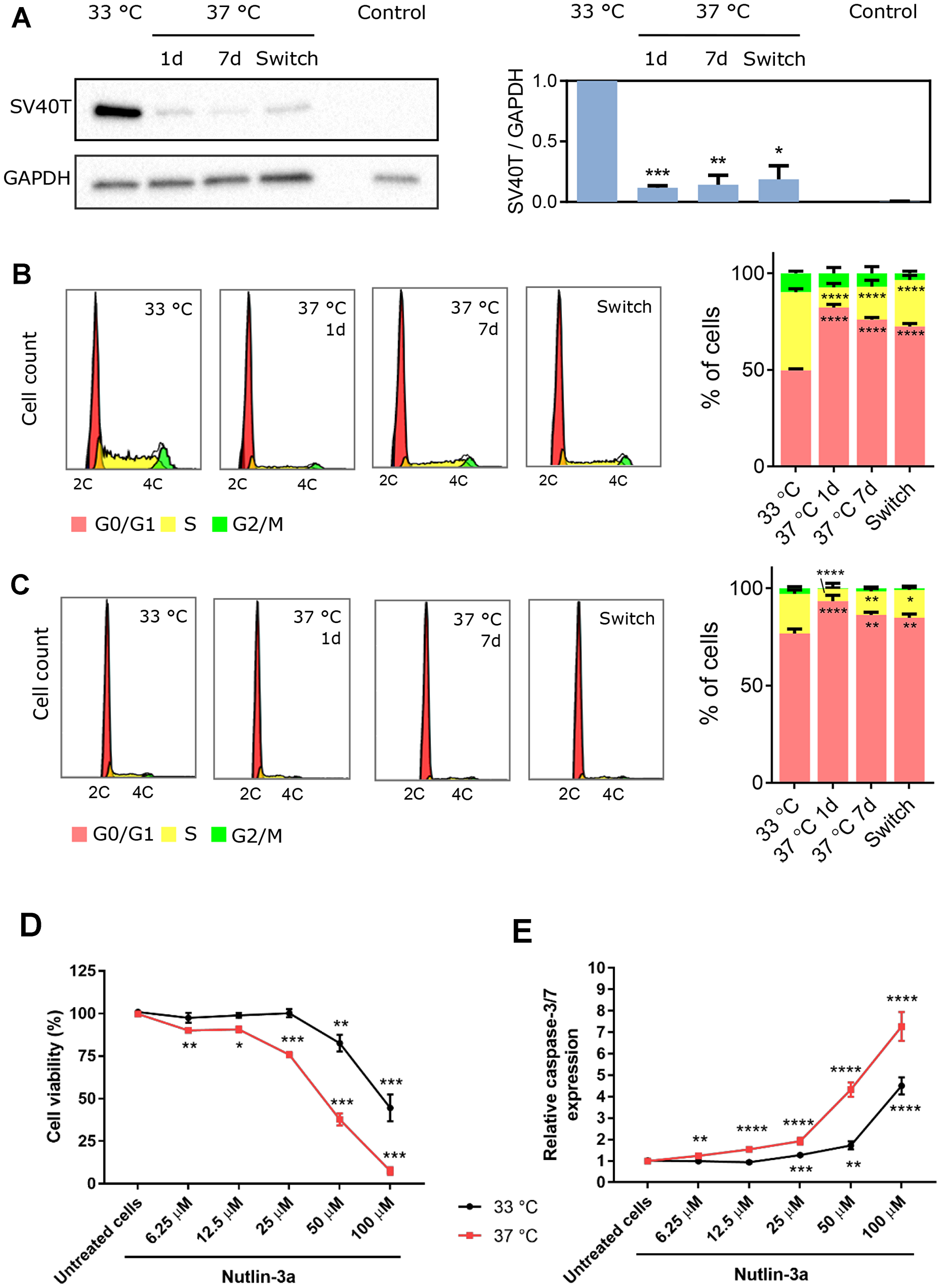
Figure 1: Temperature-dependent effect of SV40T expression on ciPTEC-OAT1 proliferation and apoptosis-sensitivity. (A) Western blot analysis of SV40T levels in ciPTEC-OAT1 cultured at the permissive (33° C) temperature and the non-permissive (37° C) temperature for 1 day, 7 days or 7 days followed by a 4 h incubation at 33° C (switch). Intensity of the bands was normalized to GAPDH and quantification is depicted in the bar graph. Human kidney tissue protein sample served as control. Representative histograms and analysis of cell cycle distribution of ciPTEC-OAT1 cultured at (B) subconfluent and (C) confluent levels at 33° C and 37° C for 1 day, 7 days or 7 days followed by 4 h at 33° C (switch). (D) Cell viability analysis and (E) caspase-3/7 expression in ciPTEC-OAT1 cultured at 33° C and 37° C and exposed to increasing concentrations of nutlin-3a for 24 h. All values are expressed as the mean ± SEM of three independent experiments performed in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001 (unpaired two-tailed Student’s t-test and one-way ANOVA followed by Dunnett’s multiple comparison test).
In a subconfluent state a higher proportion of cells was found in S-phase at the permissive temperature (Figure 1B; 40.5% ± 1.7%) compared to 37° C for 1 day (10.5% ± 2.0%) or 7 days (17.0% ± 3.4%), confirming that SV40T protein expression is directly related to cell proliferation. At full confluency (Figure 1C), the same trend was observed with a higher proportion of proliferating cells at the permissive (20.4% ± 1.9%) compared to the non-permissive temperature (6.3% ± 6.9% after 1 day at 37° C and 12.1% ± 1.5% after 7 days at 37° C).
Following 24 h of exposure to nutlin-3a to trigger p53-mediated apoptosis, one of the main targets of SV40T [20], matured cells expressing lower levels of SV40T displayed higher sensitivity to nutlin-3a compared to cells cultured at permissive temperature. Indeed, cells at 33° C were resistant to nutlin-3a-induced cell death, whereas cells at 37° C showed reduced cell viability and higher expression of caspase-3/7 even at lower nutlin-3a concentrations (Figure 1D–1E).
CiPTEC-OAT1 obey the rule of contact-inhibition
Further, ciPTEC-OAT1 growth did not extend beyond a confluent epithelial monolayer regardless of permissive or non-permissive temperature (Figure 2A), indicating contact inhibition. In contrast, HeLa cells presented multi-layered growth with >55% of growth surface covered with multiple cell layers when cultured at 33° C, and ~70% of multi-layered cell growth at 37° C (Figure 2B–2C). When ciPTEC-OAT1 were cultured on HFM, multi-layered growth was also not observed at both temperatures (Figure 2D).
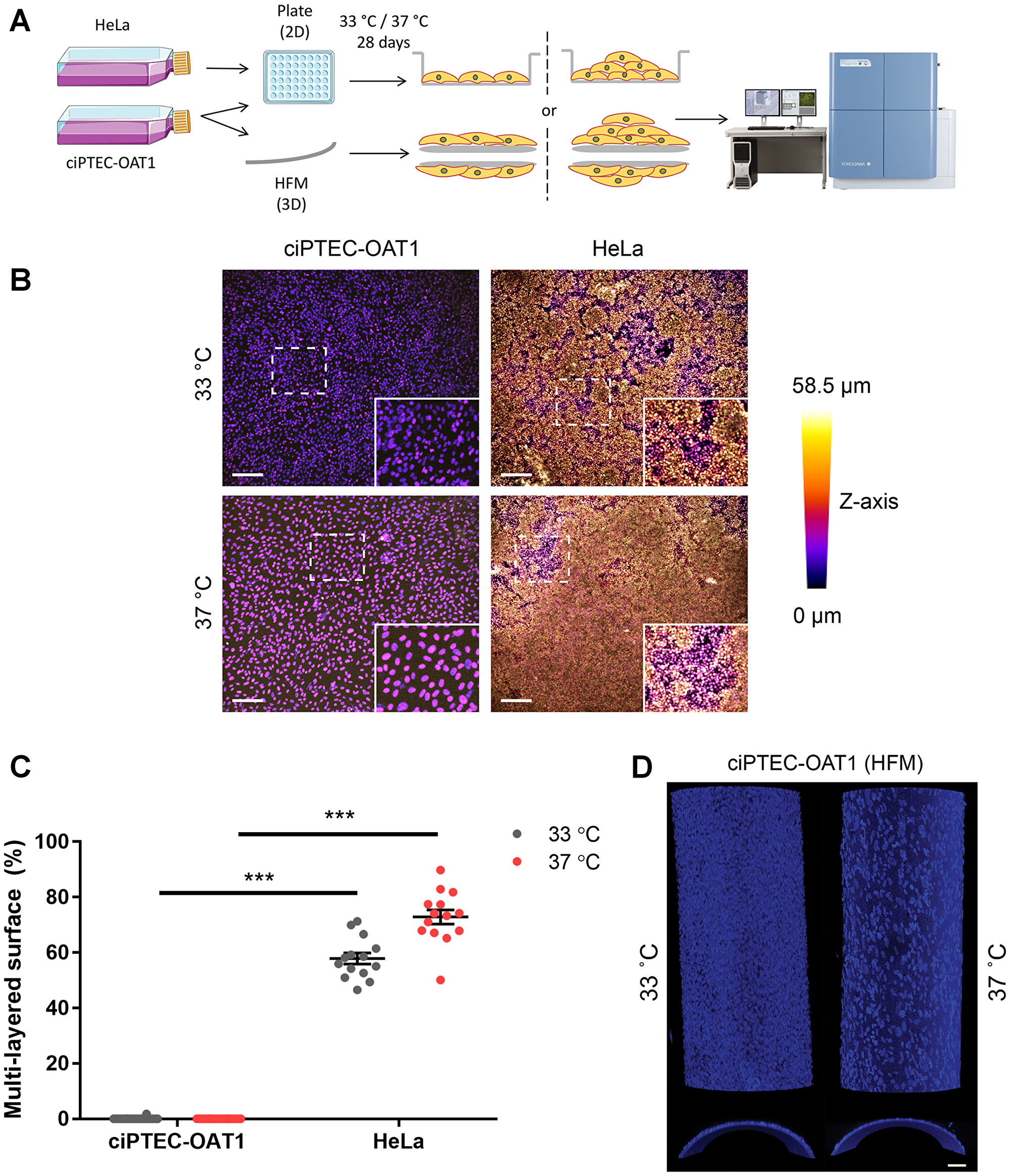
Figure 2: Contact inhibition in ciPTEC-OAT1. (A) Schematic diagram of focus formation assay. CiPTEC-OAT1 were cultured in 2D (96-well microplate) and 3D (hollow fiber membranes; HFM) for 28 days at 33° C and 37° C. HeLa cells were cultured in 2D in same conditions. Foci (multi-layered growth) formation was detected by nuclear staining and confocal imaging and (B) representative depth-coded images of nuclei-stained ciPTEC-OAT1 and HeLa cells after 28 days of culture at both permissive and non-permissive temperature are shown. Scale bars denote 200 μm in the original image and 100 μm in the zoom-in. (C) Quantification of the surface area covered by multi-layered proliferation). ND = not detected. (D) Representative confocal images of nuclei stained ciPTEC-OAT1 cultured on double-coated HFM at 33° C and 37° C, x-y confocal planes on the upper part and y-z confocal planes on the bottom part. Images taken with 10× magnification. Scale bar: 50 μm. Values are expressed as the mean ± SEM of three independent experiments performed in triplicate. ***p < 0.001 (unpaired two-tailed Student’s t-test).
CiPTEC-OAT1 require anchorage for proliferation
Anchorage-independent growth is a hallmark of cancer cells [41, 42], and it is assessed using the soft agar assay (Figure 3A). Single cells were encapsulated in semi-solid agarose medium thereby creating an environment lacking anchorage means (i.e. cell-cell and cell-extracellular matrix (ECM) interactions). After 4 weeks of culture, sporadic colony formation was observed for ciPTEC-OAT1 (0.87 ± 0.21 colonies per field (CPF)) compared to control HeLa cells (12.70 ± 0.58 CPF). Similarly, colonies were absent in cultures of mature ciPTEC-OAT1 (0.07 ± 0.05 CPF) while abundantly present in cultures of HeLa cells (15.10 ± 0.95 CPF) (Figure 3B). Qualitatively, colonies incidentally observed in ciPTEC-OAT1 cultures were much smaller than in HeLa cell cultures (Figure 3C), as also illustrated by the macroscopic images (Figure 3D), where only HeLa cells formed colonies at 37° C large enough to be visible by eye.
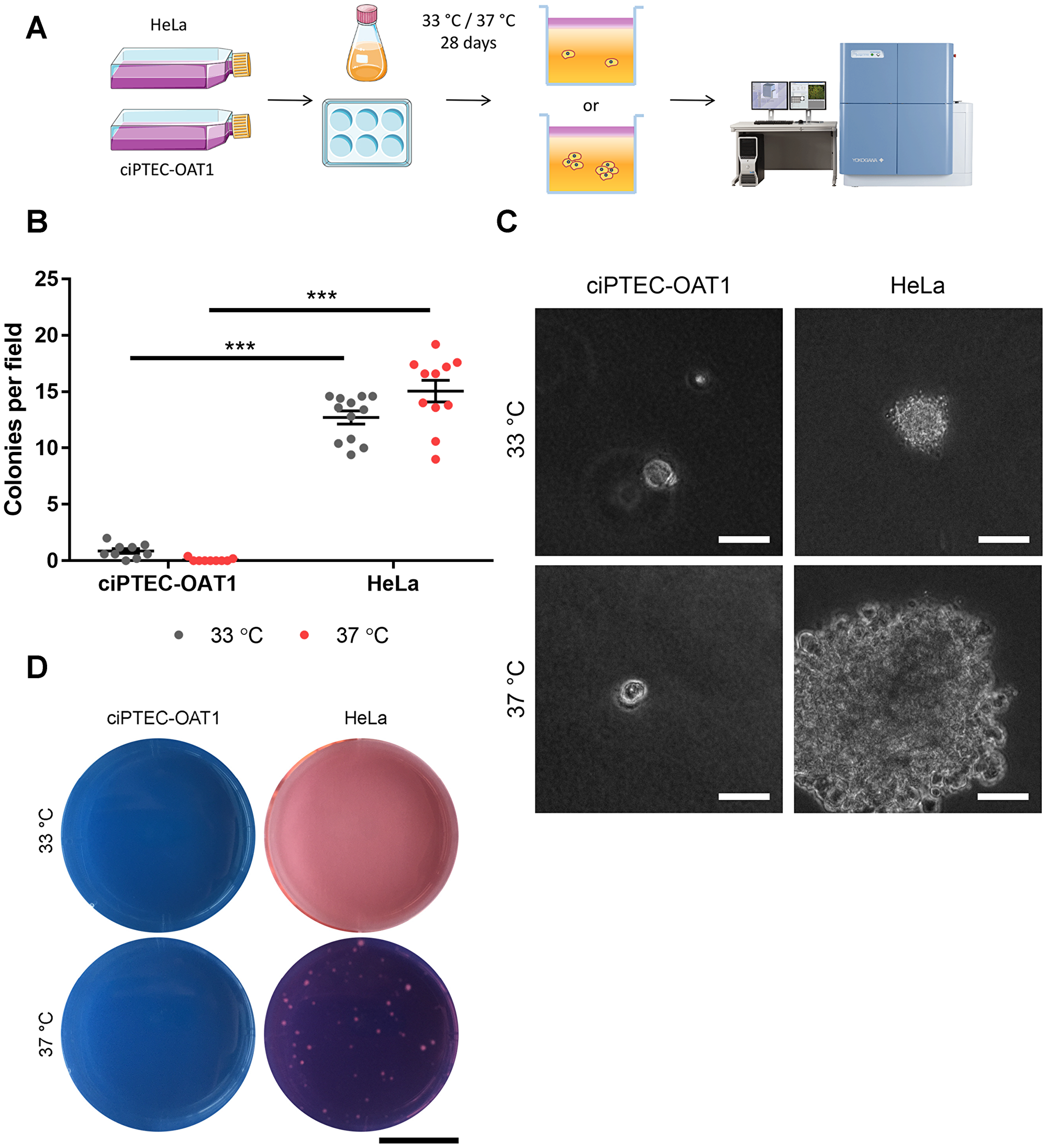
Figure 3: Anchorage-independent growth at permissive and non-permissive temperatures. (A) Schematic diagram of soft agar assay. Single ciPTEC-OAT1 and HeLa cells were seeded in agarose-containing medium (0.3% (w/v)) and incubated for 28 days at either 33° C or 37° C. Cell growth and colony formation was detected by confocal imaging. (B) Quantification of colonies detected for ciPTEC-OAT1 and HeLa cells presented as number of colonies per field. (C) Representative microscopic pictures of cell colonies formed by ciPTEC-OAT1 HeLa cells after 28 days culture at 33° C and 37° C. Scale bars denote 50 μm. (D) Representative macroscopic pictures of ciPTEC-OAT1 and HeLa cell colonies. Scale bar denotes 1 cm. Values are expressed as the mean ± SEM of three independent experiments performed in triplicate. ***p < 0.001 (unpaired two-tailed Student’s t-test).
CiPTEC-OAT1 are predominantly non-invasive
A cell-tracking experiment in growth factor reduced Matrigel™ was performed to model invasive and metastatic capacity of matured ciPTEC-OAT1 in vitro. During a 24 h time-lapse, the majority of cells (89.0%) did not migrate and remained non-invasive (Figure 4A, Supplementary Figure 1). However, a small population was able to migrate through the ECM. Morphologically, these cells showed mesenchymal cell-like features and moved accordingly with a speed higher than 6 μm/h, whereas non-invading cells remained round-shaped with minimal movement within their own space at a speed below 6 μm/h (Figure 4B, 4C).
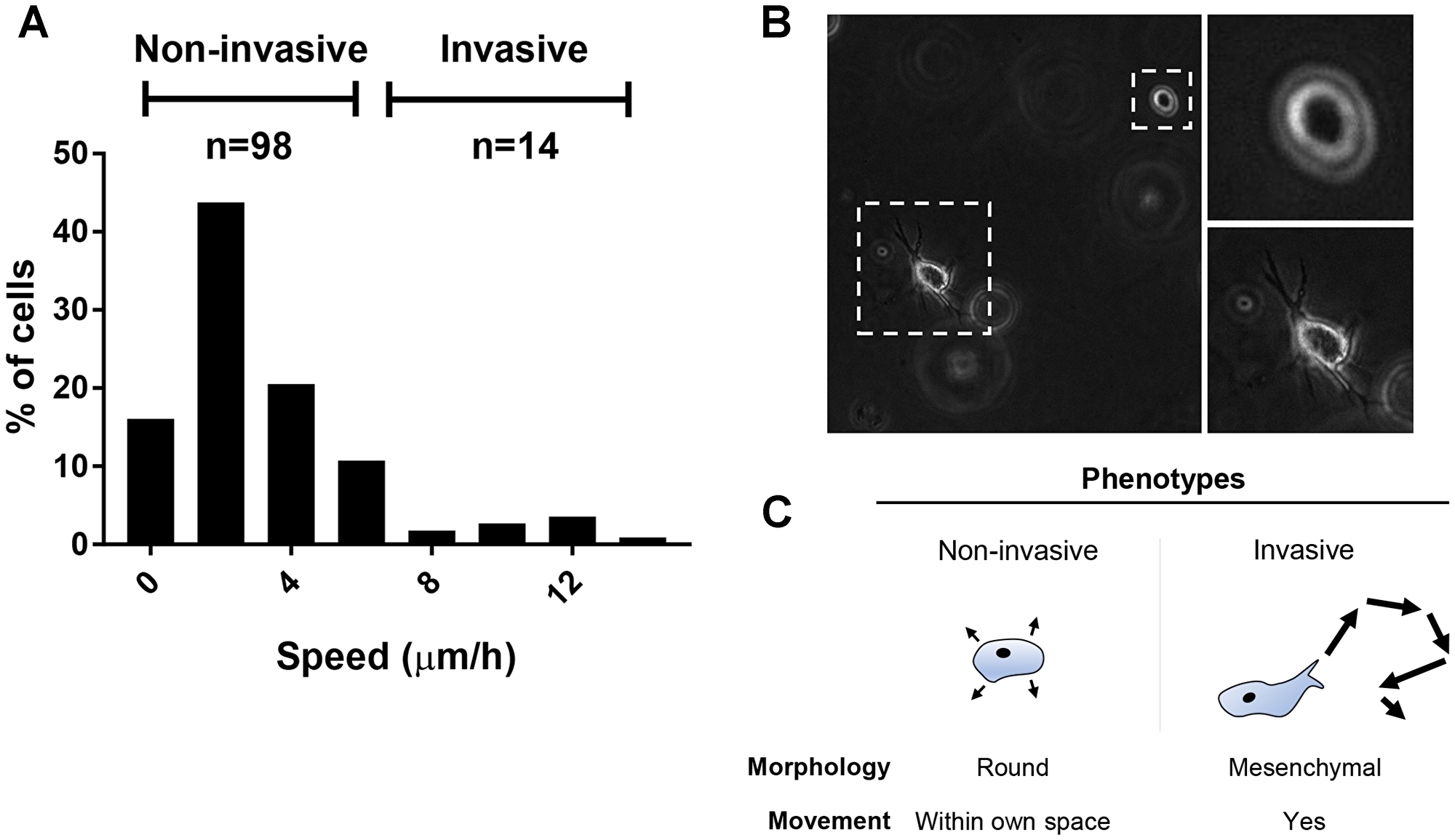
Figure 4: Migration and invasive potential of ciPTEC-OAT1. (A) Histogram showing the average speed (μm/h) at which the cells moved through Matrigel™ basement membrane matrix during the 24 h time-lapse imaging. Obtained values arose from 112 cells analyzed from two independent experiments. (B) Representative pictures showing morphological differences between the invasive and non-invasive phenotypes (20× magnification). (C) Invasive cells displayed mesenchymal cell-like movement, while non-invasive cells were round and static, wobbled only within their own space.
Transgene integration sites and genomic stability
Next, we evaluated the integration sites of the genes encoding for SV40T, hTERT and OAT1 and their consequences for ciPTEC-OAT1. For this, TLA was utilized to amplify the transgenes and their surrounding regions, from which precise integration sites were mapped (Figure 5A). We identified integrations sites located in the intronic regions of six endogenous genes. SV40T was integrated in GNA12 (chromosome 7) (Figure 5B; Supplementary Figure 2A, 2C) and BCL2L1 (chromosome 20) (Supplementary Figure 2B, 2C). The hTERT was stably integrated in the CAMTA1 gene (chromosome 1) (Supplementary Figure 3), whereas SLC22A6 (encoding OAT1) was integrated in WDR90 (chromosome 16), KIAA1958 (chromosome 9) and EEA1 (chromosome 12) (Supplementary Figure 4). Several predictions were made regarding the functional consequences of these insertion sites (Figure 5C). Except for BCL2L1 and EEA1, all endogenous genes are transcribed in the opposite direction compared to the inserted gene. A moderate degree of conservation was found for BCL2L1 and CAMTA1, with phastCons 100-way scores of 0.149 and 0.273, respectively. The other sites were classified as not conserved. All integration sites were analysed with Ensembl Variant Effect Predictor. Only BCL2L1 insertion site was found to be of relevance, containing both a regulatory-active site as well as being part of an antisense sequence (Figure 5C). The remaining transcript types were classified as low probability of being functionally relevant as they are either designated for degradation, such as nonsense-mediated decay (NMD), or the integrated sequence resides in a location where it would be spliced out.
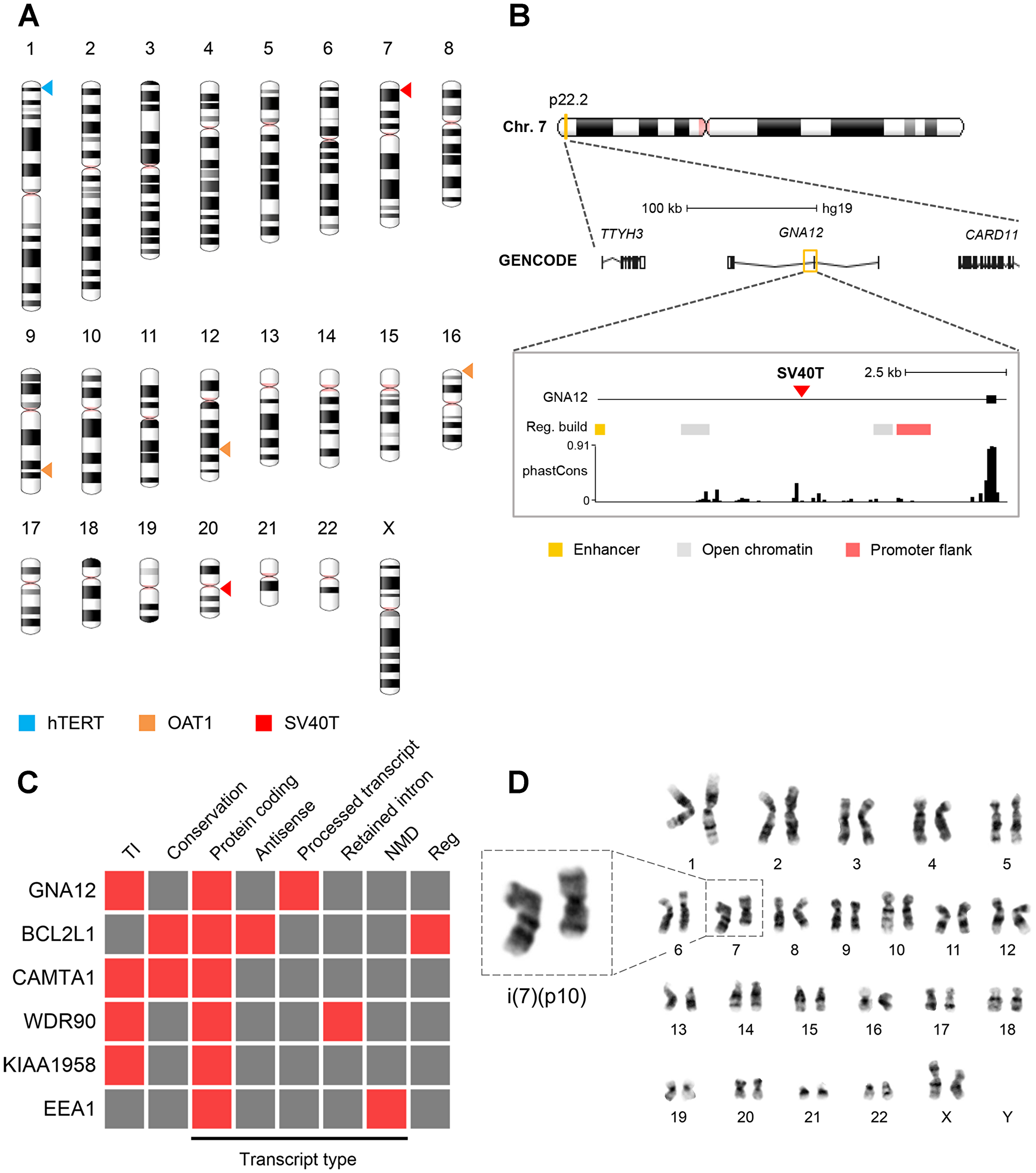
Figure 5: Viral integration sites and chromosomal stability. (A) Chromosomal distribution of the viral integration sites of the SV40T, hTERT and OAT1 transgenes. (B) Schematic representation of the integration of SV40T gene into GNA12. See Supplementary Figures 2–4 for schematic representation of remaining affected genes. Gene legend: untranslated region (empty box), exon (filled box), intron (line). (C) Functional consequence prediction of the viral integration sites. Presence and absence of a specific feature is shown in red and grey, respectively. Legend: TI = transcriptional interference, NMD = nonsense-mediated decay, reg = regulatory features. The phastCons P100 database was used to identify evolutionary conserved regions. (D) Cytogenetic analysis of ciPTEC-OAT1 at passage number 52. Representative female karyotype of a diploid cell showing an isochromosome for the short arm of chromosome 7.
For a final assessment of genomic abnormalities, ciPTEC-OAT1 at passage 52 were subjected to karyotype analysis. The cell population examined consisted of 68.2% near-tetraploid cells with the remainder being diploid. In-depth analysis of the diploid subpopulation (Figure 5D) shows the prevalence of an isochromosome abnormality concerning the p-arm of chromosome 7, i(7)(p10), which occurred in 2 out of 20 diploid metaphase spreads examined. Further, cells were re-examined at passage 62, where a complete shift towards near-tetraploidy was observed, with the presence of various complex aberrances among the entire cell population.
Transgene integration does not affect endocytosis
One of the SLC22A6 integration sites may have affected the function of one copy of EEA1, of which product, early endosome antigen (EEA1), is involved in endosomal trafficking (Figure 6A), a crucial function in PTEC [43]. However, endosomal clusters of EEA1, referred to as spots (Figure 6B–6C), did not differ between ciPTEC-OAT1 and the control parent cell line lacking the EEA1-affecting integration site. This maintained EEA1 expression was accompanied by preserved endocytosis function, as assessed by the uptake of fluorescently labelled BSA in ciPTEC-OAT1 (Figure 6B). No differences between EEA1 expression levels and BSA spot intensity were found (Figure 6D).
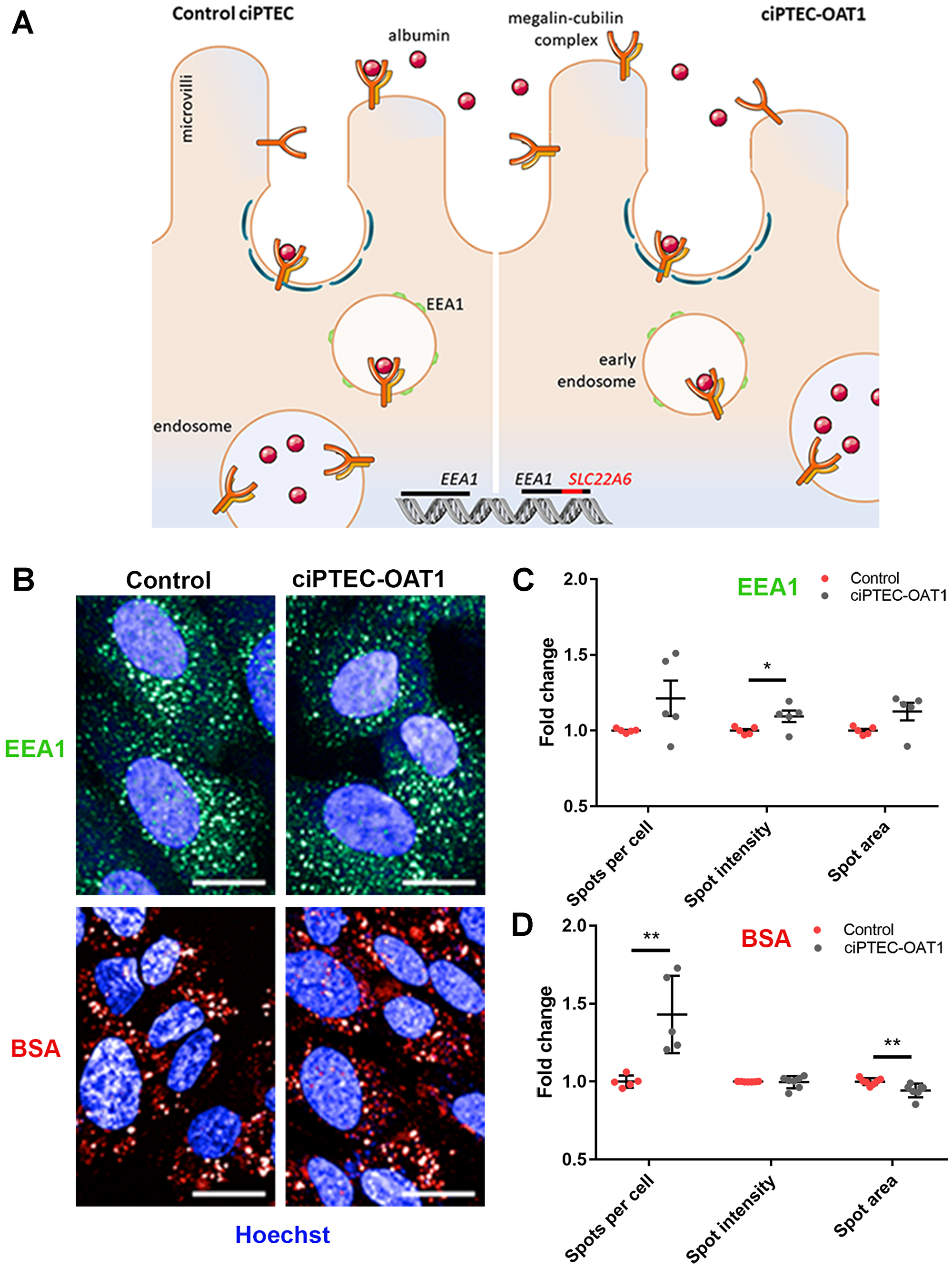
Figure 6: Endocytotic capacity of ciPTEC-OAT1. (A) Schematic representation of receptor-mediated endocytosis of albumin by control ciPTEC and ciPTEC-OAT1, showing the integration of OAT1-encoding gene (SLC22A6) within EEA1. (B) Representative immunofluorescence images of EEA1 expression and endocytotic uptake of Alexa Fluor 647 labelled bovine serum albumin (BSA). Scale bars denote 20 μm. Quantification of the (C) EEA1 expression and (D) BSA uptake by ciPTEC-OAT1 compared to control (parent ciPTEC). Values are normalized against control and expressed as the mean ± SEM of three independent experiments performed in triplicate. *p < 0.05, **p < 0.01 (unpaired two-tailed Student’s t-test).
ciPTEC-OAT1 are not tumorigenic or oncogenic in vivo
Finally, the tumorigenic and/or oncogenic potential ciPTEC-OAT1 were studied in vivo using nude athymic rats, and performed according to the World Health Organization (WHO) guidelines [44]. Tumorigenicity was evaluated by subcutaneous injection of 107 living cells, and oncogenicity was assessed by injection of cell lysates originating from the same number of cells (Figure 7A). HeLa cells produced palpable but not measurable nodules within the first week of injection in all 10 rats. Over time, in 6 out of 10 animals the nodules progressed into larger neoplastic masses that, by histological analysis, were confirmed to be anaplastic carcinomas in 5 out of 6 cases (Figure 7B, 7D). In the remaining animal, we could not histologically confirm carcinoma formation, most likely attributable to a small-sized, non-measurable neoplastic mass at necropsy. PCR analysis confirmed the presence of human specific Alu elements in all identified carcinomas, and also in the rat without histological evidence of carcinoma (Figure 7G). Importantly, the subcutaneous injection of HBSS (vehicle), ciPTEC-OAT1 or the cell lysate did not lead to nodule formation (Figure 7B–7F) and did not compromise animal well-being (Supplementary Figure 5). Analysis showed no histological lesion in the skin, adnexa, subcutaneous tissue and lymph nodes at the site of injection, nor any neoplastic formations in other major organs including liver, lungs, colon, spleen, mesenteric lymph nodes, and kidneys. This suggests that rats were not susceptible to spontaneous tumour formation and that ciPTEC-OAT1, in the given animal model, do not exert tumorigenic or oncogenic potential.
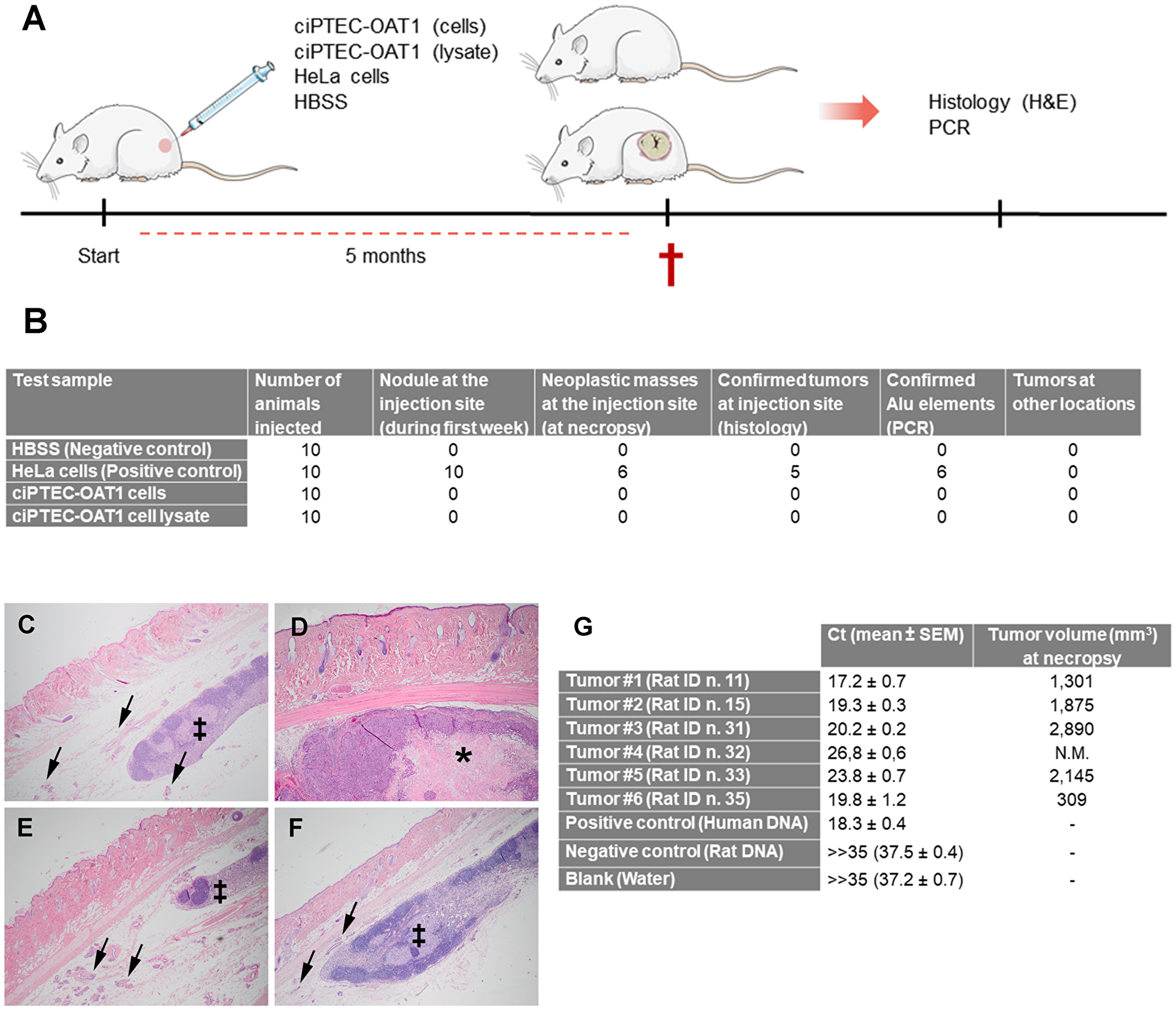
Figure 7: Tumorigenicity and oncogenicity of ciPTEC-OAT1. (A) Schematic representation of tumorigenicity and oncogenicity study in vivo. A total of 107 cells (ciPTEC-OAT1 or HeLa) resuspended in 100 μl of HBSS were injected subcutaneously in the flank of the rats. In an additional group cell lysate derived from 107 ciPTEC-OAT1 cells and resuspended in 100 μl of HBSS was injected per animal. In the negative control group rats received 100 μl of the vehicle (HBSS). Following the observational period of 5 months, animals were sacrificed (†) and histopathological and molecular (PCR) analyses were performed to confirm tumor formation and origin, respectively. (B) Summary of ciPTEC-OAT1 tumorigenicity and oncogenicity study results. (C–F) Representative pictures of histopathological analysis of the injection sites, performed by eosin and hematoxylin staining. (C) Negative control group injection site showing normal skin with subcutaneous lymph node (‡) and mammary tissue (arrows). (D) Positive control group injected with HeLa cells, showing the presence of anaplastic carcinoma with central area of necrosis (*) expanding in subcutaneous tissue at the site of injection. (E) Experimental tumorigenicity group injected with ciPTEC-OAT1 cells, presenting normal skin with subcutaneous lymph node (‡) and mammary tissue (arrows). (F) Oncogenicity group injected with ciPTEC-OAT1 cell lysate, showing normal skin with subcutaneous lymph node (‡) and mammary tissue (arrows). Pictures taken at magnification 2×. (G) PCR analysis of human-specific Alu elements confirming human origin of identified tumors and tumor volume (mm3) at necropsy. Ct values of all tumor samples were similar to that obtained for positive control human genomic DNA. Both the negative control (rat genomic DNA) and blank sample produced Ct values higher than 35. Limit of the blank [74], defined as the highest signal expected to be found when a blank sample containing no human DNA is tested, had a Ct value of 37.2 ± 0.71. N.M. (not measurable).
DISCUSSION
In the present study, we mechanistically and functionally confirmed that ciPTEC-OAT1 behave in accordance with conditional immortalization. After culture at non-permissive temperature they lose proliferative capacity and show contact inhibition. We found no indication for an important effect of transgene genomic integrations on endogenous gene expression or function. Although chromosomal aberrations could be demonstrated after multiple cell culture passages, there were no signs of in vivo tumorigenicity or oncogenicity.
The presence of residual SV40T, even after 7 days of culture at the non-permissive temperature, is in line with the known thermolabile characteristics of the SV40T mutant, as it will only be completely inactivated above 39.5° C [18]. Furthermore, considering BAK application and in case of a potential temperature drop during hemodialysis treatment, we observed a slight, but not significant, rebound in SV40T expression after re-exposing fully matured cells for 4 h at 33° C, however, this has no apparent effect on cell proliferation. Moreover, we showed that cell proliferation is dependent on SV40T expression as cells maintained at permissive temperature were highly proliferative compared to cells incubated at non-permissive temperature. This is in line with the mechanism of action of SV40T, which is known to interfere with Rb and p53 pathways thus driving cell proliferation [20]. Finally, the susceptibility of cells to undergo p53-mediated apoptosis at non-permissive temperature was confirmed using nutlin-3a, a compound that selectively induces p53 by inhibiting its degradation via Mdm2 [45, 46], suggesting normal p53 activity and apoptosis regulation at non-permissive temperature.
We examined the presence of contact inhibition in ciPTEC-OAT1 for two reasons. Firstly, non-transformed epithelial cells are expected to be responsive to growth-regulatory signals and undergo contact inhibition. Failure to display this would reflect cancerous cell behaviour and thus raise safety concerns [42]. Secondly, eyeing the potential use of ciPTEC-OAT1 in a BAK device [27], overgrowth of the cell monolayer could result in clogging of the tubules of the BAK device or affecting the epithelial barrier function. Our results showed that ciPTEC-OAT1 undergo contact inhibition and do not grow beyond the expected monolayer, even when they are cultured for long periods of time at permissive temperature. The observed absence of multi-layered growth of ciPTEC-OAT1 could also be explained by the presence of a crowding-induced live cell extrusion mechanism that helps maintaining homeostatic cell numbers in the epithelium [47].
Employing a soft-agar assay, known to correlate closely to in vivo tumour-forming ability of cells [48], we demonstrated that ciPTEC-OAT1 do not proliferate in an anchorage-independent manner. Small colonies observed sporadically were not progressively growing and exceeding 35 μm in diameter, indicating small cell clumps. In addition, we observed that the number of single cells present in the agarose at the beginning of the experiment decreased over the 4 weeks period of culturing, suggesting that some cells were dying, possibly due to anoikis [49]. However, this observation will be confirmed in future studies. In contrast to ciPTEC-OAT1, HeLa cells grew in absence of anchorage as described for cancer cell types [42].
Many assays have been described to study metastatic behaviour of malignant cells in vitro [50]. A requirement for metastasis is that cells are capable of invasion, i.e. migration through an ECM barrier in which movement is primarily limited to the cell’s ability to proteolytically degrade its surroundings, though amoeboid motility has also been reported depending on the (micro-)environment [51, 52]. Here, single ciPTEC-OAT1 cells were confined to a 3D environment that consisted of growth factor-reduced Matrigel™, using FCS-containing complete medium as a chemoattractant. The majority of cells lacked signs of invasive behaviour, though in a small subset we observed mesenchymal cell-like movement during the 24 h incubation. But studies have shown that this type of movement also plays an important role in tissue repair [53]. Furthermore, it should be emphasized that metastasis is a complex multi-step process, involving detachment from the cell bulk, intravasation into the systemic circulation, survival in a relatively harsh environment (e.g. lacking anchorage and presence of immune surveillance) and finally, extravasation into a distant tissue or organ [52]. No single in vitro assay fully recapitulates the complete chain of these events [50]. However, our in vivo results further support an absence of tumorigenic and metastatic potential of the cells.
The transgenes integration mediated by retroviral transduction could potentially lead to cell transformation and oncogenesis [37, 54]. Understanding functional consequences of the viral integration of the SV40T gene, hTERT and SLC22A6 transgenes is far from trivial. In contrast to protein-coding sequences, the function of non-coding DNA remains largely unknown and the annotation of regulatory elements is often based on predictive models. Ensembl’s Regulatory Build is a good example, taking into account epigenetic markers, transcription-factor binding sites and DNase I hypersensitive sites (DHS), amongst other features, to define regulatory regions [55]. The limitations become clear when considering the integration site of BCL2L1, a gene involved in both pro- and anti-apoptotic signaling through its two protein isoforms, Bcl-xS and Bcl-xL [56]. The Regulatory Build categorizes this area as a promoter-flanking region, despite its location being approximately 27 kb downstream from the actual promoter. The activity of regulatory elements tends to be cell-type specific, which makes the impact estimation through an in-silico approach particularly challenging. Our results demonstrating that ciPTEC-OAT1 remain subjected to the intrinsic apoptosis pathway, of which Bcl2l1 is a key regulator [56], suggest that the viral insertion did not reduce the cell’s capacity to undergo apoptosis. Moreover, four out of six transgenes are integrated in the opposite DNA strand. A difference in orientation can cause transcription machineries to converge and collide, a process termed transcriptional interference (TI). This generally manifests itself in decreased transcript levels [57, 58]. However, this type of integration also has a protective effect as it allows the transgene to be spliced out, resulting in an intact messenger RNA (mRNA) of the endogenous gene [57]. On the other hand, the transgenes of which the orientation matches that of the endogenous gene, in case of BCL2L1 and EEA1, could potentially lead to premature halt of endogenous genes transcription due to the presence of a termination signal in the long-terminal repeats (LTRs) of the viral vectors, leading to a truncated transcript of the endogenous gene [38]. However, it should be noted that one healthy allele remained for each affected gene, possibly limiting the impact of integration. In support, we demonstrated an intact endocytotic capacity in ciPTEC-OAT1 proving the unaltered expression and function of EEA1.
Karyotyping of ciPTEC-OAT1 at passages 52 and 62 showed the presence of a growing subpopulation of near-tetraploid cells with various complex chromosomal aberrations. Through its interaction with Bub-1, a spindle assembly checkpoint protein, SV40T can breach genomic integrity and induce tetraploidy [59]. Although this interaction appears to be unnecessary for immortalization, it has been demonstrated to trigger oncogenic transformation [60]. While the latter seemed absent in ciPTEC-OAT1, chromosomal abnormalities could have been avoided [18]. Still, SV40T as a tool for immortalization requires additional scrutiny, warranting a case-by-case evaluation of its impact on the chromosomal stability. This is especially important from a clinical perspective, as chromosomal instability could affect safety characteristics of a cell line. The creation of a SV40T mutant (U19dl89-97tsA58) that lacks the interaction site with Bub-1 is a promising development [61].
WHO guidelines regarding cell-based therapies suggest that all cell types intended for therapeutic purposes should be genetically stable as otherwise they would impose a significant risk regarding cell function and tumorigenic potential [44]. It should be noted that ciPTEC-OAT1 have previously undergone a rigorous functional assessment, showing that, at least from the functional perspective, cells remain stable over a wide range of passages [26]. Despite chromosomal aberrations, these cells differentiate in mature cell monolayers exhibiting mulitple PTEC-related functions, including epithelial barrier formation, protein uptake, vitamin D activation, and transport of uremic metabolites [26, 27, 62].
Further, in accordance to the WHO regulations [44], we addressed tumorigenic and oncogenic potential of ciPTEC-OAT1 in vivo. During the 5 months follow-up, our negative control group (vehicle control) confirmed the absence of spontaneous tumour formation. The guidelines also suggest that 90% of the animals within the positive control group should develop progressively growing tumours at the injection site. In all animals of the positive control group nodules appeared within the first week after HeLa cell injections, but only 50% of animals developed histologically and 60% PCR-confirmed HeLa derived tumours, indicating susceptibility of the animal model to grow tumour xenografts. Moreover, according to the guidelines, at least 20% of the animals within the test group should develop tumours in order to consider a particular cell type to be tumorigenic or oncogenic. Given that none of the animals developed tumours in the two test groups, we carefully conclude that in the athymic nude rat model, ciPTEC-OAT1 did not possess tumorigenic or oncogenic potential confirming our in vitro results.
Finally, considering that possible clinical use of these cells would only be in a context of an extracorporeal medical device and not direct transplantation, the altered karyotype and rare events of invasion observed in vitro, provided proper cell function, should not pose an extreme safety threat.
In conclusion, by showing that ciPTEC-OAT1 do not portray fundamental characteristics of oncogenically transformed cells, do not present negative consequences of viral transductions and genomic transgene integrations, such as insertional mutagenesis, nor possess tumorigenic capacity in vivo, the present study lays an important foundation towards validating the safety of a conditionally immortalized cell line for clinical application as cell-based renal replacement therapy.
Materials and Methods
Materials
All reagents were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands) unless stated otherwise.
Cell culture
Parent cell line (ciPTEC) and its OAT1-overexpressing derivate (ciPTEC-OAT1) were maintained in culture as described previously [26]. Culture of HeLa cells (ECACC, cat. nr. 93021013) is described in Supplementary Detailed Methods.
Western blot analysis of SV40T
Protein expression of SV40T was analysed by Western blotting as described [16] and in Supplementary Detailed Methods.
Cell cycle analysis
Cells fixed in ice-cold 70% (v/v) ethanol were stained with 40 μg/mL propidium iodide (PI) solution for 30 min and DNA content of >10 000 cells per condition was measured using FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA), as described in Supplementary Detailed Methods.
Apoptosis evaluation
Cell viability and caspase-3/7 expression, as indicators of apoptosis, were determined using PrestoBlue® cell viability (Life Technologies, Paisly, UK) and CellEvent™ Caspase-3/7 Green detection (Invitrogen, Eugene, OR, USA) reagents, respectively, following manufacturer’s instructions. Details are reported in Supplementary Detailed Methods.
Contact inhibition
Contact inhibition and multi-layered cell growth was determined by performing a z-stack imaging of Hoechst 33342 (1 μM) labelled cells by means of Cell Voyager 7000 (CV7000) confocal microscope (Yokogawa Electric Corporation, Tokyo, Japan). An ImageJ plugin was developed to quantify the surface area covered by cell multi-layers (Supplementary Method 1), as described in Supplementary Detailed Methods.
CiPTEC-OAT1 culture on hollow fiber membranes
Cell proliferation in a 3D environment was assessed by culturing cells for 28 days on L-DOPA (2 mg/ml) and collagen IV (25 μg/ml) double-coated microPES hollow fiber membranes (HFM; Membrana GmbH, Wuppertal, Germany), as described previously [27, 63, 64] and in Supplementary Detailed Methods.
Soft agar assay
Colony-forming ability of cells in anchorage-independent conditions was assayed in a similar manner as described by Borowicz et al. [41]. Procedure is described in Supplementary Detailed Methods.
Single cell invasion assay
The invasion assay was based on the protocol described by Zaman et al. [51]. Cell tracking analysis was performed using Fiji’s TrackMate plugin to determine the speed of motion across acquisitions, as well as the average speed throughout the experiment [65, 66]. Cells having a speed of > 6 μm/h were classified as invasive. We manually validated this threshold for optimum discrimination between invasive and non-invasive cells. Inclusion and exclusion criteria were applied as previously described [51]. All data were processed in MySQL 5.6.17 (Oracle, Redwood City, CA, USA). Procedure is described in Supplementary Detailed Methods.
Targeted locus amplification for viral integration sites
The ciPTEC-OAT1 cell line was stably transduced using three viral vectors [16, 26], warranting an investigation into the occurrence of cell behavior-altering insertional mutagenesis. Determination of the exact location of the integrated transgenes was performed by Cergentis B.V. (Utrecht, the Netherlands) using targeted locus-amplification (TLA) technology as described [67]. Data was analysed using Ensembl’s genome browser in conjunction with the regulatory build [55, 68]. To predict the functional consequences of the integration sites, the Ensembl Variant Effect Predictor was utilized [69].
Cytogenetic analysis
Metaphase spreads of ciPTEC-OAT1 were G-banded and analysed for abnormalities (Cell Guidance Systems, Cambridge, UK). Approximately 20 metaphase spreads were analysed per experiment. Sample preparation is described in Supplementary Detailed Methods.
Endocytosis
Endocytosis was evaluated by evaluating early endosome antigen (EEA1) expression and bovine serum albumin (BSA) uptake, similarly to [70] and reported in Supplementary Detailed Methods.
Tumorigenicity and oncogenicity evaluation in vivo
Animal procedures were approved by the Ethics Committee of Animal Research of Utrecht University, Utrecht, The Netherlands (CCD approval number AVD108002017879). Male (n = 20; 4 weeks old, weighing between 103 and 172 g) and female (n = 20; 4 weeks old, weighing between 94 and 133 g) athymic nude rats (Hsd:RH-Foxn1rnu; Envigo, Horst, Netherlands) were maintained in the Central Laboratory Animal Research Facility (GDL, Utrecht, Netherlands), and housed in individually ventilated cage units at RT under a 12 h light/dark cycle. Food and water were provided ad libitum. All animals were treated according to IVD and CCD guidelines and all efforts were made to minimize suffering. Animals were euthanized by pentobarbital (Faculty of Veterinary Medicine, Utrecht, the Netherlands) overdose via intraperitoneal injection, followed by cervical dislocation as soon as animals became unconscious. Details are reported in Supplementary Detailed Methods.
Histopathological analysis
First, formalin-fixed tissues and organs were examined macroscopically for presence of abnormalities. Afterwards, formalin-fixed tissues were subjected to further microscopical histological analysis as described [71, 72]. Examination was performed by board-certified veterinary pathologists of the Dutch Molecular Pathology Centre (Department of Pathobiology, Faculty of Veterinary Medicine, Utrecht, the Netherlands). Representative images were taken using Olympus BX45 microscope equipped with DP25 camera (Leiderdorp, the Netherlands) with 2× magnification.
DNA extraction and PCR analysis
Human origin of observed tumors was confirmed by detection of human specific Alu elements [73], using Real-Time PCR, following manufacturer’s instructions. Details regarding sample preparation, reaction protocol and primers used are described in Supplementary Detailed Methods.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, Inc., La Jolla, CA, USA), unless stated otherwise. Data are presented as mean ± standard error of the mean (SEM) of three independent experiments performed in triplicate, unless stated otherwise. Significance was evaluated using the unpaired two-tailed Student’s t-test or one-way ANOVA followed by Dunnett’s multiple comparison test where appropriate. P-values < 0.05 were considered as significant. Where appropriate, significance is denoted as *(p < 0.05), **(p < 0.01) and ***(p < 0.001).
Abbreviations
BAK: bioartificial kidney; BCL2L1: BCL2 Like 1; BSA: bovine serum albumin; ciPTEC: conditionally immortalized proximal tubule epithelial cells; ECM: extracellular matrix; EEA1: early endosome antigen; ESKD: end-stage kidney disease; HFM: hollow fiber membranes; hTERT: human telomerase; L-DOPA: levodopa; LLC-PK1: Lilly Laboratories cell porcine kidney 1; LTR: long-terminal repeats; MDCK: Madin-Darby canine kidney; OAT1: organic anion transporter 1; PTEC: proximal tubule epithelial cells; SV40T: SV40 large T antigen; TLA: targeted locus amplification.
ACKNOWLEDGMENTS
Milos Mihajlovic and Koen Westphal would like to thank Monika Verdouw for her help with animal handling during subcutaneous injections.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
FUNDING
This research was supported by the Marie Curie ITN project BIOART (grant no. 316690, EU-FP7-PEOPLE-ITN-2012).
References
1. Jansen J, Fedecostante M, Wilmer MJ, van den Heuvel LP, Hoenderop JG, Masereeuw R. Biotechnological challenges of bioartificial kidney engineering. Biotechnol Adv. 2014; 32:1317–27. https://doi.org/10.1016/j.biotechadv.2014.08.001. [PubMed].
2. Liyanage T, Ninomiya T, Jha V, Neal B, Patrice HM, Okpechi I, Zhao MH, Lv J, Garg AX, Knight J, Rodgers A, Gallagher M, Kotwal S, et al. Worldwide access to treatment for end-stage kidney disease: a systematic review. Lancet. 2015; 385:1975–82. https://doi.org/10.1016/S0140-6736(14)61601-9. [PubMed].
3. Thomas R, Kanso A, Sedor JR. Chronic kidney disease and its complications. Prim Care. 2008; 35:329–44. https://doi.org/10.1016/j.pop.2008.01.008. [PubMed].
4. Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJ, Jardine M, Kasiske B, Köttgen A, Kretzler M, et al, and ISN Global Kidney Health Summit participants. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017; 390:1888–917. https://doi.org/10.1016/S0140-6736(17)30788-2. [PubMed].
5. Humes HD, Buffington D, Westover AJ, Roy S, Fissell WH. The bioartificial kidney: current status and future promise. Pediatr Nephrol. 2014; 29:343–51. https://doi.org/10.1007/s00467-013-2467-y. [PubMed].
6. Vesey DA, Qi W, Chen X, Pollock CA, Johnson DW. Isolation and primary culture of human proximal tubule cells. Methods Mol Biol. 2009; 466:19–24. https://doi.org/10.1007/978-1-59745-352-3_2. [PubMed].
7. Weiland C, Ahr HJ, Vohr HW, Ellinger-Ziegelbauer H. Characterization of primary rat proximal tubular cells by gene expression analysis. Toxicol In Vitro. 2007; 21:466–91. https://doi.org/10.1016/j.tiv.2006.10.008. [PubMed].
8. Humes HD, Buffington DA, MacKay SM, Funke AJ, Weitzel WF. Replacement of renal function in uremic animals with a tissue-engineered kidney. Nat Biotechnol. 1999; 17:451–55. https://doi.org/10.1038/8626. [PubMed].
9. Humes HD, MacKay SM, Funke AJ, Buffington DA. Tissue engineering of a bioartificial renal tubule assist device: in vitro transport and metabolic characteristics. Kidney Int. 1999; 55:2502–14. https://doi.org/10.1046/j.1523-1755.1999.00486.x. [PubMed].
10. Fissell WH, Lou L, Abrishami S, Buffington DA, Humes HD. Bioartificial kidney ameliorates gram-negative bacteria-induced septic shock in uremic animals. J Am Soc Nephrol. 2003; 14:454–61. https://doi.org/10.1097/01.ASN.0000045046.94575.96. [PubMed].
11. Fujita Y, Terashima M, Kakuta T, Itoh J, Tokimasa T, Brown D, Saito A. Transcellular water transport and stability of expression in aquaporin 1-transfected LLC-PK1 cells in the development of a portable bioartificial renal tubule device. Tissue Eng. 2004; 10:711–22. https://doi.org/10.1089/1076327041348383. [PubMed].
12. Ozgen N, Terashima M, Aung T, Sato Y, Isoe C, Kakuta T, Saito A. Evaluation of long-term transport ability of a bioartificial renal tubule device using LLC-PK1 cells. Nephrol Dial Transplant. 2004; 19:2198–207. https://doi.org/10.1093/ndt/gfh399. [PubMed].
13. Sato Y, Terashima M, Kagiwada N, Tun T, Inagaki M, Kakuta T, Saito A. Evaluation of proliferation and functional differentiation of LLC-PK1 cells on porous polymer membranes for the development of a bioartificial renal tubule device. Tissue Eng. 2005; 11:1506–15. https://doi.org/10.1089/ten.2005.11.1506. [PubMed].
14. Fey-Lamprecht F, Albrecht W, Groth T, Weigel T, Gross U. Morphological studies on the culture of kidney epithelial cells in a fiber-in-fiber bioreactor design with hollow fiber membranes. J Biomed Mater Res A. 2003; 65:144–57. https://doi.org/10.1002/jbm.a.10328. [PubMed].
15. Fujita Y, Kakuta T, Asano M, Itoh J, Sakabe K, Tokimasa T, Saito A. Evaluation of Na+ active transport and morphological changes for bioartificial renal tubule cell device using Madin-Darby canine kidney cells. Tissue Eng. 2002; 8:13–24. https://doi.org/10.1089/107632702753503018. [PubMed].
16. Wilmer MJ, Saleem MA, Masereeuw R, Ni L, van der Velden TJ, Russel FG, Mathieson PW, Monnens LA, van den Heuvel LP, Levtchenko EN. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010; 339:449–57. https://doi.org/10.1007/s00441-009-0882-y. [PubMed].
17. Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O’Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006; 69:1633–40. https://doi.org/10.1038/sj.ki.5000277. [PubMed].
18. O’Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, Moody J, Silver AR, Davies DC, Alsop AE, Neville AM, Jat PS. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci U S A. 2001; 98:646–51. https://doi.org/10.1073/pnas.98.2.646. [PubMed].
19. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279:349–52. https://doi.org/10.1126/science.279.5349.349. [PubMed].
20. Ahuja D, Sáenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005; 24:7729–45. https://doi.org/10.1038/sj.onc.1209046. [PubMed].
21. Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006; 20:2373–82. https://doi.org/10.1101/gad.1456306. [PubMed].
22. Jansen J, Schophuizen CM, Wilmer MJ, Lahham SH, Mutsaers HA, Wetzels JF, Bank RA, van den Heuvel LP, Hoenderop JG, Masereeuw R. A morphological and functional comparison of proximal tubule cell lines established from human urine and kidney tissue. Exp Cell Res. 2014; 323:87–99. https://doi.org/10.1016/j.yexcr.2014.02.011. [PubMed].
23. Schophuizen CM, Wilmer MJ, Jansen J, Gustavsson L, Hilgendorf C, Hoenderop JG, van den Heuvel LP, Masereeuw R. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch. 2013; 465:1701–14. https://doi.org/10.1007/s00424-013-1307-z. [PubMed].
24. Mutsaers HA, van den Heuvel LP, Ringens LH, Dankers AC, Russel FG, Wetzels JF, Hoenderop JG, Masereeuw R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS One. 2011; 6:e18438. https://doi.org/10.1371/journal.pone.0018438. [PubMed].
25. Mutsaers HA, Wilmer MJ, Reijnders D, Jansen J, van den Broek PH, Forkink M, Schepers E, Glorieux G, Vanholder R, van den Heuvel LP, Hoenderop JG, Masereeuw R. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim Biophys Acta. 2013; 1832:142–50. https://doi.org/10.1016/j.bbadis.2012.09.006. [PubMed].
26. Nieskens TT, Peters JG, Schreurs MJ, Smits N, Woestenenk R, Jansen K, van der Made TK, Röring M, Hilgendorf C, Wilmer MJ, Masereeuw R. A Human Renal Proximal Tubule Cell Line with Stable Organic Anion Transporter 1 and 3 Expression Predictive for Antiviral-Induced Toxicity. AAPS J. 2016; 18:465–75. https://doi.org/10.1208/s12248-016-9871-8. [PubMed].
27. Jansen J, Fedecostante M, Wilmer MJ, Peters JG, Kreuser UM, van den Broek PH, Mensink RA, Boltje TJ, Stamatialis D, Wetzels JF, van den Heuvel LP, Hoenderop JG, Masereeuw R. Bioengineered kidney tubules efficiently excrete uremic toxins. Sci Rep. 2016; 6:26715. https://doi.org/10.1038/srep26715. [PubMed].
28. Mihajlovic M, van den Heuvel LP, Hoenderop JG, Jansen J, Wilmer MJ, Westheim AJ, Allebes WA, Stamatialis D, Hilbrands LB, Masereeuw R. Allostimulatory capacity of conditionally immortalized proximal tubule cell lines for bioartificial kidney application. Sci Rep. 2017; 7:7103. https://doi.org/10.1038/s41598-017-07582-1. [PubMed].
29. Chevtchik NV, Fedecostante M, Jansen J, Mihajlovic M, Wilmer M, Rüth M, Masereeuw R, Stamatialis D. Upscaling of a living membrane for bioartificial kidney device. Eur J Pharmacol. 2016; 790:28–35. https://doi.org/10.1016/j.ejphar.2016.07.009. [PubMed].
30. Mihajlovic M, Wever KE, van der Made TK, de Vries RB, Hilbrands LB, Masereeuw R. Are cell-based therapies for kidney disease safe? A systematic review of preclinical evidence. Pharmacol Ther. 2019; 197:191–211. https://doi.org/10.1016/j.pharmthera.2019.01.004. [PubMed].
31. Manfredi JJ, Prives C. Binding of p53 and p105-RB is not sufficient for oncogenic transformation by a hybrid polyomavirus-simian virus 40 large T antigen. J Virol. 1990; 64:5250–59. [PubMed].
32. Sachsenmeier KF, Pipas JM. Inhibition of Rb and p53 is insufficient for SV40 T-antigen transformation. Virology. 2001; 283:40–48. https://doi.org/10.1006/viro.2001.0866. [PubMed].
33. Wai LK. Telomeres, telomerase, and tumorigenesis—a review. MedGenMed. 2004; 6:19. [PubMed].
34. Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999; 21:111–14. https://doi.org/10.1038/5056. [PubMed].
35. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997; 91:25–34. https://doi.org/10.1016/S0092-8674(01)80006-4. [PubMed].
36. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003; 4:346–58. https://doi.org/10.1038/nrg1066. [PubMed].
37. Yi Y, Hahm SH, Lee KH. Retroviral gene therapy: safety issues and possible solutions. Curr Gene Ther. 2005; 5:25–35. https://doi.org/10.2174/1566523052997514. [PubMed].
38. Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005; 24:7656–72. https://doi.org/10.1038/sj.onc.1209043. [PubMed].
39. Ayoub A, Finlayson M. Effect of cool temperature dialysate on the quality and patients’ perception of haemodialysis. Nephrol Dial Transplant. 2004; 19:190–94. https://doi.org/10.1093/ndt/gfg512. [PubMed].
40. Sakkas GK, Krase AA, Giannaki CD, Karatzaferi C. Cold dialysis and its impact on renal patients’ health: an evidence-based mini review. World J Nephrol. 2017; 6:119–22. https://doi.org/10.5527/wjn.v6.i3.119. [PubMed].
41. Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, Winn RA. The soft agar colony formation assay. J Vis Exp. 2014; 92:e51998. https://doi.org/10.3791/51998. [PubMed].
42. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013. [PubMed].
43. Eshbach ML, Weisz OA. Receptor-Mediated Endocytosis in the Proximal Tubule. Annu Rev Physiol. 2017; 79:425–48. https://doi.org/10.1146/annurev-physiol-022516-034234. [PubMed].
44. WHO Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks. Geneva, World Health Organization (WHO Expert Committee on Biological Standardization Sixty-first report; WHO Technical Report Series, No 978, Annex 3). 2010. Available from: https://www.who.int/biologicals/expert_committee/TRS_978_61st_report.pdf?ua=1.
45. Kojima K, Konopleva M, McQueen T, O’Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006; 108:993–1000. https://doi.org/10.1182/blood-2005-12-5148. [PubMed].
46. Hui W, Liu S, Zheng J, Fang Z, Ding Q, Feng C. Nutlin-3a as a novel anticancer agent for adrenocortical carcinoma with CTNNB1 mutation. Cancer Med. 2018; 7:1440–49. https://doi.org/10.1002/cam4.1431. [PubMed].
47. Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012; 484:546–49. https://doi.org/10.1038/nature10999. [PubMed].
48. Rotem A, Janzer A, Izar B, Ji Z, Doench JG, Garraway LA, Struhl K. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc Natl Acad Sci U S A. 2015; 112:5708–13. https://doi.org/10.1073/pnas.1505979112. [PubMed].
49. Gilmore AP. Anoikis. Cell Death Differ. 2005 (Suppl 12); (2):1473–77. https://doi.org/10.1038/sj.cdd.4401723. [PubMed].
50. Kramer N, Walzl A, Unger C, Rosner M, Krupitza G, Hengstschläger M, Dolznig H. In vitro cell migration and invasion assays. Mutat Res. 2013; 752:10–24. https://doi.org/10.1016/j.mrrev.2012.08.001. [PubMed].
51. Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci U S A. 2006; 103:10889–94. https://doi.org/10.1073/pnas.0604460103. [PubMed].
52. van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011; 728:23–34. https://doi.org/10.1016/j.mrrev.2011.05.002. [PubMed].
53. Krakhmal NV, Zavyalova MV, Denisov EV, Vtorushin SV, Perelmuter VM. Cancer Invasion: patterns and Mechanisms. Acta Naturae. 2015; 7:17–28. https://doi.org/10.32607/20758251-2015-7-2-17-28. [PubMed].
54. Desfarges S, Ciuffi A. Retroviral integration site selection. Viruses. 2010; 2:111–30. https://doi.org/10.3390/v2010111. [PubMed].
55. Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The ensembl regulatory build. Genome Biol. 2015; 16:56. https://doi.org/10.1186/s13059-015-0621-5. [PubMed].
56. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010; 37:299–310. https://doi.org/10.1016/j.molcel.2010.01.025. [PubMed].
57. Han Y, Lin YB, An W, Xu J, Yang HC, O’Connell K, Dordai D, Boeke JD, Siliciano JD, Siliciano RF. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe. 2008; 4:134–46. https://doi.org/10.1016/j.chom.2008.06.008. [PubMed].
58. Shearwin KE, Callen BP, Egan JB. Transcriptional interference—a crash course. Trends Genet. 2005; 21:339–45. https://doi.org/10.1016/j.tig.2005.04.009. [PubMed].
59. Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol. 2009; 83:117–27. https://doi.org/10.1128/JVI.01515-08. [PubMed].
60. Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci U S A. 2004; 101:947–52. https://doi.org/10.1073/pnas.0308006100. [PubMed].
61. Brandt A, Löhers K, Beier M, Leube B, de Torres C, Mora J, Arora P, Jat PS, Royer-Pokora B. Establishment of a Conditionally Immortalized Wilms Tumor Cell Line with a Homozygous WT1 Deletion within a Heterozygous 11p13 Deletion and UPD Limited to 11p15. PLoS One. 2016; 11:e0155561. https://doi.org/10.1371/journal.pone.0155561. [PubMed].
62. Mihajlovic M, Fedecostante M, Oost MJ, Steenhuis SK, Lentjes EG, Maitimu-Smeele I, Janssen MJ, Hilbrands LB, Masereeuw R. Role of Vitamin D in Maintaining Renal Epithelial Barrier Function in Uremic Conditions. Int J Mol Sci. 2017; 18. https://doi.org/10.3390/ijms18122531. [PubMed].
63. Ni M, Teo JC, Ibrahim MS, Zhang K, Tasnim F, Chow PY, Zink D, Ying JY. Characterization of membrane materials and membrane coatings for bioreactor units of bioartificial kidneys. Biomaterials. 2011; 32:1465–76. https://doi.org/10.1016/j.biomaterials.2010.10.061. [PubMed].
64. Oo ZY, Deng R, Hu M, Ni M, Kandasamy K, bin Ibrahim MS, Ying JY, Zink D. The performance of primary human renal cells in hollow fiber bioreactors for bioartificial kidneys. Biomaterials. 2011; 32:8806–15. https://doi.org/10.1016/j.biomaterials.2011.08.030. [PubMed].
65. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012; 9:676–82. https://doi.org/10.1038/nmeth.2019. [PubMed].
66. Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW. TrackMate: an open and extensible platform for single-particle tracking. Methods. 2017; 115:80–90. https://doi.org/10.1016/j.ymeth.2016.09.016. [PubMed].
67. de Vree PJ, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJ, Wan Y, Teunissen H, Krijger PH, Geeven G, Eijk PP, Sie D, Ylstra B, et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol. 2014; 32:1019–25. https://doi.org/10.1038/nbt.2959. [PubMed].
68. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, et al. Ensembl 2016. Nucleic Acids Res. 2016; 44:D710–16. https://doi.org/10.1093/nar/gkv1157. [PubMed].
69. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016; 17:122. https://doi.org/10.1186/s13059-016-0974-4. [PubMed].
70. Caetano-Pinto P, Janssen MJ, Gijzen L, Verscheijden L, Wilmer MJ, Masereeuw R. Fluorescence-Based Transport Assays Revisited in a Human Renal Proximal Tubule Cell Line. Mol Pharm. 2016; 13:933–44. https://doi.org/10.1021/acs.molpharmaceut.5b00821. [PubMed].
71. Henriques U. Histological technique in routine histopathology. An opinion. Pathol Res Pract. 1981; 171:417–22. https://doi.org/10.1016/S0344-0338(81)80114-8. [PubMed].
72. McInnes EF, Scudamore CL. Review of approaches to the recording of background lesions in toxicologic pathology studies in rats. Toxicol Lett. 2014; 229:134–43. https://doi.org/10.1016/j.toxlet.2014.06.005. [PubMed].
73. Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002; 3:370–79. https://doi.org/10.1038/nrg798. [PubMed].
74. Armbruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008; 29:S49–52. [PubMed].