
Introduction
Immune checkpoint blockade has been reported to have anti-cancer effects with antibodies (Abs) against programmed cell death 1 (PD-1) and programmed death-ligand 1 (PD-L1) being effective in types of several cancer [1–3]. The expression level of PD-1 ligands is increased in various cancers, resulting in cancer cell resistance to elimination by tumor-specific T cells [1]. The anti-tumor activity of an anti-PD-1 Ab has been observed in non-small cell lung cancer, melanoma, and renal cell cancer; however, no response was observed in colorectal cancer [2]. Similarly, the anti-tumor activity of an anti-PD-L1 Ab was observed in melanoma, non-small cell lung cancer, renal cell cancer, and ovarian cancer, but no response was observed in colorectal cancer [4]. However, in most cases, anti-PD-1/PD-L1 monotherapies result in only partial tumor regression [1]. Therefore, combination therapies based on PD-1/PD-L1 blockade may be required to increase the therapeutic effects of these agents. In melanoma, tumor regression based on the anti-tumor effects of an anti-PD-1 Ab requires the presence of CD8+ T-cells in the tumor [5] and activation of the β-catenin pathway in the tumor is inversely correlated with the extent of CD8+ T-cell infiltration [6]. Moreover, activation of β-catenin induces the expression of the transcriptional repressor AMP-dependent transcription factor-3 (ATF3), which suppresses the production of C-C motif chemokine ligand-4 (CCL4), leading to T-cell exclusion [6].
Wnt/β-catenin signaling is involved in virtually every aspect of embryonic development, as well as homeostatic self-renewal in adult tissues and is also associated with the pathogenesis of many human diseases such as colorectal cancer [7] and liver fibrosis [8, 9]. Moreover, immunosuppressive roles of activated β-catenin signaling have been reported in melanoma cells [10]. Following activation by upstream signaling from Wnt, β-catenin translocates to the nucleus. Nuclear β-catenin then recruits the Kat3 transcriptional co-activators cAMP-response element-binding protein (CREB)-binding protein (CBP) or EP300. Accordingly, the CBP/β-catenin antagonist ICG-001 has been shown to reduce the growth of cholangiocarcinoma in an animal model [11]. In addition, ICG-001 was found to increase sensitivity to cisplatin in platinum-resistant ovarian cancer cells [12]. PRI-724 is a second-generation specific CBP/β-catenin antagonist developed by Prism Pharma and is known to increase the expression of several chemokines in the liver during the resolution of liver fibrosis [8]. Colon cancer, one of the most common malignancies, frequently metastasizes to the liver and is often resistant to anti-PD-1 or anti-PD-L1 Abs [2, 4]. Therefore, in effort to increase the anti-tumor efficacy of PD-1/PD-L1 immune checkpoint blockade, we investigated the in vivo effects of PRI-724 treatment in combination with an anti-PD-L1 Ab on the progression of liver metastasis from colon cancer using an mouse model.
Results
An anti-PD-L1 Ab combined with a CBP/β-catenin inhibitor decreased metastatic tumor growth in liver
To determine if the mouse colon cancer cell line SL4 expressed PD-L1, we performed immunohistochemical analysis and FACS using an anti-PD-L1 Ab. As shown in Supplementary Figure 1, the SL-4 cells did express PD-L1. To examine the anti-tumor effect of PD-1/PD-L1 immune checkpoint blockade on metastasis liver tumors, liver lesions were induced by the intrasplenic injection of SL4 cells and then PRI-724 (0.4 mg/mouse) and/or anti-PD-L1 Ab (200 μg/mouse) were administrated to these animals. Two weeks post inoculation, the liver weight and Ki67-positive tumor area were found to be increased in the PBS-treated control group (Figure 1A, 1B). Moreover, individual treatment with either PRI-724 or PD-L1 Ab had no anti-tumor effect as these treatments failed to reduce liver weight or Ki67-positive area. However, in contrast, the combination treatment with both agents significantly reduced liver weight and Ki67-positive area (Figure 1A, 1B). These results suggested that the co-administration of PRI-724 and PD-L1 Ab was able to exert an anti-tumor effect on SL4 cell metastasis to the liver. Consistent with these data, the combination therapy also improved the survival rate after the inoculation of colon cancer cells (Figure 1C). Importantly, the combination therapy did not increase serum alanine aminotransferase (ALT) levels (Figure 1D), indicating that there was less adverse effect on hepatocytes.
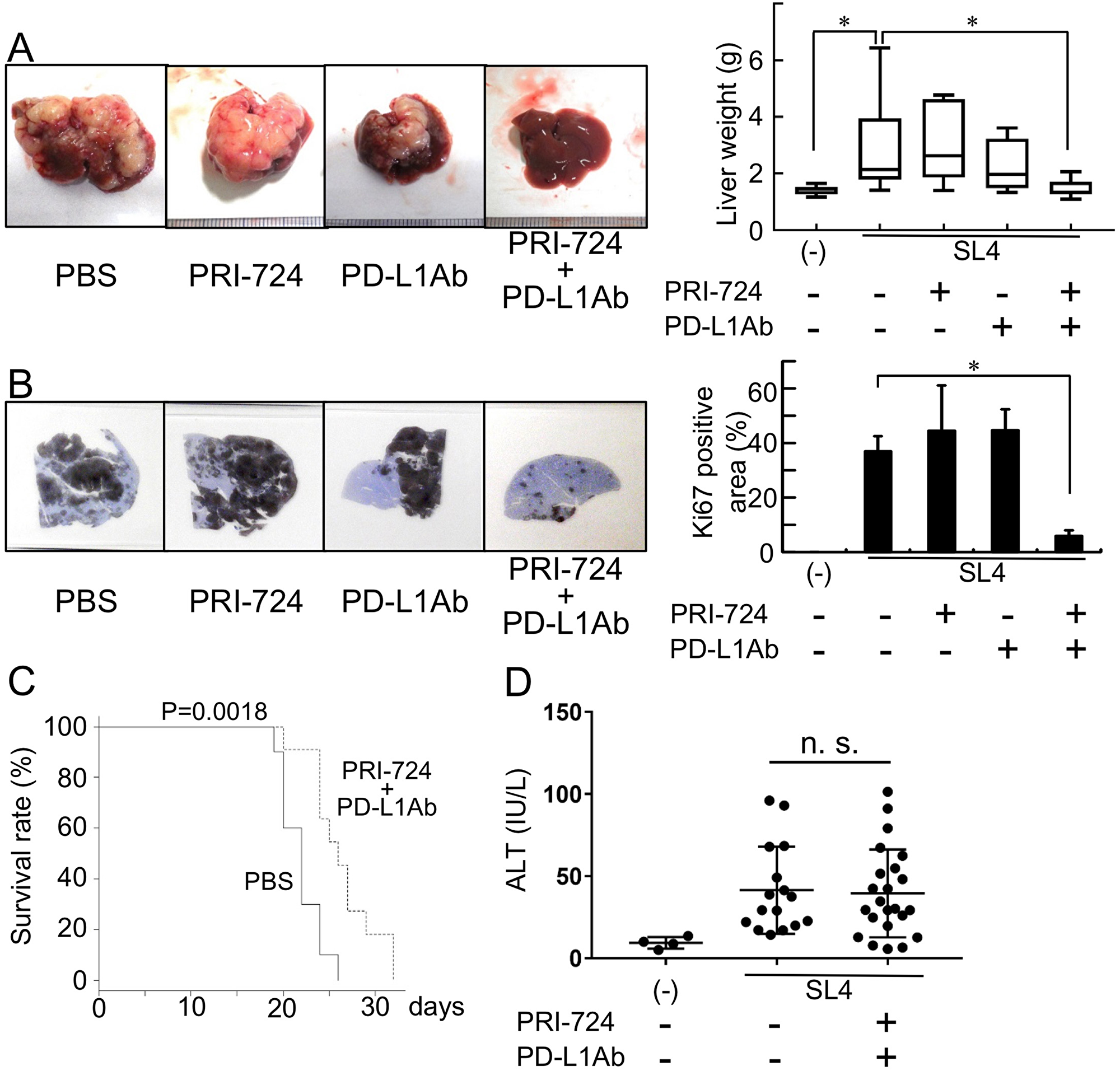
Figure 1: Anti-PD-L1 antibody (Ab) with a CBP/β-catenin inhibitor decreases metastatic tumor growth in the liver. Male C57BL/6J mice were intrasplenically injected with SL4 cells (5 × 105 cells) and treated with or without anti-PD-L1 Ab and/or PRI-724. The animals were humanely sacrificed 14 days post inoculation (A, B, D). (A) The livers were excised and photographed (left panels). Liver weights were measured (right panel). (B) Expression of Ki67 in the metastatic liver tumor (loupe magnification) was examined by immunohistochemistry using an anti-Ki67 Ab. Intrahepatic tumor load is presented as Ki67-positive areas based on the measurement of two non-sequential for each animal (graph on right panel). The pictures shown are representative of at least four independent experiments. Results are provided as box-and-whisker plot or as means ± SD of data collected from at least four independent experiments. *P < 0.05 based on the Kruskal-Wallis test (A) and one-way ANOVA test (B). (C) The survival rates of the animals are shown. Statistically significant differences were determined by performing a log-rank test. (D) Serum ALT levels were determined and the results are provided as means ± SD. n. s.; not significant.
PRI-724 treatment reduced mRNA expression of β-catenin target genes in SL4-inoculated livers
To examine whether Wnt/β-catenin signaling was activated in livers of mice inoculated with SL4 cells, we analyzed the expression levels of Wnt/β-catenin target genes (Figure 2). Inoculation of SL4 cells resulted in increased expression of Wnt/β-catenin target-genes in the livers of mice, which was decreased following PRI-724 treatment, indicating that PRI-724 could inhibit β-catenin signaling in the metastatic liver. These results suggest that effective tumor regression after anti-PD-L1 Ab administration required CBP/β-catenin inhibition in the metastatic liver tumors of colon cancer.
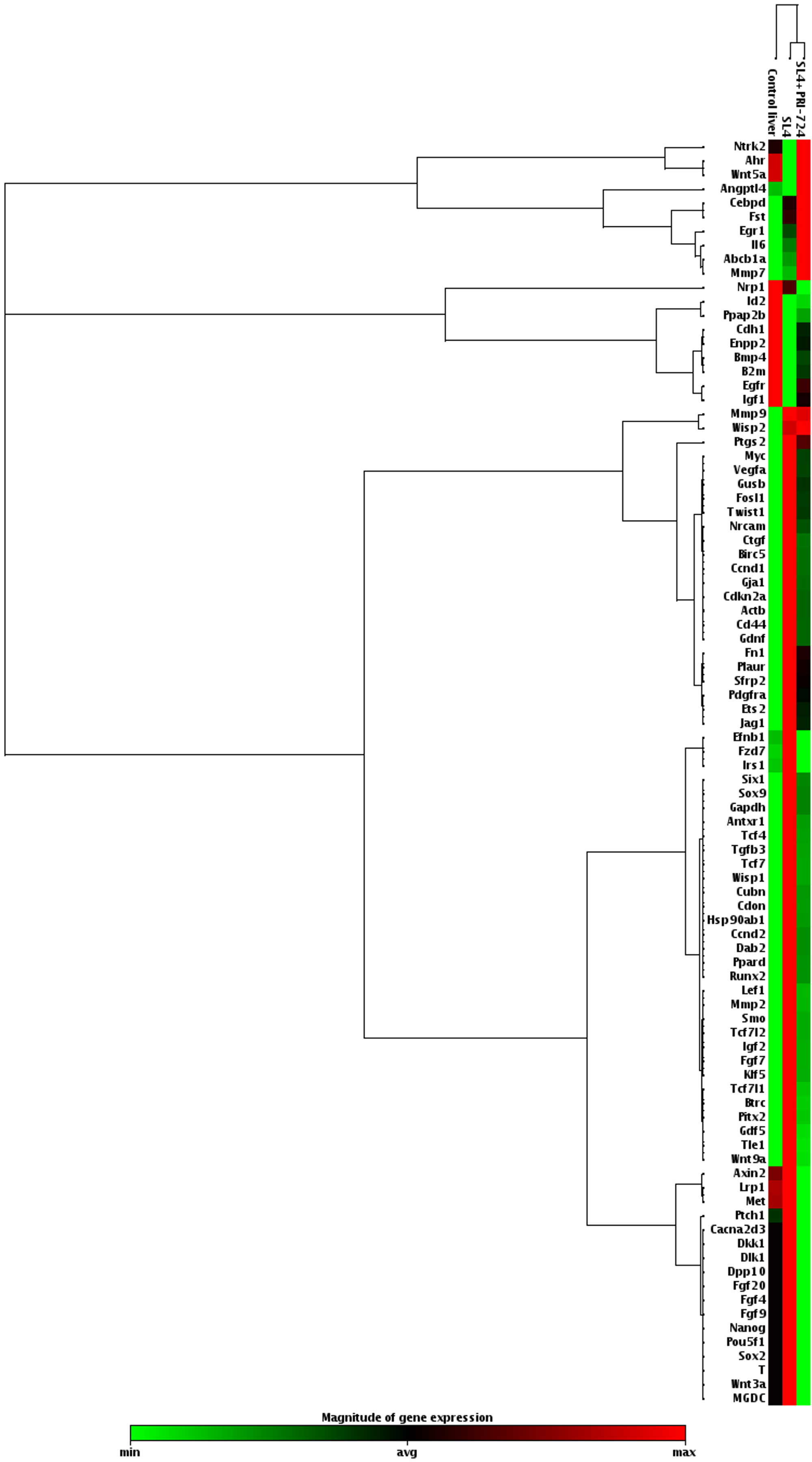
Figure 2: PRI-724 treatment decreased the mRNA expression of β-catenin target genes in the livers of mice inoculated with SL4 cells. After male C57BL/6J mice were intrasplenically injected with SL4 cells (5 × 105 cells) and treated with PRI-724 (0.4 mg/mouse) or PBS three times per week, the animals were humanely sacrificed 14 days after inoculation. Expression of the indicated mRNA expression levels of β-catenin target genes in the liver was determined using an RT2 Profiler™ PCR Array.
PRI-724 increased T-lymphocyte infiltration into metastatic liver tumors
In melanoma, tumor regression after PD-1 blockade requires pre-existing CD8+ T-cells in the tumor [5]. Immunohistochemical analysis was performed using SL4-inoculated liver tissue 14 days after PRI-724 administration to determine whether CD3+ T-cells would invade the tumor site. The mean number of CD3+ cells per high-power microscopic field in metastatic liver tumors was comparable to that observed in the normal control livers (12 ± 3.5 vs. 15 ± 7.9 cells/high power field, respectively). However, the number of CD3+ cells in the tumors increased following PRI-724 treatment (Figure 3). Flow cytometric analysis of isolated intrahepatic leukocytes (IHLs) at 14 days after injection revealed that the relative percentages of CD4+, CD8+, and NK1.1+ cells were not altered by PRI-724 treatment with or without anti-PD-L1 Ab administration (Table 1, Supplementary Figure 2), suggesting that the subsets of T lymphocytes were not altered. The increased number of T lymphocytes observed in the PRI-724-treated group consisted of CD8+ T-cells and CD8+ T-cells, as well as other subsets of T lymphocytes. Since PRI-724 monotherapy failed to suppress tumor growth, the increase in CD8+ T-cell infiltration alone was not sufficient to induce tumor regression. PD-1 is expressed at low levels on naïve T-cells and is upregulated during their activation [13]. In addition, activation of Wnt/β-catenin signaling maintains T cells in an undifferentiated state [14]. The proportion of CD44−CD62L+ naïve T-cells in among IHLs was not increased by PRI-724 treatment (Table 1), suggesting that the tumor lymphocyte population, which was found to increase, contained PD-1-expressing cells. Furthermore, the anti-PD-L1 Ab increased the percentage of CD69+ lymphocytes, indicating the activation of these cells (Table 1). Based on the previous reports and our current findings, the increase in CD8+ T-cell infiltration mediated by PRI-724 and their activation induced by anti-PD-L1 Ab are both required for the regression of liver tumors caused by metastatic colon cancer.
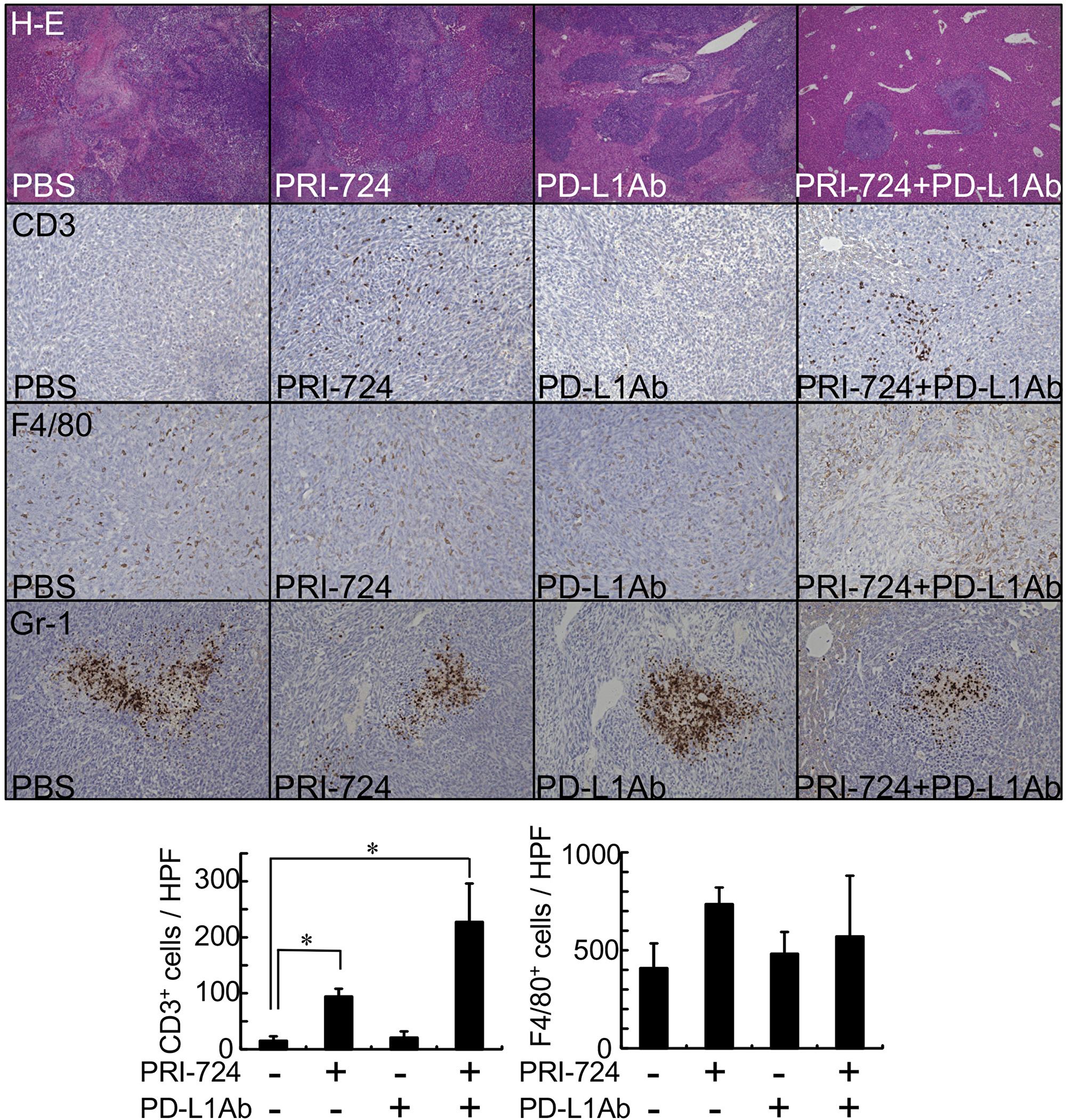
Figure 3: CBP/β-catenin inhibition increased lymphocyte infiltration to metastatic tumors in the liver. Male C57BL/6J mice were intrasplenically injected with SL4 cells (5 × 105 cells). The animals were treated with or without anti-PD-L1 antibody (Ab) and/or PRI-724 and were humanely sacrificed 14 days after inoculation. Liver sections were stained with H&E (original magnification: 40×). Expression of CD3, F4/80, and Gr-1 in the metastatic tumor was examined by immunohistochemistry with an anti-CD3, anti-F4/80, and anti-Gr-1 Abs (graph on lower panel) to assess the number of lymphocytes, macrophages, and neutrophils, respectively (original magnification: 200×). Results are provided as means ± SD of data collected from at least four independent experiments. *P < 0.05 based on one-way ANOVA test.
Table 1: Changes in lymphocyte surface marker profiles in SL4 cell-inoculated mice
| Markers | SL4 | |||
|---|---|---|---|---|
| PBS | PRI-724 | PD-L1Ab | PRI-724 + PD-L1Ab | |
| CD3+ CD4+ | 4.33 ± 1.35 | 5.57 ± 2.51 | 5.27 ± 1.54 | 4.98 ± 1.13 |
| CD3+ CD8+ | 7.18 ± 1.64 | 10.30 ± 3.64 | 8.30 ± 1.32 | 8.15 ± 1.66 |
| CD3+ NK1.1+ | 2.49 ± 0.62 | 1.82 ± 0.63 | 1.85 ± 0.18 | 2.47 ± 0.64 |
| CD4+ CD44- CD62L+ | 46.73 ± 5.80 | 51.13 ± 6.67 | 38.8 ± 5.11 | 37.3 ± 7.88* |
| CD8+ CD44- CD62L+ | 37.22 ± 1.85 | 40.58 ± 9.14 | 28.15 ± 3.30* | 30.11 ± 7.76 |
| CD4+ CD44- CD62L- | 10.22 ± 1.82 | 11.60 ± 2.15 | 10.80 ± 1.29 | 10.89 ± 3.29 |
| CD8+ CD44- CD62L- | 9.25 ± 4.58 | 11.64 ± 2.09 | 10.55 ± 1.43 | 8.51 ± 2.71 |
| CD4+ CD69+ | 1.06 ± 0.40 | 1.20 ± 0.26 | 1.67 ± 0.71 | 1.89 ± 0.57* |
| CD8+ CD69+ | 1.25 ± 0.26 | 1.69 ± 0.26 | 2.36 ± 0.21* | 1.80 ± 0.30* |
| CD4+ Foxp3+ | 0.05 ± 0.05 | 0.11 ± 0.05 | 0.24 ± 0.14* | 0.46 ± 0.18* |
Role of T cells in the anti-tumor effect in liver
Because we found that PRI-724 and PD-L1 Ab could induce the recruitment of CD3+ T-cells to the liver at 14 days post treatment, we next assessed which T-cell population was essential for the anti-tumor effects. Although results for IHLs were observed at 14 days after PRI-724 administration, it was possible that the immune response to the tumor had already peaked by this time point. Therefore, IHLs were collected and analyzed 5 days after co-administration of PRI-724 and PD-L1 Ab, which is the time at which tumor is found to exist in the liver. IHLs isolated from SL4 cell-inoculated mice were stained with Abs against PD-1, CD44, and CD62L and the effect of the combined treatment was evaluated. The expression of PD-1 on CD8+ T-cells was significantly decreased in the combined-treatment group compared to that in the control group. In addition, the proportion of effector CD44lowCD62Llow cells was significantly increased among the CD4+ and CD8+ T-cells in the liver (Figure 4A, 4B).
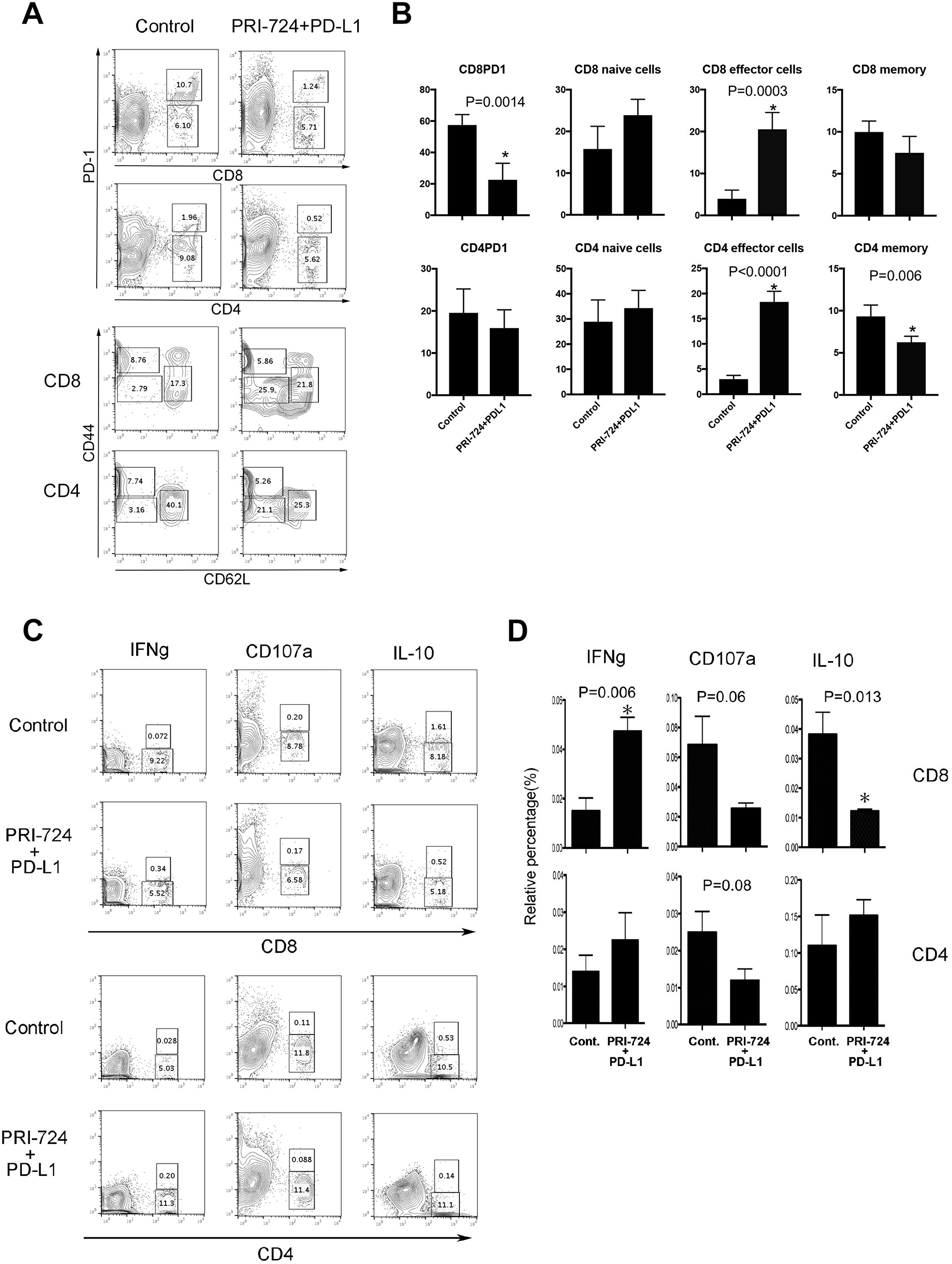
Figure 4: Role of T cells in the anti-tumor effect of anti-PD-L1 antibody (Ab) and/or PRI-724. Male C57BL/6J mice were intrasplenically injected with SL4 cells (5 × 105 cells). The animals were treated with or without anti-PD-L1 Ab and PRI-724 and were humanely sacrificed 5 days after inoculation. (A) Intrahepatic leukocytes (IHLs) isolated from mice inoculated with SL4 cells were stained for PD-1, CD44, and CD62L in CD8 and CD4 T-cells. The numbers represent the percentage of PD-1-, CD44-, and CD62L-positive and negative cells among the CD8 and CD4 T-cells. (B) Results are representative of three independent experiments. Significant relationships are indicated by P-values based on a two-tailed Student’s t-test. *P < 0.05. (C) The numbers represent the percentage of IFN-γ-, CD107a-, and IL-10-positive and negative cells among CD8 and CD4 T-cells. (D) Results are representative of three independent experiments. Significant relationships are indicated by P-values based on a two-tailed Student’s t-test. *P < 0.05.
Next, to determine the functional activity of CD4+ and CD8+ T-cells, we performed a CD107a mobilization assay and intracellular interferon (IFN)-γ staining. IHLs isolated from mice inoculated with SL4 cells were co-cultured with SL4 cells and recombinant IL-2 to analyze the cytokine/chemokine production. Cytolytic cell activation was then measured based on the degranulation marker CD107a. As shown in Figure 4C and 4D, the ratio of CD4+ and CD8+ CD107a+-cells relative to all T-cells decreased in IHLs isolated from SL4-inoculated mice after PRI-724 and PD-L1 Ab treatment. This indicated that cytolytic activity, such as perforin and granzyme B release, was not involved in the anti-tumor effects. In contrast, the ratio of CD8+ cells, but not CD4+IFN-γ+ cells, relative to all T cells following PRI-724 and PD-L1 Ab treatment was significantly increased compared to that in control treated mice, suggesting that CD8+ cells were activated in the livers of mice treated with the combination therapy. Interestingly, the proportion of CD8+ interleukin (IL)-10+ cells was also significantly reduced compared to that in the control group after co-injection. Together, these findings indicated that PRI-724 and PD-L1 Ab treatment resulted in the induction of an effective CD8+ T-cell immune response against SL4 cells in the liver.
CD8+ T-cells were required for the anti-tumor effect of combined anti-PD-L1 Ab and PRI-724 treatment
To evaluate the role of CD8+ T-cells in the anti-tumor effect of the combination treatment, a neutralizing Ab against CD8+ T-cells was injected into SL4-inoculated mice prior to administering the PRI-724 and PD-L1 Ab combination treatment. As shown in Figure 5, the anti-CD8 Ab, which eliminated CD8+ cells in the tumor (Supplementary Figure 3), inhibited the reductions in liver weight and Ki67-positive area that was mediated by the combined treatment. In contrast, based on liver weight and Ki67-positive area, an anti-CD4 Ab did not alter the anti-tumor effect. These results suggested that the anti-tumor activity of combined PRI-724 and anti-PD-L1 Ab treatment was dependent on CD8+ T-cells.
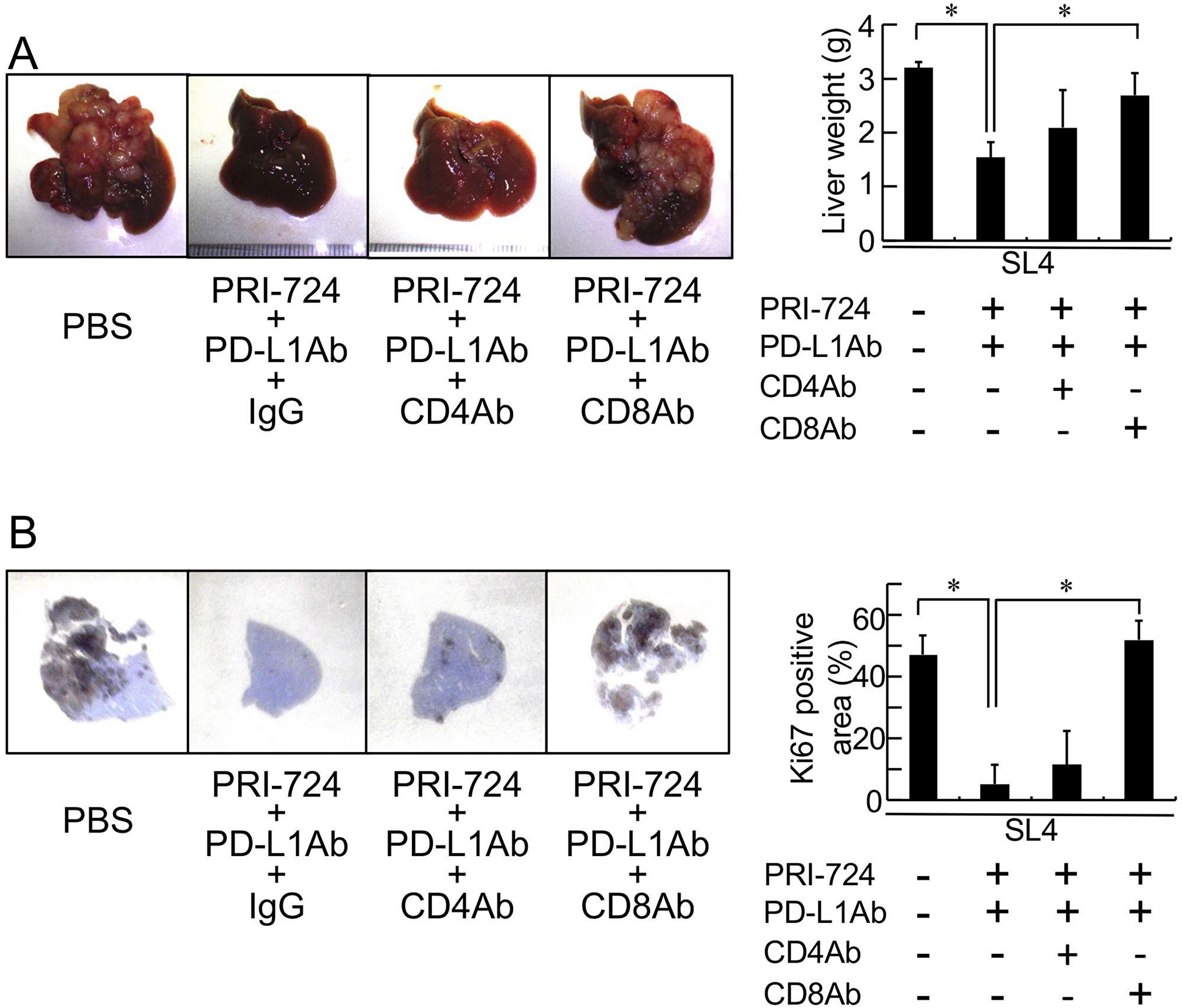
Figure 5: Anti-CD8 antibody (Ab) mitigated the inhibitory effects of an anti-PD-L1 antibody combined with a CBP/β-catenin inhibitor on tumor growth in the liver. Male C57BL/6J mice were intrasplenically injected with SL4 cells (5 × 105 cells). The animals were treated an anti-PD-L1 Ab and PRI-724 with or without an anti-CD4 or anti-CD8 Ab and were humanely sacrificed 14 days after inoculation. (A) The livers were excised and photographed (left panels). Liver weights were measured (right panel). (B) Expression of Ki67 in the metastatic liver tumor (loupe magnification) was examined by immunohistochemistry using an anti-Ki67 Ab. Intrahepatic tumor load is presented as Ki67-positive area for each animal (graph on right panel). The pictures shown are representative of at least four independent experiments. Results are given as means ± SD of data collected from at least four independent experiments. *P < 0.05 based on a one-way ANOVA test.
Inflammatory cytokine and chemokine expression in the livers of SL4-inoculated mice
Activation of β-catenin induces the expression of the transcriptional repressor ATF3, which suppresses CCL4 production leading to T-cell exclusion [6]. Indeed, mRNA expression levels of various chemokines in the livers of SL4-inoculated mice were increased following PRI-724 treatment (Table 2). In addition, serum levels of several chemokines in SL4-inoculated mice were also increased by PRI-724 administration (Supplementary Table 1). ATF3 mRNA expression was increased in vitro in SL4 cells cultured with C-82, an active form of PRI-724 (Supplementary Table 2). However, the expression levels of these chemokines in SL4 cells were low (Supplementary Table 1). Especially for levels of serum CCL4, which were not increased by SL4 cell inoculation or PRI-724 treatment (Supplementary Table 1). In contrast, mRNA expression levels of several chemokines were increased in IHLs isolated from the PRI-724-treated mice (Supplementary Table 3), suggesting that the source of the chemokines was not likely to be SL4 cells, but rather inflammatory cells. These increases in chemokines may have induced the CD8+ cells in the tumors.
Table 2: Changes in the mRNA profiles of chemokines, cytokines, and biomarkers of classically activated and regulatory macrophages in SL4 cell-inoculated mice
| PBS | PRI-724 | SL4 | ||
|---|---|---|---|---|
| PBS | PRI-724 | |||
| CCL2 | 1.00 ± 0.59 | 5.21 ± 1.74 | 14.15 ± 6.73 | 28.41 ± 11.47* |
| CCL3 | 1.00 ± 0.49 | 4.10 ± 2.23 | 18.12 ± 8.30 | 35.98 ± 5.81* |
| CCL4 | 1.00 ± 0.51 | 2.48 ± 0.69 | 11.05 ± 5.50 | 27.96 ± 10.63* |
| CCL5 | 1.00 ± 0.57 | 0.82 ± 0.13 | 2.40 ± 1.59 | 3.41 ± 1.06 |
| CCL7 | 1.00 ± 0.36 | 8.03 ± 1.80 | 28.44 ± 14.36 | 49.65 ± 21.70 |
| CCL8 | 1.00 ± 1.11 | 0.41 ± 0.21 | 10.59 ± 6.66 | 16.66 ± 6.19 |
| CXCL1 | 1.00 ± 0.47 | 18.84 ± 6.51 | 1.28 ± 1.03 | 2.21 ± 1.63 |
| CXCL2 | 1.00 ± 0.24 | 5.05 ± 2.63 | 30.99 ± 22.82 | 39.03 ± 17.59 |
| CXCL3 | 1.00 ± 0.41 | 5.08 ± 2.33 | 58.41 ± 49.23 | 76.54 ± 48.01 |
| CXCL9 | 1.00 ± 0.41 | 1.21 ± 0.09 | 1.30 ± 0.65 | 7.57 ± 1.32* |
| CXCL10 | 1.00 ± 0.37 | 2.04 ± 0.24 | 0.66 ± 0.19 | 3.44 ± 1.02* |
| CXCL12 | 1.00 ± 0.18 | 0.81 ± 0.13 | 0.32 ± 0.09 | 0.32 ± 0.13 |
| CXCL13 | 1.00 ± 0.16 | 1.23 ± 0.14 | 1.15 ± 0.71 | 0.25 ± 0.67 |
| TNF-α | 1.00 ± 0.47 | 2.38 ± 0.71 | 3.11 ± 1.57 | 8.09 ± 1.62* |
| IL-1β | 1.00 ± 0.26 | 2.65 ± 1.05 | 3.19 ± 1.84 | 8.87 ± 2.20* |
| IL-6 | 1.00 ± 0.64 | 3.66 ± 1.63 | 10.59 ± 4.41 | 21.79 ± 15.09 |
| CD11c | 1.00 ± 0.68 | 0.53 ± 0.16 | 31.64 ± 12.49 | 85.24 ± 30.64* |
| IFN-γ | 1.00 ± 1.31 | 0.47 ± 0.04 | 3.12 ± 4.20 | 31.52 ± 17.98* |
| CD163 | 1.00 ± 0.24 | 1.07 ± 0.10 | 1.13 ± 0.59 | 0.77 ± 0.35 |
| Mannose receptor | 1.00 ± 0.48 | 0.80 ± 0.34 | 1.00 ± 0.36 | 1.26 ± 0.64 |
| IL-10 | 1.00 ± 0.70 | 1.96 ± 1.01 | 26.57 ± 12.50 | 31.93 ± 7.84 |
DISCUSSION
In the current study, we investigated the effect of a CBP/β-catenin inhibitor combined with PD-L1/PD-1 blockade on the suppression of tumor growth in a mouse model of colon cancer liver metastasis. Our results indicated that inhibition of CBP/β-catenin increased the therapeutic effect of PD-L1/PD-1 blockade through the accumulation of CD8+ T-cells in the tumors. These results suggest therapeutic potential for treating metastatic liver tumors derived from colon cancer.
Previously, poor anti-tumor efficacy was reported for anti-PD-1/PD-L1 monotherapies with colorectal cancer [2, 4]. Similarly, anti-PD-L1 Ab monotherapy demonstrated no anti-tumor activity in our mouse model. However, an inverse correlation between PD-L1 expression and disease-free survival in stage III colorectal cancer has been reported [15]. Therefore, the immune checkpoint appears to contribute to colon cancer progression. Importantly, in vitro evaluation of SL4 cells treated with C-82 and an anti-PD-L1 Ab revealed no cytotoxic effects (data not shown). In melanoma, tumor regression mediated by the anti-tumor effects of an anti-PD-1 Ab requires the presence of CD8+ T-cells in the tumor [5]. In colon cancer, tumor T-lymphocytes correlate with patient survival [16–18] and enhanced CD8+ T-cell infiltration to the invasive margin of liver metastasis is predictive of a better response to chemotherapy [19]. In our model, the number of CD3+ cells in metastatic tumors was comparable to that in normal liver tissues. Moreover, anti-PD-L1 Ab monotherapy did not increase the number of CD3+ cells. In such states of diminished T-lymphocytes in tumors, the anti-tumor effects of the anti-PD-L1 Ab might not be apparent. In contrast, the number of CD3+ cells did increase in metastatic tumors following PRI-724 treatment and anti-tumor activity by anti-PD-L1 Ab was demonstrated in these mice. These anti-tumor effects were mitigated by an anti-CD8+ Ab, indicating that an increase in CD8+ cells was essential for the anti-tumor activity of the anti-PD-L1 Ab in this model. Because PRI-724 monotherapy failed to show anti-tumor effects, despite the increase in CD3+ cells, T-cell accumulation was essential but not sufficient for the inhibition of metastatic tumor growth. Following T-cell activation, PD-1 is expressed and receptor signals limit the expansion and activation of T-cell receptor-triggered T cells [20]; indeed, anti-PD-L1 therapy increased lymphocyte activation in our model. These results suggest that, in addition to the increase in T-cell infiltration mediated by PRI-724, T-cell activation via anti-PD-L1 agents was required for anti-tumor activity. Moreover, the accumulation of T cells in the tumors was more evident in animals treated with the combination of these drugs (Figure 3). Furthermore, the combined treatment lowered expression of PD-1 on CD8+ T-cells in the liver, which resulted in infiltration of effector CD44lowCD62Llow cells among the CD4+ and CD8+ T-cells. Consistent with this result, the CD8+ T-cells in the liver exhibited enhanced IFN-γ production, and conversely, IL-10 production was suppressed when CD8+ T-cells were co-culture with SL4 cells. These findings revealed that the combined treatment induced a robust CD8+ T-cell immune response to the tumor. The anti-tumor activity exerted by CD8+ T-cells was not a result of the degranulation of CD8+ cells, and thus, apoptosis pathways via TNF-α or Fas may have been involved in the anti-tumor effects. Unlike that observed in melanoma, the accumulation of T lymphocytes was not due to the production of CCL4 by the tumors. Instead, the levels of several chemokines were increased in IHLs isolated from PRI-724-treated mice. We previously reported that the depletion of liver macrophages via alendronate liposome treatment increases SL4 cell tumor growth in the liver, which is accompanied by a decrease in CD3+ cells in metastatic liver tumors [21], suggesting that liver macrophages promote T-cell accumulation.
It has been demonstrated that liver macrophages modulate the host immune response to cancer cells by releasing cytotoxic products and immune-stimulating factors [22]. Macrophage depletion increases the severity of tumor growth in the livers of SL4-inoculated mice [21], which is accompanied by a decrease in CD3+ T-cell infiltration. Although the number of F4/80+ cells per high-power microscopic field of metastatic liver tumors was not altered (Figure 3), the expression of inflammatory cytokines TNF-α and IL-1β, which are mainly produced by macrophages in the liver, was increased in the livers of SL4-inoculated mice following PRI-724 treatment (Table 2). In addition, TNF-α serum levels were increased by PRI-724 treatment in these animals (Supplementary Table 2). Moreover, mRNA expression levels of the classically activated M1 macrophage markers CD11c and IFN-γ were increased by PRI-724 treatment, whereas expression levels of the genes encoding markers of regulatory M2 macrophages, such as CD163, mannose receptor, and IL-10, were not affected (Table 2). M1 macrophages are considered to be cytotoxic macrophages that inhibit cancer growth while repair-type M2 macrophages stimulate tumor growth [23]. Thus, PRI-724 also appears to contribute to tumor regression by altering macrophage properties and promoting a cytotoxic phenotype, which might induce T-cell infiltration to the tumor. In an autoimmune hepatitis mouse model induced by neonatal thymectomy and PD-1 knockout, the migration of T cells was found to be triggered by hepatic macrophages that produce c-x-c motif ligand (CXCL) 9 [24]. M1 and M2 macrophages have different chemokine-production profiles with M1 macrophages thought to produce T-cell-attracting chemokines such as CXCL9 and CXCL10 [25]. In our model, it appeared that PRI-724 altered macrophage properties toward a cytotoxic M1 phenotype since CXCL9 and CXCL10 mRNA expression were increased by PRI-724 in the livers of mice inoculated with SL4 cells. In addition to macrophages, dendritic cells also express CD11c and may also contribute to the activity. Thus, we believe that the accumulation of T lymphocytes caused by PRI-724 might occur through M1 macrophages and dendritic cells. This is supported by the fact that β-catenin is well known to contribute to macrophage motility and adhesion [26]; however, its roles in chemokine production in this model remains unclear.
In contrast, Gr-1-positive cells accumulated locally in the central part of the tumor (Figure 3), suggesting that neutrophils did not contribute to the observed tumor regression. Regulatory T (Treg) lymphocytes are known to inhibit anti-tumor immunity by inhibiting the effector T-cell response. Overexpression of stabilized β-catenin in Treg lymphocytes was previously found to increase the survival of these cells [27]. However, Treg lymphocytes in our study were not decreased by treatment with PRI-724 and anti-PD-L1 Ab (Table 1), suggesting that these cells were not involved in the observed anti-tumor effects.
Unfortunately, our data failed to identify the source of chemokines found to be induced upon PRI-724 treatment; however, the effect of the anti-PD-L1 Ab on macrophages has not yet been investigated. It is not clear which chemokines were involved in the PRI-724-induced CD8+ cell infiltration into the tumors. Moreover, the potential roles of other liver cells currently remains unclear as does the mechanism through which CBP/β-catenin stimulates the activation of macrophages. The effects of combination therapy were not sufficient to induce the complete elimination of the tumors. It is clear that further studies are needed to resolve these uncertainties.
In conclusion, inhibition of CBP/β-catenin increased the anti-tumor effects of anti-PD-L1 Ab therapy against metastatic liver tumors derived from colon cancer by promoting the accumulation of CD8+ T-cells and inducing a shift in the properties of macrophages toward a cytotoxic phenotype. Thus, approaches targeting CBP/β-catenin combined with PD-1/PD-L1 immune checkpoint blockade represents a novel therapeutic strategy for treating liver metastasis during colon cancer.
Materials and Methods
Study approval
The experiments were conducted in accordance with institutional guidelines (Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences) and the protocol was approved by the Research Committee of Komagome Hospital. All surgery was performed under anesthesia and all efforts were made to minimize suffering.
Information regarding PRI-724 and C82
C-82 is a second-generation specific CBP/β-catenin antagonist developed by Prism Pharma, which inhibits the binding between β-catenin and CBP and increases the binding between β-catenin and p300. PRI-724 is the phosphorylated form of C-82 and is rapidly hydrolyzed in vivo to its active form C-82. The chemical name of PRI-724 is 4-(((6S,9S,9aS)-1-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro-1H-pyrazino[2,1-c][1,2,4]triazin-6-yl)methyl)phenyldihydrogen phosphate. The chemical name of C-82 is 4-(((6S,9S,9aS)-1-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro-1H-pyrazino[2,1- c][1,2,4]triazin-6-yl)methyl)phenyl.
Cell culture
A green fluorescent protein (GFP)-expressing SL4 mouse colon adenocarcinoma cell line (Anti-Cancer Japan, Osaka, Japan) was maintained as a monolayer culture in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) supplemented with penicillin and streptomycin (Invitrogen). For in vitro experiments, the cells were treated with or without C-82 (1 μM) for 24 h. For cell inoculation into mice, SL4 cells were harvested using trypsin and EDTA, washed with PBS, and then resuspended in PBS at a concentration of 5 × 106 cells/mL.
Liver metastasis model
Male wild-type C57BL/6J mice 8-weeks of age were obtained from Japan SLC (Shizuoka, Japan). After making a small incision under anesthesia to expose the spleen, 0.1 mL of a viable cell suspension containing 5 × 106 cells/mL was injected into the spleen. We chose SL4 cells because the cells grow rapidly, even in the liver of wild-type mice [21]. The animals were then each intraperitoneally injected with or without 0.4 mg PRI-724 (Prism Pharma, Yokohama, Japan) and/or 200 μg of an anti-PD-L1 Ab (10F.9G2; Bio X Cell, NH, USA) three times per week. In addition, some mice treated with PRI-724 and the anti-PD-L1 Ab were administrated anti-mouse CD4 or CD8 Ab (250 μg/mouse; Bio X Cell) three times per week. After the course of treatment, the mice were anesthetized and humanely sacrificed by exsanguination 14 days post cell-inoculation. The livers of the animals were immediately removed, washed in ice-cold PBS, and weighed before a portion of the dissected liver tissue was frozen in liquid nitrogen. Additional animals were maintained and used for survival analysis.
Histological analysis
The mouse livers were fixed with 10% formalin, sectioned, and stained with hematoxylin and eosin (H-E). Ki67, CD3, F4/80, and Gr-1 immunostaining was performed using anti-Ki67 (SP6, NeoMarkers, Fremont, CA, USA), anti-CD3 (SP7, Abcam, Cambridge UK), anti-F4/80 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or Gr-1 (eBioscience, San Diego, CA, USA) Abs, respectively, and a VECTASTAIN Elite ABC Kit. Diaminobenzidine tetrahydrochloride (DAB) was used as the peroxidase substrate and sections were counterstained with hematoxylin. The positive immunostained area was analyzed using ImageJ software (National Institutes of Health [NIH], Bethesda, MD, USA; http://rsb.info.nih.gov/ij/) and the percentage of positive-stained tissue relative to the total area of the tissue section was determined.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
RNeasy and DNase Kits from Qiagen (Valencia, CA, USA) were used for RNA extraction from liver tissue and cultured cells, respectively, and a High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems (Foster City, CA, USA) was used for reverse transcription of the RNA into complementary DNA (cDNA). Real time RT-qPCR was performed using the SYBR Premix Ex Taq (Takara, Shiga, Japan) for genes encoding CCL-2, CCL-3, CCL-4, CCL-5, CCL-7, CCL-8, CXCL-1, CXCL-2, CXCL-3, CXCL-9, CXCL-10 CXCL-12, CXCL-13, ATF-3, CD11c, IFN-γ, CD163, mannose receptor, and IL-10 (primer sequences are shown in Supplementary Table 4). Probe and primer sets from Applied Biosystems was used for PCR amplification of 18S rRNA (Hs99999901s1), tumor necrosis factor (TNF)-α (Mm00443258m1), IL-1β (Mm00434228m1), and IL-6 (Mm99999064m1) using Thunderbird Probe qPCR mix (Toyobo, Tokyo, Japan) and a LightCycler 480 (Roche Applied Science, Mannheim, Germany). The results were normalized to 18S rRNA expression values. For mRNA expression analysis of Wnt target genes, an RT2 Profiler™ PCR Array (Mouse Wnt Signaling Targets, QIAGEN Sciences, MD, USA) was used and real time RT-qPCR was performed using RT² SYBR Green qPCR Mastermix and an RT2 First Strand Kit (QIAGEN).
Isolation of mouse IHLs
Single-cell suspensions were prepared from the liver median lobe by digesting the tissue in RPMI-1640 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) containing 0.02% collagenase IV and 0.002% DNase I (Sigma-Aldrich, St. Louis, MO, USA) for 40 min at 37°C [28]. The cells were overlaid onto Lympholyte-M (Cedarlane, Westbury, NY, USA) in PBS. After density separation, the isolated IHLs were evaluated by FACS analysis.
FACS analysis
The cells were surface-stained with fluorochrome-conjugated Abs for 20 min on ice using the following Abs: anti-CD3, anti-CD4, anti-CD8, anti-NK1.1, anti-CD69 (eBioscience), and anti-Foxp3 (BioLegend, San Diego, CA, USA). To perform intracellular cytokine staining, IHLs were co-cultured with SL4 cells (1 × 105 cells/well) for 4 h at 37°C in 96-well round-bottom plates containing 200 μL/well RPMI-1640 medium. Mouse recombinant IL-2 (50 units) and 0.2 μL of BD GolgiPlug protein transport inhibitor (BD Biosciences) were added to each well. After incubation, the cells were harvested, washed in PBS containing 1% FBS, and incubated for 10 min on ice with unlabeled anti-mouse CD16/32 Ab (BD Biosciences) to block FcγRII/III binding. The cells were then surface-stained for 20 min on ice with the indicated Abs. Following staining, the cells were washed to remove unbound Ab and fixed using a Cytofix/Cytoperm Kit (BD Biosciences). Cells were then subjected to secondary staining with reagents obtained from BioLegend except as noted, including the following: FITC-conjugated CD107a, PE-conjugated anti-interferon gamma, and anti-IL-10 (BD Biosciences). Data were acquired using a FACSCalibur flow cytometer and analyzed using CellQuest (BD Immunocytometry Systems, San Jose, CA, USA) and FlowJo software (Tree Star, San Carlos, CA, USA).
Measurement of serum cytokines and chemokines
Serum ALT levels were measured using a Wako Transaminase CII-test Kit (FUJIFILM Wako). Serum cytokines and chemokines were measured using a Luminex MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay (MCYTOMAG-70K; Merck Millipore, Darmstadt, Germany). This procedure was performed in accordance with the manufacturer’s instructions.
Statistical analysis
Results are expressed as the mean ± standard deviation (SD) of the data collected from at least three independent experiments. Data between groups were analyzed by one-way ANOVA, the Kruskal-Wallis test, or a two-tailed Student’s t-test. Kaplan–Meier survival analysis was performed using the log-rank test. A P-value less than 0.05 was considered statistically significant. The software used for all statistical analyses was Graph Pad Prism version 7 (Graph Pad Software Inc., San Diego, CA, USA).
Abbreviations
PD-L1: programmed cell death ligand 1; CREB: cAMP-response element-binding protein; CBP: (CREB)-binding protein; PD-1: programmed cell death 1; IFN: interferon; Ab: antibody; CCL: chemokine ligand; ATF: AMP-dependent transcription factor; IHL: intrahepatic leukocytes; IL: interleukin; CXCL: c-x-c motif ligand; T-reg: regulatory T; TNF: tumor necrosis factor.
ACKNOWLEDGMENTS
The authors would like to thank Kei Kozuka, Fumi Wakui, and Yukiko Hayashi for technical assistance.
CONFLICTS OF INTEREST
There is no conflicts of interest.
FUNDING
This work was supported by the following sponsors and grants: the Research Program on Hepatitis (15fk0210025h003, 15fk0210034h0001), Development of the Translational Research Network Program (16lm0103008j0005), and Acceleration Transformative Research for Medical Innovation (ACT-M) (16im0210105h0001) from the Japan Agency for Medical Research and Development (AMED).
References
1. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015; 27:450–61. https://doi.org/10.1016/j.ccell.2015.03.001. [PubMed].
2. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. https://doi.org/10.1056/NEJMoa1200690. [PubMed].
3. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596. [PubMed].
4. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366:2455–65. https://doi.org/10.1056/NEJMoa1200694. [PubMed].
5. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568–71. https://doi.org/10.1038/nature13954. [PubMed].
6. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015; 523:231–5. https://doi.org/10.1038/nature14404. [PubMed].
7. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469–80. https://doi.org/10.1016/j.cell.2006.10.018. [PubMed].
8. Osawa Y, Oboki K, Imamura J, Kojika E, Hayashi Y, Hishima T, Saibara T, Shibasaki F, Kohara M, Kimura K. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/beta-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine. 2015; 2:1751–8. https://doi.org/10.1016/j.ebiom.2015.10.010. [PubMed].
9. Tokunaga Y, Osawa Y, Ohtsuki T, Hayashi Y, Yamaji K, Yamane D, Hara M, Munekata K, Tsukiyama-Kohara K, Hishima T, Kojima S, Kimura K, Kohara M. Selective inhibitor of Wnt/beta-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model. Sci Rep. 2017; 7:325. https://doi.org/10.1038/s41598-017-00282-w. [PubMed].
10. Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, Kudo-Saito C, Fujita T, Sumimoto H, Kawakami Y. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J Immunol. 2012; 189:2110–7. https://doi.org/10.4049/jimmunol.1102282. [PubMed].
11. Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, Wigmore SJ, Sansom OJ, Forbes SJ. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015; 125:1269–85. https://doi.org/10.1172/jci76452. [PubMed].
12. Nagaraj AB, Joseph P, Kovalenko O, Singh S, Armstrong A, Redline R, Resnick K, Zanotti K, Waggoner S, DiFeo A. Critical role of Wnt/beta-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 2015; 6:23720–34. https://doi.org/10.18632/oncotarget.4690. [PubMed].
13. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008; 26:677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331. [PubMed].
14. Staal FJ, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol. 2008; 8:581–93. https://doi.org/10.1038/nri2360. [PubMed].
15. Koganemaru S, Inoshita N, Miura Y, Miyama Y, Fukui Y, Ozaki Y, Tomizawa K, Hanaoka Y, Toda S, Suyama K, Tanabe Y, Moriyama J, Fujii T, et al. Prognostic value of Programmed death-ligand 1 expression in patients with stage III colorectal cancer. Cancer Sci. 2017; 108:853–858. https://doi.org/10.1111/cas.13229. [PubMed].
16. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005; 353:2654–66. https://doi.org/10.1056/NEJMoa051424. [PubMed].
17. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006; 313:1960–4. https://doi.org/10.1126/science.1129139. [PubMed].
18. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12:298–306. https://doi.org/10.1038/nrc3245. [PubMed].
19. Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B, Feyen N, Martens UM, Beckhove P, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011; 71:5670–7. https://doi.org/10.1158/0008-5472.can-11-0268. [PubMed].
20. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009; 27:591–619. https://doi.org/10.1146/annurev.immunol.021908.132706. [PubMed].
21. Osawa Y, Suetsugu A, Matsushima-Nishiwaki R, Yasuda I, Saibara T, Moriwaki H, Seishima M, Kozawa O. Liver acid sphingomyelinase inhibits growth of metastatic colon cancer. J Clin Invest. 2013; 123:834–43. https://doi.org/10.1172/jci65188. [PubMed].
22. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010; 141:39–51. https://doi.org/10.1016/j.cell.2010.03.014. [PubMed].
23. Mills CD, Lenz LL, Harris RA. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016; 76:513–6. https://doi.org/10.1158/0008-5472.can-15-1737. [PubMed].
24. Ikeda A, Aoki N, Kido M, Iwamoto S, Nishiura H, Maruoka R, Chiba T, Watanabe N. Progression of autoimmune hepatitis is mediated by IL-18-producing dendritic cells and hepatic CXCL9 expression in mice. Hepatology. 2014; 60:224–36. https://doi.org/10.1002/hep.27087. [PubMed].
25. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006; 177:7303–11. https://doi.org/10.4049/jimmunol.177.10.7303. [PubMed].
26. Amini-Nik S, Cambridge E, Yu W, Guo A, Whetstone H, Nadesan P, Poon R, Hinz B, Alman BA. β-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J Clin Invest. 2014; 124:2599–610. https://doi.org/10.1172/jci62059. [PubMed].
27. Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008; 14:162–9. https://doi.org/10.1038/nm1707. [PubMed].
28. Kimura K, Nagaki M, Takai S, Satake S, Moriwaki H. Pivotal role of nuclear factor kappaB signaling in anti-CD40-induced liver injury in mice. Hepatology. 2004; 40:1180–9. https://doi.org/10.1002/hep.20432. [PubMed].