
Introduction
AXL, a member of the mammalian TYRO3, AXL, and MER (TAM) receptor kinase family, is a cell-surface transmembrane receptor that exerts regulated kinase activity through its cytoplasmic domain. It plays important roles in migration, invasion, cell cycle, and drug sensitivity in malignant cells [1–3]. The transforming gene AXL (derived from the Greek word “anexelekto,” which means uncontrolled) was originally isolated from chronic myelogenous leukemia cells, is located on chromosome 19q13.2, and encodes 20 exons [4]. AXL is ubiquitously expressed in various organs and cells and is overexpressed in several human cancers, including lung [5–8], colon [9, 10], esophageal [11, 12], breast [13, 14], astrocytoma-glioblastoma [15], and hematological cancers [16–18]. AXL is therapeutic target in some cancers [8, 17, 19]. GAS6 is the major ligand for TAM receptor tyrosine kinases and, particularly, is the sole ligand for AXL [20]. The binding of GAS6 to its receptors promotes cancer cell proliferation, survival, and migration in vitro [1, 2, 21].
AXL expression is upregulated in certain tumors resistant to molecular targeting [16, 22–25] and chemotherapeutic drugs [25–28], and AXL siRNA- or shRNA-mediated knockdown improved the sensitivity of resistant cells. AXL and MER receptor tyrosine kinases reportedly play key roles in the resistance to multiple anticancer therapies [29]. Brand et al. identified AXL as a key mediator of cetuximab resistance, providing a rationale for the clinical evaluation of AXL-targeting drugs to treat cetuximab-resistant cancers [30]. AXL expression can bypass resistance to targeted agents and specifically, to inhibitors of other RTKs, by maintaining pathway activity via alternative effectors or by inducing the activation of other signaling networks [29]. Increased AXL expression has been correlated with resistance to both chemotherapeutic drugs and targeted agents [31].
Human non-small cell lung cancer (NSCLC), with activating mutations in epidermal growth factor receptor (EGFR), responds very well to treatment with EGFR-targeted tyrosine kinase inhibitors (TKIs), such as erlotinib and gefitinib; however, these responses are reduced by acquired resistance [32]. Several mechanisms underlie the acquired resistance, and among them, the amino acid alteration from threonine790 to methionine790 (T790M) in EGFR and amplification of MET as a secondary genetic alteration are found in more than 50% of resistant tumors [33]. In addition, recent studies have reported that approximately 20% of patients who develop resistance to erlotinib show enhanced AXL expression, suggesting that AXL-mediated signaling may be involved in acquired resistance [5, 7]. Furthermore, it has been recently reported that AXL confers intrinsic resistance to osimertinib, a third-generation EGFR-TKI, and accelerates the emergence of tolerant NSCLC cells [34]. Thus, AXL inhibition may prevent or overcome acquired resistance to EGFR TKIs [5].
In this study, we evaluated the effect of DS-1205b, a novel, specific, small-molecule inhibitor of AXL kinase, on tumor growth and resistance to EGFR TKIs. Our findings strongly suggest that combination treatment with DS-1205b can prolong the therapeutic benefit of EGFR TKIs. Phase I clinical trials of DS-1205c in combination with osimertinib or gefitinib are underway, and information on these studies can be found at https://clinicaltrials.gov/ (ID: NCT03255083; NCT03599518). Both DS-1205b and DS-1205c are sulfate hydrates, with similar stoichiometries.
Results
DS-1205b is a potent and highly selective AXL inhibitor
The chemical structure of DS-1205b is shown in Figure 1A. DS-1205b is a novel compound discovered and synthesized by Daiichi Sankyo Co., Ltd. This report is the first disclosure of the compound structure; experimental details of synthesis are described in the Supplementary Methods.
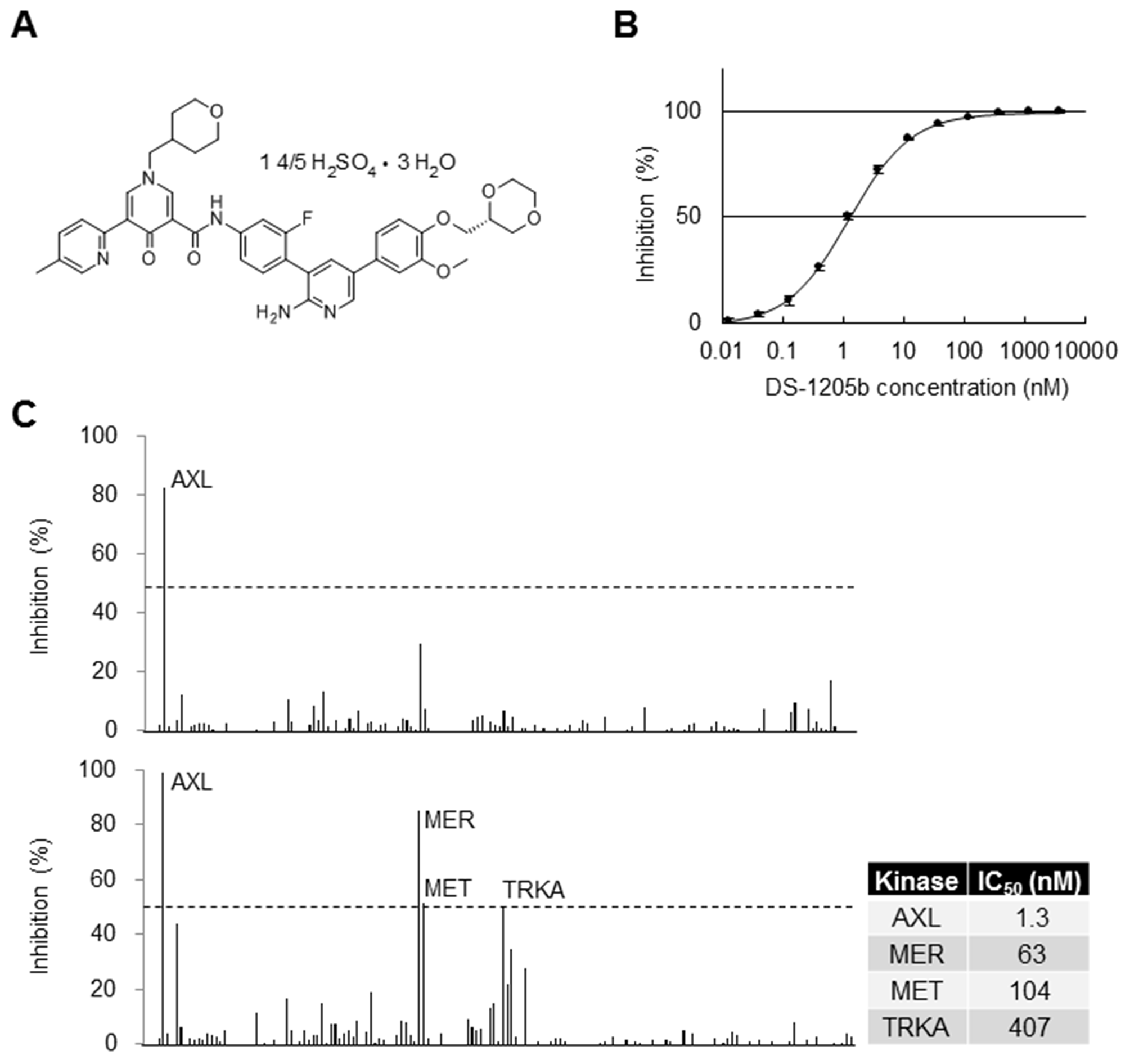
Figure 1: Selectivity of DS-1205b in biochemical assays. (A) Structural formula of DS-1205b. (B) Inhibition curve of DS-1205b against AXL kinase in a mobility shift assay using recombinant AXL protein. The graph shows mean inhibition at each concentration point with the SD (N = 4). (C) Kinase selectivity of DS-1205b was evaluated by mobility shift assay using 161 kinases (Supplementary Table 1), in the presence of 1 mM ATP. Upper panel: 13 nM (approximately IC80), lower panel: 200 nM. The IC50 values of DS-1205b toward AXL, MER, MET, and TRKA kinases were determined by mobility shift assay in the presence of 1 mM ATP using recombinant human AXL, MER, MET, and TRKA proteins.
The AXL inhibition curve of DS-1205b is shown in Figure 1B; the 50% inhibitory concentration (IC50) was 1.3 nM. The kinase selectivity of DS-1205b was examined for 161 kinases by mobility shift assay in the presence of 1 mM ATP, which is a near-physiological condition. None of the kinases were inhibited by more than 30% at 13 nM (the IC80 value for AXL which is potent inhibition concentration; upper panel in Figure 1C), and MER, MET, and TRKA kinases were inhibited by more than 50% at 200 nM DS-1205b (lower panel in Figure 1C). The IC50 values of MER, MET, and TRKA were 48-, 80-, and 313-fold that of AXL, and clear difference was confirmed. Thus, DS-1205b is a highly selective and potent AXL kinase inhibitor. DS-1205b showed potent and highly selective inhibitory activities toward AXL kinase.
In vitro and in vivo activities of DS-1205b in NIH3T3-AXL cells
To clarify the potency of DS-1205b as a selective AXL inhibitor, protein phosphorylation, growth, and migration were assessed in artificially AXL-overexpressing mouse embryonic fibroblast cells NIH3T3 (NIH3T3-AXL cells). BGB324, a forefront runner of AXL kinase inhibitors in clinical stage, was used for comparison [35]. Phosphorylation of AXL was completely inhibited upon 2 h treatment with DS-1205b at concentrations above 10 nM, whereas phosphorylation of AKT serine/threonine kinase, its downstream factor, was slightly inhibited, in a dose-dependent manner (Figure 2A). AXL phosphorylation was notably enhanced by BGB324 at 10–100 nM and inhibited at concentrations above 1,000 nM. After 24 h treatment with 10,000 nM BGB324, there were no signals for AXL and AKT, as most cells were killed. Cell proliferation decreased gradually upon DS-1205b treatment, but growth was not obviously inhibited (GI50: >10,000 nM; Figure 2B). However, the GI50 of BGB324 for AXL was 642 nM in NIH3T3-AXL cells, and cell viability was clearly affected at concentrations above 1,000 nM. The similar results were obtained when AXL ligand hGAS6 was added to examine AXL and the downstream signaling activation (Supplementary Figure 1). We also observed cell morphological changes upon drug treatments. Cytoplasmic vacuoles were observed within 24 h after treatment with 1,000 nM BGB324, whereas treatment with 1,000 nM DS-1205b induced no clear changes (Figure 2C). After 24 h, no viable cells were observed by microscopy (data not shown).
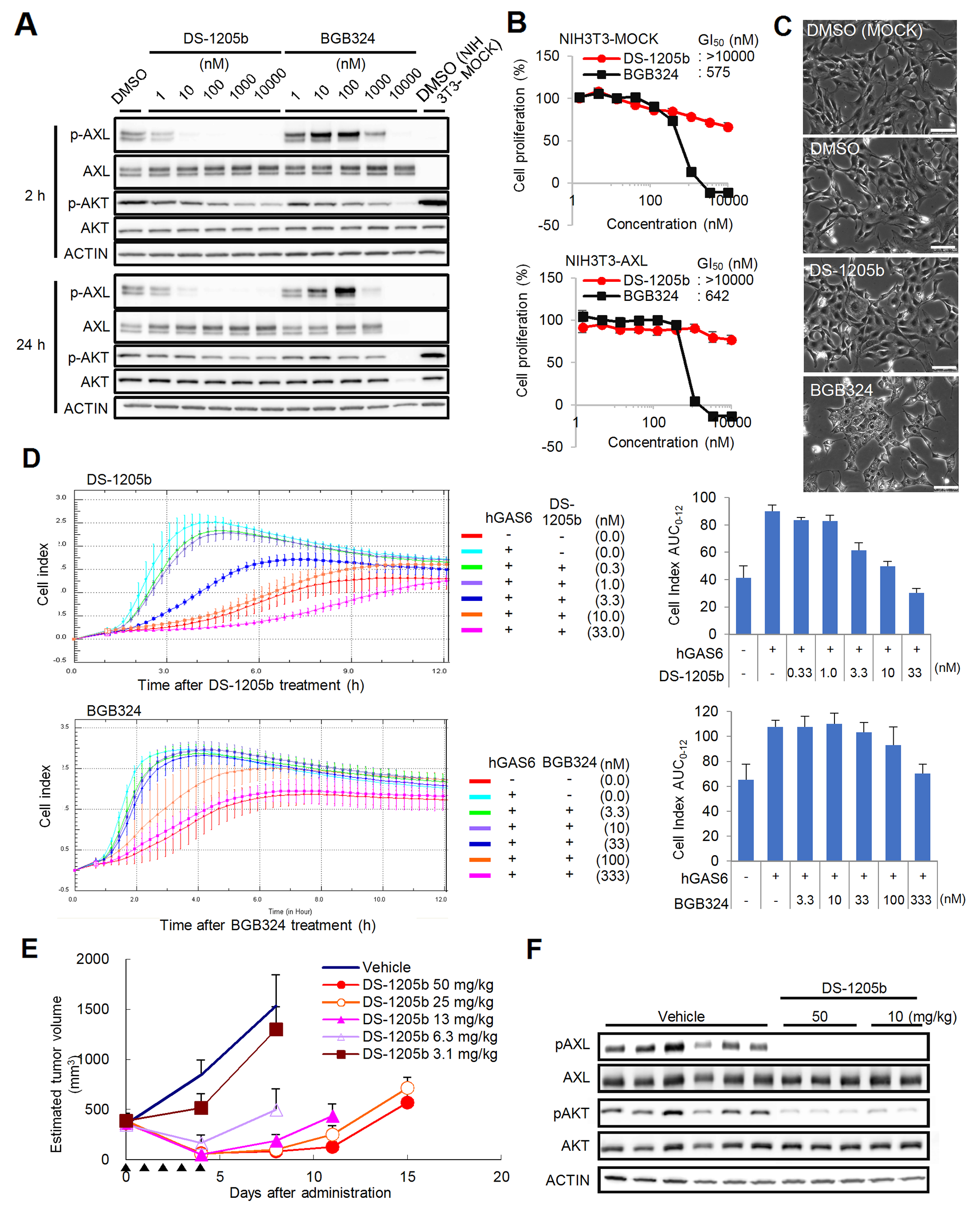
Figure 2: Effects of DS-1205b and/or BGB324 in NIH3T3-AXL cells in vitro and in vivo. (A) In vitro phosphorylation inhibition assay of AXL and AKT. NIH3T3-AXL cells were treated with DS-1205b or BGB324 for 2 h or 24 h and then subjected to western blot analysis. Detailed information on the antibodies used is given in the Materials and Methods. (B) Growth inhibitory activities as assessed by ATP assay. The graph shows mean inhibition at each concentration point with the SD (N = 3). (C) Photographs show cell morphological changes after 24 h of treatment with DMSO, 1 μM DS-1205b, and 1 μM BGB324, respectively. Scale bar represents 100 μm. (D) Inhibitory activities of DS-1205b or BGB324 on hGAS6-induced migration in NIH3T3-AXL cells. The cell index (CI; a dimensionless parameter) was derived as the relative change in measured electrical impedance to represent cell migration. hGAS6: 500 ng/mL. Bars indicate the SD (N = 4). The CI AUC0–12 indicates the sum of each CI for 12 h, and ΔAUC0–12 indicates the difference between the CI AUC0–12 of the DMSO-treated control group and the experimental group. (E) Effects on growth of NIH3T3-AXL cells in vivo. Data are the mean ± SE (N = 5; NOD-SCID). Arrowhead: administration day (po, bid). (F) In vivo phosphorylation inhibition assay of AXL and AKT. Tumor samples taken after 2 h of DS-1205b administration were subjected to western blot analysis. Detailed information on the antibodies used is given in the Materials and Methods.
To evaluate the effects of DS-1205b and BGB324 on NIH3T3-AXL cell migration, a cell index (CI) was calculated as the relative change in measured electrical impedance to represent cell migration. DS-1205b significantly suppressed the CI at 3.3–33 nM, and the half maximal effective concentration (EC50) was 2.7 nM (Figure 2D). BGB324 significantly suppressed the CI at concentrations above 333 nM, and the EC50 was 132.3 nM.
Antitumor effects of DS-1205b were assessed in mice bearing subcutaneously implanted NIH3T3-AXL cells. Tumor growth was inhibited by 39–94% at doses of 3.1–50 mg/kg, and a statistically significant antitumor effect was observed at doses above 6.3 mg/kg (3.1 mg/kg: P < 0.01; 6.3–50 mg/kg: P < 0.001 vs. vehicle-treated control by parametric Dunnett’s test using the data on day 4; Figure 2E). The dose-dependent antitumor effect of DS-1205b on day 4 was confirmed by Spearman’s rank correlation testing (P < 0.0001). In addition, DS-1205b induced tumor regression by 54–86% at doses of 6.3–50 mg/kg. We also examined the effect of DS-1205b on AXL and AKT phosphorylation in tumors. Phosphorylation of both proteins was clearly reduced, suggesting that the antitumor effect of DS-1205b monotherapy in this model is mediated by pAXL inhibition (Figure 2F).
Generation of erlotinib- or osimertinib-resistant cells and AXL expression
We generated HCC827 human NSCLC cells with acquired resistance to erlotinib or osimertinib by treating them for 80 days with 1,000 nM erlotinib or osimertinib, and we examined AXL expression by western blotting to evaluate the potential relation between acquired resistance and AXL upregulation. In parental HCC827 cells, the IC50 values of erlotinib and osimertinib were 11.3 nM and 9.2 nM, respectively, whereas in erlotinib- and osimertinib-resistant HCC827 cells, the IC50 values were 4,278.4 nM and 3,975.9 nM, respectively (Figure 3A). AXL expression was obviously upregulated in both erlotinib- and osimertinib-resistant HCC827 cells, and the phosphorylation was inhibited by DS-1205b treatment in a dose-dependent manner (Figure 3B). DS-1205b treatment tended to suppress AKT phosphorylation in osimertinib-resistant HCC827 cells. It is also important to examine the other putative molecular alterations present in these resistance cells. We had an additional experiment for understanding of potential other alterations using RTK array (Proteome Profiler Human Phospho-RTK Array Kit by R&D, #ARY001B). The reduction of phosphorylation levels of EGFR, ERBB2, ERBB3, MET, and RET, or the increase of INSR and IGF1R were observed in both resistant cells (Supplementary Figure 2). The possibility that dominant pathway on growth and survival of the NSCLC cells shifts from EGFR pathway partially to bypass signal pathways such as AXL by EGFR inhibitor treatment is considered, however, it is not clear.
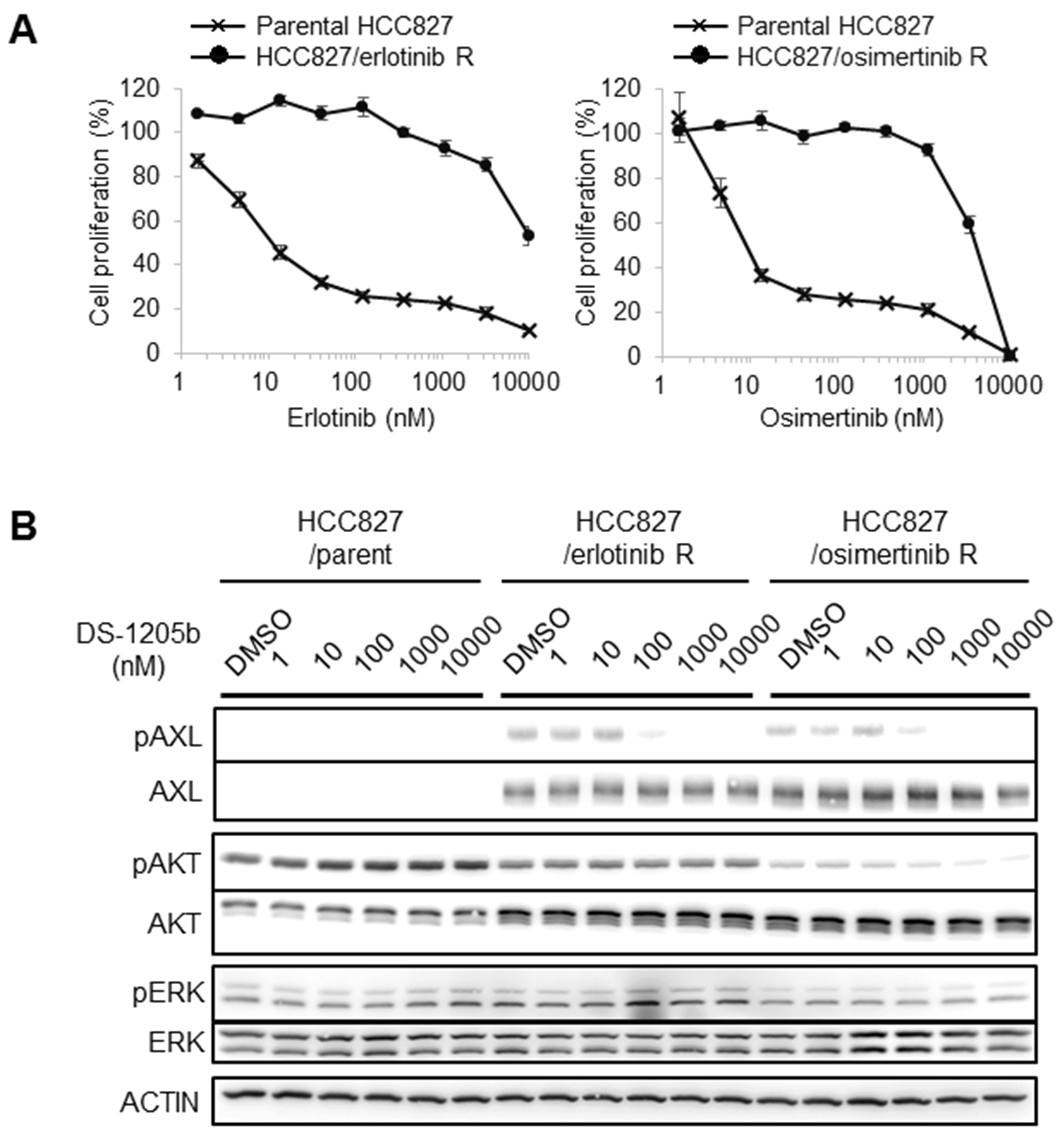
Figure 3: Generation of erlotinib- or osimertinib-resistant cells and AXL expression. (A) HCC827 cells were treated with erlotinib or osimertinib at 1,000 nM to generate acquired resistance. IC50 values were determined on day 80 after starting drug treatments. (B) Parental, erlotinib-, or osimertinib-resistant HCC827 cells were treated with 1, 10, 100, 1,000, or 10,000 nM DS-1205b for 2 h and subjected to western blot analysis on day 52 (for the erlotinib resistant cells) or day 54 (for the osimertinib resistant cells). Detailed information on the antibodies used is given in the Materials and Methods.
Combination treatment with erlotinib or osimertinib and DS-1205b inhibits AXL signaling in vitro
We examined the effects of combined treatment with erlotinib or osimertinib and DS-1205b on EGFR and AXL downstream signaling with the aim to elucidate the mechanism of action of the compounds, using a cell-based assay. As shown in Figure 4, when HCC827 cells were treated with erlotinib or osimertinib at 16, 80, 400, or 2,000 nM, EGFR, AKT, and extracellular signal-regulated kinase (ERK) phosphorylation was reduced. However, erlotinib or osimertinib alone could not completely inhibit phosphorylation, even at 2,000 nM. Cotreatment with erlotinib or osimertinib and DS-1205b significantly inhibited phosphorylation compared to each monotreatment, especially the combination with osimertinib. In particular, AKT phosphorylation was completely inhibited at the lowest doses of osimertinib and DS-1205b (16 nM). The co-treatment seems to be efficient only in osimertinib resistant cells by inhibition of AKT pathway.
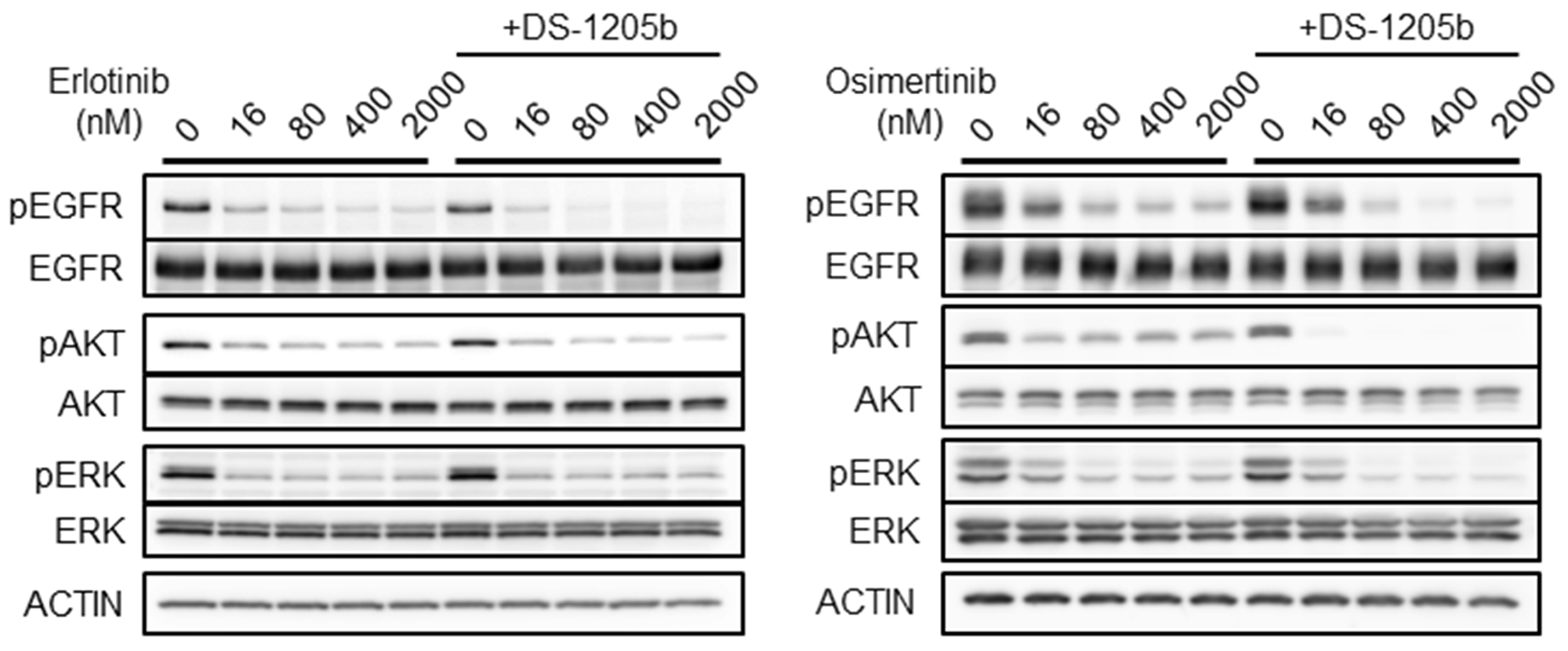
Figure 4: Combination treatment with erlotinib or osimertinib and DS-1205b inhibits AXL signaling in vitro. HCC827 cells were treated with erlotinib or osimertinib at 16, 80, 400, or 2,000 nM. In case of cotreatment, DS-1205b was used at 1 μM in this assay. After 2 h treatment, the HCC cells were collected and evaluated by western blot analysis. Detailed information on the antibodies used is given in the Materials and Methods.
Antitumor effects of DS-1205b in an acquired erlotinib resistance subcutaneous xenograft model
The antitumor effect of DS-1205b in combination with erlotinib was evaluated in nude mice subcutaneously inoculated with HCC827 cells. The mean tumor volumes on day 100 for groups administered erlotinib combined with DS-1205b at doses of 50, 25, or 12.5 mg/kg twice daily (bid) were 216.8, 321.0, and 541.7 mm3, respectively, whereas the mean tumor volume of the erlotinib monotherapy group (erlotinib: 25 mg/kg qd) was 814.5 mm3. Tumor volumes in the erlotinib plus high-dose DS-1205b group on days 59–80, 98, and 100 were significantly (P = 0.0257–0.0469 by parametric Dunnett’s test) lower than those in the erlotinib group (Figure 5A). As DS-1205b showed no antitumor effect, these results suggested that combined treatment with DS-1205b and erlotinib may delay the onset of acquired resistance to erlotinib.
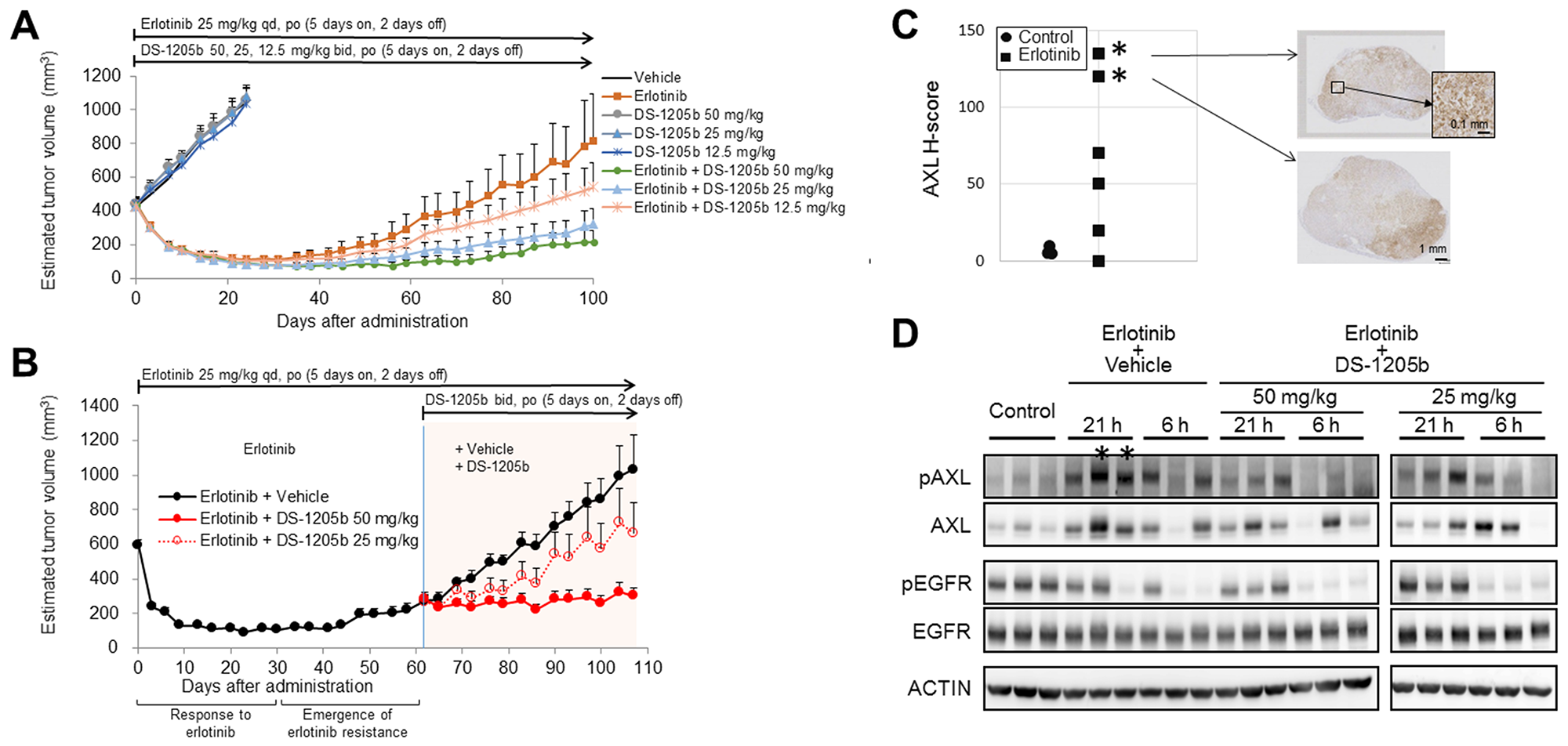
Figure 5: DS-1205b delays erlotinib acquired resistance and restores the treatment effect of erlotinib in an HCC827 sc xenograft model. (A) Addition of DS-1205b to erlotinib delays the onset of drug resistance in a human EGFR-mutant NSCLC xenograft model. Mice with subcutaneous xenograft human HCC827 tumors were treated as indicated. Data are the mean ± SE (N = 8; nude mice). (B) Antitumor activity of erlotinib was restored by addition of DS-1205b in an erlotinib acquired resistance xenograft model. Mice with subcutaneous xenograft human HCC827 tumors were treated as indicated. Data are the mean ± SE (N = 6). (C) AXL immunostaining images and AXL signal intensities and H scores for HCC827 xenograft tumors are shown. Immunohistochemistry of AXL is described in detail in the Materials and Methods. (D) AXL and EGFR expression was analyzed by western blotting in tumor samples obtained 24 days after tumor implantation for the control group or day 108 for the erlotinib-treated group in Figure 5B. “21 h” and “6 h” indicate the time in hours after the last treatment with DS-1205b. *same tumor sources.
Next, we examined the effect of DS-1205b plus erlotinib in HCC827 tumors in vivo after acquired resistance to erlotinib was established. Tumors started to regrow after 48 days of treatment with erlotinib, and DS-1205b treatment was initiated at day 62. DS-1205b inhibited tumor growth by 97% and 47% at doses of 50 and 25 mg/kg, respectively. A statistically significant antitumor effect was observed at each measurement point for DS-1205b at 50 mg/kg (P = 0.0100) (Figure 5B). DS-1205b restored the antitumor activity of erlotinib in a dose-dependent manner, which was confirmed by Spearman’s rank correlation coefficient testing (P = 0.0002). These results suggested that DS-1205b restores the antitumor activity of erlotinib in combination treatment.
Immunostaining for AXL expression and H scores for HCC827 xenograft tumors are shown in Figure 5C. The erlotinib-treated groups had higher H scores than the non-treated group (mean ± standard deviation (SD) for H score, 6.7 ± 2.9 for the non-treated group (control); 65.8 ± 53.7 for erlotinib monotherapy). AXL expression was heterogeneously distributed in the tumor cells, although AXL-expressing and non-AXL-expressing tumors were not morphologically distinct.
AXL expression was analyzed by western blotting using tumors sampled at 24 days after tumor implantation for the control group and at 108 days after administration for the erlotinib-treated group. AXL was markedly upregulated in erlotinib-treated tumors, though expression was not observed in all tumor samples because of the high heterogeneity. AXL phosphorylation was inhibited at 6 h after the last DS-1205b administration (Figure 5D). It was reduced after 21 h of the last administration, however, the antitumor effects were maintained.
Antitumor effects of DS-1205b in an acquired osimertinib resistance subcutaneous xenograft model
Using the same mouse model, delay of HCC827 tumor growth by DS-1205b upon acquired resistance to osimertinib was evaluated. On day 100, the mean tumor volume in the group administered osimertinib monotherapy was 727.5 mm3. The mean tumor volumes of the groups administered osimertinib in combination with DS-1205b at 12.5, 25, or 50 mg/kg were 205.1, 211.3, and 145.8 mm3, respectively. In all groups receiving osimertinib in combination with DS-1205b, tumor volumes were lower than those in the osimertinib monotherapy group. DS-1205b significantly delayed the tumor volume increase and days to reach initial tumor volume in a dose-dependent manner at doses of 12.5–50 mg/kg in the osimertinib acquired resistance model (Figure 6A).
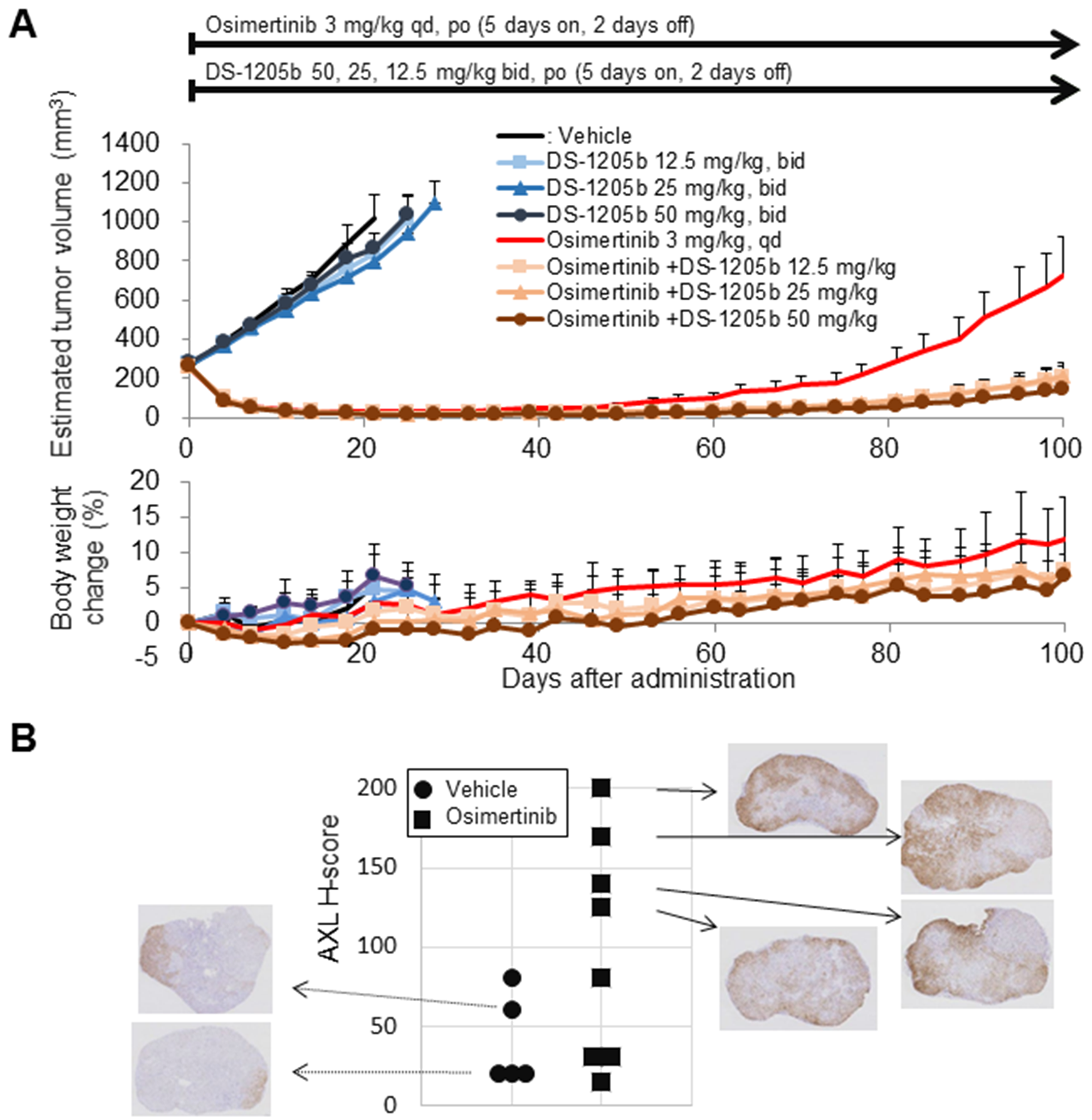
Figure 6: DS-1205b delays osimertinib-acquired resistance in an HCC827 sc xenograft model, and the underlying mechanism. (A) Addition of DS-1205b to osimertinib delays the onset of drug resistance in a human EGFR-mutant NSCLC xenograft model. Mice with subcutaneous xenograft human HCC827 tumors were treated as indicated. For vehicle and DS-1205b monotherapy groups, data are the mean ± SE (N = 5); for osimertinib monotherapy and the combination groups, data are the mean ± SE (N = 8), and body weight change data are the mean ± SD. (B) AXL immunostaining images and H scores for HCC827 xenograft tumors are shown (sampling; day 100). AXL immunostaining images and AXL signal intensities and H scores (see Materials and Methods) for HCC827 xenograft tumors are shown.
Immunostaining for AXL expression and H scores for HCC827 xenograft tumors are shown in Figure 6B (tumors were sampled on day 100). The osimertinib-treated groups had higher H scores for AXL expression than the vehicle control group (mean ± SD for H score, 40.0 ± 28.3 for vehicle control; 98.8 ± 70.2 for osimertinib monotherapy), suggesting that enhanced AXL expression may contribute to acquired resistance to osimertinib, at least in this model. AXL expression was heterogeneously distributed in the tumors.
Our results indicated that AXL upregulation works, at least in part, as a bypass signal of EGFR-TKI acquired resistance in NSCLC cells harboring EGFR mutations. DS-1205b inhibited AXL phosphorylation and consequently led to a delay in drug resistance or combination effects with EGFR TKIs (Figure 7).
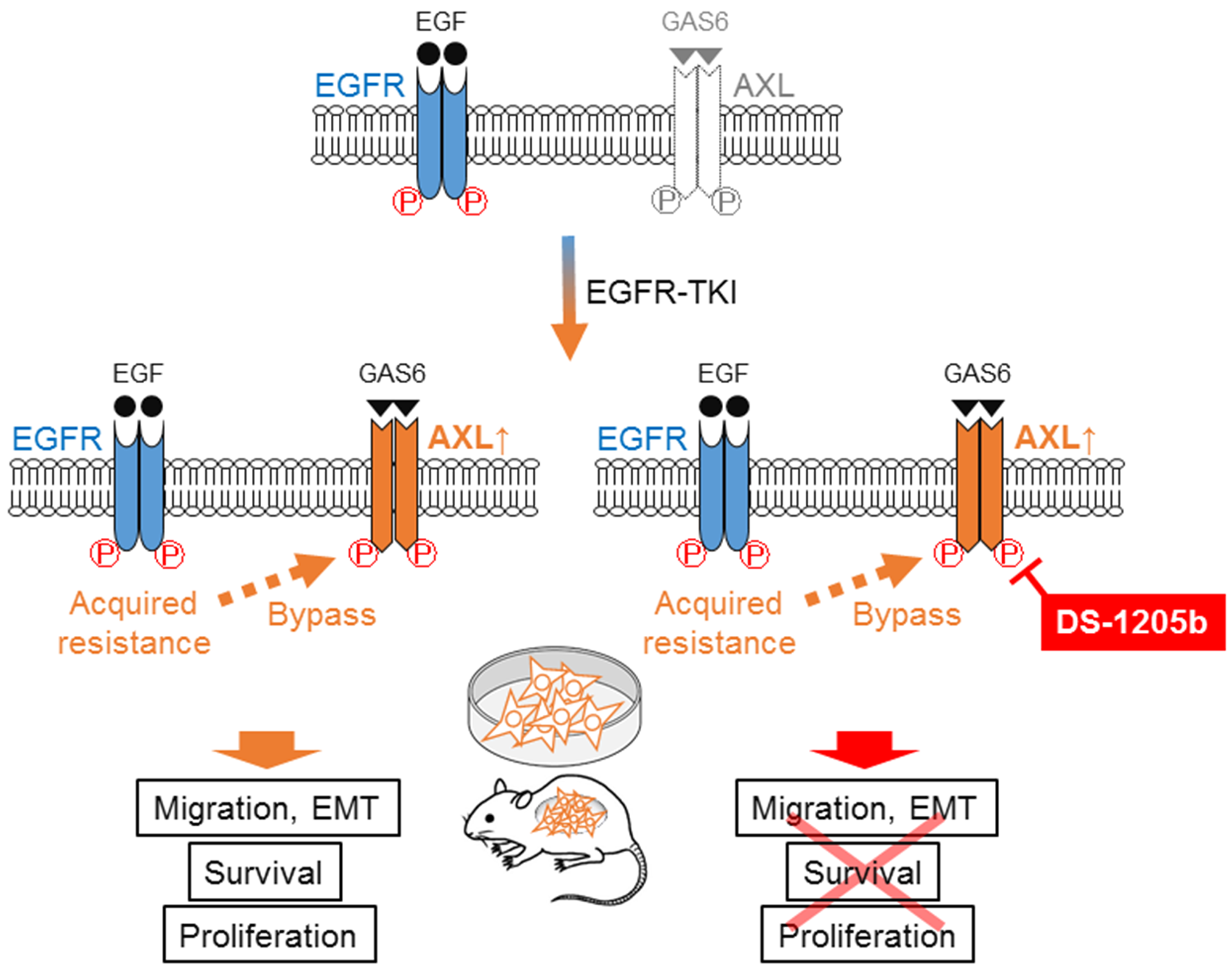
Figure 7: Scheme of mechanism of DS-1205b inhibition. Schematic diagram showing the mechanism of EGFR-TKI acquired resistance because of AXL activation in NSCLC cells harboring EGFR mutations. AXL upregulation works as a bypass signal of EGFR-TKI acquired resistance in EGFR-mutated NSCLC. DS-1205b inhibits AXL phosphorylation, thus delaying drug resistance or inducing combination effects with EGFR-TKI.
DISCUSSION
This study showed that DS-1205b delayed the onset of resistance and overcame acquired resistance to EGFR TKIs in a human EGFR-mutated NSCLC (T790M-negative) xenograft model. In clinic, EGFR TKIs significantly improve the overall survival rates of patients with advanced EGFR-mutant NSCLC. However, despite the high rates of initial response to targeted therapy, patients uniformly develop treatment resistance. Accordingly, TKI resistance has become a major therapeutic challenge in the care of patients with EGFR mutations.
There are several mechanisms of resistance to EGFR TKIs [36], and T790M as a gatekeeper mutation is the most frequent cause of acquired resistance to first-generation EGFR TKIs [37, 38]. While osimertinib conquered resistance induced by this mutation, the C797S mutation, which induces resistance to this drug, has emerged [38, 39]. In addition to acquired mutations in EGFR, several other mechanisms, such as bypass mechanisms in EGFR-mediated signaling [40], epithelial-mesenchymal transition (EMT) [32, 41, 42], pathological transformation to small cell lung cancer [43], immune escape [44], and downstream activation of the RAS or PI3K pathways [31] have been reported. Induced AXL expression may be one of the bypass mechanisms. In the current study, it was demonstrated that after treatment with erlotinib or osimertinib, AXL expression was increased in the HCC827 xenograft. Namba et al. recently reported AXL upregulation in a xenograft model upon long-term osimertinib treatment [45]; however, there is currently no evidence of the involvement of AXL upregulation in acquired resistance in vivo. When erlotinib was administered in combination with DS-1205b, its activity was restored, suggesting that AXL overexpression indeed contributes to resistance to EGFR TKIs. Byers et al. developed a robust EMT signature that predicts resistance to EGFR and PI3K/AKT inhibitors, highlighted different patterns of drug responsiveness in epithelial and mesenchymal cells, and identified AXL as a potential therapeutic target for overcoming EGFR TKI resistance associated with the mesenchymal phenotype [32, 42]. The current study showed that DS-1205b in combination with osimertinib more effectively inhibited phosphorylation than osimertinib monotreatment, and AKT phosphorylation was completely inhibited, even at the lowest dose (16 nM), under cotreatment. Thus, combination treatment with DS-1205 might prolong the therapeutic benefit of this EGFR-TKI by inhibiting the bypass pathway.
As AXL has numerous physiological functions, it is important to know its exact biological functions and to evaluate the most beneficial use of inhibitors. Recently, Mak et al. developed a patient-derived, pan-cancer EMT signature using 11 distinct tumor types from The Cancer Genome Atlas [46]. Mesenchymal tumors showed similar patterns of gene, protein, and miRNA expression, regardless of the cancer type. Tumors with mesenchymal EMT scores not only had higher AXL expression, but also expressed high levels of multiple immune checkpoints, including PD1, PD-L1, PD-L2, CTLA4, OX40L, CXCR4, CXCR6, and CXCR16 [47]. This finding, which was validated in an independent patient cohort, highlights the potential for utilizing EMT status, regardless of cancer type, as an additional selection tool to select patients who may benefit from immune checkpoint blockade. With regard to the role of AXL in immune evasion, emerging data show that increased AXL expression is involved in the anti-PD-1 or PD-L1 resistance program in non-responders [48–50]. Therefore, it is important to further clarify the biological roles of AXL in the context of anti-PD-1 or PD-L1 therapy, to develop novel approaches in cancer therapy.
We generated an AXL-overexpressing cell line, NIH3T3-AXL [4, 51], as an artificial model of tumorigenesis driven by AXL, and we used it to evaluate the activities of DS-1205b in vitro and in vivo. There are several lines of evidence that AXL is involved in tumor metastatic processes, such as cell migration and invasion [13, 52], and we confirmed that DS-1205b inhibited hGAS6-dependent cell migration in vitro. In the NIH3T3-AXL xenograft model, DS-1205b showed a potent tumor growth-inhibitory effect in vivo. This result strongly suggested that DS-1205b clearly inhibits AXL function in vivo when it is systemically administered. Several AXL inhibitors are under development, including selective and multi-targeted kinase inhibitors [19, 53–55]. It should be noted that multi-kinase inhibitors might have insufficient AXL inhibitory activity because of their broad spectrum or might demonstrate off-target toxicity. In this regard, a selective AXL inhibitor would be more convenient and efficacious in mono- or combination therapy. Bemcentinib, also known as BGB324 or R428, is an experimental oral small-molecule AXL kinase inhibitor [35]. Bemcentinib was licensed from Rigel Pharmaceuticals by BerGenBio and is currently under investigation in phase II trials in various solid and hematological tumors as monotherapy and in combination with immunotherapy, chemotherapy, and targeted therapeutics as a forefront runner in development (ClinicalTrials.gov IDs: NSCLC; NCT03184571, NCT02424617, triple negative breast cancer; NCT03184558, metastatic melanoma; NCT02872259, and acute myelogenous leukemia or myelodysplastic syndromes; NCT02488408). Recently, BGB324 in addition to its ability to inhibit AXL has been reported [56]. However, the molecular mechanisms of BGB324 in regulating cancer cell growth and metastasis have not been thoroughly investigated. It has been reported that BGB324 induced cancer cell apoptosis [56–58], but the role of AXL inhibition in BGB324-induced apoptosis has not been clarified. We evaluated the potency of BGB324 to inhibit hGAS6-dependent NIH3T3-AXL cell migration, AXL phosphorylation, and cell growth in comparison with DS-1205b. The EC50 of BGB324 was 132.3 nM, which was a 49-fold lower activity based on the area under concentration curve (AUC) than that of DS-1205b. Furthermore, its AXL phosphorylation-inhibitory activity was less potent. However, the lower GI50 and the apoptosis phenotype was observed at a similar concentration, consistent with a recent report [58]. In terms of AXL inhibition, DS-1205b can be thus expected to show better efficacy than BGB324 in future clinical studies. As DS-1205b is a potent and highly selective AXL inhibitor, the outcome of clinical trials of DS-1205b is highly anticipated. A phase I study in combination with osimertinib is underway (ClinicalTrials.gov IDs: NCT03255083, NCT03599518).
Materials and Methods
Test compounds
DS-1205b (chemical name: N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ylmethoxy]-3-methoxyphenyl}pyridin-3-yl)-3-fluorophenyl]-5-methyl-4′-oxo-1′-(tetrahydro-2H-pyran-4-ylmethyl)-1′,4′-dihydro-2,3′-bipyridine-5′-carboxamide 1 4/5 sulfate trihydrate, molecular formula: C41H42FN5O7·1 4/5H2SO4·3H2O) was synthesized by Daiichi Sankyo Co., Ltd. (Tokyo, Japan). The molecular formula was redefined as a non-stoichiometric sulfate hydrate, DS-1205c, C41H42FN5O7·xH2SO4·yH2O (where x is approximately 1 4/5 and y is approximately 3) for clinical trials. Erlotinib and osimertinib were provided by LC Laboratories (Woburn, MA, USA) and Shanghai Sun-shine Chemical Technology Corporation Ltd. (Shanghai, China), respectively. BGB324 was synthesized by Daiichi Sankyo Co., Ltd. (Tokyo, Japan).
Kinase inhibition assay
AXL, MER, MET, and TRKA kinase inhibition by DS-1205b was determined using the LabChip EZ Reader (PerkinElmer Inc., Waltham, MA, USA) in the presence of 1 mM ATP. DS-1205b dimethyl sulfoxide (DMSO) solution was added to kinase solution and the mixture was incubated at room temperature for 20 min. After the addition of ATP, the reaction plates were incubated at 28°C for 45 min. The kinase panel was evaluated by mobility shift assay (Carna Biosciences, Inc., Natick, MA, USA). The IC50 was determined.
In vitro growth inhibition assay
NIH3T3 mouse embryonic fibroblast cells were purchased from CLS Cell Lines Service GmbH (Eppelheim, Germany). Full-length AXL cDNA was inserted into a pLXSN retroviral vector (Clontech Laboratories, Inc., Mountain View, CA, USA) and the construct was transfected into NIH3T3 cells (NIH3T3-AXL). The cells were plated in 96-well plates (Corning, New York, NY, USA) and allowed to attach for 24 h. Then, the cells were treated with DS-1205b or BGB324 at concentrations in the range 1.52–10,000 nM in DMEM with 0.2% DMSO and incubated under 5% CO2 at 37°C for 3 days. Growth inhibition was determined by an ATP assay using the CellTiter-Glo® 2.0 Assay (Promega, Madison, WI, USA). The data were analyzed using GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA, USA) to derive a dose-response curve and the GI50 values. Each treatment was tested in triplicate at least three times independently, and the results represent the average of the independent experiments.
Cell morphology evaluation
NIH3T3-AXL cells were plated at 1 × 105 cells/well in 6-well plates and were incubated overnight (5% CO2, 37°C). DMSO, DS-1205b, or BGB324 were added to the culture medium at a final concentration 1 μM (for DS-1205b and BGB324 treatments) for 24 h. Phase-contrast images were captured with Leica DMI6000B microscope, using a Leica N PLAN L 20×/0.35 objective lens.
Migration inhibition assay
Inhibition of NIH3T3-AXL cell migration by DS-1205b or BGB324 was evaluated using a real-time cell analyzer (RTCA) DP instrument (xCELLigence; Roche Diagnostics, Basel, Switzerland), and the CI, a dimensionless parameter, was calculated as a relative change in measured electrical impedance to represent cell migration. DS-1205b or BGB324 was added to the lower chamber of the RTCA well, together with hGAS6 as the chemoattractant. The cell migration kinetics across the membrane separating the chambers of the RTCA well were continuously recorded by the RTCA DP instrument for 12 h in an incubator set at 37°C, with 5% CO2. The CI AUC0–12 indicates the sum of each CI for 12 h, and the EC50 was calculated based on the CI AUC0–12.
In vivo growth inhibition assay
NIH3T3-AXL tumor blocks (2 mm × 3 mm) were implanted in the right flanks of female NOD/Shi-scid IL-2Rγ KO Jic mice. Eighteen days after implantation, the mice were orally administered vehicle (0.5% methylcellulose) or DS-1205b at 50, 25, 13, 6.3, or 3.1 mg/kg bid for 5 days. The long and short tumor diameters and body weight were assessed at 3–4- and 2–4-day intervals, respectively. Tumor dimensions were measured with a digital caliper and tumor volumes were calculated as long diameter × short diameter2/2. Mice were sacrificed before the growing tumors reached 2,000 mm3, or when the once-shrunken tumors regrew to the size on day 0. All animal experimental procedures conformed to the guiding principles of the Daiichi Sankyo Co., Ltd. Institutional Animal Care and Use Committee.
Evaluation of the delaying effect of DS-1205b on acquired erlotinib or osimertinib resistance in a xenograft model in vivo
HCC827 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were inoculated subcutaneously into 6-week-old female CAnN.Cg-Foxn1nu/CrlCrlj mice. Erlotinib (25 mg/kg), osimertinib (3 mg/kg), or 0.5 w/v% methyl cellulose 400 solution were administered orally once per day, and DS-1205b (50, 25, or 12.5 mg/kg bid) was administered orally in accordance with a 5 days on/2 days off schedule. In resistance delay studies, erlotinib and osimertinib were started from day 56 and 45 after tumor inoculation, respectively. Vehicle administration and DS-1205b monotherapy were continued until the tumor volume reached 1,000 mm3. Erlotinib and osimertinib monotherapy and the combination with DS-1205b were continued until day 100. Body weight and tumor diameters were measured twice per week.
Evaluation of the effect of DS-1205b on acquired erlotinib resistance in a xenograft model in vivo
Erlotinib-resistant tumors were generated in female CAnN.Cg-Foxn1nu/CrlCrlj mice by subcutaneous inguinal inoculation of HCC827 human NSCLC cells, followed by once-daily erlotinib administration (25 mg/kg) for approximately 9 weeks with a 5 days on/2 days off schedule. After the generation of erlotinib-resistant tumors in the mice, DS-1205b was administered twice per day at a dose of 25 or 50 mg/kg for approximately 7 weeks in combination with erlotinib, which was administered once per day. Tumor length and width and body weight were measured every 3 or 4 days. Tumor volumes on the day of the last administration of erlotinib and DS-1205b were used to evaluate antitumor effects indicated as total growth inhibition by net (TGIn) %. The measurements were continued until the mean tumor volume of the vehicle-treated group reached a volume not exceeding 1,500 mm3.
Western blot analysis of drug-treated cells and tissue samples
Polyvinylidene difluoride membranes containing electrophoretically separated proteins from whole-cell lysates and tumor tissues were probed with antibodies. All antibodies, except anti-actin and rabbit IgG antibodies and Can Get Signal Immunoreaction Enhancer Solution 1 (Can Get Solution 1; Toyobo Co., Ltd., Osaka, Japan), were mixed at 1:1,000. Anti-actin antibody and Can Get Solution 1 were mixed at 1:2,000. For the secondary antibody, anti-rabbit IgG antibody and Can Get Signal Immunoreaction Enhancer Solution 2 (Can Get Solution 2; Toyobo Co., Ltd.) were mixed at 1:2,000. All antibodies were stored at –20°C. The following antibodies were used: anti-AXL (C44G1) rabbit monoclonal antibody (Cell Signaling Technology, Inc., Danvers, MA, USA, #4566), anti-phospho AXL (Tyr702) (D12B2) rabbit monoclonal antibody (Cell Signaling Technology, Inc., #5724), anti-EGFR rabbit polyclonal antibody (Cell Signaling Technology, Inc., #2232), anti-phospho EGFR (Tyr1173) (53A5) rabbit monoclonal antibody (Cell Signaling Technology, Inc., #4407), anti-AKT rabbit polyclonal antibody (Cell Signaling Technology, Inc., #9272), anti-phospho AKT (Ser473) (D9E) rabbit monoclonal antibody (Cell Signaling Technology, Inc., #4060), anti-p44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) rabbit polyclonal antibody (Cell Signaling Technology, Inc., #9102), anti-phospho 44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E) rabbit monoclonal antibody (Cell Signaling Technology, Inc., #4370), anti-β-actin (13E5) rabbit monoclonal antibody (Cell Signaling Technology, Inc., #4970), anti-rabbit IgG, and HRP-linked goat polyclonal antibody (Cell Signaling Technology, Inc., #7074).
Immunohistochemical analysis of tumor samples
Primary AXL antibody (C89E7, rabbit mAb; Cell Signaling Technology, #8661) was diluted to a final concentration of 3.72 μg/mL with Dako antibody diluent (DAKO REAL Antibody Diluent K3468; Dako Denmark A/S, Glostrup, Denmark). Approximately 4-μm-thick sections were deparaffinized and heat-pretreated with Target Retrieval Solution (pH 9) (Dako Denmark A/S) in a microwave oven for antigen retrieval. Subsequently, the sections were subjected to endogenous peroxidase blocking and protein blocking. The sections were sequentially incubated with the primary antibody for 60 min, EnVision + System-HRP Labelled Polymer Anti-Rabbit (Dako Denmark A/S) as a secondary antibody for 30 min, and Liquid DAB + Substrate Chromogen System (Dako Denmark A/S) for 5 min. The sections were washed after each step. Finally, the sections were counterstained with hematoxylin for 5 min and mounted.
AXL signal intensities were scored on the whole tumor area (excluding necrotic and stromal areas) according to a semi-quantitative grading system, in which the staining intensity was graded as 0 (no staining), 1+ (faint to weak), 2+ (moderate), or 3+ (strong). Representative images for each grade are shown in Figures 3D and 4B. The AXL-stained sections were observed under a BX51 microscope (Olympus Corporation, Tokyo, Japan). The AXL-positive staining area (percent positive) was calculated as the sum of percentages for each score. H scores were calculated in Microsoft Excel 2010 according to the following equation: H score = (3 × % of cells with strong staining) + (2 × % of cells with moderate staining) + (1 × % of cells with weak staining). Group means and SDs were also calculated with regard to percent positive and H scores for each group.
Statistical analysis
Data are presented as the mean ± SD or mean ± standard error (Excel 2013, Microsoft Corporation, Redmond, WA, USA). Treatment groups were compared with controls by parametric Dunnett’s tests. Spearman’s rank correlation coefficient testing was used to analyze dose-dependent effects. P-values <0.05 were considered statistically significant. EC50 was calculated using a sigmoid Emax model [59, 60]. SAS system release 9.2 was used for statistical analyses and to generate the Emax model (SAS Institute, Inc., Cary, NC, USA).
Abbreviations
AKT: AKT serine/threonine kinase; ATP: adenosine triphosphate; AUC0-t: area under the plasma concentration-time curve to time t; AXL: AXL receptor tyrosine kinase; bid: twice daily; CI: cell index; DMSO: dimethyl sulfoxide; EGFR: epidermal growth factor receptor; EMT: epithelial-mesenchymal transition; ERK: extracellular signal-regulated kinase; hGAS6: human growth arrest-specific gene 6; MAPK: mitogen-activated protein kinase; NSCLC: non-small cell lung cancer; qd: once a day; TAM: TYRO-3, AXL, and MER; TKI: tyrosine kinase inhibitor.
ACKNOWLEDGMENTS
The authors would like to thank Marie Hisamoto, Makiko Uno, Channing Yu, Eric Slosberg for critical review, comments, and materials, and acknowledge all researchers and staff members who are not listed as authors but worked to make this study possible.
CONFLICTS OF INTEREST
Disclosure of potential conflicts of interest: M.O. is an employee of Daiichi Sankyo RD Novare Co., Ltd., Tokyo, Japan. All other authors are employees of Daiichi Sankyo Co., Ltd., Tokyo, Japan.
FUNDING
This study was funded by Daiichi Sankyo Co., Ltd.
References
1. Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets. 2010; 14:1073–90. https://doi.org/10.1517/14728222.2010.515980. [PubMed].
2. Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011; 10:1763–73. https://doi.org/10.1158/1535-7163.MCT-11-0116. [PubMed].
3. Axelrod H, Pienta KJ. Axl as a mediator of cellular growth and survival. Oncotarget. 2014; 5:8818–52. https://doi.org/10.18632/oncotarget.2422. [PubMed].
4. O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R 3rd, Le Beau MM, Earp HS, Liu ET. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991; 11:5016–31. https://doi.org/10.1128/MCB.11.10.5016. [PubMed].
5. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012; 44:852–60. https://doi.org/10.1038/ng.2330. [PubMed].
6. Ishikawa M, Sonobe M, Nakayama E, Kobayashi M, Kikuchi R, Kitamura J, Imamura N, Date H. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann Surg Oncol. 2013; 20:S467–76. https://doi.org/10.1245/s10434-012-2795-3. [PubMed].
7. Ji W, Choi CM, Rho JK, Jang SJ, Park YS, Chun SM, Kim WS, Lee JS, Kim SW, Lee DH, Lee JC. Mechanisms of acquired resistance to EGFR-tyrosine kinase inhibitor in Korean patients with lung cancer. BMC Cancer. 2013; 13:606–13. https://doi.org/10.1186/1471-2407-13-606. [PubMed].
8. Okimoto RA, Bivona TG. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer (Auckl). 2015; 6:27–34. https://doi.org/10.2147/LCTT.S60438. [PubMed].
9. Mudduluru G, Vajkoczy P, Allgayer H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol Cancer Res. 2010; 8:159–69. https://doi.org/10.1158/1541-7786.MCR-09-0326. [PubMed].
10. Dunne PD, McArt DG, Blayney JK, Kalimutho M, Greer S, Wang T, Srivastava S, Ong CW, Arthur K, Loughrey M, Redmond K, Longley DB, Salto-Tellez M, et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res. 2014; 20:164–75. https://doi.org/10.1158/1078-0432.CCR-13-1354. [PubMed].
11. Belkhiri A, El-Rifai W. Advances in targeted therapies and new promising targets in esophageal cancer. Oncotarget. 2015; 6:1348–58. https://doi.org/10.18632/oncotarget.2752. [PubMed].
12. Hsieh MS, Yang PW, Wong LF, Lee JM. The AXL receptor tyrosine kinase is associated with adverse prognosis and distant metastasis in esophageal squamous cell carcinoma. Oncotarget. 2016; 7:36956–70. https://doi.org/10.18632/oncotarget.9231. [PubMed].
13. Zhang YX, Knyazev PG, Cheburkin YV, Sharma K, Knyazev YP, Őrfi L, Szabadkai I, Daub H, Kéri G, Ullrich A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008; 68:1905–15. https://doi.org/10.1158/0008-5472.CAN-07-2661. [PubMed].
14. Wang X, Saso H, Iwamoto T, Xia W, Gong Y, Pusztai L, Woodward WA, Reuben JM, Warner SL, Bearss DJ, Hortobagyi GN, Hung MC, Ueno NT. TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase. Cancer Res. 2013; 73:6516–25. https://doi.org/10.1158/0008-5472.CAN-13-0967. [PubMed].
15. Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, Behrndt S, von Tengg-Kobligk H, Kiessling F, Eichelsbacher U, Essig M, Read TA, Erber R, Ullrich A. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006; 103:5799–804. https://doi.org/10.1073/pnas.0510923103. [PubMed].
16. Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, Cassuto JP, Raynaud S, Auberger P. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget. 2011; 2:874–85. https://doi.org/10.18632/oncotarget.360. [PubMed].
17. Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS, Riecken K, Binder M, Schewe D, Sawall S, Witzke V, Cubas-Cordova M, Janning M, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013; 122:2443–52. https://doi.org/10.1182/blood-2013-03-491431. [PubMed].
18. Boysen J, Sinha S, Price-Troska T, Warner SL, Bearss DJ, Viswanatha D, Shanafelt TD, Kay NE, Ghosh AK. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: implications for therapy in p53-defective CLL patients. Leukemia. 2014; 28:451–55. https://doi.org/10.1038/leu.2013.298. [PubMed].
19. Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014; 5:9546–63. https://doi.org/10.18632/oncotarget.2542. [PubMed].
20. Park IK, Giovenzana C, Hughes TL, Yu J, Trotta R, Caligiuri MA. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood. 2009; 113:2470–77. https://doi.org/10.1182/blood-2008-05-157073. [PubMed].
21. D’Arcangelo D, Gaetano C, Capogrossi MC. Acidification prevents endothelial cell apoptosis by Axl activation. Circ Res. 2002; 91:e4–12. https://doi.org/10.1161/01.RES.0000036753.50601.E9. [PubMed].
22. Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, Nagle R, Bearss D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007; 26:3909–19. https://doi.org/10.1038/sj.onc.1210173. [PubMed].
23. Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009; 69:6871–78. https://doi.org/10.1158/0008-5472.CAN-08-4490. [PubMed].
24. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010; 468:968–72. https://doi.org/10.1038/nature09627. [PubMed].
25. Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, Lai GM, Chuang SE. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008; 268:314–24. https://doi.org/10.1016/j.canlet.2008.04.017. [PubMed].
26. Keating AK, Kim GK, Jones AE, Donson AM, Ware K, Mulcahy JM, Salzberg DB, Foreman NK, Liang X, Thorburn A, Graham DK. Inhibition of Mer and Axl receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther. 2010; 9:1298–307. https://doi.org/10.1158/1535-7163.MCT-09-0707. [PubMed].
27. Hong J, Peng D, Chen Z, Sehdev V, Belkhiri A. ABL regulation by AXL promotes cisplatin resistance in esophageal cancer. Cancer Res. 2013; 73:331–40. https://doi.org/10.1158/0008-5472.CAN-12-3151. [PubMed].
28. Linger RM, Cohen RA, Cummings CT, Sather S, Migdall-Wilson J, Middleton DH, Lu X, Barón AE, Franklin WA, Merrick DT, Jedlicka P, DeRyckere D, Heasley LE, Graham DK. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene. 2013; 32:3420–31. https://doi.org/10.1038/onc.2012.355. [PubMed].
29. Schoumacher M, Burbridge M. Key Roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr Oncol Rep. 2017; 19:19–32. https://doi.org/10.1007/s11912-017-0579-4. [PubMed].
30. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N, Toulany M, Gill PS, Salgia R, Kimple RJ, Wheeler DL. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014; 74:5152–64. https://doi.org/10.1158/0008-5472.CAN-14-0294. [PubMed].
31. Scaltriti M, Elkabets M, Baselga J. Molecular Pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. 2016; 22:1313–17. https://doi.org/10.1158/1078-0432.CCR-15-1458. [PubMed].
32. Gibbons DL, Byers LA. A HER 1-2 punch: dual EGFR targeting deals resistance a deadly blow. Cancer Discov. 2014; 4:991–94. https://doi.org/10.1158/2159-8290.CD-14-0791. [PubMed].
33. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012; 2:922–33. https://doi.org/10.1158/2159-8290.CD-12-0108. [PubMed].
34. Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, Tanimoto A, Takeuchi S, Araujo LH, Boroni M, Yoshimura A, Shiotsu S, Matsumoto I, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019; 10:259. https://doi.org/10.1038/s41467-018-08074-0. [PubMed].
35. Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, Zhang J, Ding P, Apatira A, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010; 70:1544–54. https://doi.org/10.1158/0008-5472.CAN-09-2997. [PubMed].
36. Husain H, Scur M, Murtuza A, Bui N, Woodward B, Kurzrock R. Strategies to overcome bypass mechanisms mediating clinical resistance to EGFR tyrosine kinase inhibition in lung cancer. Mol Cancer Ther. 2017; 16:265–72. https://doi.org/10.1158/1535-7163.MCT-16-0105. [PubMed].
37. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005; 2:e73. https://doi.org/10.1371/journal.pmed.0020073. [PubMed].
38. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017; 17:637–58. https://doi.org/10.1038/nrc.2017.84. [PubMed].
39. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560–62. https://doi.org/10.1038/nm.3854. [PubMed].
40. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014; 25:282–303. https://doi.org/10.1016/j.ccr.2014.02.025. [PubMed].
41. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–43. https://doi.org/10.1126/science.1141478. [PubMed].
42. Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013; 19:279–90. https://doi.org/10.1158/1078-0432.CCR-12-1558. [PubMed].
43. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, Howe E, Fulton LE, Mulvey HE, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015; 6:6377–86. https://doi.org/10.1038/ncomms7377. [PubMed].
44. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013; 3:1355–63. https://doi.org/10.1158/2159-8290.CD-13-0310. [PubMed].
45. Namba K, Shien K, Takahashi Y, Torigoe H, Sato H, Yoshioka T, Takeda T, Kurihara E, Ogoshi Y, Yamamoto H, Soh J, Tomida S, Toyooka S. Activation of AXL as a preclinical acquired resistance mechanism against osimertinib Treatment in EGFR-mutant non-small cell lung cancer cells. Mol Cancer Res. 2019; 17:499–507. https://doi.org/10.1158/1541-7786.MCR-18-0628. [PubMed].
46. Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, Rodriguez-Canales J, Wistuba II, Heymach JV, Weinstein JN, Coombes KR, et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin Cancer Res. 2016; 22:609–20. https://doi.org/10.1158/1078-0432.CCR-15-0876. [PubMed].
47. Tsukita Y, Fujino N, Miyauchi E, Saito R, Fujishima F, Itakura K, Kyogoku Y, Okutomo K, Yamada M, Okazaki T, Sugiura H, Inoue A, Okada Y, Ichinose M. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol Cancer. 2019; 18:24–29. https://doi.org/10.1186/s12943-019-0953-y. [PubMed].
48. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016; 165:35–44. https://doi.org/10.1016/j.cell.2016.02.065. [PubMed]. Erratum in: Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. [Cell. 2017]. https://doi.org/10.1016/j.cell.2017.01.010. [PubMed].
49. Skinner HD, Giri U, Yang LP, Kumar M, Liu Y, Story MD, Pickering CR, Byers LA, Williams MD, Wang J, Shen L, Yoo SY, Fan YH, et al. Integrative analysis identifies a novel AXL-PI3 kinase-PD-L1 signaling axis associated with radiation resistance in head and neck cancer. Clin Cancer Res. 2017; 23:2713–22. https://doi.org/10.1158/1078-0432.CCR-16-2586. [PubMed].
50. Aguilera TA, Giaccia AJ. Molecular Pathways: Oncologic Pathways and Their Role in T-cell Exclusion and Immune Evasion-A New Role for the AXL Receptor Tyrosine Kinase. Clin Cancer Res. 2017; 23:2928–33. https://doi.org/10.1158/1078-0432.CCR-17-0189. [PubMed].
51. Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, Bartram CR. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991; 6:2113–20. [PubMed].
52. Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, Kallop D, Ludlam MJ, Pei L. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009; 28:3442–55. https://doi.org/10.1038/onc.2009.212. [PubMed].
53. Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013; 31:775–76. https://doi.org/10.1038/nbt0913-775a. [PubMed].
54. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017; 116:415–23. https://doi.org/10.1038/bjc.2016.428. [PubMed].
55. Shen Y, Chen X, He J, Liao D, Zu X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018; 198:99–111. https://doi.org/10.1016/j.lfs.2018.02.033. [PubMed].
56. Chen F, Song Q, Yu Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am J Cancer Res. 2018; 8:1466–82. [PubMed].
57. Smolock EM, Korshunov VA. Pharmacological inhibition of Axl affects smooth muscle cell functions under oxidative stress. Vascul Pharmacol. 2010; 53:185–92. https://doi.org/10.1016/j.vph.2010.07.002. [PubMed].
58. Ghosh AK, Secreto C, Boysen J, Sassoon T, Shanafelt TD, Mukhopadhyay D, Kay NE. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011; 117:1928–37. https://doi.org/10.1182/blood-2010-09-305649. [PubMed].
59. Levy G, Ebling WF, Forrest A. Concentration- or effect-controlled clinical trials with sparse data. Clin Pharmacol Ther. 1994; 56:1–8. https://doi.org/10.1038/clpt.1994.93. [PubMed].
60. Dutta S, Matsumoto Y, Ebling WF. Is it possible to estimate the parameters of the sigmoid Emax model with truncated data typical of clinical studies? J Pharm Sci. 1996; 85:232–39. https://doi.org/10.1021/js950067y. [PubMed].