
Introduction
Sézary syndrome (SS) is an aggressive leukemic form of cutaneous T-cell lymphoma (CTCL), which constitutes a heterogeneous group of extranodal non-Hodgkin lymphomas [1]. SS typically manifests as erythroderma and leukemic peripheral blood involvement by atypical malignant T cells named Sézary cells [2, 3]. SS patients experience heavy suffering from severe pruritus, desquamation and accompanying infectious complications [4]. The etiology is unknown, and genetic, epigenetic, and environmental factors have all been implicated in CTCL. Notably, a recent study on CTCL in a Danish cohort of twins showed no evidence for heredity playing a role in CTCL [5]. In parallel, an increasing body of evidence suggests that environmental factors may have both an etiological and a pathogenic role in CTCL [6–9]. Indeed, eradication of skin colonization by staphylococcus aureus through aggressive treatment with antibiotics is associated with a decrease in the fraction of malignant T cells in skin lesions [10] and staphylococcal alpha-toxin induces an increased ration between malignant and non-malignant T cells [11]. Importantly, the pathogenesis of CTCL including SS is closely associated with chronic inflammation and aberrant activation of nuclear factor-κB (NF-κB) pathway, nuclear factor of activated T cells (NFAT) pathway, and Janus kinase / signal transducer of activation (Jak/STAT) pathway in lesional skin and blood [12–15]. Inflammatory macrophages as well as dendritic cells are abundant in the CTCL tumor microenvironment [16, 17]. Recent genomic studies have reported activating mutations in genes such as PLCG1 promoting Ca2+/Calcineurin-NFAT signaling, leading to malignant proliferation in a fraction of CTCL patients [18]. However, the overall low recurrence of pathogenic small-scale mutations suggests that these are not the primary drivers of the disease. Notably, a recent study of single-cell heterogeneity showed a high degree of heterogeneity when comparing malignant T cells in different SS patients and even a considerable heterogeneity within the malignant T cell population in each individual patient [19, 20] suggesting that genetic instability may play a key role in CTCL. MicroRNAs are also important in the pathogenesis of CTCL, for some MicroRNAs (e.g., miR-21, miR-22, miR-155) have been reported to target cell-cycle regulators, signaling molecules and tumor suppressors, leading to enhanced proliferation, cytokine release, and resistance to apoptosis in malignant T cells [21–25]. Interestingly, loss of regulatory control through deletion, epigenetic silencing, or functional deviation of Jak/STAT signaling inhibitors such as SOCS1, SOCS3, SHP-1, and HNRNPK also appears to be implicated in aberrant cytokine signaling [26–30] indicating that a dynamic, multifactorial interplay between genetic, epigenetic and environmental triggers drive disease progression in SS and other variants of CTCL.
Current therapeutic strategies for SS involve multidisciplinary combinations of skin directed therapies and systemic chemotherapy, as well as immune-modulators (e.g., Interferon α, Bexarotene) and histone deacetylase inhibitors (HDACi, e.g., Vorinostat, Romidepsin). In recent years targeted therapies are emerging and a number of monoclonal antibodies have shown various efficacy in SS [31]. For instance, Mogamulizumab targeting C-C chemokine receptor 4 (CCR4) has been approved in 2018 for clinical treatment of SS [32, 33]. Alemtuzumab against CD52 gained considerable efficacy [34, 35], and Pembrolizumab against programmed cell death protein-1 (PD-1) showed activity among SS patients in clinical trials [36, 37]. Brentuximab vedotin as an antibody-drug conjugate targeting CD30 also demonstrated encouraging results in CTCL phase II/III trials [38, 39]. In addition, a newly developed oligonucleotide inhibitor of miR-155 named Cobomarsen is being evaluated in a phase II trial for CTCL [40]. However, despite all these therapeutic options, the prognosis of SS remains poor, and novel therapeutics are highly needed [4, 41].
The Kv1.3 ion channel was discovered on T lymphocytes over three decades ago [42]. It is a voltage-gated potassium channel residing in the plasma membrane that plays an important role in T cell activation by regulating membrane potential and calcium signaling. Kv1.3 channel-mediated K+ efflux maintains a negative membrane potential during T cell activation providing a driving force for calcium influx and a high concentration of cytoplasmic calcium (~ 1 μM) [43, 44]. The elevated cytosolic calcium following T cell receptor stimulation activates the phosphatase calcineurin that in turn dephosphorylates NFAT. Once dephosphorylated, NFAT translocates to the nucleus and initiates gene transcription, cytokine secretion, and T cell proliferation [45, 46]. Blockade of Kv1.3 influences Ca2+ homeostasis and inhibits T cell activation and proliferation. Kv1.3 channels have therefore been proposed to be a promising therapeutic target [45, 47]. In past years, Kv1.3 has been intensively investigated which has provided a series of encouraging results. For instance, in many autoimmune diseases, disease-associated autoreactive cells are found having high expression of Kv1.3 protein in the plasma membrane including multiple sclerosis [48], rheumatoid arthritis [49], psoriasis [50], and type-1 diabetes mellitus [49]. Moreover, selective Kv1.3 inhibitors have shown efficacy in preclinical animal models [48–50], and one inhibitor, Dalazatide, is now in clinical trials for psoriasis [51], suggesting that Kv1.3 may indeed be a promising therapeutic target in chronically activated T-cell disorders.
Malignant T cells in SS derive from chronically activated T memory cells [52]. Yet, it has to date not been investigated if malignant SS cells express Kv1.3. In this study, we explored the expression and possible functions of Kv1.3 in SS. We demonstrate that Kv1.3 is expressed on primary malignant cells of SS patients, and that Kv1.3 blockade inhibits malignant cell proliferation and cytokine signaling, without inducing apoptosis.
Results
Kv1.3 is expressed by ex vivo isolated malignant cells from SS patients
Kv1.3 has been reported to be highly expressed in many chronically activated T-cell diseases [48–50]. Yet, no studies have investigated the expression of Kv1.3 in SS. Accordingly we obtained peripheral blood mononuclear cells (PBMCs) from six SS patients and analyzed Kv1.3 expression using flow cytometry. Malignant cells were defined as CD26- within the CD3+CD4+ T cell population (Figure 1A). As shown in Figure 1B, Kv1.3 protein was expressed on the cell surface of malignant T cells from all six SS patients, although at varying levels.
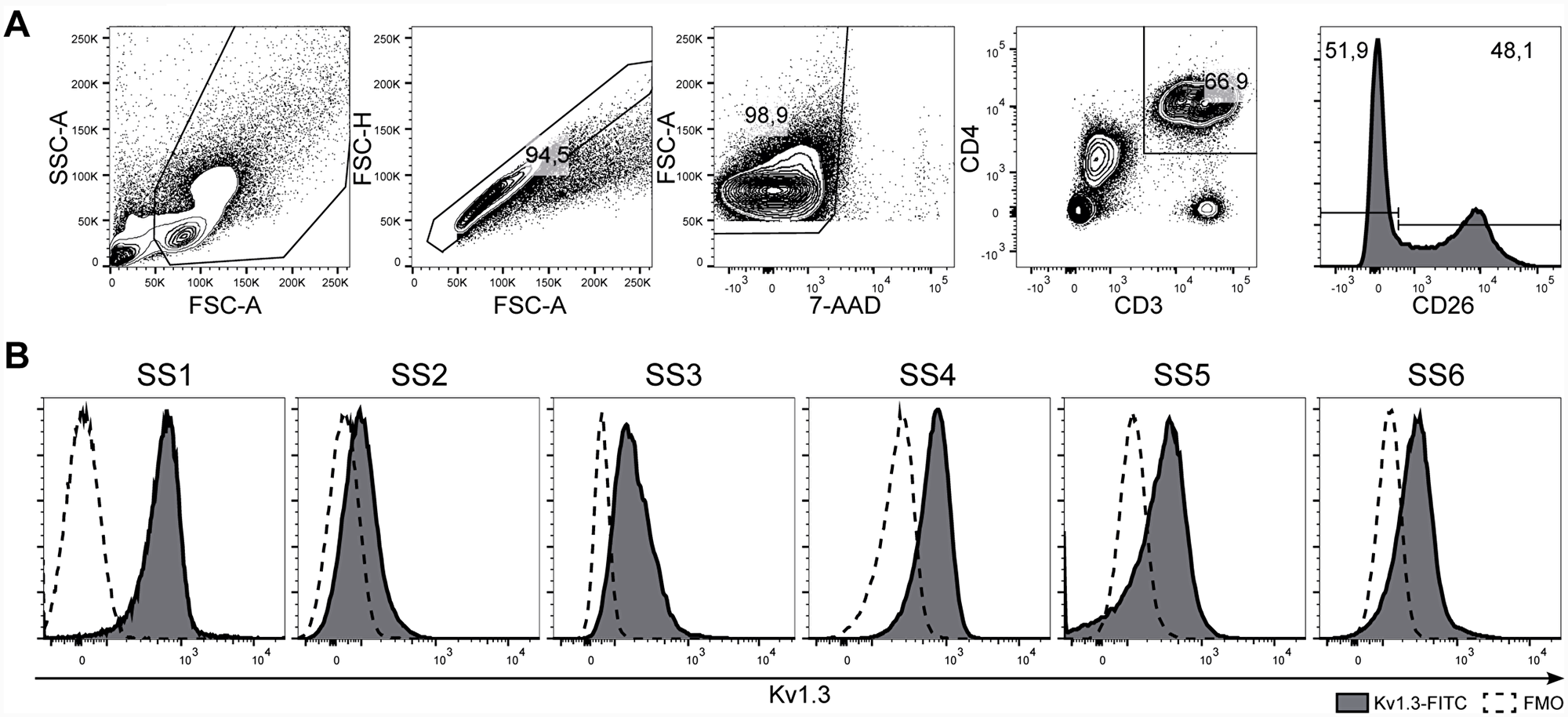
Figure 1: Kv1.3 is expressed in malignant cells from SS patients. PBMCs from six SS patients (SS1–SS6) were stained with anti-Kv1.3, CD3, CD4, CD26 antibodies, and 7-AAD and analyzed by flow cytometry. (A) Representative gating strategy for the identification of malignant T cells (CD3+CD4+CD26–). (B) Histograms of Kv1.3 protein expression on the malignant T cell population.
Kv1.3 blockade inhibits activation-induced proliferation of malignant T cells from SS patients
Since Kv1.3 channels showed to be expressed by malignant T cells from SS patients, we next aimed to determine whether Kv1.3 augments cell proliferation. We used three well characterized Kv1.3 inhibitors with varied potency: two potent ones called ShK and Vm24, and a 150 times less potent one called [N17A/F32T]-AnTx, see Supplementary Table 1 [53–55]. We treated PBMCs from SS patients with 10-fold increasing concentrations of ShK (0, 10, 100 and 1000 nM), with or without anti-CD3/CD28 beads for 72 hours and measured the incorporation of 3H-thymidine during the last 24 hours of culture. While no 3H-thymidine incorporation was observed in the absence of bead stimulation, the 3H-thymidine incorporation of SS cells stimulated with anti-CD3/CD28 beads was inhibited in a dose-dependent manner by the potent Kv1.3 inhibitors, ShK and Vm24, suggesting that blockade of Kv1.3 inhibits the proliferation of SS cells (Figure 2). Likewise, ShK inhibited activation induced proliferation in another three patients (Figure 3, left), whereas [N17A/F32T]-AnTx had little effect (Figure 3).
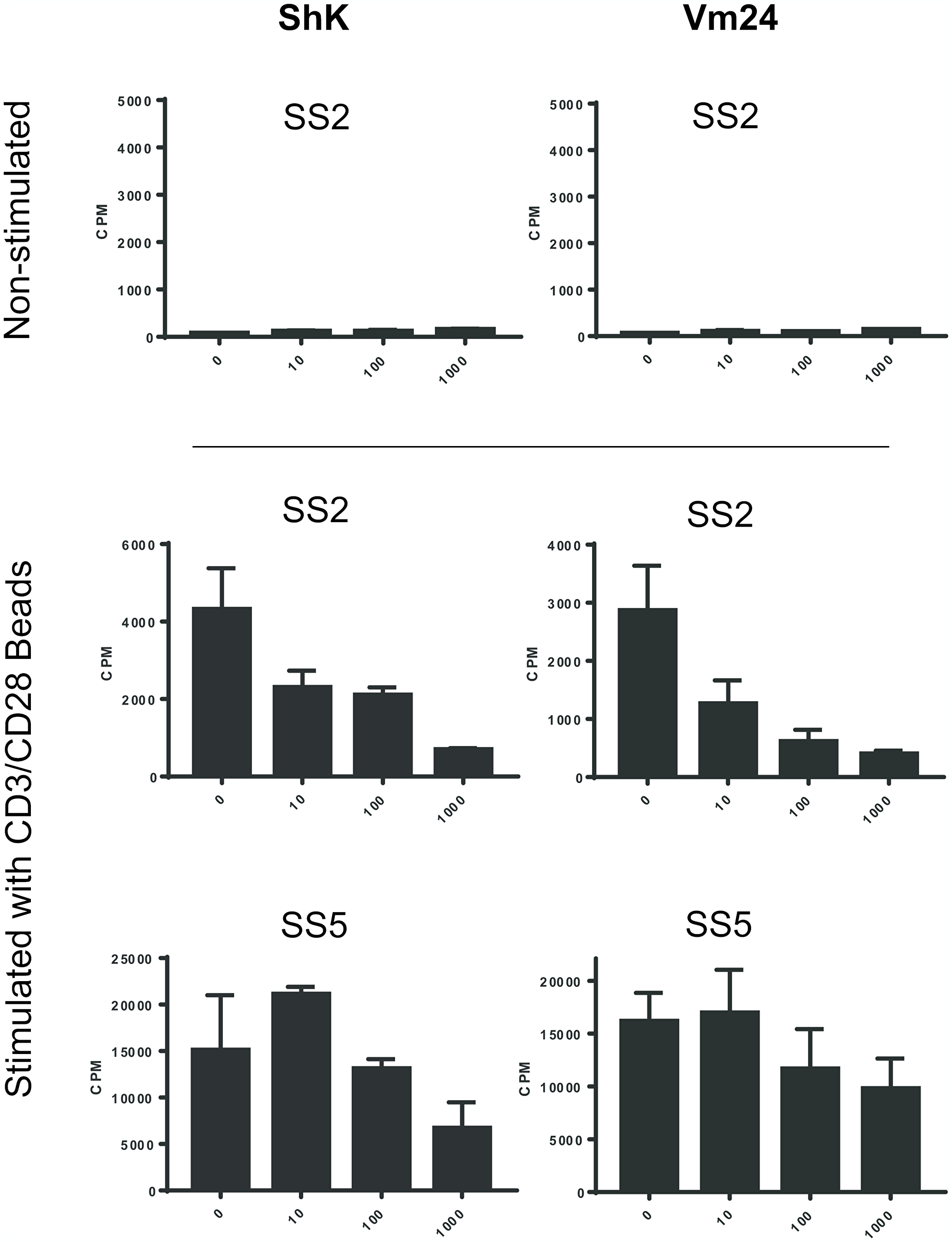
Figure 2: Kv1.3 inhibitors ShK and Vm24 inhibit activation-induced proliferation of PBMCs from SS patients. PBMCs from two SS patients were incubated with Kv1.3 inhibitors, ShK and Vm24, in 10-fold increasing concentrations (0, 10, 100 and 1000 nM) for 72 hours, with or without anti-CD3/CD28 beads. 3H-thymidine incorporation of the last 24 hours was measured as counts per minute (CPM). Data of two experiments with bead stimulation and one representative experiment without bead stimulation are shown. Error bars represent standard deviation (SD) of 3 technical replicates.
![Kv1.3 inhibitor ShK inhibits activation-induced proliferation of PBMCs from SS patients, whereas [N17A/F32T]-AnTx shows little inhibition.](https://oncotarget-cdn.atl1.digitaloceanspaces.com/article/27122/figure/1034694/27122-1034694-1-PB.png)
Figure 3: Kv1.3 inhibitor ShK inhibits activation-induced proliferation of PBMCs from SS patients, whereas [N17A/F32T]-AnTx shows little inhibition. PBMCs from three SS patients were incubated with Kv1.3 inhibitors, ShK and [N17A/F32T]-AnTx, in 10-fold increasing concentrations (0, 10, 100 and 1000 nM) for 72 hours, with or without anti-CD3/CD28 beads. 3H-thymidine incorporation of the last 24 hours was measured as counts per minute (CPM). Data of three experiments with bead stimulation and one representative experiment without bead stimulation are shown. Error bars represent standard deviation (SD) of 3 technical replicates.
To elucidate whether the reduction of 3H-thymidine incorporation in SS cells was caused by increased cell death, we analyzed the proportion of apoptotic and dead cells in response to ShK treatment by flow cytometry. PBMCs from three SS patients were treated with ShK and stained with Annexin V and propidium iodide (PI). As shown in Figure 4A and 4B, we observed high viability (Annexin V- and PI-) and low apoptosis (Annexin V+) in unstimulated malignant T cells from all three patients. Following anti-CD3/CD28 stimulation, some degree of activation-induced cell death (AICD) was observed in one patient (SS8) (Figure 4B, lower part), whereas little AICD was observed in the other two patients (Figure 4B, upper and middle part). The difference between the patients could be explained by the functional heterogeneity among SS patients [19]. Regardless, increasing ShK concentrations did not increase the proportion of apoptotic (Annexin V+) or dead (PI+) cells (Figure 4A and 4B), indicating that ShK-mediated blockade of Kv1.3 does not induce cell death. This finding is in line with previous reports indicating that some Kv1.3 inhibitors, including ShK, do not induce apoptosis [56, 57].
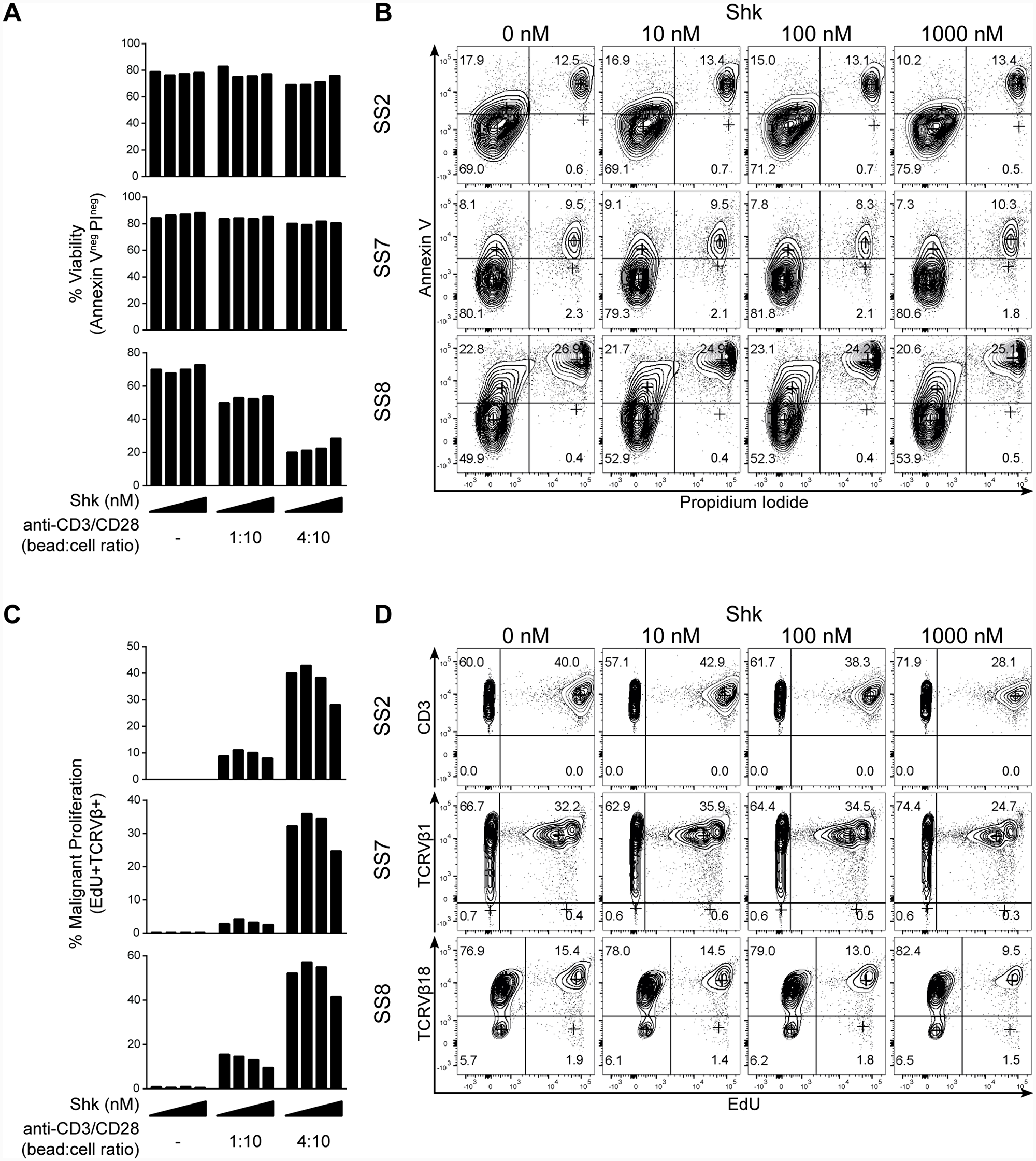
Figure 4: Kv1.3 blockade inhibits activation-induced proliferation of malignant T cells from SS patients. PBMCs from three SS patients were incubated with 10-fold increasing concentrations of ShK (0, 10, 100 and 1000 nM) for 72 hours in the presence or absence of anti-CD3/CD28 beads. EdU was added during the last 24 hours of culture. Anti-CD3, CD4, TCRVβ, EdU, Annexin V and PI were stained followed by flow cytometric analysis. Quantification of viability and representative flow cytometric plots are shown in (A) and (B). Quantification of malignant proliferation and representative flow cytometric plots are shown in (C) and (D).
To confirm that ShK inhibited the proliferation of malignant T cells per se, we further analyzed the proliferative response at the single-cell level by measuring EdU (5-ethynyl-2′-deoxyuridine) incorporation using flow cytometry during the last 24 hours of 72 hour long ShK treatment. We found that, in the presence of anti-CD3/CD28 stimulation, the population of malignant T cells that had proliferated (marked by TCRVβ+EdU+) was decreased by ShK in a dose-dependent manner (Figure 4C and 4D). For patient SS2, CD3 was used as a surrogate of TCRVβ8 in the EdU staining and the feasibility was confirmed in a different staining showing that 99% of CD3+CD4+ cells were TCRVβ8+ (see Figure 5B). Collectively, these findings strongly suggest that Kv1.3 blockade inhibits the proliferation of malignant T cells in SS patients.
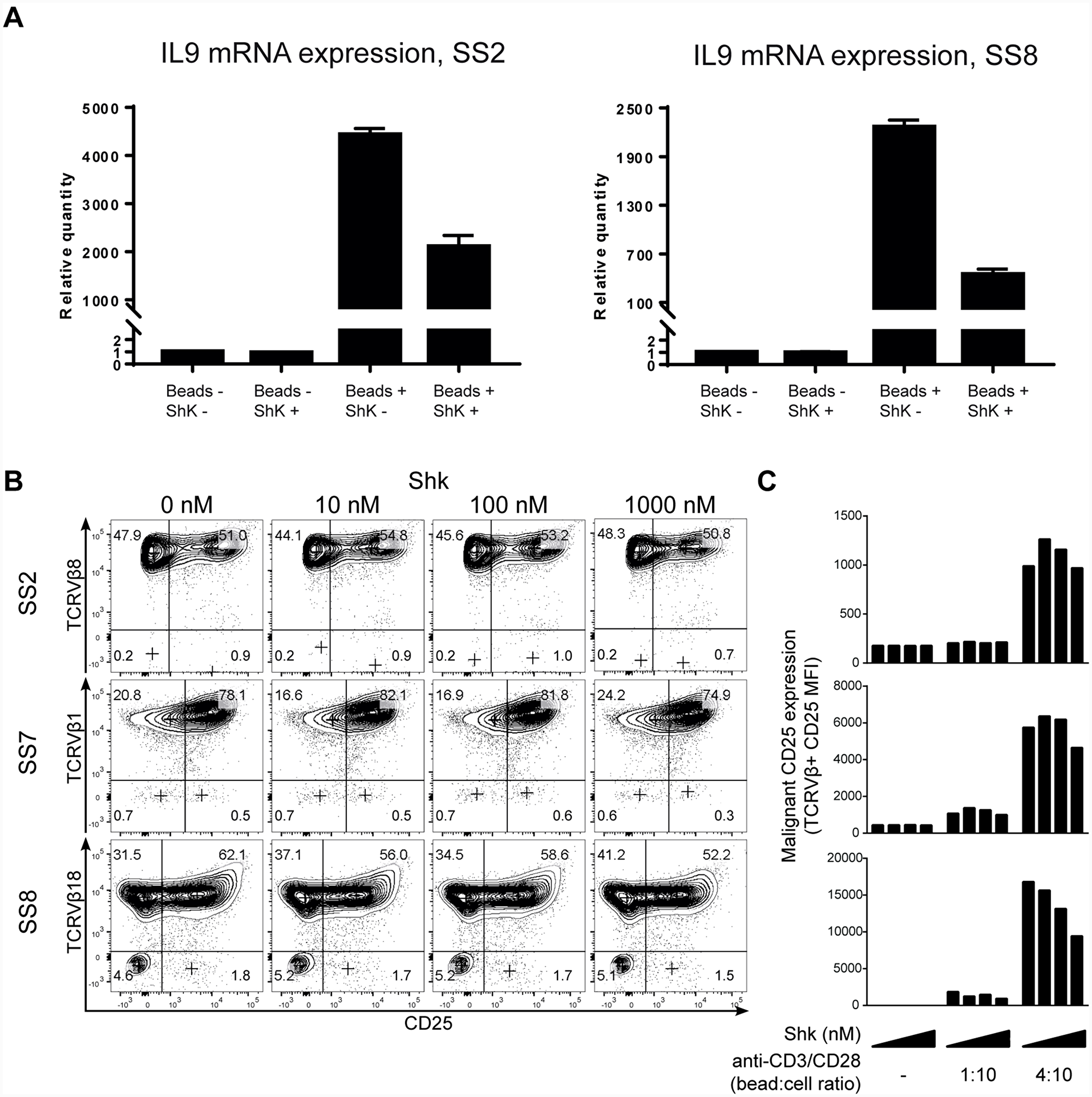
Figure 5: Kv1.3 blockade inhibits activation-induced cytokine and cytokine receptor expression in malignant T cells from SS patients. (A) PBMCs from two SS patients were treated with 1 µM ShK in the presence or absence of anti-CD3/CD28 beads for 72 hours. IL-9 mRNA expression was analyzed by quantitative PCR, with GAPDH as normalization control. Error bars represent standard deviation (SD) of 3 technical replicates. (B and C) PBMCs from three SS patients were incubated with different concentrations of ShK (0, 10, 100 and 1000 nM) for 72 hours in the presence or absence of anti-CD3/CD28 beads. Anti-CD3, CD4, CD25 and TCRVβ were stained followed by flow cytometric analysis. Representative plots of CD25 expression (B) and mean fluorescence intensity (MFI) of CD25 in malignant populations (C) are shown.
Kv1.3 blockade inhibits activation-induced cytokine and cytokine receptor expression in malignant T cells from SS patients
As the T cell receptor (TCR)-mediated increase of intracellular free calcium is a key trigger of NFAT activation and expression of growth factors such as cytokines in healthy T cells [43, 44], we next addressed whether Kv1.3 blockade inhibited TCR-induced cytokine expression. Cytokines have been implicated in the pathogenesis of SS (reviewed in Ref. [15]) and recently, interleukin 9 (IL-9) was shown to promote T cell proliferation in healthy and malignant T cells [58]. To determine if Kv1.3 inhibition altered the cytokine expression in SS cells, we next determined the expression of IL-9 by quantitative PCR following treatment with or without 1 µM ShK in the presence or absence of anti-CD3/CD28 beads. As shown in Figure 5A, anti-CD3/CD28 stimulation strongly increased the mRNA expression of IL-9, compared to unstimulated cells. Notably, the CD3/CD28-mediated IL-9 induction was significantly inhibited by ShK, indicating that Kv1.3 plays an important role in TCR-mediated cytokine expression in SS T cells. This is consistent with our findings that ShK inhibits TCR-driven proliferation.
The high-affinity IL-2 receptor alpha chain (CD25, IL-2Rα) is a direct target of calcium activated NFAT in T cells and its aberrant expression has been reported in many lymphoid malignancies [59–61]. To determine if blocking Kv1.3 affected TCR-induced CD25 expression in malignant T cells, we examined CD25 expression on malignant T cells from three SS patients. Malignant T cells displayed pronounced CD25 expression following stimulation by anti-CD3/CD28 beads, which was partly inhibited by Kv1.3 blockade (Figure 5B and 5C). Thus, ShK induced a dose-dependent decrease of CD25 surface expression.
Taken together, we provide the first evidence that blocking Kv1.3 inhibits activation-induced expression of the cytokine receptor CD25, IL-9, and proliferation of malignant T cells.
DISCUSSION
The present study provides the first evidence that Kv1.3 potassium channels are expressed and play a functional role in SS cells. Primary malignant T cells from investigated SS patients displayed a modest surface expression of Kv1.3. Blockade of Kv1.3 with the potent Kv1.3 inhibitors ShK and Vm24 inhibited TCR-mediated activation and proliferation of malignant T cells from SS patients. Notably, we observed some degree of heterogeneity in Kv1.3 expression among SS patients supporting recent reports on inter-individual heterogeneity in SS [19]. However, despite varied Kv1.3 expression, the TCR-induced proliferation of malignant T cells was suppressed by Kv1.3 blockade in all investigated patients. These findings show that Kv1.3 channels were not only expressed on the surface, but also functional in SS T cells. Interestingly, other studies have shown that malignant cells of B and T cell origin also express functional Kv1.3 channels [62, 63], implying that Kv1.3 expression and function is not lost during neoplastic transformation of B and T cells. On the contrary, Kv1.3 expression may provide a growth advantage to malignant cells. In support of this, Kv1.3 blockade inhibits activation-induced proliferation of malignant T cells (as shown in Figures 2–4), and inhibits cell proliferation in hematological cancers of B cell origin, as shown by others [62, 63].
It is well established that a key event in TCR-mediated activation of T cells involves the release of calcium from internal stores and an influx of extracellular calcium resulting in a steep increase in intracellular free calcium, which activates the protein phosphatase calcineurin to dephosphorylate and activate the transcription factor NFAT. In turn, this triggers nuclear translocation of NFAT and transcription of genes encoding cytokines and their receptors, eventually driving mitogenesis and T cell proliferation [43–46]. The mechanism by which Kv1.3 blockade inhibits cell proliferation has been elucidated in previous studies [44, 45, 48, 64]: Kv1.3 inhibitors including ShK block potassium efflux by attaching to and obstructing the central pore of Kv1.3 channel, which results in a reduced influx of extracellular calcium. Gradually, the reduced calcium influx decelerates calcineurin activation and NFAT signaling leading to a relative decrease in gene transcription, cytokine secretion and cell proliferation. In our study, we found that the Kv1.3 blocker ShK inhibited anti-CD3-CD28 bead-induced expression of the high-affinity IL-2 receptor CD25, transcription of the cytokine and malignant growth factor IL-9 [65], and T cell proliferation. Our data thus support the hypothesis that Kv1.3 channels are expressed and functional in malignant T cells.
While Kv1.3 inhibition has been reported to induce apoptosis in B-CLL cells [62], we did not observe increased apoptosis following Kv1.3 blockade in malignant T cells (Figure 4A and 4B), which is consistent with previous reports showing that ShK does not induce apoptosis in healthy T cells [57].
It is generally believed that SS and other variants of CTCL arise from chronically activated T cells as also reflected by their memory T cell phenotype [52]. However, it is unknown what drives this chronic stimulation of malignant T cells. Both endogenous and exogenous factors are believed to be involved. For instance, gain-of-function mutations affect signaling pathways to drive T cell activation, and the loss of regulatory control due to deletions in SOCS1 or epigenetic silencing of SHP-1, lower the activation threshold and may drive aberrant activation and proliferation [9, 26, 27, 29]. Exogenous factors such as bacteria and their toxins may also trigger or amplify chronic activation of malignant T cells [7]. Notably, malignant T cells induce structural changes in the skin, which compromises skin barrier function [66] and possibly paves the way for bacterial colonization of lesional skin by toxin producing Staphylococcus aureus, which is a characteristic feature of SS [67]. Moreover, Staphylococcal toxins induce activation of proto-oncogenes such as STAT3 and a release of inflammatory and immune regulatory cytokines [7]. Thus, it is likely that multiple factors – endogenous and exogenous – act in concert to induce proliferation of malignant T cells. In this context, Kv1.3 could be a potential target for therapy. Due to the moderate inhibitory effect of ShK on activation and proliferation of malignant T cells, Kv1.3 inhibitors may, at first glance, not appear as ideal therapeutic candidates for mono-therapy of SS. However, as SS patients notoriously develop drug resistance, it is feasible that Kv1.3 inhibition might have a role in combination-therapy together with other drugs targeting different disease mechanisms such as resistance to apoptosis. HDAC inhibitors (HDACi), such as vorinostat and romidepsin, induce apoptosis in SS cells [68, 69], but they may not be able to completely eliminate all malignant subpopulations in these patients [19], potentially leading to cancer recurrence and treatment resistance. Thus, it is likely that multiple drugs directed against different targets are needed to obtain high treatment efficacy in each individual patient. As such, Kv1.3 blockers might have a role in a personalized treatment approach to SS. Accordingly, studies are in progress to investigate the effect on primary SS cells ex vivo by ShK in combination with approved treatments such as HDACi, retinoids, and interferon-α.
In conclusion, the present study provides first evidence that malignant T cells from SS patients express functional Kv1.3 channels, and that Kv1.3 blockade inhibits activation induced CD25 and IL-9 expression and proliferation of SS cells, suggesting that Kv1.3 is a potential therapeutic target in SS.
Materials and Methods
Patient cells
In accordance with the Declaration of Helsinki, the samples were obtained with written informed consent from all patients after approval by the Committee on Health Research Ethics. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of Sézary syndrome (SS) patients by density-gradient centrifugation, using LymphoPrep and Sepmate-50 tubes (Stem Cell Technologies, Cat# 07851 and Cat# 85460, Cambridge, UK). Malignant SS T cells typically lack the expression of cell surface marker CD26 and express a common TCRVβ chain [1]. Accordingly, T cells were identified as malignant (TCRVβ+ or CD26–) and non-malignant (TCRVβ- or CD26+).
Kv1.3 inhibitors
ShK (a toxin from sea anemone Stichodactyla helianthus), Vm24 (a toxin from scorpion Vaejovis mexicanus smithi) and [N17A/F32T]-AnTx (a mutant toxin from scorpion Anuroctonus phaiodactylus) were synthesized and tested by Zealand Pharma A/S (Copenhagen, Denmark). A CHO cell line stably expressing human Kv1.3 channel (hKv1.3) was purchased from Perkin Elmer. hKv1.3 activities in different concentrations of Kv1.3 inhibitors were measured using the FluxOR Potassium Ion Channel Assay from Invitrogen as described by the manufacturer. The flux of thallium ions into the cells as a response to Kv1.3 activation was quantified using a Fluorometric Imaging Plate Reader (FLIPR®) instrument from Molecular Devices, shown in Supplementary Table 1.
[Methyl-3H]-thymidine proliferation assay
Assays were performed in RPMI-1640 medium containing 2 mM L-glutamine, 0.5 units/mL penicillin, 0.1 mg/mL streptomycin (Sigma-Aldrich, Darmstadt, Germany) and 10% human serum (Bloodbank, Rigshospitalet, Denmark).
25,000 PBMCs were seeded per well in 96-well round bottom plates in different concentrations of Kv1.3 inhibitors, stimulated or non-stimulated with anti-CD3/CD28 beads (Cat# 11132D; Thermo Fisher) and incubated for 72 hours. 3H-thymidine (6.7 Ci; Perkin Elmer, Skovlunde, Denmark) was added 24 hours before cells were harvested onto glass fiber filter plates (Uni-Filter-96 GF/C; Perkin Elmer) using FilterMate cell harvester (Unifilter-96; Perkin Elmer). Filter plates were dried over night at room temperature, and scintillation fluid was added (MicroScint-O; Perkin Elmer), then 3H-thymidine incorporation was measured as counts per minute (CPM) in a scintillation counter (TopCount NXT; Parkard, Meriden, CT, USA).
Flow cytometry
Antibodies against CD3, CD4, CD25, CD26, TCRVβ1, TCRVβ8 and TCRVβ18 as well as Annexin V were purchased from BD Biosciences, BioLegend, Miltenyi Biotec or Beckman Coulter. Dead cells were excluded using propidium iodide (PI). FITC conjugated Kv1.3 antibody was purchased from Sigma-Aldrich (Cat# P4247-50UL).
Single cell suspension was assured by filtering cells through a 100 µm cell strainer. For all stainings, antibodies were diluted in BD Brilliant Stain Buffer (Cat# 563794; BD Biosciences) and stained for 30 minutes at room temperature protected from light. For Annexin V staining, surface stained cells were resuspended in 50 µl Annexin V binding buffer (Cat# 556454; BD Biosciences) containing Annexin V and PI and incubated at room temperature for 15 minutes protected from light followed by addition of 150 µl Annexin V binding buffer and analyzed immediately. EdU (5-ethynyl-2′-deoxyuridine) incorporation was analyzed using the Click-iT® Plus EdU Alexa Fluor® 488 Flow Cytometry Assay Kit (Cat# C10632; Thermo Fisher) according to manufacturer’s instructions. PBMCs were seeded 100,000 cells per well in 96-well round-bottom plates in different concentrations of ShK, with or without anti-CD3/CD28 bead stimulation, and incubated for 72 hours. EdU was added to the wells 24 hours before cells were harvested.
Flow cytometric analysis was conducted on a 5 laser BD LSR-Fortessa at the Core Facility for Flow Cytometry at the University of Copenhagen. Data were analyzed using the FlowJo software (Treestar, Ashland, OR, USA).
RNA purification and quantitative PCR
Total RNA was purified with miRNeasy Mini Kit (Cat# 217004; Qiagen), and cDNA was transcribed from 450 ng RNA using High-Capacity cDNA Reverse Transcription Kit (Cat# 4368813; Applied Biosystems). All samples were DNAse treated before cDNA synthesis using DNase 1 (Cat# AMPD1-1KT; Sigma-Aldrich). Quantitative PCR (qPCR) was performed using Taqman Gene Expression assay probes/primers (Thermo Fisher) and LightCycler 480 probes master mix. Amplification was performed using a LightCycler 480 II (Roche, Hvidovre, Denmark). mRNA expression is presented as relative quantity to the control determined by ddCt method. IL-9 (Cat# Hs00174125-m1) and GAPDH (Cat# Hs02786624-g1) probes were purchased from Thermo Fisher.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research was funded by LEO Foundation, The Danish Cancer Society (Kræftens Bekæmpelse), the Fight Cancer Program (Knæk Cancer), Novo Nordisk Research Foundation, Novo Nordic Foundation Tandem Program (grant number NNF14OC0012345), Lundbeck Foundation, The Danish Council for Independent Research (Danmarks Frie Forskningsfond, 2 project grants for N.Ø., a Sapera Aude Talent Grant [DFF-4092-00122] for T.K.), and Dansk Kræftforsknings Fond.
References
1. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005; 105:3768–85. https://doi.org/10.1182/blood-2004-09-3502. [PubMed].
2. Taswell HF, Winkelmann RK. Sézary Syndrome— A Malignant Reticulemic Erythroderma. JAMA. 1961; 177:465–72. https://doi.org/10.1001/jama.1961.03040330001001. [PubMed].
3. Lutzner MA, Emerit I, Durepaire R, Flandrin G, Grupper C, Prunieras M. Cytogenetic, cytophotometric, and ultrastructural study of large cerebriform cells of the Sézary syndrome and description of a small-cell variant. J Natl Cancer Inst. 1973; 50:1145–62. https://doi.org/10.1093/jnci/50.5.1145. [PubMed].
4. Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): Part I. Diagnosis: Clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014; 70:205.e1–205.e16. https://doi.org/10.1016/j.jaad.2013.07.049. [PubMed].
5. Odum N, Lindahl LM, Wod M, Krejsgaard T, Skytthe A, Woetmann A, Iversen L, Christensen K. Investigating heredity in cutaneous T-cell lymphoma in a unique cohort of Danish twins. Blood Cancer J. 2017; 7:e517. https://doi.org/10.1038/bcj.2016.128. [PubMed].
6. Ghazawi FM, Netchiporouk E, Rahme E, Tsang M, Moreau L, Glassman S, Provost N, Gilbert M, Jean SE, Pehr K, Sasseville D, Litvinov IV. Comprehensive analysis of cutaneous T-cell lymphoma (CTCL) incidence and mortality in Canada reveals changing trends and geographic clustering for this malignancy. Cancer. 2017; 123:3550–67. https://doi.org/10.1002/cncr.30758. [PubMed].
7. Willerslev-Olsen A, Krejsgaard T, Lindahl LM, Litvinov IV, Fredholm S, Petersen DL, Nastasi C, Gniadecki R, Mongan NP, Sasseville D, Wasik MA, Bonefeld CM, Geisler C, et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood. 2016; 127:1287–96. https://doi.org/10.1182/blood-2015-08-662353. [PubMed].
8. Krejsgaard T, Willerslev-Olsen A, Lindahl LM, Bonefeld CM, Koralov SB, Geisler C, Wasik MA, Gniadecki R, Kilian M, Iversen L, Woetmann A, Odum N. Staphylococcal enterotoxins stimulate lymphoma-associated immune dysregulation. Blood. 2014; 124:761–70. https://doi.org/10.1182/blood-2014-01-551184. [PubMed].
9. Fanok MH, Sun A, Fogli LK, Narendran V, Eckstein M, Kannan K, Dolgalev I, Lazaris C, Heguy A, Laird ME, Sundrud MS, Liu C, Kutok J, et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2018; 138:1116–25. https://doi.org/10.1016/j.jid.2017.10.028. [PubMed].
10. Lindahl LM, Willerslev-Olsen A, Gjerdrum LMR, Nielsen PR, Blümel E, Rittig AH, Celis P, Herpers B, Becker JC, Stausbøl-Grøn B, Wasik MA, Gluud M, Fredholm S, et al. Antibiotics inhibit tumor and disease activity in cutaneous T cell lymphoma. Blood. 2019 Jun 22. https://doi.org/10.1182/blood.2018888107. [Epub ahead of print]. [PubMed].
11. Blümel E, Willerslev-Olsen A, Gluud M, Lindahl LM, Fredholm S, Nastasi C, Krejsgaard T, Surewaard BGJ, Koralov SB, Hu T, Persson JL, Bonefeld CM, Geisler C, et al. Staphylococcal alpha-toxin tilts the balance between malignant and non-malignant CD4+ T cells in Cutaneous T-cell lymphoma. Oncoimmunology. 2019 Jul 11. https://doi.org/10.1080/2162402x.2019.1641387. [Epub ahead of print].
12. Sors A, Jean-Louis F, Pellet C, Laroche L, Dubertret L, Courtois G, Bachelez H, Michel L. Down-regulating constitutive activation of the NF-κB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood. 2006; 107:2354–63. https://doi.org/10.1182/blood-2005-06-2536. [PubMed].
13. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, Vermeer MH, Rabadan R, Ferrando A, Palomero T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genet. 2015; 47:1465–70. https://doi.org/10.1038/ng.3442. [PubMed].
14. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, Bailey NG, Betz BL, Miranda RN, Porcu P, Byrd JC, Medeiros LJ, Kunkel SL, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in Sézary syndrome. Nat Commun. 2015; 6:8470. https://doi.org/10.1038/ncomms9470. [PubMed].
15. Krejsgaard T, Lindahl LM, Mongan NP, Wasik MA, Litvinov IV, Iversen L, Langhoff E, Woetmann A, Odum N. Malignant inflammation in cutaneous T-cell lymphoma—a hostile takeover. Semin Immunopathol. 2017; 39:269–82. https://doi.org/10.1007/s00281-016-0594-9. [PubMed].
16. Wilcox RA, Wada DA, Ziesmer SC, Elsawa SF, Comfere NI, Dietz AB, Novak AJ, Witzig TE, Feldman AL, Pittelkow MR, Ansell SM. Monocytes promote tumor cell survival in T-cell lymphoproliferative disorders and are impaired in their ability to differentiate into mature dendritic cells. Blood. 2009; 114:2936–44. https://doi.org/10.1182/blood-2009-05-220111. [PubMed].
17. Schlapbach C, Ochsenbein A, Kaelin U, Hassan AS, Hunger RE, Yawalkar N. High numbers of DC-SIGN+ dendritic cells in lesional skin of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2010; 62:995–1004. https://doi.org/10.1016/j.jaad.2009.06.082. [PubMed].
18. Vaqué JP, Gómez-López G, Monsálvez V, Varela I, Martínez N, Pérez C, Domínguez O, Graña O, Rodríguez-Peralto JL, Rodríguez-Pinilla SM, González-Vela C, Rubio-Camarillo M, Martín-Sánchez E, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood. 2014; 123:2034–43. https://doi.org/10.1182/blood-2013-05-504308. [PubMed].
19. Buus TB, Willerslev-Olsen A, Fredholm S, Blümel E, Nastasi C, Gluud M, Hu T, Lindahl LM, Iversen L, Fogh H, Gniadecki R, Litvinov IV, Persson JL, et al. Single-cell heterogeneity in Sézary syndrome. Blood Adv. 2018; 2:2115–26. https://doi.org/10.1182/bloodadvances.2018022608. [PubMed].
20. Litvinov IV, Tetzlaff MT, Thibault P, Gangar P, Moreau L, Watters AK, Netchiporouk E, Pehr K, Prieto VG, Rahme E, Provost N, Gilbert M, Sasseville D, et al. Gene expression analysis in Cutaneous T-Cell Lymphomas (CTCL) highlights disease heterogeneity and potential diagnostic and prognostic indicators. Oncoimmunology. 2017; 6:e1306618. https://doi.org/10.1080/2162402X.2017.1306618. [PubMed].
21. Sandoval J, Díaz-Lagares A, Salgado R, Servitje O, Climent F, Ortiz-Romero PL, Pérez-Ferriols A, Garcia-Muret MP, Estrach T, Garcia M, Nonell L, Esteller M, Pujol RM, et al. MicroRNA Expression Profiling and DNA Methylation Signature for Deregulated MicroRNA in Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2015; 135:1128–37. https://doi.org/10.1038/jid.2014.487. [PubMed].
22. Sibbesen NA, Kopp KL, Litvinov IV, Jønson L, Willerslev-Olsen A, Fredholm S, Petersen DL, Nastasi C, Krejsgaard T, Lindahl LM, Gniadecki R, Mongan NP, Sasseville D, et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget. 2015; 6:20555–69. https://doi.org/10.18632/oncotarget.4111. [PubMed].
23. Lindahl LM, Fredholm S, Joseph C, Nielsen BS, Jønson L, Willerslev-Olsen A, Gluud M, Blümel E, Petersen DL, Sibbesen N, Hu T, Nastasi C, Krejsgaard T, et al. STAT5 induces miR-21 expression in cutaneous T cell lymphoma. Oncotarget. 2016; 7:45730–44. https://doi.org/10.18632/oncotarget.10160. [PubMed].
24. Kohnken R, Wen J, Mundy-Bosse B, McConnell K, Keiter A, Grinshpun L, Hartlage A, Yano M, McNeil B, Chakravarti N, William B, Bradner JE, Caligiuri MA, et al. Diminished microRNA-29b level is associated with BRD4-mediated activation of oncogenes in cutaneous T-cell lymphoma. Blood. 2018; 131:771–81. https://doi.org/10.1182/blood-2017-09-805663. [PubMed].
25. Kopp K, Ralfkiaer U, Gjerdrum LM, Helvad R, Pedersen I, Litman T, Jønson L, Hagedorn P, Krejsgaard T, Gniadecki R, Bonefeld C, Skov L, Geisler C, et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle. 2013; 12:1939–47. https://doi.org/10.4161/cc.24987. [PubMed].
26. Ferrara G, Pancione M, Votino C, Quaglino P, Tomasini C, Santucci M, Pimpinelli N, Cusano F, Sabatino L, Colantuoni V. A specific DNA methylation profile correlates with a high risk of disease progression in stage I classical (Alibert-Bazin type) mycosis fungoides. Br J Dermatol. 2014; 170:1266–75. https://doi.org/10.1111/bjd.12717. [PubMed].
27. Witkiewicz A, Raghunath P, Wasik A, Junkins-Hopkins JM, Jones D, Zhang Q, Odum N, Wasik MA. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma. Hum Pathol. 2007; 38:462–7. https://doi.org/10.1016/j.humpath.2006.09.012. [PubMed].
28. Brender C, Lovato P, Sommer VH, Woetmann A, Mathiesen AM, Geisler C, Wasik M, Ødum N. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNα. Leukemia. 2005; 19:209–13. https://doi.org/10.1038/sj.leu.2403610. [PubMed].
29. Bastidas Torres AN, Cats D, Mei H, Szuhai K, Willemze R, Vermeer MH, Tensen CP. Genomic analysis reveals recurrent deletion of JAK-STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer. 2018; 57:653–64. https://doi.org/10.1002/gcc.22679. [PubMed].
30. Fredholm S, Willerslev-Olsen A, Met Ö, Kubat L, Gluud M, Mathiasen SL, Friese C, Blümel E, Petersen DL, Hu T, Nastasi C, Lindahl LM, Buus TB, et al. SATB1 in Malignant T Cells. J Invest Dermatol. 2018; 138:1805–15. https://doi.org/10.1016/j.jid.2018.03.1526. [PubMed].
31. Welborn M, Duvic M. Antibody-Based Therapies for Cutaneous T-Cell Lymphoma. Am J Clin Dermatol. 2019; 20:115–22. https://doi.org/10.1007/s40257-018-0402-5. [PubMed].
32. Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, Whittaker S, Tokura Y, Vermeer M, Zinzani PL, Sokol L, Morris S, Kim EJ, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018; 19:1192–204. https://doi.org/10.1016/S1470-2045(18)30379-6. [PubMed].
33. Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019; 11:219–38. https://doi.org/10.1080/19420862.2018.1556465. [PubMed].
34. de Masson A, Guitera P, Brice P, Moulonguet I, Mouly F, Bouaziz JD, Battistella M, Madelaine I, Roux J, Ram-Wolff C, Cayuela JM, Bachelez H, Bensussan A, et al. Long-term efficacy and safety of alemtuzumab in advanced primary cutaneous T-cell lymphomas. Br J Dermatol. 2014; 170:720–4. https://doi.org/10.1111/bjd.12690. [PubMed].
35. Querfeld C, Mehta N, Rosen ST, Guitart J, Rademaker A, Gerami P, Kuzel TM. Alemtuzumab for relapsed and refractory erythrodermic cutaneous T-cell lymphoma: a single institution experience from the Robert H. Lurie Comprehensive Cancer Center. Leuk Lymphoma. 2009; 50:1969–76. https://doi.org/10.3109/10428190903216770. [PubMed].
36. Dai J, Almazan T, Kim Y, Khodadoust M. Pembrolizumab in systemic and cutaneous T-cell lymphoma. Ann Lymphoma. 2018; 2:3. https://doi.org/10.21037/aol.2018.03.02.
37. Khodadoust M, Rook AH, Porcu P, Foss FM, Moskowitz AJ, Shustov AR, Shanbhag S, Sokol L, Shine R, Fling SP, Li S, Rabhar Z, Kim J, et al. Pembrolizumab for Treatment of Relapsed/Refractory Mycosis Fungoides and Sezary Syndrome: Clinical Efficacy in a Citn Multicenter Phase 2 Study. Blood. 2016; 128:181–181.
38. Kim YH, Tavallaee M, Sundram U, Salva KA, Wood GS, Li S, Rozati S, Nagpal S, Krathen M, Reddy S, Hoppe RT, Nguyen-Lin A, Weng WK, et al. Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome With Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J Clin Oncol. 2015; 33:3750–8. https://doi.org/10.1200/JCO.2014.60.3969. [PubMed].
39. Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, Zinzani PL, Wolter P, Sanches JA, Ortiz-Romero PL, Akilov OE, Geskin L, Trotman J, et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. The Lancet. 2017; 390:555–66. https://doi.org/10.1016/S0140-6736(17)31266-7. [PubMed].
40. Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, Jackson AL. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br J Haematol. 2018; 183:428–44. https://doi.org/10.1111/bjh.15547. [PubMed].
41. Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017; 92:1085–102. https://doi.org/10.1002/ajh.24876. [PubMed].
42. DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984; 307:465–8. https://doi.org/10.1038/307465a0. [PubMed].
43. Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000; 275:37137–49. https://doi.org/10.1074/jbc.M003941200. [PubMed].
44. Fanger CM, Rauer H, Neben AL, Miller MJ, Rauer H, Wulff H, Rosa JC, Ganellin CR, Chandy KG, Cahalan MD. Calcium-activated potassium channels sustain calcium signaling in T lymphocytes. Selective blockers and manipulated channel expression levels. J Biol Chem. 2001; 276:12249–56. https://doi.org/10.1074/jbc.M011342200. [PubMed].
45. Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004; 25:280–9. https://doi.org/10.1016/j.tips.2004.03.010. [PubMed].
46. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005; 5:472–84. https://doi.org/10.1038/nri1632. [PubMed].
47. Wulff H, Pennington M. Targeting effector memory T-cells with Kv1.3 blockers. Curr Opin Drug Discov Devel. 2007; 10:438–45. [PubMed].
48. Rangaraju S, Chi V, Pennington MW, Chandy KG. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin Ther Targets. 2009; 13:909–24. https://doi.org/10.1517/14728220903018957. [PubMed].
49. Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJ, S Andrews B, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006; 103:17414–9. https://doi.org/10.1073/pnas.0605136103. [PubMed].
50. Kundu-Raychaudhuri S, Chen YJ, Wulff H, Raychaudhuri SP. Kv1.3 in psoriatic disease: PAP-1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis--xenograft model. J Autoimmun. 2014; 55:63–72. https://doi.org/10.1016/j.jaut.2014.07.003. [PubMed].
51. Tarcha EJ, Olsen CM, Probst P, Peckham D, Muñoz-Elías EJ, Kruger JG, Iadonato SP. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS One. 2017; 12:e0180762. https://doi.org/10.1371/journal.pone.0180762. [PubMed].
52. Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sézary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010; 116:767–71. https://doi.org/10.1182/blood-2009-11-251926. [PubMed].
53. Castañeda O, Sotolongo V, Amor AM, Stöcklin R, Anderson AJ, Harvey AL, Engström Å, Wernstedt C, Karlsson E. Characterization of a potassium channel toxin from the Caribbean sea anemone Stichodactyla helianthus. Toxicon. 1995; 33:603–13. https://doi.org/10.1016/0041-0101(95)00013-C. [PubMed].
54. Varga Z, Gurrola-Briones G, Papp F, Rodríguez de la Vega RC, Pedraza-Alva G, Tajhya RB, Gaspar R, Cardenas L, Rosenstein Y, Beeton C, Possani LD, Panyi G. Vm24, a Natural Immunosuppressive Peptide, Potently and Selectively Blocks Kv1.3 Potassium Channels of Human T Cells. Mol Pharmacol. 2012; 82:372–82. https://doi.org/10.1124/mol.112.078006.
55. Bartok A, Fehér K, Bodor A, Rákosi K, Tóth GK, Kövér KE, Panyi G, Varga Z. An engineered scorpion toxin analogue with improved Kv1.3 selectivity displays reduced conformational flexibility. Sci Rep. 2015; 5:18397. https://doi.org/10.1038/srep18397. [PubMed].
56. Leanza L, Trentin L, Becker KA, Frezzato F, Zoratti M, Semenzato G, Gulbins E, Szabo I. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia. 2013; 27:1782–5. https://doi.org/10.1038/leu.2013.56. [PubMed].
57. Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabò I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med. 2012; 4:577–93. https://doi.org/10.1002/emmm.201200235. [PubMed].
58. Vieyra-Garcia PA, Wei T, Naym DG, Fredholm S, Fink-Puches R, Cerroni L, Odum N, O’Malley JT, Gniadecki R, Wolf P. STAT3/5-Dependent IL9 Overexpression Contributes to Neoplastic Cell Survival in Mycosis Fungoides. Clin Cancer Res. 2016; 22:3328–39. https://doi.org/10.1158/1078-0432.CCR-15-1784. [PubMed].
59. Taniguchi T, Minami Y. The IL -2 IL-2 receptor system: A current overview. Cell. 1993; 73:5–8. https://doi.org/10.1016/0092-8674(93)90152-G. [PubMed].
60. Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, Nissen MH, Röpke C, Wasik MA, Odum N. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001; 15:787–93. https://doi.org/10.1038/sj.leu.2402093. [PubMed].
61. Waldmann TA, Goldman CK, Bongiovanni KF, Sharrow SO, Davey MP, Cease KB, Greenberg SJ, Longo DL. Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2. Blood. 1988; 72:1805–16. [PubMed].
62. Szabo I, Trentin L, Trimarco V, Semenzato G, Leanza L. Biophysical Characterization and Expression Analysis of Kv1.3 Potassium Channel in Primary Human Leukemic B Cells. Cell Physiol Biochem. 2015; 37:965–78. https://doi.org/10.1159/000430223. [PubMed].
63. Groessinger E, Weiss L, Chen M, Wulff H, Greil R, Kerschbaum H. Malignant Lymphoblasts in T Cell Acute Lymphoblastic Leukemia Express High Levels of Kv1.3. Biophys J. 2014; 106:551a. https://doi.org/10.1016/j.bpj.2013.11.3069.
64. Zhao Y, Huang J, Yuan X, Peng B, Liu W, Han S, He X. Toxins Targeting the KV1.3 Channel: Potential Immunomodulators for Autoimmune Diseases. Toxins. 2015; 7:1749–64. https://doi.org/10.3390/toxins7051749. [PubMed].
65. Goswami R, Kaplan MH. A Brief History of IL-9. J Immunol. 2011; 186:3283–8. https://doi.org/10.4049/jimmunol.1003049. [PubMed].
66. Thode C, Woetmann A, Wandall HH, Carlsson MC, Qvortrup K, Kauczok CS, Wobser M, Printzlau A, Ødum N, Dabelsteen S. Malignant T Cells Secrete Galectins and Induce Epidermal Hyperproliferation and Disorganized Stratification in a Skin Model of Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2015; 135:238–46. https://doi.org/10.1038/jid.2014.284. [PubMed].
67. Blaizot R, Ouattara E, Fauconneau A, Beylot-Barry M, Pham-Ledard A. Infectious events and associated risk factors in mycosis fungoides/Sézary syndrome: a retrospective cohort study. Br J Dermatol. 2018; 179:1322–8. https://doi.org/10.1111/bjd.17073. [PubMed].
68. Duvic M, Vu J. Vorinostat in cutaneous T-cell lymphoma. Drugs Today (Barc). 2007; 43:585–99. https://doi.org/10.1358/dot.2007.43.9.1112980. [PubMed].
69. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH. Final Results From a Multicenter, International, Pivotal Study of Romidepsin in Refractory Cutaneous T-Cell Lymphoma. J Clin Oncol. 2010; 28:4485–91. https://doi.org/10.1200/JCO.2010.28.9066. [PubMed].