
INTRODUCTION
Myocyte enhancer factor (MEF)-2 is a member of the MCM+Agamous+Deficiens+Serum response factor (MADS) box group of transcription factors. MEF-2 transcription factors have been well studied in the context of various developmental programs, including those of skeletal muscle, cardiac muscle, and neural tissue [1]. There are 4 isoforms of MEF-2, namely MEF-2A, MEF-2B, MEF-2C, and MEF-2D. These isoforms play an important role in embryogenesis and other epigenetic modifications that control gene expression [2–14]. Initially studied in the context of muscle development; involvement of MEF-2 is now well established in B-cell development, T-cell differentiation and thymocyte selection. MEF-2 regulates calcium-dependent transcription of the Interleukin-2 (IL-2) gene in T lymphocytes, and down-modulating MEF-2 by siRNA in primary human T cells leads to the inhibition endogenous IL-2 transcription. Due to intrinsic involvement of MEF-2 in T-cell development and IL-2 transcription, this transcription factor has been associated with T-cell leukemias/lymphomas (TCLs) in addition to a variety of other cancers as will be discussed later for each isoform. There are four major subtypes of TCLs: human T-Cell Acute Lymphoblastic Leukemia/Lymphoma (T-ALL), Adult T-Cell Leukemia/Lymphoma and Peripheral T-cell Lymphoma/unspecified (PTCL/U) [15]. MEF-2 isoforms are transcriptionally dysregulated in T-ALL and possess non-rearranged T-cell receptors [15, 16]. Moreover, MEF-2 has been implicated in HTLV-1-induced ATLL [17], and is under clinical investigation in various trials of CTCL and PTCL by targeting via HDACi [18].
Over the years, research progress has altered our perception of MEF-2 as less of a conventional transcription factor involved in development, and more as a vital player in tumor and leukemogenesis. MEF-2 is not only implicated in development, regulation and carcinogenesis, but also in chronic viral infections such as Epstein-Barr virus (EBV) [19–22], Human immunodeficiency virus-1 (HIV-1) [23] and Human T-cell leukemia virus-1 (HTLV-1) [17]. In addition, MEF-2 has been implicated in muscle related parasitic infections such as Trichinella [24]. It can also act as an immune-metabolic switch that can regulate infection susceptibility in drosophila models [25]. Our investigations into MEF-2 and HTLV-1 [26–40] are now providing rationale towards potential therapeutic strategies against virally induced ATLL based upon MEF-2: HDAC interactions. Our lab was the first to describe MEF-2A as a regulator of viral gene expression in the pathogenesis of the retrovirus HTLV-1. HTLV-1 encodes an oncogenic protein in the pX region of the viral genome known as Tax. Tax directly binds to MEF-2A and together with various co-activating transcription complexes hijacks the host transcriptional machinery to increase viral gene expression [17, 28]. With the exception of this study, the roles of MEF-2 and its isoforms in the course of HTLV-1 infection remain largely unexplored. MEF-2 isoforms were reported as the direct gene target of EBV nuclear antigen 1 (EBNA1), which supports the survival of EBV infected B-lymphocytes. EBNA-1 bound to B-cell specific transcription factors can inhibit MEF-2B and also suppresses cell growth as well as the viability of latently infected EBV cells [21, 22]. Additionally, other groups reported that MEF-2B enhances the activity of the promoter Zp of viral trans-activator protein BZLF1 by approximately 17.8-fold and promotes viral reactivation from latency [21].
MEF-2 contains a highly conserved MADS domain in the N-terminus, approximately 55 amino acids. The adjacent MEF-2 domain (29 amino acids) is also evolutionarily conserved, and serves as the DNA binding domain, interaction region for other proteins, and a site for dimerization. The MEF-2 domain acts as a docking site for various coactivators such as p300, CREB, etc., and corepressors including CABIN1, class IIa HDACs (histone deacetylases) and some kinases like GSK3β and other selective regulators like Protein Kinase A (PKA) [6, 8, 41, 42]. The transcriptional program of MEF-2 involves interactions with various coactivators such as p300, CBP, cFOS, cJUN, and ATF [43–52]. Another negative regulator of MEF-2 is Glycogen synthase kinase 3β (GSK3β) which has an indirect role of regulation of suppression of MEF-2 by modulation of p38MAPK. Pharmacological inhibition of GSK3β resulted in the increased activity of MEF-2(A-D) expression in myoblasts and cardiac myocytes. In-silico analysis showed consensus sites for GSK3β on MEF-2 as (S/T) XXX(S/T), although in vitro kinase assay stated that MEF-2A is only a weak substrate. Heart specific knockout of GSK3β in mice resulted in the upregulation of p38MAPK activity, suggesting the GSK3β as a negative regulator of MEF-2 isoforms and suggested crosstalk between the P38MAPK and GSK3β [53]. In cultured cerebellar neurons, a non-competitive inhibitor of GSK3β, inhibited caspase-3 activation and chromatin condensation but removing the depolarizing potassium and serum. Also, Lithium decreased MEF-2D hyperphosphorylation and apoptosis induced by calcineurin inhibition under depolarizing conditions. This suggests that GSK-3β phosphorylates and inhibits pro-survival activity of MEF-2D in cerebellar granular neurons [54]. GSK3β has been implicated in neuronal death and increase in its activity can induce the neurodegeneration and Alzheimer’s disease. It has been stated that phosphorylation of MEF-2D at three specific residues in the transactivation domain inhibits MEF-2D transcriptional activity. Overexpressing a MEF-2 mutant resistant to GSK3β inhibition protected cerebellar granular neurons survival, stating the more suppressive role of GSK3β role in MEF-2 transcriptional activity [55]. In cardiomyocytes, CaMKII promotes hypertrophy and pathological remodeling by phosphorylating HDAC4 and subsequent activation of MEF-2. Protein kinase A (PKA) overcomes CaMKII mediated activation and selectively activates MEF-2 by regulated proteolysis of HDAC4. PKA degrades the N-terminal of HDAC4(HDAC-NT), which selectively inhibits the MEF-2 domain but not the SRF, thereby antagonizing the prohypertropic activity of CaMKII, without causing any effect on the cardiomyocyte survival and aiding in the cardio-protection and other cellular processes [56]. Although in certain studies it has been claimed that activation of PKA elevates the intracellular levels of cyclic AMP (cAMP) and inhibits skeletal myogenesis and this suggests MEF-2D as primary target of PKA and represses the transactivation of MEF-2D, but enhanced accumulation of HDAC4-MEF-2 complex inhibits the skeletal muscle differentiation [57]. It has been also shown that in embryonic day 18 (E18), Sprague Dawley hippocampal neurons, with the experimental induction of cAMP/PKA signaling promoted apoptosis. Also, Krüppel- like factor 6 (KLF6) was a transcriptional target of MEF-2 hippocampal neurons and knockdown of KLF6 antagonized the pro-survival role of MEF-2D and caused neuronal cell death [58]. HDACs are important and well characterized transcriptional partners of MEF-2, which have been exploited for therapeutic intervention using HDAC inhibitors (HDACi) to modulate the transcriptional machinery via the HDAC: MEF-2 axis. There are 18 categories of HDACs classified on the basis of their homology with yeast transcriptional regulator RPD3 and other biochemical properties [59]. The histone tails and their interactions with the DNA that control their modifications lead to activation or repression of gene transcription. Of these HDACs, classes I, II, and IV are zinc dependent and class III is NAD+ dependent. Class I HDACs [1–3, 8] are expressed ubiquitously in human cell lines and tissues, and are predominantly expressed in the nucleus. The class II HDACs can be defined into two subgroups IIA and IIB, which comprise HDAC4, 5, 7, and 9, and HDAC6, and 10 respectively, and they tend to shuttle between the nucleus and the cytoplasm. The third major group of HDACs consist of Class III HDACs and are also known as Sirtuins (SIRT1-7); at present their subcellular localization and tissue-specific properties if any, are not fully known. Class IV HDACs consists of only the most recently discovered HDAC11, and shows homology with both classes I and II [22, 60–70]. Class IIa HDACs are involved in the direct binding and suppression of MEF-2 proteins through the MADS/MEF-2 domains in the N-terminus. Association of class II HDACs with MEF-2 results in the deacetylation of histones in the vicinity of MEF-2 DNA-binding sites and subsequent suppression of MEF-2 target genes.
Many HDACs are a part of multiprotein complexes and interact with various proteins that affect their activity and specificity, and are controlled by various post-translational modifications such as phosphorylation, acetylation, and sumoylation [71, 72]. HDACs are constitutively bound with co-repressor complexes including HDAC1: SIN3, NURD and HDAC3: NCOR1 (nuclear receptor co-repressor 1) and SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) [73]. The class IIa HDACs form multiprotein complexes with MEF-2, NCOR1 and SMRT along with association with HDAC3 for regulation of its deacetylase activity. The nuclear co-repression of the various transcription factors is regulated by phosphorylation events which promotes the binding of 14-3-3 via G-protein coupled receptors, and results in their shuttling back to the cytosol thus allowing for reactivation of the target genes [74]. HDACs not only target histone moieties but target non-histone targets as well, thereby enhancing the diverse regulatory properties of PTMs. There are lysine acetylation signatures that have been identified not only on nuclear proteins but also on cytosolic and mitochondrial proteins across many different species [75]. More recently, the MEF-2: HDAC axis has also been implicated in various cancers. Increased expression of class IIa HDACs has been correlated with suppression of MEF-2 transcriptional activity and poor prognosis of estrogen receptor-positive (ER+) breast tumors as well as increased proliferation of mammary epithelial cells [76]. Overexpression of HDAC9 has been linked to poor prognosis of oral squamous cell carcinoma by targeting MEF-2D and repressing expression of MEF2-dependent genes [77]. Class IIa HDACs in conjunction with MEF-2 have been implicated in leiomyosarcomas as well [78].
Because of their widespread influence on oncogenesis, HDACs have emerged as the target of an entire class of anticancer drugs collectively known as HDACi (Histone deacetyalase inhibitors) that encompass a diverse group of small molecule drugs that can induce apoptosis, cell cycle arrest, differentiation, and autophagy of cancer cells and promote anti-angiogenic effects [79–86]. These drugs are mostly efficacious for treatment of leukemias, and have not shown great success against solid tumors [83, 86]. MEF-2 proteins also play a key role in T-cell survival by interacting with calcium-regulated NFAT family members, p300, and p/CAF [87–91]. This review will provide a detailed discussion on the signaling pathways that control the activation of different isoforms of MEF-2 via various signaling molecules and cascades, and their implications in various disease phenotypes. Furthermore, this review will also provide a glimpse of the potential of HDACi in the context of MEF-2-based therapeutics.
REGULATION OF MEF-2
MEF-2 family proteins act as effector molecules for multiple signaling cascades, with the major regulators of their signaling being activated and guided by multiple pathways through various second messenger and signaling molecules. Various activators of MEF-2 activity include Calcium (Ca2+)-mediated signaling, Mitogen activated protein signaling (MAPK), Wnt signaling and the PI3K/AKT signaling pathways. The basal expression in various tissues and cells will be illustrated in Figure 1 and a detailed outline of the key regulatory molecules as well as the signaling pathways are provided in Figures 2 and 3 of this review.
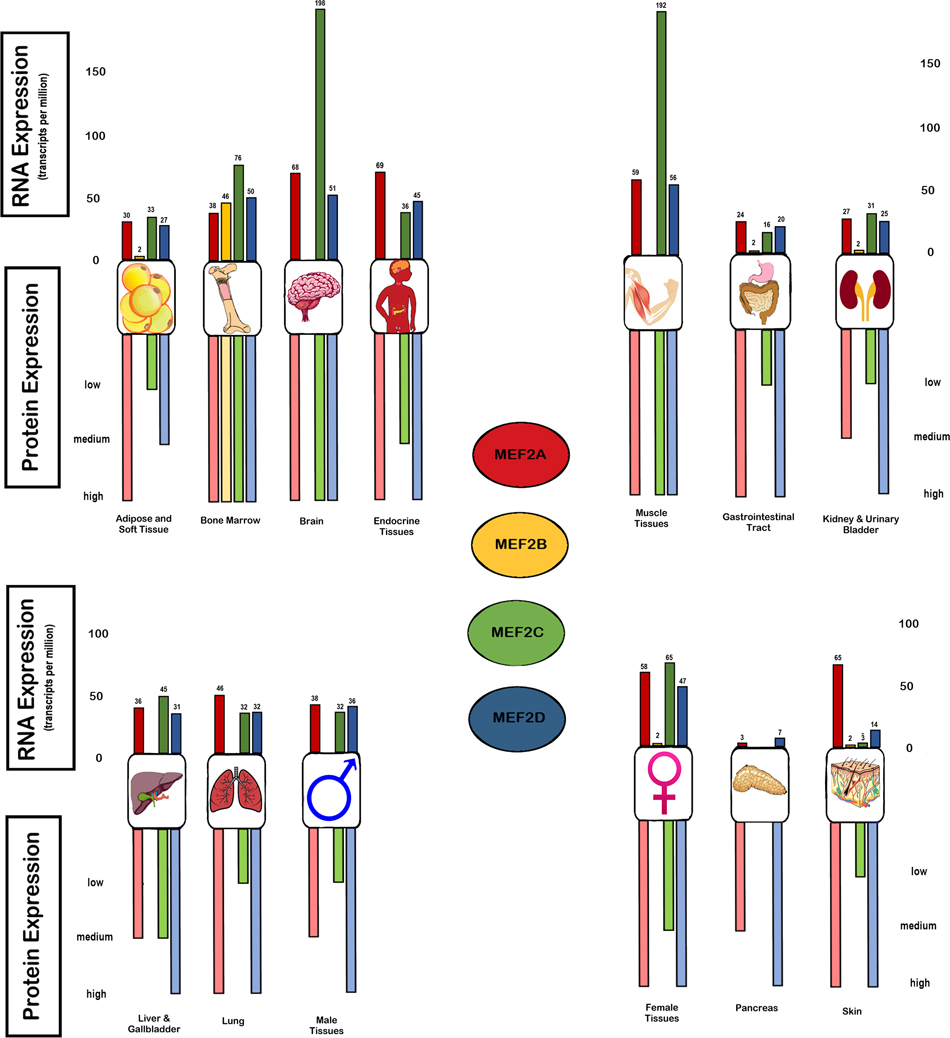
Figure 1: Basal expression of MEF-2 isoforms in organ systems and tissues in RNA and protein. The expression of MEF-2 isoforms are illustrated in the various organ systems at the transcriptome level in TPM (transcripts for kilobase million) via RNA-seq expression and the protein expression from high to low levels which is obtained from the immune-cytochemistry quantification (https://www.proteinatlas.org/).
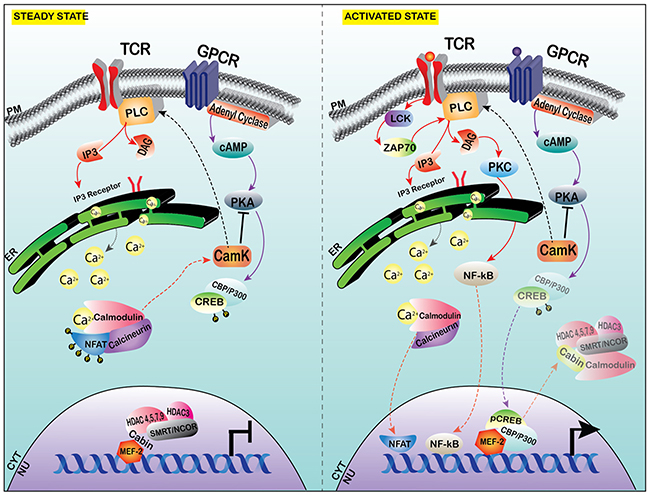
Figure 2: Calcium signaling affecting MEF-2 transcription at steady state versus the activated state in T cells. MEF-2 is constitutively bound with co-repressor complex, which comprises of CABIN-1, HDAC3 and Class II HDACs along with SMRT/NCOR in steady state. With the activation of stimulus, calcium stores from the ER are released into the cytoplasm via the IP3 receptor activation. The Ca2+ activates the calmodulin-calcineurin complex, which dephosphorylates the inactive NFAT in the cytoplasm into activated NFAT and translocates into the nucleus (this has been observed in depolarized neurons and skeletal myoblasts in specific isoforms of MEF-2). In T-lymphocytes, Calcium influx activates the CamK and PKA, which allows the transcription of NF-ĸB. PKA phosphorylates CREB and pCREB/P300 complex translocates to the nucleus and the co-repressor shuttles out of the nucleus and allows the activation of MEF-2, in the proliferation of T cells (shown in Jurkat T-cell line in-vitro).
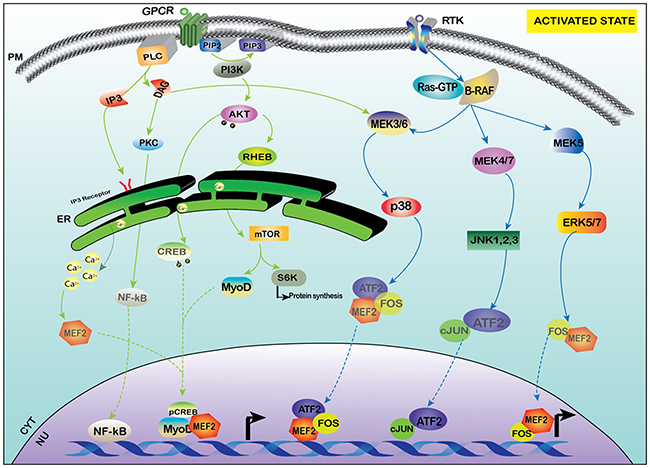
Figure 3: Activation of MEF-2 by PI3K-AKT and MAPK signaling. MEF-2 is also activated by PI3K/AKT pathway which activates mTOR as a downstream target and also increases the transcription of MyoD and MEF-2 along with other transcription factors. The receptor tyrosine kinase (RTK) under specific stimulus activates various different transcription factors of AP-1 family like c-JUN and FOS and aids in the activation of MEF-2 via the MAPK family of MEK/ERK.
Calcium mediated regulation
Calcium is a critical second messenger in various cellular processes in the cell. The major calcium/calcineurin responsive elements present in the NUR77/NR4A1 promoter act as docking sites for MEF-2 transcription factors. NFAT (Nuclear Factor in activated T cells) is activated by calcium influx, and in turn interacts with MEF-2D for its activation via calcineurin. NF-κB becomes activated when stimulation of the T-cell receptor results in calcium and protein kinase-mediated signaling that activates the IκB kinase (IKK) complex which then phosphorylates inhibitory κB proteins (IκBs), triggering their degradation by the proteasome and thus allowing NF-κB to translocate to the nucleus. The calcineurin-mediated dephosphorylation of MEF-2 at residue S408 activates the switch from sumoylation to acetylation at lysine-408, leading to the inhibition of the differentiation of the dendritic claw [92]. Furthermore, Calcineurin binding protein 1 (CABIN1), MEF-2-interacting transcriptional repressor (MITR), and HDAC4 are repressors of MEF-2 activity. CABIN-1, together with MITR and HDAC4, have been shown to repress MEF-2 transcriptional activation by binding to MEF-2 constitutively. HDAC4 and MITR contain calmodulin binding domains that are similar to the MEF-2 binding domain; in response to an influx of calcium, calmodulin binds to Cabin-1 and HDAC4 in turn releases MEF-2 and derepresses its transcriptional activity [47]. T cells expressing a truncated mutant of CABIN1, devoid of the C-terminus, serves as the binding domain for MEF-2 and calcineurin, showed significant increase in the release of cytokines such as IL-2, IL-13, IL-4 and IFN-γ [93]. Calreticulin represents another chaperone protein in the calcium signaling to MEF-2. Calreticulin is located in the ER/SR (endoplasmic/sarcoplasmic reticulum), and modulates the intracellular Ca2+ stores and associated signaling events. Calreticulin is known to control myocardial development; in the absence of Calreticulin (Crt–/–), there is impaired nucleocytoplasmic transport of the cardiac transcription factor MEF-2C [94], and myofibrillogenesis is impeded in a calcium dependent manner. The Ca2+/calmodulin-dependent protein kinase IIδ2 (CaMKIIδ2) is a positive regulator of vascular smooth muscle cells (VSMCs) dedifferentiation from contractile to synthetic phenotype in chronic vascular diseases such as atherosclerosis, that contribute to the growth of neointima. MEF-2 is a transcriptional activator of the VSM phenotype and regulated in an HDAC-dependent manner, and suppression of CaMKIIδ2 decreased the DNA binding affinity of MEF-2 and down regulation of its target genes Nur77 and MCP1n [90]. Calpain-3 (Capn-3) is a calcium-dependent proteolytic enzyme which also regulates certain non-proteolytic functions. MEF-2A has been implicated in regulating Capn3 gene expression by occupying the promoter region of the Capn3 gene in rat denervated gastrocnemius muscle tissue, positively correlated with the increase of MEF-2A expression in protein and RNA expression in L6 myoblasts [95]. Calcium is known to exert an important role in the survival and proliferation of ATLL cells and NFAT family members have been found to be activated in HTLV-1 transformed cells [96]. In HTLV-1-infected cells, the viral transactivator protein Tax interacts with a number of transcription factors and proteins to stimulate T-cell proliferation, many of them regulated by calcium [97].
In HTLV-1-infected cells, Tax-mediated activation of histone acetylase p300, CRE-binding protein (CBP), and p300/CBP associated factor (p/CAF), among others, results in transcriptional activation and T-cell proliferation [17]. For instance, Tax-mediated activation of NF-κB results in expression of interleukin-2 (IL-2)/IL-2 receptor (IL-2R), IL-9, IL-13, IL-15/IL-15R, and receptors of the tumor necrosis factor family, thereby promoting proliferation as well as transformation of T cells [97]. MEF-2 proteins also play a key role in regulating T-cell survival by associating with proteins including p300, p/CAF, and NFAT and are regulated by calcium [17]. Cabin1, MITR, and HDAC4 are repressors of MEF-2 activity that are displaced by the calcium/calmodulin complex [98]. Hypercalcemia is observed in 31% of ATL patients at disease onset [99]. About 70% of ATL patients develop hypercalcemia, particularly in the aggressive stage of the disease [100]. Calcium levels are often elevated in acute and lymphoma forms of ATL, but not in chronic and smoldering forms [101]. Hypercalcemia is present in about 50% of acute type ATLL patients at diagnosis, and it may develop in another third of patients as the disease progresses [101]. In contrast to other leukemia cells, ATL cells show enhanced growth by addition of calcium in a dose-dependent manner and inhibited growth in response to calcium antagonists and calmodulin inhibitors [102].
MAPK (P38/ERK/JNK) signaling
The other signaling pathways associated with MEF-2 proteins are mitogen-activated protein kinase (MAPK) signaling pathways which control various cellular processes such as proliferation, differentiation [103, 104], cell cycle, survival and apoptosis as reviewed in [105]. MAPKs are serine/threonine kinases that can target both cytoplasmic and nuclear substrates. In mammals, there are four canonical MAPK pathways: P38 mediated signaling, extracellular signal regulated kinase signaling (ERK1/2/5), non-mitogen mediated signaling and c-Jun kinase/stress-activated protein kinase (JNK/SAPK) signaling [103, 104, 106]. The important phosphorylation nuclear targets of p38 include ATF1/2/6, p53, MEF-2, and ELK-1 [103, 104]. p38-MEF-2 signaling is involved in the proliferation and differentiation of B cells [107, 108], and also mediates the survival of cerebellar granular neurons [109, 110]. It has been further implicated in various types of developmental pathways in the context of regulation with the MEF-2 family and regulates various signaling cascades in cardiomyocyte biology [111]. Studies have shown close correlation between p38 MAPKs, calcium-calmodulin dependent protein kinases and calcineurin signaling pathways that activate the expression of myogenesis via activating MEF-2 transcription factors [112]. The presence of high glucose significantly increased p38 MAPK signaling and escalated cardiac hypertrophy induced by hyperglycemia, whereas miR-373 inhibits MEF-2 activity by down regulating p38 signaling and reducing glucose induced hypertrophy [113]. The activation of p38 MAPK results in the degradation of HDAC4 followed by the release of RUNX2 and activation of MEF-2 genes in modulating chondrocyte hypertrophy [114]. The phosphorylation of p38 MAPK results in the interaction of β-catenin and MEF-2 in primary vascular smooth muscle cells (VSMCs) via increased β-catenin nuclear retention and increased activation of MEF-2 mediated Wnt/β-catenin signaling for cell proliferation [115]. The activation of MEF-2 via a p38-dependent pathway causes vascular inflammation through the activation of MCP-1 in VSMCs and allows the infiltration of macrophages into the endothelium [116]. MEF-2 also plays an important role in the Alzheimers disease, specifically amyloid precursor protein (APP)-mediated signaling pathway by inhibiting neuronal apoptosis by mediating the p38 MAPK-dependent pathway and activation of MEF-2-dependent gene transcription; alterations in this pathway results in the neuronal degeneration that occurs in Alzheimer’s disease (AD) [117].
There is a unique MEF-2 interacting domain in the C-terminus of ERK5, and mice lacking ERK5 or MEK5 exhibit cardiovascular defects during embryogenesis [118]. ERK5 also activates members of the MEF-2 family during the process of monocytic differentiation of myeloid leukemia cells [119]. Mouse embryonic fibroblasts that lack ERK 1/2 completely fail to proliferate [120]. The sheer stress-induced factor Krüppel like factor- 2 (KLF-2) is transcriptionally induced by a MEK5/ERK5/MEF-2-MAPK pathway which indirectly activates RhoA which causes the formation of actin shear fibers that control the alignment of flow and also inhibits the JNK/c-Jun/ATF signaling pathway in the vascular endothelial homeostasis [121]. It has also been shown that MEF-2 is activated via the MEK5-ERK5 pathway by Hepatitis C non-enveloped core like particles (HCVne) for altering biological activity [122]. MEF-2 and miR-193b-3p and miR-203a have direct biological association in downstream p38 signaling and are implicated as dysregulated genes in colorectal carcinoma [123, 124]. DNA damage induced apoptosis suppressor (DDAIS) is a protein that promotes cancer cell survival via its anti-apoptotic activity and is activated by NFAT. In response to EGF (Epidermal growth factor) stimulation, the ERK5/MEF-2 pathway subsequently induces DDIAS expression to promote the invasion of cancer cells by activation of various β-catenin target genes [125].
Post-translational modifications associated with MEF-2
The regulation of MEF-2 is also associated with the degradation pathways involved in the cell cycle. It has been reported that MEF-2C is degraded in the mitotic phase of proliferating cells by Anaphase Promoting complex/Cyclosome (APC/C), this downregulation is necessary for the efficient progression of the cell cycle checkpoints. This mechanism requires the presence of 2 phospho-motifs, pSer98 and pSer110, which mediate the interaction of CDC20 and MEF-2C for this process to occur and can mediate the cell proliferation [126]. In primary cerebellar granular neurons, Cyclin-dependent kinase5(CDK5) induces neurotoxic effects by phosphorylating and degrading MEF-2 via a caspase dependent manner. Neurotoxic conditions results in the nuclear activation of Cdk5 activity, which in-turn resulted in the activation of caspase3 and subsequently apoptosis [127]. Moreover, in other studies it has been reported that CDK5 also stimulated the sumoylation. It has been shown that Ser-444 of MEF-2D is required for the sumoylation of Lys-439. HDAC4 stimulated the modification by acting through Ser444. Opposing this inhibitory function over MEF-2D, Calcineurin also known as protein phosphatase 2B (PP2B) dephosphorylated Ser-444 and inhibited sumoylation of Lys-439 thereby activation of MEF-2 [128]. The transcriptional activity of MEF-2A is highly repressed by sumoylation, there are SENPs (Sumo specific proteases) which acts as an de-sumoylation enzyme of MEF-2A. The expression of SENP2 markedly increased the activity of MEF-2A in neuroblastoma cell lines and it mediated the activity dependent regulation of MEF-2A based on the stimuli, thus playing an important role in the activation dynamics of MEF-2A [129]. Neuronal survival and activity are controlled by MEF-2 isoforms and they exhibit a differential stress response to various stimuli and certain isoforms of MEF-2 are activated more than the others. It has been shown that MEF-2A but not MEF-2C or MEF-2D was modified in dopaminergic neuronal cell lines. It has been shown that MEF-2A is the only isoform that is ubiquitinated in the N’ terminus and also markedly reduced the DNA binding activity and transcriptional activation. Moreover, interfering the degradation of ubiquitinated MEF-2A induced neurotoxins associated with Parkinson’s disease (PD in model animals suggesting the selective regulation of MEF-2 in ubiquitin-proteasome pathway [130]. Redox-mediated post translational modifications can act has a molecular switch in MEF-2 function. Nitric oxide (NO) mediates redox reaction called S-nitrosylation of MEF-2 which acts a redox switch to inhibit both neurogenesis and survival. Structural analysis showed the dimerization of MEF-2 creates a pocket and nitrosylation occurs on the evolutionarily conserved cysteine residue in the DNA binding domain, which disrupts the binding and transcriptional activity of MEF-2 and leads to impairment of neuronal survival and neurogenesis both in-vitro and in-vivo [131]. MEF-2 suppresses the excitatory synapse number by the degradation of the synaptic scaffold protein or post synaptic density protein 95 (PSD-95) degradation, a gene that is defective in the rodent model of Fragile X syndrome. MEF-2 induces a PP2A mediated dephosphorylation of murine double minute-2 (Mdm-2), which is an ubiquitin ligase for PSD-95 which translocates to the nucleus and degrades PSD-95 and causes synapse elimination [132]. MEF-2D is activated by calcium/calmodulin-dependent protein Calcineurin (CaN or PP2B). Both MEF-2D and CaN bind to the scaffold of muscle A-kinase anchoring protein β (mAKAPβ) which is localized in the nuclear envelope. This phenomenon decreases skeletal myoblast differentiation and inhibits neonatal rat ventricular myocyte hypertrophy. The formation of signalosomes by mAKAPβ is required for the dephosphorylation and desumoylation of MEF-2D in C2C12 cells. The decrease in MEF-2D phosphorylation switched the activation of p300 instead of type IIA histone deacetylases and caused ventricular myocyte hypertrophy, suggesting the role of post-translational modifications of MEF-2 in the formation of active and repressive transcriptional complexes with signalosomes [133].
MEF-2 and its functions in mitochondria
MEF-2 and its isoforms play a critical role in mitochondrial maintenance and biogenesis and the phenomenon has been established in the various in-vitro and in-vivo models. A conserved region of MEF-2 gene, the MADS-box phosphorylation domain regulates the mitochondrial function in many cell types such as skeletal, cardiac and smooth muscle cells [134]. MEF-2 along with Serum response factor (SRF) regulate the expression of miR-133a and mitochondrial function via translational inhibition of mitophagy and cell modulating protein called NIX. In mice, MEF-2 also plays an important role in mitochondrial biogenesis. It has been shown that specific mi-RNAs, such as miRNA-27B regulates mitochondrial biogenesis in myocytes via targeting the fork-head box j3 (FoxJ3) and downregulated the expression levels of downstream targets like MEF-2c, PGC1α, NRF-1and mtTFA [135]. In xenopus models, mitochondria have been linked to the maintenance of synaptic and neuronal plasticity by integrated functions of MEF-2 and mitochondria. MEF-2 serves as a target of mitochondrial caspases and in-turn MEF-2 regulates the mitochondrial gene transcription essential for the production of hydrogen peroxide and superoxidase controlling the spatio-temporal neuronal plasticity [136]. It has also been shown that MEF-2D has a neuro-protective ability with neurotoxicity associated with the MPTP in Parkinson’s disease [137]. This sort of neuroprotective activity is induced by agents like Methylene blue (MB) via MEF-2D and can ameliorate the neurochemical/neuropathological impairments in animal models of various neurodegenerative disorders. MB upregulated the expression of mitochondrial NADH dehydrogenase -6 (ND6) in a MEF-2D dependent manner, and knockdown of MEF-2D abolished the MB mediated increase of ND6 and the neuroprotective effect. Moreover, MB also induced the activation of AKT pathway and suppressed the GSK-3β activity which is an inhibitor of MEF-2D function [138]. Some anti-cancer agents like SU4312, yielded an unexpected neuroprotective effect by potentiating the pro-survival effect of PI3-K/AKT pathway to downregulate the inhibitory effect of MEF-2D inhibitor glycogen synthase kinase-3 beta (GSK3β) and ameliorated parkinsonian motor defects and restored levels of MEF-2D [137]. It has also been established that there is an association between PD and mitochondrial toxins, other environmental pesticides. In this study it has been shown that mitochondrial-toxin induced nitrosative/oxidative stress results in the S-nitrosylation of MEF-2C in A53T mutated cells compared to isogenic controls [139], via inhibiting the MEF-2C-PGC1α pathway and leading to mitochondrial dysfunction and apoptotic cell death. In certain murine models, it has also been shown that the activation of conventional Tcells and Tregs (T regulatory cells) led to the increase in gene expression of OXPHOS (Oxidative phosphorylation). The deletion of HDAC9, which is an inhibitor of MEF-2 increased the Treg suppressive activity as well as expression of Pgc1α and Sirt3 and improved mitochondrial respiration. The study shows that key regulators of OXPHOS are required for optimal functioning of Tregs and Treg dependent allograft acceptance [140].
Mutations in MEF-2 in the context of diseases
Numerous studies have investigated the genetic aberrations of MEF-2 in various cancers [1, 141]. Mutations in the N-terminal domain lead to defects in DNA binding activity and impairs MEF-2 transcriptional activity [142]. The C-terminal domain, which acts as the regulatory or the catalytic domain, has 2 transactivation domains (TADs) which are highly divergent between the isoforms; they are also known to contain specific phosphorylation sites for multiple kinases, allowing for tight regulatory activity of the MEF-2 proteins [143]. The final few amino acids of the C-terminal region comprise the nuclear localization signal (NLS) for all the isoforms which is absent in the MEF-2B isoform, although MEF-2B can be found both in the nucleus and the cytoplasm [144]. The defects associated with the absence and/or aberrations of MEF-2 have been well characterized in murine models and humans. MEF-2 activity is coordinated by multiple layers of regulation and all the isoforms are subjected to alternative splicing events, post-translational modifications in the C-terminus, and dimerization capability with other transcription factors at the N-terminus. Mutations in these respective domains result not only in developmental abnormalities, but may also cause tumors, leukemias and aberrations in the transcriptional machinery. MEF-2a null mice exhibit pre/post-natal defects in cardiac development and muscle differentiation [145, 146]. However, mice deficient in MEF-2B do not have significant observable phenotypes, since the other isoforms can compensate for the loss of its function [147]. Several mouse models have been generated to study the abnormalities associated with loss of MEF-2C. In the absence of MEF-2C there were defects associated with B-cell proliferation and BCR stimulation, and a decrease in B-cell numbers in the bone marrow [148]. There was also a considerable loss of granulocyte progenitor function with a loss of function allele of MEF-2c [149]. Preliminary studies of MEF-2d-null mice indicated that these mice exhibited normal viability and life span, but heterozygous alleles of MEF-2c mouse showed debilitating effects in bone differentiation, and MEF-2d knock-out mice had blindness in photoreceptor cells within the retina [150].
Isoforms of MEF-2
Table 1 summarizes some key information on MEF-2A, MEF-2B, MEF-2C, and MEF-2D. These isoforms play an important role in embryogenesis and other epigenetic modifications that control gene expression [13, 151–153]. The chromosomal coordinates of MEF-2 are 15, 19, 5 and 1 for the MEF-2A, 2B, 2C, and 2D, respectively [154]. The perception of MEF-2 as a developmental transcription factor has gradually shifted to encompass emerging findings of MEF-2 in more pathological processes such as tumorigenesis and involvement in viral pathogenesis/gene expression in HTLV-1 and activation of latent virus in the case of EBV [17, 19–22]. The basal RNA and protein expression and various aberrations of each isoform will be discussed in detail in the following sections and is illustrated in Figure 1.
Table 1: Expression, transcriptional activity and associated diseases of MEF-2 isoforms
Isoforms |
Expression |
Development |
Mutations |
Activity |
Gene-targets |
Disease |
MEF-2A |
Expressed in brain, endocrine tissue, lung, skin and soft tissue. Expressed in cardiac, skeletal and smooth muscle. Highly overexpressed in glioma, lymphoma, and melanoma. |
Required for proliferation, differentiation and maturation of neural stem cells into neurons and glial cells [164, 165]. Muscle-regeneration and tissue repair [95]. Myofibroblast differentiation [158]. |
MEF-2A mutations is associated with premature myocardial infarction and coronary heart disease [172]. Also associated with reduced myofibroblast proliferation and differentiation [152]. |
Downregulation of Wnt signaling via miRNA/Gtl2-Dio3 loci [155]. Regulates miRNA-143 [156]. Regulates Calpain 3 gene expression [95]. |
MEF-2A is located on chromosome 15q26 [154]. GSK3beta and Xirp2 are transcriptional target of MEF-2A [53–55, 161]. Cited2 is a possible transcriptional target [155]. |
MEF-2A is involved in the pathogenesis of HTLV-1 and ATLL [18, 26–40]. Accelerates the progression of atherosclerosis [184]. |
MEF-2B |
Expressed in tonsil, appendix, germinal center of lymph node, and bone marrow. Highly-expressed in lung carcinoma and lymphoma. Expressed in cardiac and skeletal muscle. |
MEF-2B is required for the development of cardiac and skeletal muscle [189]. |
Mutation causes dysregulation of proto-oncogene BCL6 [193, 194]. |
Promotes tumorigenesis [125]. |
MEF-2B is located on chromosome 19p12 [154]. ROS-induced NOX-1 signaling [188]. Binds in the promoter of DDAIS [125]. |
Involved in follicular lymphoma, Burkitt’s lymphoma, mantle cell lymphoma, splenic marginal zone lymphoma, and diffuse large B cell lymphoma [196, 197]. Hypertension in VSMCs [188] and TGFβ-induced EMT and tumorigenesis [125, 191]. |
MEF-2C |
Expressed in nucleoplasm and neuroplasmic vesicles. Expressed in cardiac, skeletal muscle and brain tissue (especially in cortical excitatory neurons). Expressed in both common myeloid & lymphoid progenitors. Maturing hematopoietic stem cells and B cells. |
It is involved in the development of cardiac and skeletal muscle [152]. Development of lymphoid and myeloid progenitor cells [153]. Development of the cerebrocortex in its role in corticostriatal circuit [202]. |
Associated with decline in cognitive ability as well as in development of autism, schizophrenia, and epilepsy [10, 202–204]. Implicated in congenital heart defect and forelimb malformations [210, 221]. |
It possesses differential activity in membrane depolarization [201]. Required for neuronal differentiation and commitment of embryonic stem cells [199]. |
MEF-2C is located on chromosome 5q14 [154]. Target genes include GPM6A, DLx6, Hand2, Myogenin, and Myokinase [200, 207, 233]. |
MEF-2C implicated in DiGeorge syndrome [209]. |
MEF-2D |
Highly expressed in cerebrocortex, endocrine system, skin, kidney, and breast. Present in hematopoietic stem cells and germinal centers of lymph node. Expressed in cardiac and skeletal muscles. |
It is required for the development of neuronal and muscle cells [53, 54]. Terminal differentiation of neonatal cardiomyocytes and human macrophages [163, 52]. |
Mutation implicated in the inhibited differentiation of common lymphoid progenitors (differentiation into CD19 positive B-cells) [248]. |
Promotes the survival of dopaminergic neurons [242]. |
MEF-2D is located on chromosome 1q12-q23 [154]. DYRK1A is a target gene [240]. |
MEF-2D is involved in cancer of the colorectal tissue, breast, prostate, lung, liver, and thyroid [124, 125]. Implicated in melanoma [279–281], and pathobiology of B-cell acute lymphoblastic leukemia (B-ALL) [248]. Also, Parkinson’s disease, cardiac myxoma, and lung cancer [239, 244, 250, 251]. |
MEF-2A
MEF-2 genes are expressed in very distinct and specific temporo-spatial patterns. MEF-2A is transcriptionally autoregulated and is activated by Erk5 and p38 MAPK signaling; the MEF-2a protein product regulates transcription either positively or negatively [53]. MEF-2A is a DNA binding transcription factor which induces muscle development, neuronal differentiation, cell growth and apoptosis specific genes. It is located on chromosome 15q26 [130]. MEF-2A is present ubiquitously in all tissues but enriched in the brain, endocrine tissues, muscle, lungs, skin and soft tissues. In the CNS, at the transcriptome level, it is highly expressed at 68.3 TPM (transcript per kilobase million) cerebral cortex, but expressed at low levels in the hippocampal, caudate or cerebellar regions. However, elevated expression of MEF-2 protein is observed in the cerebral cortex and cerebellar region. In muscle tissues, such as cardiac, skeletal, and smooth muscle, MEF-2 has medium to high level expression at 59.2 TPM, 21.6 TPM, and 52.4 TPM, respectively. Higher levels of MEF-2A protein expression are seen in cardiac and skeletal muscles, but considerably less observed in the smooth muscle. In the bone marrow and immune system, it is also expressed considerably less (medium level) at both at the transcriptome and the protein level (12 TPM). MEF-2A was found to be highly overexpressed in gliomas, lymphomas and melanomas as well as lung cancers but not to a great extent (https://www.proteinatlas.org/).
MEF-2A in muscle development
It has been suggested that MEF-2A is absolutely indispensable for proper myoblast differentiation compared to other isoforms. In bovine myoblasts, via regulation through myozenin2 (MyoZ2) [2], it has been shown to be a transcriptional regulator of Calpain3 (Capn3) gene expression, which in turn controls L6 rat myoblast differentiation. Defects in this pathway might result in atrophy of the proximal limb muscles [95]. Skeletal muscle regeneration and repair are crucial for regeneration of diseased muscle. Injured MEF-2a knockout mice are impaired by myofiber defects and necrosis, a process controlled by MEF-2A via the microRNA (miRNA) Gtl2-Dio3 locus that downregulates Wnt signaling activity. This step is critical for proper muscle regeneration and tissue repair and may be dysregulated in muscular dystrophy. The same locus is involved in regulating Cited2 in cardiomyocyte proliferation via MEF-2A through increased VEGFA activity [155]. Also, miR-143 is regulated by MEF-2A in hydrogen peroxide-induced senescence in vascular smooth muscle cells (VSMCs); overexpression of MEF-2A along with miR-143 shows synergy in senescence induction whereas the knockdown has opposite effects [156]. MEF-2A and AP-1 provide antagonistic regulation of Heat-shock protein B7 (HspB7) which has been implicated in muscular atrophy [157].
In diabetic mouse models, MEF-2A knockdown in cardiac fibroblasts led to significant reduction of hyperglycemia-induced cardiofibroblast proliferation, myofibroblast differentiation, and matrix metalloprotease (MMP) and collagen activity, as well as improved cardiac function and collagen deposition. This process was achieved via downregulation of Akt and TGF-β/Smad signaling, which can serve as a potential treatment modality in diabetic-induced cardiac remodeling. MEF-2A:MEF-2D heterodimers, responsible for GLUT-4 (Glucose transporter-4) transcriptional activity, were shown to be selectively decreased in insulin-deficient diabetic rats [158]. Protein Kinase B2 (AKT2) is involved in cardiomyocyte signaling, heart development and systemic blood pressure. AKT2 via EndoG, a mitochondrial-specific nuclease, regulates MEF-2A in the myocardium, and the deficiency of AKT2 results in defective myocyte development through the EndoG:MEF-2A signaling pathway [159]. Acute knockdown of MEF-2A results in defective integrity of costameres, a macromolecular complex responsible for transmission of contractile force through the straited muscle cells. This costamere defect results in abstruse malformations in myofibrils, defects in focal adhesion and adhesion-dependenT-cell death [160].
Xirp2 (also called CMYA3) is a cardiomyopathy-associated gene induced by Angiotensin-ii (Ang-ii), and a direct transcriptional target of MEF-2A. Both MEF-2A and Xirp2 serve as the important downstream regulators of Ang-II in mediating pathological cardiac remodeling [161]. MEF-2A is also implicated in cardiac hypertrophy and myopathy due to arsenic exposure in rats [162]. Another study suggests that glycogen synthase kinase3β (GSK3β), a suppressor of both myogenesis and cardiac hypertrophy, is a transcriptional target of MEF-2A. Inhibition of GSK3β results in the activation of the P38 MAPK pathway and subsequently MEF-2A activity that controls skeletal and muscle gene expression [53]. Also, inhibition of MEF-2A or MEF-2D results in activation of cell cycle genes, but the downregulation of terminal differentiation of neonatal rat cardiomyocytes [163].
MEF-2A in neuronal development
All the MEF-2 isoforms are extensively but differentially expressed during phases of pre- and post-natal development. They follow distinct roles at various stages of the maturation of neurons, in neural stem cells that have an ability to proliferate and differentiate into various neuronal and glial phenotypes. In vivo analysis has implicated various MEF-2 transcription factors in synapse regulation and neuronal survival [164]. Studies indicate that MEF-2A is present in undifferentiated neural stem cells, and after subsequent differentiation exhibit higher amounts in neuronal cells. MEF-2A plays a critical role in differentiation and maturation of neural stem cells into neurons in rat models [165]. MEF-2 transcriptional activity is also activated by neurotrophins such as brain-derived neurotrophic factor (BDNF) via Nur77 and ARC through a novel pathway in rat cortical neurons [166]. The glucocorticoid receptor (GR) and MEF-2A are the two key transcription factors involved in neuronal plasticity, and they both modulate c-JUN, a transcriptional target regulating the synapse strength and number. MEF-2A is hyper-phosphorylated by GR activation and in turn regulates c-JUN, which increases MEF-2A DNA binding ability. This molecular cross-talk controls genes of neuronal plasticity [167]. SENP2 (Sumo specific protease-2) post-translationally regulates MEF-2A in post-synaptic differentiation, which is de-SUMOylated and acetylated at Lys403. This modification inhibits dendritic claw differentiation via activation of MEF-2A. SENP2 is highly expressed in the cortex and hippocampus and regulates sumoylation status of MEF-2A for transcriptional activation [129]. Interestingly, sumoylated-MEF-2A is a transcriptional repressor which drives the suppression of orphan presynaptic sites. Synaptogamin (Syt1) was identified as the direct repressed target of sumoylated MEF-2A in neurons. Syt1 eliminates the orphan presynaptic sites and aids in the accumulation of presynaptic material in the maturing boutons, which may be implicated in the neuronal connectivity, brain development, and disease [168]. Formal thought disorder is an important feature of schizophrenia and other psychotic disorders. In a cohort study of high-risk family subjects, microsatellite linkage analysis and whole genome sequencing identified causative variants in the linkage region and reported a MEF-2A binding site that was located between two genes associated with schizophrenia; the locus 6q25-26 has been implicated in formal thought disorder via dysregulated MEF-2A signaling [169]. Also, it has been implicated in oxidative stress during aging and other neurodegenerative disorders that result in neuronal death. Chaperone mediated autophagy (CMA) targets MEF-2A to the lysosomes for CMA degradation, and stress-induced destabilization of the lysosome results in the disruption of MEF-2A and its function thereby leading to neuronal damage in various neurodegenerative diseases [170]. MEF-2A was detected in patient subjects with temporal lobe epilepsy (TLE) and it was significantly downregulated in the temporal neocortex of both humans and rats with TLE compared to control groups, suggesting a role in the pathogenesis of TLE [171].
MEF-2A mutations and activity control
Contrasting mutational and functional analysis of MEF-2A have been reported in many studies which identify MEF-2A as a susceptibility gene for premature myocardial infarction and coronary artery disease (CAD). Specifically, a 21-bp deletion in exon 12 and 3 and other missense mutations which affect its transcriptional activity have been implicated, but most studies have not demonstrated significance for supporting this hypothesis [172]. Cohort studies performed for Exon 11 deletion examined in CAD subjects reveal that only 0.09% of the population in the Sicilian cohort was reported to have CAD-associated genes compared to healthy subjects [173]. Similarly, in a Chinese-HAN cohort, they found 4 single nucleotide polymorphisms in Exons 9 and 11, one insertion mutation in Exon 11, one deletion mutation in Exon 11, and one STR in Exon 11; however, these structural changes in Exon 11 of MEF-2A did not relate to the sporadic CAD [174]. Similar cohort studies in Chinese Han subjects claim that the 3’ UTR may consist of functionally relevant nucleotide changes, and 2 single nucleotide polymorphisms were detected but deemed not significant, but a further haplotype carrier of rs325380-rs325381 was claimed to be associated with CAD risk, suggesting variance in the 3’ UTR of MEF-2A [175]. Another study in the Saudi population revealed MEF-2A as the susceptible gene for the risk of CAD, upon analysis of Exon 11 which revealed several substitution polymorphisms, insertions/deletions at the 11 CAG trinucleotide loci, which introduced premature stop codons at 4 nucleotide sequences nt146637, nt146647 and nt146780 or nt146783 [176].
Exogenous control of MEF-2A can be regulated by exercise. Many genes are upregulated during exercise and confer protection against diseases such as type 2 diabetes. Studies have shown that NRF-1, known to regulate mitochondrial oxidative genes, can also affect MEF-2A and GLUT-4. Exercise-induced CAMKII activation induced hyperacetylation of histones of both NRF-1 and MEF-2A genes and increased glucose transport by upregulation of GLUT4 in rats [177]. Similar exercise regimens performed in mice, where muscle-type carnitine palmitoyl l1 (CPT1b) is involved in skeletal muscle mitochondrial β-oxidation, suggest binding of MEF-2A to the Cpt1b promoter, which elevates the growth of the quadricep muscles and development of skeletal muscle; binding activity was decreased with exercise training and increased expression of HDAC5 which correlates to the decrease in MEF-2A activity [178]. Additionally, other studies indicate that exercise increased nuclear MEF-2A content and increased binding of MEF-2A to the GLUT-4 gene in a AMP activated protein kinase (AMPKα2)-dependent mechanism [179].
Infection, immunity and cancer
MEF-2A has been implicated in various disease models ranging from neuronal to tumorigenic. It has contrasting roles in various tumor types and has been linked to various disorders such as hepatocellular carcinoma (HCC), gastric cancer, atherosclerosis and also in immunity. TGF-β1 (transforming growth factor β1) is involved in HCC invasion, and MEF-2A along with other isoforms are overexpressed in HCC cells in a PI3K/AKT-dependent manner. By modulating TGF- β1 signaling, MEF-2A promotes epithelial to mesenchymal transition (EMT) [180]. TGF-β is also a transcriptional activator of Matrix metalloprotease-10 (MMP-10) via activation of MEF-2A and downregulation of Class IIa HDACs [181]. In gastric cancer MEF-2A is phosphorylated by P38 to activate glycolysis via the GLUT-4 transporter [182]; moreover, MEF-2A mRNA is overexpressed in 10% of tumors of patients with gastric carcinomas. Recently, MEF-2A has been identified as a proapoptotic factor in therapeutics of HCC via activation of caspase-3 and caspase-7, and thereby inhibiting growth of HCC xenografts in nude mice [183]. MEF-2A has been implicated in the progression of pre-existing atherosclerotic lesions. The inflammation patterns of apolipoprotein E-deficient mice following the knockdown of MEF-2A showed accelerated atherosclerosis [184]. Our lab was the first to implicate MEF-2A as a regulator of viral gene expression in the pathogenesis of HTLV-1. HTLV-1 encodes the oncogenic trans-activating Tax protein, which directly binds to MEF-2A and hijacks the host transcriptional machinery to increase viral gene expression [17]. MEF-2A expression was upregulated in the peripheral blood of HTLV-1-infected individuals. We are currently studying other isoforms of MEF-2 and their implications in HTLV-1-induced ATLL (unpublished observations). MEF-2A has been reported in the differentiation of many cell types. MEF-2A and MEF-2D play dual roles in human macrophage terminal differentiation by forming heterodimers with each other and activating P300 binding and c-JUN expression in differentiated macrophages [8].
MEF-2B
MEF-2B is the most distant member of the MEF-2 family of transcription factors based on phylogenetic analysis, and it also lacks the HJURP_C (Holliday junction recognition protein C-terminal) region and other duplication events [185]. In the apo structure of MEF-2B (only MADS-box/MEF-2 domain) without DNA or cofactor binding, it showed a preformed DNA binding interface that may be important for recognizing the DNA from the minor groove side and the C-terminal helix which can serve as the docking site for MEF-2 transcription co-factors suggesting new interaction patterns [186]. This gene undergoes considerable alternative splicing events. MEF-2B is the only isoform that is not ubiquitously expressed, but is enriched only in bone marrow and immune cells and the gene is located on chromosome 19p12. At the transcriptome level it is expressed in the appendix, bone marrow, and tonsils at 17.6 tpm, 45.7 tpm and 27.2 tpm, respectively. High protein expression of MEF-2B is only found in the germinal centers of lymph nodes and tonsils, but not in non-germinal centers or in other cell types. Low or no expression of MEF-2B is observed in many cancer types and is significantly downregulated in lymphomas and lung carcinomas (https://www.proteinatlas.org/).
MEF-2B in development
MEF-2B is also involved in developmental programs and its mutations have been implicated in the genesis of leukemias and carcinomas. In smooth muscle specific gene expression, the SMHC (smooth muscle myosin heavy chain) gene consists of an A/T rich region which provides a binding site for MEF-2B, and overexpression of MEF-2B increased SMHC activity suggesting its role in SMHC gene regulation [187]. MEF-2B modulates inducible expression of NOX-1 in superoxide production in various cardiovascular tissues via ATF-1:MEF-2B signaling [45]. During cyclic stretch (CS) in VSMCs, ROS (reactive oxygen species) are produced via MEF-2B:NOX1 signaling causing dysfunction of VSMCs by switching from contractile to synthetic phenotype leading to hypertension [188]. During mouse embryogenesis, MEF-2B transcripts were expressed in cardiac and skeletal muscle and its gene mapped to chromosome 8, and its potent transactivation was controlled in the C-terminal domain [189].
MEF-2B is widely implicated in cancer phenotypes and poor prognosis based on its mutations and the pathways it regulates. In the P19 teratocarcinoma murine stem cell line, MEF-2B homologue is differentially expressed during development and is predominantly found in non-committed cells [190]. In a kinome-wide siRNA screen, the MEK5-ERK5 signaling axis was implicated in the activation of MEF-2B, thereby leading to EMT, lung metastasis and invasion in breast cancer cells. This MEK5-ERK5:MEF-2 axis promoted TGFβ-induced EMT in murine and human breast cancer cells. MEF-2B drastically affects the morphology of the cell and gene expression patterns during EMT [191]. Similarly, EGF activates the ERK5:MEF-2B pathway which induces the expression of DDAIS, an anti-apoptotic protein activated during DNA damage, and promotes tumor invasion by activating the β-catenin signaling pathway. MEF-2B binds to sequence specific promoter binding sites in DDAIS which activates the ERK5/MEF-2B pathway for promoting tumorigenesis [125].
Extensive MEF-2B mutations in lymphomas
Mutations in MEF-2B have been shown to have fatal consequences in different disease phenotypes. MEF-2B is a target for somatic mutations in various lymphomas, most of these affecting the MADS-box/MEF-2 domain, and the most frequent being the D83V mutation in cancer. Crystallographic analysis revealed that most mutations are non-synonymous substitutions altering the structure and function of the protein, causing changes in the MEF-2 domain from an α-helix to β strand with resulting binding defects in various transcriptional co-factors. This conformational switch is a key mechanism in NHL (Non-Hodgkin’s lymphoma) in driving tumorigenesis [192]. MEF-2B acts as a transcriptional activator and is mutated in 11% of Diffuse large B-cell lymphomas (DLBCL), and 12% of follicular lymphomas. BCL6, which is a direct target of MEF-2B, controls germinal center B cells. The oncogenic mutations in MEF-2B largely impact the dysregulation of the BCL6 proto-oncogene which controls the cell cycle and B-cell differentiation. Mutation in the N-terminal domain results in a gain of function phenotype by preventing the binding of CABIN-1, a corepressor of MEF-2B. C-terminal truncations, nonsense, or frameshift mutations might affect the PTMs and transcriptional activity of MEF-2B [193]. BCL6 is also regulated by AhR (Aryl hydrocarbon receptor)/ARNT complex and wild type MEF-2B and if mutated may cause DLBCL [194]. Also, similar studies indicate hotspot mutations in K4E, Y69H and D83V in DLBCL affecting DNA binding and functional activation respectively [195]. Comparative analysis of MEF-2B with other germinal center antigens revealed that in differential diagnosis of B cell non-Hodgkin’s lymphoma there was positive expression of MEF-2B in all FL (follicular lymphomas) and BL (Burkitt’s Lymphoma), 8/9 of mantle cell lymphomas and 2/24 of Splenic marginal zone lymphomas (MZL) and 38/44 DLBCLs but negative expression in extranodal MZL and B-lymphoblastic lymphomas [196]. Genetic alterations were reported in FL and revealed 85% translocations [197], 96% mutations in the BCL2 gene and about 15% frequency of genetic alterations in MEF-2B. In an extensive analysis of various B and T-cell lymphomas to validate MEF-2B as an immunohistochemical marker, the expression revealed a statistically significant association with BCL6 in DLBCL and indicates MEF-2B as a valuable marker for differential diagnosis of various types of lymphomas [193]. Clinical interventions via HDACi that function as major repressors of MEF-2B activity have been reported. Pabinostat which is a Pan-HDACi was used to treat relapsing DBLCL subjects in a phase II trial along with/without rituximab showed durable responses in 28% of the patients and early responses were predicted for MEF-2B and significant increase in the ct-DNA was a surrogate for subsequent treatment failure [198].
MEF-2C
MEF-2C is one of the most characterized genes in the MEF-2 family and has shown to be involved in various neural, cardiac, skeletal, muscle, lymphoid and myeloid developmental programs, and has also been implicated in various types of T-cell and B-cell lymphomas, carcinomas, many neurodegenerative diseases and vascular disorders. MEF-2C has transactivation and DNA binding domains and its gene is located on chromosome 5q14.3. It is highly expressed in the nucleoplasm and nucleoplasmic vesicles. Although it is ubiquitous in nature, its enrichment is limited to specific cell types found in the brain and muscle tissues. The highest expression of MEF-2C mRNA transcripts is found in the cerebral cortex of the brain (197.9 tpm), but is absent in the hippocampal and cerebellar regions. Also, similar expression is observed in skeletal muscle (192.4 tpm). MEF-2C is perhaps the only member of the family where the transcriptome expression correlates with the protein expression patterns, both in the CNS and skeletal muscle, with high levels of protein expression observed in these tissue types. Furthermore, MEF-2C protein expression is similarly abundant in the immune system where higher expression is observed in the lymph nodes, tonsils, and spleen, but surprisingly not in the bone marrow but considerably less at the transcript level at 75.6 tpm, 70.6tpm, and 60 tpm, respectively. MEF-2C is highly overexpressed in lymphomas, melanomas and gliomas (https://www.proteinatlas.org/).
MEF-2C in development
MEF-2C is the predominant isoform regulating the development of the cerebrocortex, and conditional knockout of MEF-2c in mice in neural stem cells (NSCs) abolished neuronal differentiation. These findings suggest critical roles of MEF-2C in early neuronal differentiation and proper distribution of layers in the neocortex [146]. Activation of MEF-2C-dependent Bone Morphogenetic protein2 (BMP2) by P38 MAPK controls the commitment switch between cardiomyocyte and neuronal commitment. Inhibition of P38 MAPK in ES cells (Embryonic stem cells) results in the neuronal commitment of the ES cells; similarly, treatment with BMP-2 in ES cells results in the cardiomyocyte differentiation which is regulated by MEF-2C, suggesting a molecular mechanism for ES cell commitment [199].
Proper neurite function and outgrowth are necessary for neuronal function and synapse formation. miRNAs play an important role in neuronal development and function, and miR-124 and miR-9 are abundantly expressed in the mammalian nervous system. HDAC5 inhibits neurite extension by inhibiting the neuronal membrane glycoprotein GPM6A(M6a), which is regulated by MEF-2C. miR-124 and miR-9 modulate this HDAC5:MEF-2C:M6a pathway to regulate neurite development in primary neurons [200]. BDNF is a neurotrophin which controls synaptic development and function, and it regulates many signaling pathways leading to activation of MEF-2 family members. MEF-2C splice variants which lack the γ-domain are activated by membrane depolarization, and the knock down of MEF-2C resulted in the impairment of membrane depolarization-induced expression of Bdnf exon 1, suggesting the differential activity of MEF-2C [201]. Cortico-basal ganglia circuits control language and speech in Autism spectrum disorders (ASD). FOXP2 is known to interact with several ASD related genes and have been implicated in spoken language disabilities and dysfunction in the corticostriatal circuit. MEF-2C, which is a synapse suppressor negatively interacts with Foxp2. The deletion of FoxP2 derepresses MEF-2C function, which then restores vocalization and related striatal spinogenesis suggesting MEF-2C function in corticostriatal circuit [202]. MEF-2C is abundantly expressed in cortical excitatory neurons, and conditional knockdown of MEF-2c in hippocampal and cortical excitatory neurons decreases the cortical network activity by decreasing the excitatory synaptic transmission. MEF-2c mutants showed significant overlap of synaptic and autism-linked genes in the cortex and mutant mouse models displayed autism and schizophrenia-like behavior, suggesting functional importance of MEF-2C in the neo-cortex [10]. Constitutively expressing MEF-2C in both in vivo and in vitro systems yielded pure neurons and MEF-2C-directed neuronal progenitor cells can successfully differentiate into functional neurons in mouse models of cerebral ischemia and can improve behavioral defects [146]. The haploinsufficiency of MEF-2C reported in patient and mouse models leads to severe mental retardation, epilepsy and cerebral malformations, and also the mice exhibited severe hyperkinesis. Moreover, MEF-2C was implicated in various developmental stages of dorso-ventral neuronal cell types [203]. A microdeletion involving the 5q14.3 region of the chromosome resulted in the haploinsufficient phenotype of MEF-2C [204]. Similar clinical studies reported that decreased MEF-2C mRNA transcripts in leukocytes may serve as a diagnostic marker in Alzheimer’s disease (AD), and this may also be associated with the decline in cognitive ability [205]. Rett’s syndrome is characterized by severe mental retardation, epilepsy, absence of speech and cerebral malformations, and MEF-2C polymorphisms have been found in patients with Rett’s or severe Rett-like encephalopathies [206].
MEF-2C in diseases
Various congenital diseases arise from the improper development of the neural crest cells (NC cells) which undergo various developmental changes during morphogenesis, including craniofacial defects. MEF-2c NC knockout mice exhibit delayed ossification and hypoplasia (loss of skeletal elements of the face and skull). Dlx6 and Hand2 genes are responsible for these developmental pathways, where MEF-2C is the direct transcriptional regulator of Dlx6-Hand2 and affects craniofacial development via a feed-forward transcriptional circuit mechanism in mice [207]. In addition, Endothelin, which is also essential for neural crest development, targets MEF-2C downstream of a calmodulin-CamKii histone deacetylase signaling cascade through a positive feedback mechanism [208].
TBX-1 is a T-Box transcription factor that has been implicated as a haploinsufficient gene in DiGeorge syndrome, which is characterized by congenital defects of the heart, craniofacial dysmorphism, abnormal thymus gland and hypoplasia. Tbx null mutant embryos showed higher MEF-2c expression, and conversely MEF-2c expression was decreased in the Tbx1 gain-of-function mutants, revealing the suppressive effects of Tbx on MEF-2c. It was also shown that Tbx1 interferes with the Gata4 signaling regulation of MEF-2c affecting critical developmental pathways [209]. Conversely, TBX5 plays a synergistic role with MEF-2C in the early phases of cardiomyocyte development and differentiation, and mutations in TBX5 are associated with Holt-Oram syndrome which features massive heart and forelimb abnormalities [210]. During embryogenesis, the binding of MEF-2C in the BOP gene promoter is responsible for bop expression in the anterior heart field and its cardiac derivatives [211]. Similarly, transcription factors NKX 2.5 and MEF-2C bind physically and functionally to control ventricle formation during cardiogenesis [212], and also are required for the maintenance of the cells between the ventricular and sinoatrial precursors in the primary heart field [211]. However, in certain mouse models, knockdown of MEF-2c attenuated left ventricular hypertrophy and mechanical stress via the mTOR/S6K pathway [213]. The specific isoform MEF-2cα1 is involved in dynamic phosphorylation events implicated in myoblast and skeletal muscle regeneration via Akt/mTOR/S6K signaling which may potentially be targeted for treatment of muscle wasting diseases [214]. There is evidence for miRNA regulation of MEF-2C; miR-214 suppresses the activity of MEF-2C:MYOCD:Leiomodin1(LMOD-1) signaling pathway, and excessive proliferation of smooth muscle cells causing pulmonary arterial hypertension [215]. Dysregulated expression of several miRNAs and mRNAs in a myotonic dystrophy type 1 (DM1) cell model, was rescued with the exogenous addition of MEF-2C [216]. Hormonal mediators such as testosterone and IGF (Insulin like growth factor) also modulate MEF-2C in cardiomyogenesis and pro-hypertrophic effects in cardiac gene function via HCN4 and P38-MAPK respectively [217]. MEF-2C interacts with the transcription factor c-FOS and increases its binding to an AP-1 site in the Mmp13 promoter, which is modulated by para-thyroid hormone (PTH), a transcriptional regulator of many genes in osteoblast formation [218]. Another known co-repressor of MEF-2c is HDAC-5, whose increased expression decreases osteocyte formation. Binding of MEF-2C in the sclerostin, encoded in the SOST gene, after its detachment from the repressor complex, derepresses MEF-2C function in the direct regulation of osteocyte formation via SOST gene expression [219]. Also, studies reveal that MEF-2C/NFAT pathways along with GATA4 that juncture the Ca2+ signaling have been implicated in cardiac hypertrophy and heart failure from a cohort of heart transplant patients [220]. Similarly, in a study of 200 patients with congenital heart defects (CHD), loss of function mutations in MEF-2C were reported to be associated with CHD, which also affects the activation of transcription factors such as GATA4 [221]. MEF-2C also controls circadian clock rhythms in Drosophila models [222]. Aquaporin is present in the microvascular endothelial cells and is responsible for formation of water channels that control various physiological processes; MEF-2C is a direct target of Aquaporin which controls angiogenesis and water processes in endothelial cells [223].
MEF-2C in cancer and immunity
MEF-2C is associated with the development of various cell types including myeloid and lymphoid progenitor cells, and its deficiency results in profound defects in the phenotypes of both cell types. It has been also implicated in fate commitment of the progenitor cells, determining if a cell will differentiate along the myeloid or lymphoid lineage [149]. Deficiency of MEF-2c results in delayed B-cell development with significant reduction of immature B cells; MEF-2c deficiency also alters the expression of various proteins including CD23, OX40L and Ciita [224]. Moreover, MEF-2C is required for the survival and proliferation of B-cells and B-cell signaling receptor function [148], and also modulates B-cell proliferation via P38 MAPK activity and EBF1 (Early B cell factor 1) [107]. MEF-2C regulates megakaryopoiesis [225], and via a MEF-2C/Nur77 pathway mediates activation-induced cell death of macrophages [226]. The activation of MEF-2C by MEKK-2 kinase activity induces c-JUN expression and activates cytokine production through FcἐR1 for stimulation of mast cells [227]. MEF-2C is also required for restraining age-related microglial inflammation through excessive production of IFN-beta and its loss results in brain related inflammatory syndromes [228]. Although it has been associated with exclusively B-cell restricted transcription factors [229], it also exerts multiple roles in T-cell associated function and disease. MEF-2C is activated by several mechanisms in various T-cell acute lymphoblastic/leukemia cell lines [230]. Transcriptome and molecular-cytogenic analysis, (Chromosome conformation capture on chip (4C)) of >117 pediatric patients revealed the involvement of MEF-2C and NKX2-1 as potential candidates as oncogenes in T-cell acute lymphoblastic leukemia [231]. Additionally, MEF-2C is significantly upregulated in chronic myelogenous leukemia (CML) patients and cell lines. MEF-2C and the CEBP (CCAAT-enhancer binding protein) pathway are associated with disease progression in CML [232].
Loss of skeletal muscle is a characteristic feature and leads to poor prognosis in cancer-associated cachexia; there is a downregulation of MEF-2C (at the mRNA and protein levels) in cachexia which leads to the loss of skeletal muscle architecture and mitochondrial integrity as observed by electron microscopy. This occurs due to the dysregulation of MEF-2C gene targets-myogenin and myokinase, and also downregulated oxygen carrying capacity, ATP regeneration and the calcineurin pathway, culminating in the severe diseased phenotype of contractile muscle and skeletal instability associated with muscle cachexia [233]. Knockdown of MEF-2C in endothelial cells upregulates pro-inflammatory molecules and enhances the adhesion of leukocytes to endothelial cells and activated NF-κB. This process is partially controlled by KLF2 (Krüppel-like Factor 2), and overexpression of MEF-2C has the opposite effect, establishing the function of MEF-2C as a negative regulator of inflammation in endothelial cells [234]. MEF-2C is one of the targets downregulated by miR-223, but the expression of MEF-2C is negatively correlated with miR-223 in CML patient samples, suggesting its role in CML pathogenesis [235]. Gene expression patterns also reveal that MEF-2C is highly overexpressed in pediatric patient samples of ETP-ALL (Early T-cell precursor acute lymphoblastic leukemia) [236]. Alternative splicing of MEF-2C mRNA results in myogenesis and tumorigenic phenotype in Rhabdomyosarcoma (RMS) cells. The inactive myogenic splice variant of MEF-2Cα1 is ubiquitously expressed, but only the active variant MEF-2Cα2 is required for active differentiation. The α exon is aberrantly expressed in RMS cells and overexpression of MEF-2Cα2 resulted in normal myogenesis and differentiation of RMS cells [237].
MEF-2D
Like all the members of the MEF-2 family, MEF-2D also plays a key role in neuronal cells, muscle cells and other developmental pathways and is regulated by various class II HDACs, specifically HDAC4 and 5. MEF-2D also undergoes alternative splicing which yields multiple transcript variants. The MEF-2D gene is located on chromosome 1q12- q23. It is ubiquitously expressed and exhibits high and uniform distribution of protein expression in most-cell and tissue types ranging from brain to skin. Although elevated mRNA transcripts are found in cerebral cortex, bone marrow, and skeletal muscle with 50.9, 49.8 and 55.5 tpms, respectively. All cells and tissues have high levels of protein expression of MEF-2D in cerebral cortex, endocrine system, bone marrow, hematopoietic, germinal centers and also non-germinal centers, cardiac and skeletal muscles, kidneys, breast and skin, etc. MEF-2D is mainly localized in the nucleoplasm and overexpression of MEF-2D is observed in colorectal, breast, prostate, lung and liver cancers, with 100% overexpression in lymphomas, urothelial, breast, thyroid carcinomas and melanomas (https://www.proteinatlas.org/).
MEF-2D in development
MEF-2D null mice experience suppressed cardiac hypertrophy, decrease in fibrosis, and an inability to activate fetal activation [145]. MEF-2D represses skeletal myogenesis upon phosphorylation by protein kinase A (PKA) and HDAC4 activity in the nucleus [57]. Recently, MEF-2D has been implicated in pro-tumorigenic effects, causing cardiac myxoma (CM), the most common cardiac tumor. CM tissue has upregulated expression of MEF-2D, and this is correlated with increased tumor size. MEF-2D regulates IGF-induced proliferation and control of apoptosis in the pathophysiology of CM, and knockdown of MEF-2D reduced the activity of IGF-1 and Matrix metalloprotein 9 (MMP-9) and associated tumorigenesis [238]. Similar studies reveal that MEF-2D expression positively correlates with the proliferation of CM cells; MEF-2D expression can be suppressed by miR-218, a tumor suppressor which is downregulated in myxoma cells and serves as an important target for treatment of cardiac myxoma [239].
Dual-specificity tyrosine-phosphorylation regulated kinase 1A (DYRK1A) is encoded in chromosome 21, which is a critical region associated with Down’s syndrome. MEF-2D is involved in the upregulation of this kinase and activates isoform-5 of DYRK1A. The interaction of these genes promotes neurodevelopment [240]. MEF-2D acts as a neuronal survival factor in the maintenance of dopaminergic neurons (DN), the progressive death of which leads to Parkinson’s disease (PD). PD-associated neurotoxins destabilize MEF-2D by PTMs. MPP+, a toxic metabolite, decreases the half-life of MEF-2D in DA neuronal cell lines by MEF-2D protein degradation and destabilizing MEF-2D mRNA [241]. Another study reports that modulation of transcription factor Nur77 via MEF-2D results in the DN loss in response to another neurotoxin, 1-Methl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). The neurotoxin affects the binding of MEF-2D and Nur77 under basal conditions and significantly decreases the expression of MEF-2D and Nur77. This results in the severe dopaminergic loss and increase in the post-synaptic Fos-B activity indicative of nigrostriatal damage in mice [242].
Dysfunctions in chaperone mediated autophagy (CMA) via α-synuclein are also associated with PD. Rotenone, a mitochondrial complex-1 inhibitor, has been used to study CMA substrates. The inhibitor induces the accumulation of α-synuclein and MEF-2D in the cytoplasm due to impaired lysosome-mediated degradation, and affected the protective role of MEF-2D resulting in mitochondrial dysfunction and oxidative stress [243]. The accumulation of α-synuclein in neurons of the nigral pathway reduces the protective activity of MEF-2D in idiopathic and experimental PD [244]. The expression of MEF-2D in microglia modulates neuroinflammation. MEF-2D binds to a MEF-2 binding site in the promoter region of the IL-10 gene and transcribes IL-10; the silencing of MEF-2D decreased IL-10 induction and increased the levels of TNF-α expression resulting in neuroinflammation [245]. MEF-2D also acts as a potential oncogene in the tumorigenesis of malignant glioma and is highly upregulated in grade 3 and 4 malignant glioma patients [246].
MEF-2D fusion proteins and disease
RNAseq analysis of 560 ALL patients report the rearrangements of MEF-2D along with five other genes. The rearrangements enhance the transcriptional activity of MEF-2D, activated histone acetylase-9, and promoted lymphoid transformation. Additionally, it results in de novo fusion proteins with BCL9 and FOXJ2 in a number of cases [247]. Another study with similar RNA-seq analysis of the Philadelphia chromosome reported MEF-2D translocations in the subjects and similar fusions of MEF-2D/BCL9 and MEF-2D/HRNPUL1. MEF-2D/BCL9 fusions are lethal and have the ability to cause leukemia. Many MEF-2D fusion mutations have dire prognosis: most of the mutations cause B-ALL, although some of these fusion products are not completely investigated [41, 147]. Recently, functional studies revealed the pathogenic role of another fusion product resulting in the chromosomal translocation of MEF-2D/SS18 and had the worst prognosis. The studies showed significant inhibition of mouse common lymphoid progenitors into CD19 positive B-cells and diminished B-cell development; additionally, drug sensitivity to B-cell survival is upregulated and plays an important role in the pathobiology of B-ALL [248]. Neurotrophic tyrosine kinases (NTRKs) are highly effective targets for treatment options in many cancers. Fusion proteins involving NTRK1,2, and 3 are being actively investigated in the study of rare cancers such as secretory breast carcinoma, infantile fibrosarcoma, and congenital mesoblastic nephroma. One study reveals the cytoplasmic/membranous expression of NTRK1/MEF-2D fusion protein with a strong (3+) and uniform distribution among carcinomas [249]. Clinical bioinformatic analysis reveals that patients suffering from chronic obstructive pulmonary disease have poor prognosis for developing lung cancer and have higher expression of MEF-2D in non-small cell lung carcinoma (NSCLC), because of its affects on cell proliferation, differentiation and metastasis [250]. In lung cancer, expression of miR-30a has been inversely correlated with MEF-2D, and miR-30a mimetics inhibited the growth of lung cancer by suppressing MEF-2D [251]. The study predicts that miR-30a targets the 3’UTR of MEF-2D mRNA and promotes apoptosis in lung cancer cells. Similar studies performed in osteosarcoma patients reveal that over-expression of MEF-2D and miR-30a leads to a tumor suppressor effect and suppresses osteosarcoma cell proliferation by inhibiting MEF-2D [252]. Long-coding RNAs (lncR-D63785) are emerging trends in cancer implications, and it was revealed that lncR-D63785 expression inversely correlated with miR-422a which can downregulate MEF-2D and drug sensitivity in gastric carcinoma. Knockdown of lncR-D63785 upregulates the expression of miR-422a and sensitized gastric cancer cells to apoptosis induced by doxorubicin; the long non-coding RNA served as a competitive endogenous RNA for miR-422 and promoted chemoresistance by suppressing MEF-2D [253]. MEF-2D is also implicated in angiogenesis and EMT via TGF-β in an autoregulatory mechanism in hepatocellular carcinoma (HCC). MEF-2D along with other isoforms were found to increase expression of TGF-β in HCC, resulting in the activation of the PI3K/AKT/MEF-2 signaling pathway which promoted the progression of metastasis and EMT in HCC [180].
MEF-2 based therapeutics, HDACi
Class IIa HDACs are involved in the direct binding and suppression of MEF-2 proteins through the MAD/MEF-2 domains [63]. Association of class II HDACs with MEF-2 results in the deacetylation of histones in the vicinity of MEF-2 DNA-binding sites and subsequent suppression of MEF-2 target genes [254]. Furthermore, the association of HDAC with MEF-2 can trigger sumoylation of MEF-2 and further decrease its transcriptional potential [255]. Interestingly, HDAC4 and 5 are highly expressed in the heart, skeletal muscle, and brain, the same tissues in which MEF-2 expression is highest [63]. HDACs 4, 5, 7, and 9 interact with MEF-2 to regulate the differentiation of various cells, including those in bone, brain, skeletal and cardiac muscle, and vascular endothelium [255]. Aberrations in class IIa HDACs are associated with skeletal abnormalities, cardiac defects, denervation super sensitivity, and embryonic lethality [255]. More recently, the HDAC-MEF-2 axis has been implicated in various cancers. Increased expression of class IIa HDACs has been correlated with suppression of MEF-2 transcriptional activity and poor prognosis of estrogen receptor-positive (ER+) breast tumors as well as increased proliferation of mammary epithelial cells [76]. Overexpression of HDAC9 has been linked to poor prognosis of oral squamous cell carcinoma by targeting MEF-2D and repressing expression of MEF-2-dependent genes [77]. Class IIa HDACs in conjunction with MEF-2 have been implicated in leiomyosarcomas as well [41].
Because of their widespread influence on oncogenesis, HDACs have emerged as the target of an entire class of anticancer drugs, known as histone deacetylase inhibitors (HDACi). HDAC inhibitors encompass a diverse group of small molecule drugs that can induce apoptosis, cell cycle arrest, differentiation, and autophagy of cancer cells and promote anti-angiogenic effects [256]. These drugs are predominantly efficacious for leukemias but not solid tumors [257]. Although numerous clinical trials are underway involving HDAC inhibitors, only four have received FDA approval for anticancer drugs: Vorinostat and Romidepsin for cutaneous T-cell lymphomas, Belinostat for relapsed or refractory peripheral T-cell lymphoma, and Panobinostat for multiple myeloma [257]. A fifth molecule known as Valproic acid (VPA) is a weak HDAC inhibitor with a known clinical profile that is commonly involved in clinical trials [257]. HDAC inhibitors complementary to specific amino acid residues on particular HDAC isoforms have been formulated, including HDACs 4, 6, and 7 [257]. Zinc-binding HDAC inhibitors have also been described for class I and II HDACs [257]. HDACi are grouped under 4 different classes on the basis of their chemical structures, hydroxamates, short chain fatty acids, benzamides and cyclic peptides [72].
The precise mechanisms of HDAC inhibitors have not been fully elucidated. Vorinostat (suberoylanilide hydroxamic acid, SAHA) is characterized as a pan-HDAC inhibitor, is a hydroxamic acid derivative (inhibits classes I, II, and III), and is thought to increase activity of the tumor suppressor gene RUNX3 in conjunction with the histone acetylase protein p300. Romidepsin, a depsipeptide, is a selective inhibitor of class I HDACs, specifically HDAC1 and 2 [258]. An ex vivo mechanistic study of Romidepsin action in HTLV-1-infected ATL cells found that this drug inhibited transcription factors NF-κB and AP-1 as well as decreased expression of cyclin D2 and Bcl-xL. Belinostat is another pan-HDAC inhibitor and was found to induce apoptosis by TGF-β signaling-dependent down-regulation of survivin [259, 260]. Panobinostat is also a hydroxamic acid derivative and a pan-HDAC inhibitor with ten times the inhibitory activity of vorinostat [261]. It has been shown to reactivate tumor suppressor genes such as p21 and inhibit factors such as Akt and hypoxia-inducible factor 1α [262]. VPA or Valproic acid, a short chain fatty acid derivative has been shown to inhibit class I HDACs with more efficacy than class II HDACs and selectively induce proteasomal degradation of HDAC2 by activating the Ubc E2 ubiquitin conjugase [263, 264].
HDAC inhibitors have been utilized in clinical and pre-clinical trials for treatment of ATL. Vorinostat, Romidepsin, and Panobinostat have shown promising anti-cancer effects in pre-clinical and/or clinical studies in ATL and other T-cell malignancies [99]. Aside from Romidepsin, VPA and pan-HDAC inhibitor AR-42 have been utilized in murine ATL models and shown efficacy [265]. AR-42 was found to decrease Akt, Bcl-xL, and survivin in a prostate cancer model [266]. However, in the context of case studies, due to modest response rates, high rate of cytopenias, and concerns of viral reactivation, it is advised that HDAC inhibitors be used cautiously for treatment of ATL [267]. Nevertheless, a phase 2 clinical trial using panobinostat for relapsed DLBCL found that patients with MEF-2B mutations had a higher chance (likelihood ratio 3.67, 95% CI 1.46-9.19) of a complete or partial response to treatment [198]. Peritoneal fibrosis is a pathological condition that alters peritoneal morphology, causing inflammation. The peritoneum is a continuous monolayer of mesothelial cells. When fibrosis occurs, there is a mesothelial to mesenchymal transition (MMT) which causes invasion. MS-275 is a specific class-I HDAC inhibitor which inhibits cellular invasion and promotes MMT reversal in peritoneal fibrosis [268]. HDACi are also used in various preclinical models of immunological diseases such as EAE (experimental autoimmune encephalomyelitis), asthma, and rheumatoid arthritis [269, 270]. Also, HDACi are used as a “shock and kill” mechanism to reactivate latent HIV and to deplete the viral reservoir [271], and recently Class-I specific HDACi were shown to enhance viral latency reversal and also preserve activity of HDAC isoforms for maximal HIV gene expression to target the virus [272]. The final class of HDACi are the Sirtuins inhibitors, which primarily target Sirt 1 and Sirt 2; nicotinamide is in phase III clinical trial for laryngeal cancer. Sirtinol and Cambinol are drugs in the preclinical stage of investigation, and EX-527 is in phase I and II trial for Huntington’s disease and glaucoma [86]. Certain studies demonstrate effects of HDACi in anti-parasitic activity against Plasmodium and Trypanosoma [273]. Recently, HDACi have evolved into chimeric HDACi (chi-HDACi) which simultaneously modulate both HDAC and other targets, using molecular hybridization techniques, drugs that target protein tyrosine kinases, epidermal growth factor (EGF), BCL-ABL tyrosine kinase, or VEGFR along with HDACi were made into chi-HDACi structures which can be used to treat multiple cancer types [274].
Although there are many HDAC inhibitors being studied in preclinical and clinical trials, one of the unique class IIA HDACi, namely MC1568, is highly selective to class IIA HDACs and has been shown to activate the tumor suppressor BRAHMA, that is suppressed by HDAC9 and exhibits cytostatic effect in melanoma cells much greater than Class I inhibitors [275]. MC1568 has been shown to decrease cell proliferation and IL-8 production in human melanoma cells, suppress c-JUN which is a transcriptional target of MEF-2 transcription factors and also suppress the activity of histones 3 and 4, RNA polymerase II and TFIIB to the c-JUN promoter [276]. It is now reported as an anti-influenza drug target that inhibits the activity of HDAC6/8 and decreased expression of viral proteins and mRNAs via the early acetylation of HSP90 [277]. It is a unique molecule that utilizes a distinct mechanism by stabilizing the HDAC4-MEF-2D complex [278] and inhibiting myogenesis [279].
Our lab has been characterizing MC1568 in the context of treatment for HTLV-1-induced ATLL, which is a malignant and intractable T-cell neoplasia. Over 5% of the infected population acquire an aggressive form of non-Hodgkin’s lymphoma; and there are no preventive or treatment measures in the form of vaccines or drugs respectively. We have shown earlier the implication of MEF-2A in the viral gene expression and T-cell proliferation and transformation [17]. We are currently investigating MC1568 in the treatment of ATLL, preliminary data suggests that MC1568 is selectively cytotoxic to virus producing leukemic cells but not non-infected cells. Moreover, activated autophagic signaling and a dose-dependent inhibition of viral protein expression of Tax and HBZ was also observed (Madugula et.al unpublished observations). Further mechanistic studies are underway to delineate the involvement of MEF-2 isoforms in the pathobiology of HTLV-1-induced ATLL. MC1568 might be the first of its kind for the treatment of ATLL.
Conclusions and perspectives
Due to the high-risk disease characteristics associated with MEF-2 aberrations, the outcomes of treatment are less encouraging, the diversity of disease phenotypes caused by MEF-2 family members are very peculiar and are associated to one or more isoforms, but the treatment regimens still more uncertain. Most studies concerning MEF-2 isoforms are based on their gain and loss of function, but the basal level expression of each isoform and how much it is altered in each tissue and cell type is still an open question. Although each isoform has been classified as an oncogene or tumor suppressor, it remains unclear the genetic alterations and molecular aberrations that are occurring in neoplasms associated with MEF-2 isoforms. Due to the differences in the dynamic nature of the MEF-2 isoforms in context of their roles in infection or leukemogenesis, it is warranted to develop isoform-specific molecular targets. Moreover, due to their association with specific classes of HDACs, it would be noteworthy that MEF-2 isoforms would serve as a potential therapeutic target in the treatment of a wide array of diseases.
Epigenetic regulators in the context of therapeutics, especially in the case of cancer and more recently in infection, are a matter of intense discussion. But over time with the evolution of disease categories, HDACi have become more and more indispensable in anti-cancer therapeutics and other infections like influenza and ATLL. Because of their selective cytotoxic effects and induction of autophagy and apoptosis in tumor cells, and feasibility in combinatorial treatments, there are multiple clinical trials based on HDACi as reviewed in [280, 281] that are underway to treat various types of blood cancers and solid tumors and some have reached Phase IV of the trials. The enormous potential of these drug moieties can be tapped out for not only cancer-related syndromes but also in the treatment of infectious diseases. It will be a priority to discover more potent HDACi which are more class and isoform-specific molecules that can provide greater efficacy compared to using more broad-spectrum Pan-HDACi. In this context using Class II specific HDACi that are associated with MEF-2 isoforms may be anticipated to show greater therapeutic potential and fewer off-target effects. Therefore, MEF-2 isoforms can be harnessed by modulating these specific class of HDACs via a specific class of HDACi to open a new range of therapeutics for a wide array of diseases.
Abbreviations
MEF-2 - Myocyte enhancer factor-2
HDACs - Histone deacetylases
HDACi - Histone deacetylase inhibitors
IL-2 - Interleukin-2
TCLs - T-cell Leukemias/Lymphomas
T-ALL - T-Cell Acute Lymphoblastic Leukemia/Lymphoma
ATLL - Adult T-Cell Leukemia/Lymphoma
CTCL - Cutaneous T-Cell Lymphoma
PTCL - Peripheral T-cell Lymphoma
EBV - Epstein-Barr virus
HIV - Human immunodeficiency virus
HTLV-1 - Human T-cell Leukemia virus-1
NFAT - Nuclear Factor in activated T cells
MITR - MEF-2-interacting transcriptional repressor
IFN - Interferon
VSMCs - Vascular smooth muscle cells
AD - Alzheimer’s disease
TPM - Transcripts per kilobase million
Capn-3 - Calpain3
mi-RNA - microRNA
BDNF - Brain-derived neurotrophic factor
CMA - Chaperone mediated autophagy
TLE - Temporal lobe epilepsy
CAD - Coronary artery disease
DLBCL - Diffuse large B-cell Lymphomas
CML - Chronic myelogenous Leukemia
KLF - Krüppel-like factor
ETP-ALL - Early T-cell precursor acute Lymphoblastic Leukemia
HCC - Hepatocellular carcinoma
EMT - Epithelial to mesenchymal transformation
TGF-β - Transforming growth factor β
PKA - Protein Kinase A
GSK3 β - Glycogen synthase kinase 3β
CONFLICTS OF INTEREST
There are no conflicts of interest.
GRANT SUPPORT
This work was supported by extramural funding from NIH/NINDS to Pooja Jain (1RO1NS097147-01A1).
REFERENCES
1. Pon JR, Marra MA. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget. 2016; 7:2297–312. https://doi.org/10.18632/oncotarget.6223.
2. Wang YN, Yang WC, Li PW, Wang HB, Zhang YY, Zan LS. Myocyte enhancer factor 2A promotes proliferation and its inhibition attenuates myogenic differentiation via myozenin 2 in bovine skeletal muscle myoblast. PLoS One. 2018; 13:e0196255. https://doi.org/10.1371/journal.pone.0196255.
3. Suarez-Bregua P, Chien CJ, Megias M, Du S, Rotllant J. Promoter architecture and transcriptional regulation of musculoskeletal embryonic nuclear protein 1b (mustn1b) gene in zebrafish. Dev Dyn. 2017; 246:992–1000. https://doi.org/10.1002/dvdy.24591.
4. Sincennes MC, Brun CE, Rudnicki MA. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl Med. 2016; 5:282–90. https://doi.org/10.5966/sctm.2015-0266.
5. Sacilotto N, Chouliaras KM, Nikitenko LL, Lu YW, Fritzsche M, Wallace MD, Nornes S, García-Moreno F, Payne S, Bridges E, Liu K, Biggs D, Ratnayaka I, et al. MEF2 transcription factors are key regulators of sprouting angiogenesis. Genes Dev. 2016; 30:2297–309. https://doi.org/10.1101/gad.290619.116.
6. Rashid AJ, Cole CJ, Josselyn SA. Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav. 2014; 13:118–25. https://doi.org/10.1111/gbb.12058.
7. Moretti I, Ciciliot S, Dyar KA, Abraham R, Murgia M, Agatea L, Akimoto T, Bicciato S, Forcato M, Pierre P, Uhlenhaut NH, Rigby PW, Carvajal JJ, et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat Commun. 2016; 7:12397. https://doi.org/10.1038/ncomms12397.
8. Ichihara M, Kamiya T, Hara H, Adachi T. The MEF2A and MEF2D function as scaffold proteins that interact with HDAC1 or p300 in SOD3 expression in THP-1 cells. Free Radic Res. 2018; 52:799–807. https://doi.org/10.1080/10715762.2018.1475730.
9. Herglotz J, Unrau L, Hauschildt F, Fischer M, Kriebitzsch N, Alawi M, Indenbirken D, Spohn M, Müller U, Ziegler M, Schuh W, Jäck HM, Stocking C. Essential control of early B-cell development by Mef2 transcription factors. Blood. 2016; 127:572–81. https://doi.org/10.1182/blood-2015-04-643270.
10. Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, Raduazzo J, Guo Y, Loerwald K, Konopka G, Huber KM, Cowan CW. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife. 2016; 5:e20059. https://doi.org/10.7554/eLife.20059.
11. Di-Luoffo M, Brousseau C, Bergeron F, Tremblay JJ. The Transcription Factor MEF2 Is a Novel Regulator of Gsta Gene Class in Mouse MA-10 Leydig Cells. Endocrinology. 2015; 156:4695–706. https://doi.org/10.1210/en.2015-1500.
12. Cohen TJ, Choi MC, Kapur M, Lira VA, Yan Z, Yao TP. HDAC4 regulates muscle fiber type-specific gene expression programs. Mol Cells. 2015; 38:343–48. https://doi.org/10.14348/molcells.2015.2278.
13. Bachinski LL, Sirito M, Böhme M, Baggerly KA, Udd B, Krahe R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve. 2010; 42:856–63. https://doi.org/10.1002/mus.21789.
14. Rullman E, Fernandez-Gonzalo R, Mekjavić IB, Gustafsson T, Eiken O. MEF2 as upstream regulator of the transcriptome signature in human skeletal muscle during unloading. Am J Physiol Regul Integr Comp Physiol. 2018; 315:R799–809. https://doi.org/10.1152/ajpregu.00452.2017.
15. Nagel S, Venturini L, Meyer C, Kaufmann M, Scherr M, Drexler HG, Macleod RA. Transcriptional deregulation of oncogenic myocyte enhancer factor 2C in T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2011; 52:290–97. https://doi.org/10.3109/10428194.2010.537003.
16. Zuurbier L, Gutierrez A, Mullighan CG, Canté-Barrett K, Gevaert AO, de Rooi J, Li Y, Smits WK, Buijs-Gladdines JG, Sonneveld E, Look AT, Horstmann M, Pieters R, Meijerink JP. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica. 2014; 99:94–102. https://doi.org/10.3324/haematol.2013.090233.
17. Jain P, Lavorgna A, Sehgal M, Gao L, Ginwala R, Sagar D, Harhaj EW, Khan ZK. Myocyte enhancer factor (MEF)-2 plays essential roles in T-cell transformation associated with HTLV-1 infection by stabilizing complex between Tax and CREB. Retrovirology. 2015; 12:23. https://doi.org/10.1186/s12977-015-0140-1.
18. Cosenza M, Pozzi S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int J Mol Sci. 2018; 19:E2337. https://doi.org/10.3390/ijms19082337.
19. Tempera I, De Leo A, Kossenkov AV, Cesaroni M, Song H, Dawany N, Showe L, Lu F, Wikramasinghe P, Lieberman PM. Identification of MEF2B, EBF1, and IL6R as Direct Gene Targets of Epstein-Barr Virus (EBV) Nuclear Antigen 1 Critical for EBV-Infected B-Lymphocyte Survival. J Virol. 2015; 90:345–55. https://doi.org/10.1128/JVI.02318-15.
20. Murata T, Narita Y, Sugimoto A, Kawashima D, Kanda T, Tsurumi T. Contribution of myocyte enhancer factor 2 family transcription factors to BZLF1 expression in Epstein-Barr virus reactivation from latency. J Virol. 2013; 87:10148–62. https://doi.org/10.1128/JVI.01002-13.
21. McDonald C, Karstegl CE, Kellam P, Farrell PJ. Regulation of the Epstein-Barr virus Zp promoter in B lymphocytes during reactivation from latency. J Gen Virol. 2010; 91:622–29. https://doi.org/10.1099/vir.0.017277-0.
22. Gruffat H, Manet E, Sergeant A. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 2002; 3:141–46. https://doi.org/10.1093/embo-reports/kvf031.
23. Yndart A, Kaushik A, Agudelo M, Raymond A, Atluri VS, Saxena SK, Nair M. Investigation of Neuropathogenesis in HIV-1 Clade B and C Infection Associated with IL-33 and ST2 Regulation. ACS Chem Neurosci. 2015; 6:1600–12. https://doi.org/10.1021/acschemneuro.5b00156.
24. Wu Z, Nagano I, Takahashi Y. Candidate genes responsible for common and different pathology of infected muscle tissues between Trichinella spiralis and T. pseudospiralis infection. Parasitol Int. 2008; 57:368–78. https://doi.org/10.1016/j.parint.2008.03.005.
25. Clark RI, Tan SW, Péan CB, Roostalu U, Vivancos V, Bronda K, Pilátová M, Fu J, Walker DW, Berdeaux R, Geissmann F, Dionne MS. MEF2 is an in vivo immune-metabolic switch. Cell. 2013; 155:435–47. https://doi.org/10.1016/j.cell.2013.09.007.
26. Saito M, Jain P, Tsukasaki K, Bangham CR. HTLV-1 Infection and Its Associated Diseases. Leuk Res Treatment. 2012; 2012:123637. https://doi.org/10.1155/2012/123637.
27. Sagar D, Masih S, Schell T, Jacobson S, Comber JD, Philip R, Wigdahl B, Jain P, Khan ZK. In vivo immunogenicity of Tax(11-19) epitope in HLA-A2/DTR transgenic mice: implication for dendritic cell-based anti-HTLV-1 vaccine. Vaccine. 2014; 32:3274–84. https://doi.org/10.1016/j.vaccine.2014.03.087.
28. Rahman S, Quann K, Pandya D, Singh S, Khan ZK, Jain P. HTLV-1 Tax mediated downregulation of miRNAs associated with chromatin remodeling factors in T cells with stably integrated viral promoter. PLoS One. 2012; 7:e34490. https://doi.org/10.1371/journal.pone.0034490.
29. Mulherkar R, Karabudak A, Ginwala R, Huang X, Rowan A, Philip R, Murphy EL, Clements D, Ndhlovu LC, Khan ZK, Jain P. In vivo and in vitro immunogenicity of novel MHC class I presented epitopes to confer protective immunity against chronic HTLV-1 infection. Vaccine. 2018; 36:5046–57. https://doi.org/10.1016/j.vaccine.2018.07.002.
30. Manuel SL, Sehgal M, Khan ZK, Goedert JJ, Betts MR, Jain P. An altered maturation and adhesion phenotype of dendritic cells in diseased individuals compared to asymptomatic carriers of human T cell leukemia virus type 1. AIDS Res Hum Retroviruses. 2013; 29:1273–85. https://doi.org/10.1089/aid.2013.0054.
31. Manuel SL, Sehgal M, Connolly J, Makedonas G, Khan ZK, Gardner J, Betts MR, Jain P. Lack of recall response to Tax in ATL and HAM/TSP patients but not in asymptomatic carriers of human T-cell leukemia virus type 1. J Clin Immunol. 2013; 33:1223–39. https://doi.org/10.1007/s10875-013-9918-x.
32. Manuel SL, Schell TD, Acheampong E, Rahman S, Khan ZK, Jain P. Presentation of human T cell leukemia virus type 1 (HTLV-1) Tax protein by dendritic cells: the underlying mechanism of HTLV-1-associated neuroinflammatory disease. J Leukoc Biol. 2009; 86:1205–16. https://doi.org/10.1189/jlb.0309172.
33. Lepoutre V, Jain P, Quann K, Wigdahl B, Khan ZK. Role of resident CNS cell populations in HTLV-1-associated neuroinflammatory disease. Front Biosci. 2009; 14:1152–68. https://doi.org/10.2741/3300.
34. Jaworski E, Narayanan A, Van Duyne R, Shabbeer-Meyering S, Iordanskiy S, Saifuddin M, Das R, Afonso PV, Sampey GC, Chung M, Popratiloff A, Shrestha B, Sehgal M, et al. Human T-lymphotropic virus type 1-infected cells secrete exosomes that contain Tax protein. J Biol Chem. 2014; 289:22284–305. https://doi.org/10.1074/jbc.M114.549659.
35. Jain P, Mostoller K, Flaig KE, Ahuja J, Lepoutre V, Alefantis T, Khan ZK, Wigdahl B. Identification of human T cell leukemia virus type 1 tax amino acid signals and cellular factors involved in secretion of the viral oncoprotein. J Biol Chem. 2007; 282:34581–93. https://doi.org/10.1074/jbc.M707317200.
36. Grant C, Nonnemacher M, Jain P, Pandya D, Irish B, Williams SC, Wigdahl B. CCAAT/enhancer-binding proteins modulate human T cell leukemia virus type 1 long terminal repeat activation. Virology. 2006; 348:354–69. https://doi.org/10.1016/j.virol.2005.12.024.
37. Grant C, Jain P, Nonnemacher M, Flaig KE, Irish B, Ahuja J, Alexaki A, Alefantis T, Wigdahl B. AP-1-directed human T cell leukemia virus type 1 viral gene expression during monocytic differentiation. J Leukoc Biol. 2006; 80:640–50. https://doi.org/10.1189/jlb.1205723.
38. Ginwala R, Caruso B, Khan ZK, Pattekar A, Chew GM, Corley MJ, Loonawat R, Jacobson S, Sreedhar S, Ndhlovu LC, Jain P. HTLV-1 Infection and Neuropathogenesis in the Context of Rag1-/-γc-/- (RAG1-Hu) and BLT Mice. J Neuroimmune Pharmacol. 2017; 12:504–20. https://doi.org/10.1007/s11481-017-9740-y.
39. Alefantis T, Mostoller K, Jain P, Harhaj E, Grant C, Wigdahl B. Secretion of the human T cell leukemia virus type I transactivator protein tax. J Biol Chem. 2005; 280:17353–62. https://doi.org/10.1074/jbc.M409851200.
40. Alefantis T, Jain P, Ahuja J, Mostoller K, Wigdahl B. HTLV-1 Tax nucleocytoplasmic shuttling, interaction with the secretory pathway, extracellular signaling, and implications for neurologic disease. J Biomed Sci. 2005; 12:961–74. https://doi.org/10.1007/s11373-005-9026-x.
41. Di Giorgio E, Franforte E, Cefalù S, Rossi S, Dei Tos AP, Brenca M, Polano M, Maestro R, Paluvai H, Picco R, Brancolini C. The co-existence of transcriptional activator and transcriptional repressor MEF2 complexes influences tumor aggressiveness. PLoS Genet. 2017; 13:e1006752. https://doi.org/10.1371/journal.pgen.1006752.
42. Dietrich JB. The MEF2 family and the brain: from molecules to memory. Cell Tissue Res. 2013; 352:179–90. https://doi.org/10.1007/s00441-013-1565-2.
43. Shin HM, Han TH. CD28-mediated regulation of the c-jun promoter involves the MEF2 transcription factor in Jurkat T cells. Mol Immunol. 1999; 36:197–203. https://doi.org/10.1016/S0161-5890(99)00030-9.
44. Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol. 2005; 79:10308–29. https://doi.org/10.1128/JVI.79.16.10308-10329.2005.
45. Katsuyama M, Ozgur Cevik M, Arakawa N, Kakehi T, Nishinaka T, Iwata K, Ibi M, Matsuno K, Yabe-Nishimura C. Myocyte enhancer factor 2B is involved in the inducible expression of NOX1/NADPH oxidase, a vascular superoxide-producing enzyme. FEBS J. 2007; 274:5128–36. https://doi.org/10.1111/j.1742-4658.2007.06034.x.
46. Zheng M, Zhu J, Lu T, Liu L, Sun H, Liu Z, Tian J. p300-mediated histone acetylation is essential for the regulation of GATA4 and MEF2C by BMP2 in H9c2 cells. Cardiovasc Toxicol. 2013; 13:316–22. https://doi.org/10.1007/s12012-013-9212-4.
47. Youn HD, Liu JO. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 2000; 13:85–94. https://doi.org/10.1016/S1074-7613(00)00010-8.
48. Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000; 19:4323–31. https://doi.org/10.1093/emboj/19.16.4323.
49. Ma K, Chan JK, Zhu G, Wu Z. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol. 2005; 25:3575–82. https://doi.org/10.1128/MCB.25.9.3575-3582.2005.
50. He J, Ye J, Cai Y, Riquelme C, Liu JO, Liu X, Han A, Chen L. Structure of p300 bound to MEF2 on DNA reveals a mechanism of enhanceosome assembly. Nucleic Acids Res. 2011; 39:4464–74. https://doi.org/10.1093/nar/gkr030.
51. De Luca A, Severino A, De Paolis P, Cottone G, De Luca L, De Falco M, Porcellini A, Volpe M, Condorelli G. p300/cAMP-response-element-binding-protein (‘CREB’)-binding protein (CBP) modulates co-operation between myocyte enhancer factor 2A (MEF2A) and thyroid hormone receptor-retinoid X receptor. Biochem J. 2003; 369:477–84. https://doi.org/10.1042/bj20020057.
52. Aude-Garcia C, Collin-Faure V, Bausinger H, Hanau D, Rabilloud T, Lemercier C. Dual roles for MEF2A and MEF2D during human macrophage terminal differentiation and c-Jun expression. Biochem J. 2010; 430:237–44. https://doi.org/10.1042/BJ20100131.
53. Dionyssiou MG, Nowacki NB, Hashemi S, Zhao J, Kerr A, Tsushima RG, McDermott JC. Cross-talk between glycogen synthase kinase 3β (GSK3β) and p38MAPK regulates myocyte enhancer factor 2 (MEF2) activity in skeletal and cardiac muscle. J Mol Cell Cardiol. 2013; 54:35–44. https://doi.org/10.1016/j.yjmcc.2012.10.013.
54. Linseman DA, Cornejo BJ, Le SS, Meintzer MK, Laessig TA, Bouchard RJ, Heidenreich KA. A myocyte enhancer factor 2D (MEF2D) kinase activated during neuronal apoptosis is a novel target inhibited by lithium. J Neurochem. 2003; 85:1488–99. https://doi.org/10.1046/j.1471-4159.2003.09799.x.
55. Wang X, She H, Mao Z. Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3beta in neuronal apoptosis. J Biol Chem. 2009; 284:32619–26. https://doi.org/10.1074/jbc.M109.067785.
56. Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, Olson EN. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol. 2011; 195:403–15. https://doi.org/10.1083/jcb.201105063.
57. Du M, Perry RL, Nowacki NB, Gordon JW, Salma J, Zhao J, Aziz A, Chan J, Siu KW, McDermott JC. Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol Cell Biol. 2008; 28:2952–70. https://doi.org/10.1128/MCB.00248-08.
58. Salma J, McDermott JC. Suppression of a MEF2-KLF6 survival pathway by PKA signaling promotes apoptosis in embryonic hippocampal neurons. J Neurosci. 2012; 32:2790–803. https://doi.org/10.1523/JNEUROSCI.3609-11.2012.
59. Berger I, Bieniossek C, Schaffitzel C, Hassler M, Santelli E, Richmond TJ. Direct interaction of Ca2+/calmodulin inhibits histone deacetylase 5 repressor core binding to myocyte enhancer factor 2. J Biol Chem. 2003; 278:17625–35. https://doi.org/10.1074/jbc.M301646200.
60. Zhou X, Marks PA, Rifkind RA, Richon VM. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A. 2001; 98:10572–77. https://doi.org/10.1073/pnas.191375098.
61. Zhang CL, McKinsey TA, Olson EN. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci U S A. 2001; 98:7354–59. https://doi.org/10.1073/pnas.131198498.
62. Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007; 117:2459–67. https://doi.org/10.1172/JCI31960.
63. Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, De Luca A, Rotili D, Valente S, Mai A, Usiello A, Gronemeyer H, Altucci L. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 2009; 10:776–82. https://doi.org/10.1038/embor.2009.88.
64. McGee SL. Exercise and MEF2-HDAC interactions. Appl Physiol Nutr Metab. 2007; 32:852–56. https://doi.org/10.1139/H07-082.
65. Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000; 6:233–44. https://doi.org/10.1016/S1097-2765(00)00025-3.
66. Kim MS, Fielitz J, McAnally J, Shelton JM, Lemon DD, McKinsey TA, Richardson JA, Bassel-Duby R, Olson EN. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol. 2008; 28:3600–09. https://doi.org/10.1128/MCB.00189-08.
67. Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci U S A. 2000; 97:7835–40. https://doi.org/10.1073/pnas.140199597.
68. Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class II histone deacetylases. Biochem Cell Biol. 2001; 79:337–48. https://doi.org/10.1139/o01-116.
69. Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, Muscat GE. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J Biol Chem. 2001; 276:17007–13. https://doi.org/10.1074/jbc.M101508200.
70. Calalb MB, McKinsey TA, Newkirk S, Huynh K, Sucharov CC, Bristow MR. Increased phosphorylation-dependent nuclear export of class II histone deacetylases in failing human heart. Clin Transl Sci. 2009; 2:325–32. https://doi.org/10.1111/j.1752-8062.2009.00141.x.
71. Mathias RA, Guise AJ, Cristea IM. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol Cell Proteomics. 2015; 14:456–70. https://doi.org/10.1074/mcp.O114.046565.
72. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014; 6:a018713. https://doi.org/10.1101/cshperspect.a018713.
73. You SH, Liao X, Weiss RE, Lazar MA. The interaction between nuclear receptor corepressor and histone deacetylase 3 regulates both positive and negative thyroid hormone action in vivo. Mol Endocrinol. 2010; 24:1359–67. https://doi.org/10.1210/me.2009-0501.
74. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009; 10:32–42. https://doi.org/10.1038/nrg2485.
75. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014; 15:536–50. https://doi.org/10.1038/nrm3841.
76. Clocchiatti A, Di Giorgio E, Ingrao S, Meyer-Almes FJ, Tripodo C, Brancolini C. Class IIa HDACs repressive activities on MEF2-depedent transcription are associated with poor prognosis of ER(+) breast tumors. FASEB J. 2013; 27:942–54. https://doi.org/10.1096/fj.12-209346.
77. Rastogi B, Raut SK, Panda NK, Rattan V, Radotra BD, Khullar M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol Cell Biochem. 2016; 415:183–96. https://doi.org/10.1007/s11010-016-2690-5.
78. Di Giorgio E, Clocchiatti A, Piccinin S, Sgorbissa A, Viviani G, Peruzzo P, Romeo S, Rossi S, Dei Tos AP, Maestro R, Brancolini C. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol Cell Biol. 2013; 33:4473–91. https://doi.org/10.1128/MCB.01050-13.
79. Pécuchet N, Cluzeau T, Thibault C, Mounier N, Vignot S. [Histone deacetylase inhibitors: highlight on epigenetic regulation]. [Article in French]. Bull Cancer. 2010; 97:917–35. https://doi.org/10.1684/bdc.2010.1121.
80. Chou CW, Chen CC. HDAC inhibition upregulates the expression of angiostatic ADAMTS1. FEBS Lett. 2008; 582:4059–65. https://doi.org/10.1016/j.febslet.2008.10.048.
81. Duncan HF, Smith AJ, Fleming GJ, Cooper PR. HDACi: cellular effects, opportunities for restorative dentistry. J Dent Res. 2011; 90:1377–88. https://doi.org/10.1177/0022034511406919.
82. Singh AK, Bishayee A, Pandey AK. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients. 2018; 10:E731. https://doi.org/10.3390/nu10060731.
83. Olzscha H, Bekheet ME, Sheikh S, La Thangue NB. HDAC Inhibitors. Methods Mol Biol. 2016; 1436:281–303. https://doi.org/10.1007/978-1-4939-3667-0_19.
84. Jing B, Jin J, Xiang R, Liu M, Yang L, Tong Y, Xiao X, Lei H, Liu W, Xu H, Deng J, Zhou L, Wu Y. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis. 2018; 9:589. https://doi.org/10.1038/s41419-018-0679-6.
85. Eftekhar E, Jaberie H, Naghibalhossaini F. Carcinoembryonic Antigen Expression and Resistance to Radiation and 5-Fluorouracil-Induced Apoptosis and Autophagy. Int J Mol Cell Med. 2016; 5:80–89.
86. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017; 18:E1414. https://doi.org/10.3390/ijms18071414.
87. Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, Chang S, Ling H, Bers DM, Maier LS, Olson EN, Brown JH. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007; 282:35078–87. https://doi.org/10.1074/jbc.M707083200.
88. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002; 110:479–88. https://doi.org/10.1016/S0092-8674(02)00861-9.
89. Ojuka EO, Goyaram V, Smith JA. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am J Physiol Endocrinol Metab. 2012; 303:E322–31. https://doi.org/10.1152/ajpendo.00091.2012.
90. Ginnan R, Sun LY, Schwarz JJ, Singer HA. MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J. 2012; 444:105–14. https://doi.org/10.1042/BJ20120152.
91. Ellis JJ, Valencia TG, Zeng H, Roberts LD, Deaton RA, Grant SR. CaM kinase IIdeltaC phosphorylation of 14-3-3beta in vascular smooth muscle cells: activation of class II HDAC repression. Mol Cell Biochem. 2003; 242:153–61. https://doi.org/10.1023/A:1021158216582.
92. Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006; 311:1012–17. https://doi.org/10.1126/science.1122513.
93. Esau C, Boes M, Youn HD, Tatterson L, Liu JO, Chen J. Deletion of calcineurin and myocyte enhancer factor 2 (MEF2) binding domain of Cabin1 results in enhanced cytokine gene expression in T cells. J Exp Med. 2001; 194:1449–59. https://doi.org/10.1084/jem.194.10.1449.
94. Faustino RS, Behfar A, Groenendyk J, Wyles SP, Niederlander N, Reyes S, Puceat M, Michalak M, Terzic A, Perez-Terzic C. Calreticulin secures calcium-dependent nuclear pore competency required for cardiogenesis. J Mol Cell Cardiol. 2016; 92:63–74. https://doi.org/10.1016/j.yjmcc.2016.01.022.
95. Wu R, Wang J, Yao J, Dong Z, Liu Y, Liu M. MEF2A regulates Calpain 3 expression in L6 myoblasts. Gene. 2018; 668:204–10. https://doi.org/10.1016/j.gene.2018.05.056.
96. Good L, Maggirwar SB, Harhaj EW, Sun SC. Constitutive dephosphorylation and activation of a member of the nuclear factor of activated T cells, NF-AT1, in Tax-expressing and type I human T-cell leukemia virus-infected human T cells. J Biol Chem. 1997; 272:1425–28. https://doi.org/10.1074/jbc.272.3.1425.
97. Boxus M, Willems L. Mechanisms of HTLV-1 persistence and transformation. Br J Cancer. 2009; 101:1497–501. https://doi.org/10.1038/sj.bjc.6605345.
98. Youn HD, Grozinger CM, Liu JO. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem. 2000; 275:22563–67. https://doi.org/10.1074/jbc.C000304200.
99. Tsukasaki K, Tobinai K. Clinical Trials and Treatment of ATL. Leuk Res Treatment. 2012; 2012:101754. https://doi.org/10.1155/2012/101754.
100. Nosaka K, Miyamoto T, Sakai T, Mitsuya H, Suda T, Matsuoka M. Mechanism of hypercalcemia in adult T-cell leukemia: overexpression of receptor activator of nuclear factor kappaB ligand on adult T-cell leukemia cells. Blood. 2002; 99:634–40. https://doi.org/10.1182/blood.V99.2.634.
101. Matutes E. Adult T-cell leukaemia/lymphoma. J Clin Pathol. 2007; 60:1373–77. https://doi.org/10.1136/jcp.2007.052456.
102. Shirakawa F, Yamashita U, Oda S, Chiba S, Eto S, Suzuki H. Calcium dependency in the growth of adult T-cell leukemia cells in vitro. Cancer Res. 1986; 46:658–61.
103. Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000; 25:257–60. https://doi.org/10.1016/S0968-0004(00)01595-4.
104. Nebreda AR, Ferby I. Regulation of the meiotic cell cycle in oocytes. Curr Opin Cell Biol. 2000; 12:666–75. https://doi.org/10.1016/S0955-0674(00)00150-2.
105. Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009; 5:44–51. https://doi.org/10.7150/ijbs.5.44.
106. Dinsmore CJ, Soriano P. MAPK and PI3K signaling: At the crossroads of neural crest development. Dev Biol. 2018 (Suppl 1); 444:S79–S97. https://doi.org/10.1016/j.ydbio.2018.02.003.
107. Kong NR, Davis M, Chai L, Winoto A, Tjian R. MEF2C and EBF1 Co-regulate B Cell-Specific Transcription. PLoS Genet. 2016; 12:e1005845. https://doi.org/10.1371/journal.pgen.1005845.
108. Khiem D, Cyster JG, Schwarz JJ, Black BL. A p38 MAPK-MEF2C pathway regulates B-cell proliferation. Proc Natl Acad Sci U S A. 2008; 105:17067–72. https://doi.org/10.1073/pnas.0804868105.
109. Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J Biol Chem. 1999; 274:31102–07. https://doi.org/10.1074/jbc.274.43.31102.
110. Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999; 286:785–90. https://doi.org/10.1126/science.286.5440.785.
111. Han J, Molkentin JD. Regulation of MEF2 by p38 MAPK and its implication in cardiomyocyte biology. Trends Cardiovasc Med. 2000; 10:19–22. https://doi.org/10.1016/S1050-1738(00)00039-6.
112. Xu Q, Yu L, Liu L, Cheung CF, Li X, Yee SP, Yang XJ, Wu Z. p38 Mitogen-activated protein kinase-, calcium-calmodulin-dependent protein kinase-, and calcineurin-mediated signaling pathways transcriptionally regulate myogenin expression. Mol Biol Cell. 2002; 13:1940–52. https://doi.org/10.1091/mbc.02-02-0016.
113. Shen E, Diao X, Wang X, Chen R, Hu B. MicroRNAs involved in the mitogen-activated protein kinase cascades pathway during glucose-induced cardiomyocyte hypertrophy. Am J Pathol. 2011; 179:639–50. https://doi.org/10.1016/j.ajpath.2011.04.034.
114. Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. Bonekey Rep. 2015; 4:711. https://doi.org/10.1038/bonekey.2015.80.
115. Ehyai S, Dionyssiou MG, Gordon JW, Williams D, Siu KW, McDermott JC. A p38 Mitogen-Activated Protein Kinase-Regulated Myocyte Enhancer Factor 2-β-Catenin Interaction Enhances Canonical Wnt Signaling. Mol Cell Biol. 2015; 36:330–46. https://doi.org/10.1128/MCB.00832-15.
116. Suzuki E, Satonaka H, Nishimatsu H, Oba S, Takeda R, Omata M, Fujita T, Nagai R, Hirata Y. Myocyte enhancer factor 2 mediates vascular inflammation via the p38-dependent pathway. Circ Res. 2004; 95:42–49. https://doi.org/10.1161/01.RES.0000134631.75684.4A.
117. Burton TR, Dibrov A, Kashour T, Amara FM. Anti-apoptotic wild-type Alzheimer amyloid precursor protein signaling involves the p38 mitogen-activated protein kinase/MEF2 pathway. Brain Res Mol Brain Res. 2002; 108:102–20. https://doi.org/10.1016/S0169-328X(02)00519-3.
118. Ananieva O, Macdonald A, Wang X, McCoy CE, McIlrath J, Tournier C, Arthur JS. ERK5 regulation in naïve T-cell activation and survival. Eur J Immunol. 2008; 38:2534–47. https://doi.org/10.1002/eji.200737867.
119. Zheng R, Wang X, Studzinski GP. 1,25-Dihydroxyvitamin D3 induces monocytic differentiation of human myeloid leukemia cells by regulating C/EBPβ expression through MEF2C. J Steroid Biochem Mol Biol. 2015; 148:132–37. https://doi.org/10.1016/j.jsbmb.2014.11.016.
120. Voisin L, Saba-El-Leil MK, Julien C, Frémin C, Meloche S. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol Cell Biol. 2010; 30:2918–32. https://doi.org/10.1128/MCB.00131-10.
121. Boon RA, Horrevoets AJ. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009; 29:39–40, 41-3. https://doi.org/10.1055/s-0037-1616937.
122. Katsarou K, Tsitoura P, Georgopoulou U. MEK5/ERK5/mef2: a novel signaling pathway affected by hepatitis C virus non-enveloped capsid-like particles. Biochim Biophys Acta. 2011; 1813:1854–62. https://doi.org/10.1016/j.bbamcr.2011.06.015.
123. Slattery ML, Mullany LE, Sakoda LC, Wolff RK, Stevens JR, Samowitz WS, Herrick JS. The PI3K/AKT signaling pathway: associations of miRNAs with dysregulated gene expression in colorectal cancer. Mol Carcinog. 2018; 57:243–61. https://doi.org/10.1002/mc.22752.
124. Slattery ML, Mullany LE, Sakoda LC, Wolff RK, Samowitz WS, Herrick JS. The MAPK-Signaling Pathway in Colorectal Cancer: Dysregulated Genes and Their Association With MicroRNAs. Cancer Inform. 2018; 17:1176935118766522. https://doi.org/10.1177/1176935118766522.
125. Im JY, Yoon SH, Kim BK, Ban HS, Won KJ, Chung KS, Jung KE, Won M. DNA damage induced apoptosis suppressor (DDIAS) is upregulated via ERK5/MEF2B signaling and promotes β-catenin-mediated invasion. Biochim Biophys Acta. 2016; 1859:1449–58. https://doi.org/10.1016/j.bbagrm.2016.07.003.
126. Badodi S, Baruffaldi F, Ganassi M, Battini R, Molinari S. Phosphorylation-dependent degradation of MEF2C contributes to regulate G2/M transition. Cell Cycle. 2015; 14:1517–28. https://doi.org/10.1080/15384101.2015.1026519.
127. Tang X, Wang X, Gong X, Tong M, Park D, Xia Z, Mao Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2005; 25:4823–34. https://doi.org/10.1523/JNEUROSCI.1331-05.2005.
128. Grégoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol. 2007; 27:1280–95. https://doi.org/10.1128/MCB.00882-06.
129. Lu H, Liu B, You S, Chen L, Dongmei Q, Gu M, Lu Y, Chen Y, Zhang F, Yu B. SENP2 regulates MEF2A de-SUMOylation in an activity dependent manner. Mol Biol Rep. 2013; 40:2485–90. https://doi.org/10.1007/s11033-012-2329-x.
130. She H, Yang Q, Mao Z. Neurotoxin-induced selective ubiquitination and regulation of MEF2A isoform in neuronal stress response. J Neurochem. 2012; 122:1203–10. https://doi.org/10.1111/j.1471-4159.2012.07860.x.
131. Okamoto S, Nakamura T, Cieplak P, Chan SF, Kalashnikova E, Liao L, Saleem S, Han X, Clemente A, Nutter A, Sances S, Brechtel C, Haus D, et al. S-nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 2014; 8:217–28. https://doi.org/10.1016/j.celrep.2014.06.005.
132. Tsai NP, Wilkerson JR, Guo W, Huber KM. FMRP-dependent Mdm2 dephosphorylation is required for MEF2-induced synapse elimination. Hum Mol Genet. 2017; 26:293–304. https://doi.org/10.1093/hmg/ddw386.
133. Li J, Aponte Paris S, Thakur H, Kapiloff MS, Dodge-Kafka KL. Muscle A-kinase-anchoring protein-β-bound calcineurin toggles active and repressive transcriptional complexes of myocyte enhancer factor 2D. J Biol Chem. 2019; 294:2543–2554. https://doi.org/10.1074/jbc.RA118.005465.
134. Mughal W, Nguyen L, Pustylnik S, da Silva Rosa SC, Piotrowski S, Chapman D, Du M, Alli NS, Grigull J, Halayko AJ, Aliani M, Topham MK, Epand RM, et al. A conserved MADS-box phosphorylation motif regulates differentiation and mitochondrial function in skeletal, cardiac, and smooth muscle cells. Cell Death Dis. 2015; 6:e1944. https://doi.org/10.1038/cddis.2015.306.
135. Alexander MS, Shi X, Voelker KA, Grange RW, Garcia JA, Hammer RE, Garry DJ. Foxj3 transcriptionally activates Mef2c and regulates adult skeletal muscle fiber type identity. Dev Biol. 2010; 337:396–404. https://doi.org/10.1016/j.ydbio.2009.11.015.
136. Brusco J, Haas K. Interactions between mitochondria and the transcription factor myocyte enhancer factor 2 (MEF2) regulate neuronal structural and functional plasticity and metaplasticity. J Physiol. 2015; 593:3471–81. https://doi.org/10.1113/jphysiol.2014.282459.
137. Guo B, Hu S, Zheng C, Wang H, Luo F, Li H, Cui W, Yang X, Cui G, Mak S, Choi TC, Ma ED, Wang Y, et al. Substantial protection against MPTP-associated Parkinson’s neurotoxicity in vitro and in vivo by anti-cancer agent SU4312 via activation of MEF2D and inhibition of MAO-B. Neuropharmacology. 2017; 126:12–24. https://doi.org/10.1016/j.neuropharm.2017.08.014.
138. Chen ZW, Liu A, Liu Q, Chen J, Li WM, Chao XJ, Yang Q, Liu PQ, Mao ZX, Pi RB. MEF2D Mediates the Neuroprotective Effect of Methylene Blue Against Glutamate-Induced Oxidative Damage in HT22 Hippocampal Cells. Mol Neurobiol. 2017; 54:2209–22. https://doi.org/10.1007/s12035-016-9818-1.
139. Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell. 2013; 155:1351–64. https://doi.org/10.1016/j.cell.2013.11.009. Erratum in: Cell. 2013 Dec 19;155(7):1652-3.
140. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, Jiao J, Veasey SC, Sims CA, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. 2015; 29:2315–26. https://doi.org/10.1096/fj.14-268409.
141. Chen X, Gao B, Ponnusamy M, Lin Z, Liu J. MEF2 signaling and human diseases. Oncotarget. 2017; 8:112152–65. https://doi.org/10.18632/oncotarget.22899.
142. Molkentin JD, Black BL, Martin JF, Olson EN. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol. 1996; 16:2627–36. https://doi.org/10.1128/MCB.16.6.2627.
143. Jayathilaka N, Han A, Gaffney KJ, Dey R, Jarusiewicz JA, Noridomi K, Philips MA, Lei X, He J, Ye J, Gao T, Petasis NA, Chen L. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res. 2012; 40:5378–88. https://doi.org/10.1093/nar/gks189.
144. Borghi S, Molinari S, Razzini G, Parise F, Battini R, Ferrari S. The nuclear localization domain of the MEF2 family of transcription factors shows member-specific features and mediates the nuclear import of histone deacetylase 4. J Cell Sci. 2001; 114:4477–83.
145. Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008; 118:124–32.
146. Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, Soussou W, Nie Z, Kang YJ, Nakanishi N, Okamoto S, Roberts AJ, Schwarz JJ, Lipton SA. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc Natl Acad Sci U S A. 2008; 105:9397–402. https://doi.org/10.1073/pnas.0802876105.
147. Di Giorgio E, Hancock WW, Brancolini C. MEF2 and the tumorigenic process, hic sunt leones. Biochim Biophys Acta Rev Cancer. 2018; 1870:261–73. https://doi.org/10.1016/j.bbcan.2018.05.007.
148. Wilker PR, Kohyama M, Sandau MM, Albring JC, Nakagawa O, Schwarz JJ, Murphy KM. Transcription factor Mef2c is required for B cell proliferation and survival after antigen receptor stimulation. Nat Immunol. 2008; 9:603–12. https://doi.org/10.1038/ni.1609.
149. Stehling-Sun S, Dade J, Nutt SL, DeKoter RP, Camargo FD. Regulation of lymphoid versus myeloid fate ‘choice’ by the transcription factor Mef2c. Nat Immunol. 2009; 10:289–96. https://doi.org/10.1038/ni.1694.
150. Andzelm MM, Cherry TJ, Harmin DA, Boeke AC, Lee C, Hemberg M, Pawlyk B, Malik AN, Flavell SW, Sandberg MA, Raviola E, Greenberg ME. MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron. 2015; 86:247–63. https://doi.org/10.1016/j.neuron.2015.02.038.
151. Elgar SJ, Han J, Taylor MV. mef2 activity levels differentially affect gene expression during Drosophila muscle development. Proc Natl Acad Sci U S A. 2008; 105:918–23. https://doi.org/10.1073/pnas.0711255105.
152. Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994; 120:1251–63.
153. Subramanian SV, Nadal-Ginard B. Early expression of the different isoforms of the myocyte enhancer factor-2 (MEF2) protein in myogenic as well as non-myogenic cell lineages during mouse embryogenesis. Mech Dev. 1996; 57:103–12. https://doi.org/10.1016/0925-4773(96)00542-4.
154. Hobson GM, Krahe R, Garcia E, Siciliano MJ, Funanage VL. Regional chromosomal assignments for four members of the MADS domain transcription enhancer factor 2 (MEF2) gene family to human chromosomes 15q26, 19p12, 5q14, and 1q12-q23. Genomics. 1995; 29:704–11. https://doi.org/10.1006/geno.1995.9007.
155. Clark AL, Naya FJ. MicroRNAs in the Myocyte Enhancer Factor 2 (MEF2)-regulated Gtl2-Dio3 Noncoding RNA Locus Promote Cardiomyocyte Proliferation by Targeting the Transcriptional Coactivator Cited2. J Biol Chem. 2015; 290:23162–72. https://doi.org/10.1074/jbc.M115.672659.
156. Zhao W, Zheng XL, Peng DQ, Zhao SP. Myocyte Enhancer Factor 2A Regulates Hydrogen Peroxide-Induced Senescence of Vascular Smooth Muscle Cells Via microRNA-143. J Cell Physiol. 2015; 230:2202–11. https://doi.org/10.1002/jcp.24948.
157. Tobin SW, Yang D, Girgis J, Farahzad A, Blais A, McDermott JC. Regulation of Hspb7 by MEF2 and AP-1: implications for Hspb7 in muscle atrophy. J Cell Sci. 2016; 129:4076–90. https://doi.org/10.1242/jcs.190009.
158. Chen X, Liu G, Zhang W, Zhang J, Yan Y, Dong W, Liang E, Zhang Y, Zhang M. Inhibition of MEF2A prevents hyperglycemia-induced extracellular matrix accumulation by blocking Akt and TGF-β1/Smad activation in cardiac fibroblasts. Int J Biochem Cell Biol. 2015; 69:52–61. https://doi.org/10.1016/j.biocel.2015.10.012.
159. Chen D, Chen F, Xu Y, Zhang Y, Li Z, Zhang H, Pan T, Su Y, Wan M, Wang X, Ye J. AKT2 deficiency induces retardation of myocyte development through EndoG-MEF2A signaling in mouse heart. Biochem Biophys Res Commun. 2017; 493:1410–17. https://doi.org/10.1016/j.bbrc.2017.09.149.
160. Ewen EP, Snyder CM, Wilson M, Desjardins D, Naya FJ. The Mef2A transcription factor coordinately regulates a costamere gene program in cardiac muscle. J Biol Chem. 2011; 286:29644–53. https://doi.org/10.1074/jbc.M111.268094.
161. McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, Ohashi K, Kielbasa OM, Mathew M, Ewen EP, Walsh K, Gavras H, Naya FJ. Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. 2010; 106:952–60. https://doi.org/10.1161/CIRCRESAHA.109.209007.
162. Phan NN, Wang CY, Lin YC. The novel regulations of MEF2A, CAMKK2, CALM3, and TNNI3 in ventricular hypertrophy induced by arsenic exposure in rats. Toxicology. 2014; 324:123–35. https://doi.org/10.1016/j.tox.2014.07.010.
163. Desjardins CA, Naya FJ. Antagonistic regulation of cell-cycle and differentiation gene programs in neonatal cardiomyocytes by homologous MEF2 transcription factors. J Biol Chem. 2017; 292:10613–29. https://doi.org/10.1074/jbc.M117.776153.
164. Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, Monteggia LM. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One. 2012; 7:e34863. https://doi.org/10.1371/journal.pone.0034863.
165. Zhu B, Carmichael RE, Solabre Valois L, Wilkinson KA, Henley JM. The transcription factor MEF2A plays a key role in the differentiation/maturation of rat neural stem cells into neurons. Biochem Biophys Res Commun. 2018; 500:645–49. https://doi.org/10.1016/j.bbrc.2018.04.125.
166. Finsterwald C, Carrard A, Martin JL. Role of salt-inducible kinase 1 in the activation of MEF2-dependent transcription by BDNF. PLoS One. 2013; 8:e54545. https://doi.org/10.1371/journal.pone.0054545.
167. Speksnijder N, Christensen KV, Didriksen M, De Kloet ER, Datson NA. Glucocorticoid receptor and myocyte enhancer factor 2 cooperate to regulate the expression of c-JUN in a neuronal context. J Mol Neurosci. 2012; 48:209–18. https://doi.org/10.1007/s12031-012-9809-2.
168. Yamada T, Yang Y, Huang J, Coppola G, Geschwind DH, Bonni A. Sumoylated MEF2A coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons. J Neurosci. 2013; 33:4726–40. https://doi.org/10.1523/JNEUROSCI.4191-12.2013.
169. Thygesen JH, Zambach SK, Ingason A, Lundin P, Hansen T, Bertalan M, Rosengren A, Bjerre D, Ferrero-Miliani L, Rasmussen HB, Parnas J, Werge T. Linkage and whole genome sequencing identify a locus on 6q25-26 for formal thought disorder and implicate MEF2A regulation. Schizophr Res. 2015; 169:441–46. https://doi.org/10.1016/j.schres.2015.08.037.
170. Zhang L, Sun Y, Fei M, Tan C, Wu J, Zheng J, Tang J, Sun W, Lv Z, Bao J, Xu Q, Yu H. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy. 2014; 10:1015–35. https://doi.org/10.4161/auto.28477.
171. Huang Y, Wu X, Guo J, Yuan J. Myocyte-specific enhancer binding factor 2A expression is downregulated during temporal lobe epilepsy. Int J Neurosci. 2016; 126:786–96. https://doi.org/10.3109/00207454.2015.1062003.
172. Guella I, Rimoldi V, Asselta R, Ardissino D, Francolini M, Martinelli N, Girelli D, Peyvandi F, Tubaro M, Merlini PA, Mannucci PM, Duga S. Association and functional analyses of MEF2A as a susceptibility gene for premature myocardial infarction and coronary artery disease. Circ Cardiovasc Genet. 2009; 2:165–72. https://doi.org/10.1161/CIRCGENETICS.108.819326.
173. Maiolino G, Colonna S, Zanchetta M, Pedon L, Seccia TM, Cesari M, Vigili de Kreutzenberg S, Avogaro A, Rossi GP. Exon 11 deletion in the myocyte enhancer factor (MEF)2A and early onset coronary artery disease gene in a Sicilian family. Eur J Cardiovasc Prev Rehabil. 2011; 18:557–60. https://doi.org/10.1177/1741826710397112.
174. Dai DP, Zhou XY, Xiao Y, Xu F, Sun FC, Ji FS, Zhang ZX, Hu JH, Guo J, Zheng JD, Dong JM, Zhu WG, Shen Y, et al. Structural changes in exon 11 of MEF2A are not related to sporadic coronary artery disease in Han Chinese population. Eur J Clin Invest. 2010; 40:669–77. https://doi.org/10.1111/j.1365-2362.2010.02307.x.
175. Huang XC, Wang W. Association of MEF2A gene 3’UTR mutations with coronary artery disease. Genet Mol Res. 2015; 14:11073–78. https://doi.org/10.4238/2015.September.21.20.
176. Elhawari S, Al-Boudari O, Muiya P, Khalak H, Andres E, Al-Shahid M, Al-Dosari M, Meyer BF, Al-Mohanna F, Dzimiri N. A study of the role of the Myocyte-specific Enhancer Factor-2A gene in coronary artery disease. Atherosclerosis. 2010; 209:152–54. https://doi.org/10.1016/j.atherosclerosis.2009.09.005.
177. Joseph JS, Ayeleso AO, Mukwevho E. Exercise increases hyper-acetylation of histones on the Cis-element of NRF-1 binding to the Mef2a promoter: implications on type 2 diabetes. Biochem Biophys Res Commun. 2017; 486:83–87. https://doi.org/10.1016/j.bbrc.2017.03.002.
178. Yuan H, Niu Y, Liu X, Fu L. Exercise increases the binding of MEF2A to the Cpt1b promoter in mouse skeletal muscle. Acta Physiol (Oxf). 2014; 212:283–92. https://doi.org/10.1111/apha.12395.
179. Gong H, Xie J, Zhang N, Yao L, Zhang Y. MEF2A binding to the Glut4 promoter occurs via an AMPKα2-dependent mechanism. Med Sci Sports Exerc. 2011; 43:1441–50. https://doi.org/10.1249/MSS.0b013e31820f6093.
180. Yu W, Huang C, Wang Q, Huang T, Ding Y, Ma C, Ma H, Chen W. MEF2 transcription factors promotes EMT and invasiveness of hepatocellular carcinoma through TGF-β1 autoregulation circuitry. Tumour Biol. 2014; 35:10943–51. https://doi.org/10.1007/s13277-014-2403-1.
181. Ishikawa F, Miyoshi H, Nose K, Shibanuma M. Transcriptional induction of MMP-10 by TGF-beta, mediated by activation of MEF2A and downregulation of class IIa HDACs. Oncogene. 2010; 29:909–19. https://doi.org/10.1038/onc.2009.387.
182. Liu J, Wen D, Fang X, Wang X, Liu T, Zhu J. p38MAPK Signaling Enhances Glycolysis Through the Up-Regulation of the Glucose Transporter GLUT-4 in Gastric Cancer Cells. Cell Physiol Biochem. 2015; 36:155–65. https://doi.org/10.1159/000374060.
183. Kim JM, Hwang IH, Jang IS, Kim M, Bang IS, Park SJ, Chung YJ, Joo JC, Lee MG. Houttuynia cordata Thunb Promotes Activation of HIF-1A-FOXO3 and MEF2A Pathways to Induce Apoptosis in Human HepG2 Hepatocellular Carcinoma Cells. Integr Cancer Ther. 2017; 16:360–72. https://doi.org/10.1177/1534735416670987.
184. Zhou WP, Zhang H, Zhao YX, Liu GQ, Zhang JY. RNA interference of myocyte enhancer factor 2A accelerates atherosclerosis in apolipoprotein E-deficient mice. PLoS One. 2015; 10:e0121823. https://doi.org/10.1371/journal.pone.0121823.
185. Wu W, de Folter S, Shen X, Zhang W, Tao S. Vertebrate paralogous MEF2 genes: origin, conservation, and evolution. PLoS One. 2011; 6:e17334. https://doi.org/10.1371/journal.pone.0017334.
186. Lei X, Shi H, Kou Y, Rajashekar N, Wu F, Sen C, Xu J, Chen L. Crystal Structure of Apo MEF2B Reveals New Insights in DNA Binding and Cofactor Interaction. Biochemistry. 2018; 57:4047–51. https://doi.org/10.1021/acs.biochem.8b00439.
187. Katoh Y, Molkentin JD, Dave V, Olson EN, Periasamy M. MEF2B is a component of a smooth muscle-specific complex that binds an A/T-rich element important for smooth muscle myosin heavy chain gene expression. J Biol Chem. 1998; 273:1511–18. https://doi.org/10.1074/jbc.273.3.1511.
188. Rodríguez AI, Csányi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, Vorp DA, Pagano PJ. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2015; 35:430–38. https://doi.org/10.1161/ATVBAHA.114.304936.
189. Molkentin JD, Firulli AB, Black BL, Martin JF, Hustad CM, Copeland N, Jenkins N, Lyons G, Olson EN. MEF2B is a potent transactivator expressed in early myogenic lineages. Mol Cell Biol. 1996; 16:3814–24. https://doi.org/10.1128/MCB.16.7.3814.
190. Hidaka K, Morisaki T, Byun SH, Hashido K, Toyama K, Mukai T. The MEF2B homologue differentially expressed in mouse embryonal carcinoma cells. Biochem Biophys Res Commun. 1995; 213:555–60. https://doi.org/10.1006/bbrc.1995.2167.
191. Pavan S, Meyer-Schaller N, Diepenbruck M, Kalathur RK, Saxena M, Christofori G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene. 2018; 37:4197–213. https://doi.org/10.1038/s41388-018-0270-8.
192. Lei X, Kou Y, Fu Y, Rajashekar N, Shi H, Wu F, Xu J, Luo Y, Chen L. The Cancer Mutation D83V Induces an α-Helix to β-Strand Conformation Switch in MEF2B. J Mol Biol. 2018; 430:1157–72. https://doi.org/10.1016/j.jmb.2018.02.012.
193. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A, Pasqualucci L, Basso K, Dalla-Favera R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013; 14:1084–92. https://doi.org/10.1038/ni.2688.
194. Ding J, Dirks WG, Ehrentraut S, Geffers R, MacLeod RA, Nagel S, Pommerenke C, Romani J, Scherr M, Vaas LA, Zaborski M, Drexler HG, Quentmeier H. BCL6—regulated by AhR/ARNT and wild-type MEF2B—drives expression of germinal center markers MYBL1 and LMO2. Haematologica. 2015; 100:801–09. https://doi.org/10.3324/haematol.2014.120048.
195. Pon JR, Wong J, Saberi S, Alder O, Moksa M, Grace Cheng SW, Morin GB, Hoodless PA, Hirst M, Marra MA. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat Commun. 2015; 6:7953. https://doi.org/10.1038/ncomms8953.
196. Moore EM, Swerdlow SH, Gibson SE. Comparison of Myocyte Enhancer Factor 2B Versus Other Germinal Center-associated Antigens in the Differential Diagnosis of B-Cell Non-Hodgkin Lymphomas. Am J Surg Pathol. 2018; 42:342–50. https://doi.org/10.1097/PAS.0000000000001015.
197. Kishimoto W, Nishikori M. Molecular pathogenesis of follicular lymphoma. J Clin Exp Hematop. 2014; 54:23–30. https://doi.org/10.3960/jslrt.54.23.
198. Assouline SE, Nielsen TH, Yu S, Alcaide M, Chong L, MacDonald D, Tosikyan A, Kukreti V, Kezouh A, Petrogiannis-Haliotis T, Albuquerque M, Fornika D, Alamouti S, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016; 128:185–94. https://doi.org/10.1182/blood-2016-02-699520.
199. Wu J, Kubota J, Hirayama J, Nagai Y, Nishina S, Yokoi T, Asaoka Y, Seo J, Shimizu N, Kajiho H, Watanabe T, Azuma N, Katada T, Nishina H. p38 Mitogen-activated protein kinase controls a switch between cardiomyocyte and neuronal commitment of murine embryonic stem cells by activating myocyte enhancer factor 2C-dependent bone morphogenetic protein 2 transcription. Stem Cells Dev. 2010; 19:1723–34. https://doi.org/10.1089/scd.2010.0066.
200. Gu X, Fu C, Lin L, Liu S, Su X, Li A, Wu Q, Jia C, Zhang P, Chen L, Zhu X, Wang X. miR-124 and miR-9 mediated downregulation of HDAC5 promotes neurite development through activating MEF2C-GPM6A pathway. J Cell Physiol. 2018; 233:673–87. https://doi.org/10.1002/jcp.25927.
201. Lyons MR, Schwarz CM, West AE. Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. J Neurosci. 2012; 32:12780–85. https://doi.org/10.1523/JNEUROSCI.0534-12.2012.
202. Chen YC, Kuo HY, Bornschein U, Takahashi H, Chen SY, Lu KM, Yang HY, Chen GM, Lin JR, Lee YH, Chou YC, Cheng SJ, Chien CT, et al. Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci. 2016; 19:1513–22. https://doi.org/10.1038/nn.4380.
203. Paciorkowski AR, Traylor RN, Rosenfeld JA, Hoover JM, Harris CJ, Winter S, Lacassie Y, Bialer M, Lamb AN, Schultz RA, Berry-Kravis E, Porter BE, Falk M, et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 2013; 14:99–111. https://doi.org/10.1007/s10048-013-0356-y.
204. Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, Guichet A, Barth M, Charollais A, Journel H, Auvin S, Boucher C, Kerckaert JP, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010; 47:22–29. https://doi.org/10.1136/jmg.2009.069732.
205. Sao T, Yoshino Y, Yamazaki K, Ozaki Y, Mori Y, Ochi S, Yoshida T, Mori T, Iga JI, Ueno SI. MEF2C mRNA expression and cognitive function in Japanese patients with Alzheimer’s disease. Psychiatry Clin Neurosci. 2018; 72:160–67. https://doi.org/10.1111/pcn.12618.
206. Lambert L, Bienvenu T, Allou L, Valduga M, Echenne B, Diebold B, Mignot C, Héron D, Roth V, Saunier A, Moustaïne A, Jonveaux P, Philippe C. MEF2C mutations are a rare cause of Rett or severe Rett-like encephalopathies. Clin Genet. 2012; 82:499–501. https://doi.org/10.1111/j.1399-0004.2012.01861.x.
207. Verzi MP, Agarwal P, Brown C, McCulley DJ, Schwarz JJ, Black BL. The transcription factor MEF2C is required for craniofacial development. Dev Cell. 2007; 12:645–52. https://doi.org/10.1016/j.devcel.2007.03.007.
208. Hu J, Verzi MP, Robinson AS, Tang PL, Hua LL, Xu SM, Kwok PY, Black BL. Endothelin signaling activates Mef2c expression in the neural crest through a MEF2C-dependent positive-feedback transcriptional pathway. Development. 2015; 142:2775–80. https://doi.org/10.1242/dev.126391.
209. Pane LS, Zhang Z, Ferrentino R, Huynh T, Cutillo L, Baldini A. Tbx1 is a negative modulator of Mef2c. Hum Mol Genet. 2012; 21:2485–96. https://doi.org/10.1093/hmg/dds063.
210. Ghosh TK, Song FF, Packham EA, Buxton S, Robinson TE, Ronksley J, Self T, Bonser AJ, Brook JD. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol Cell Biol. 2009; 29:2205–18. https://doi.org/10.1128/MCB.01923-08.
211. Vong L, Bi W, O'Connor-Halligan KE, Li C, Cserjesi P, Schwarz JJ. MEF2C is required for the normal allocation of cells between the ventricular and sinoatrial precursors of the primary heart field. Dev Dyn. 2006; 235:1809–21. https://doi.org/10.1002/dvdy.20828.
212. Vincentz JW, Barnes RM, Firulli BA, Conway SJ, Firulli AB. Cooperative interaction of Nkx2.5 and Mef2c transcription factors during heart development. Dev Dyn. 2008; 237:3809–19. https://doi.org/10.1002/dvdy.21803.
213. Pereira AH, Clemente CF, Cardoso AC, Theizen TH, Rocco SA, Judice CC, Guido MC, Pascoal VD, Lopes-Cendes I, Souza JR, Franchini KG. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS One. 2009; 4:e8472. https://doi.org/10.1371/journal.pone.0008472. Erratum in: PLoS One. 2014;9(10):e112344.
214. Baruffaldi F, Montarras D, Basile V, De Feo L, Badodi S, Ganassi M, Battini R, Nicoletti C, Imbriano C, Musarò A, Molinari S. Dynamic Phosphorylation of the Myocyte Enhancer Factor 2Cα1 Splice Variant Promotes Skeletal Muscle Regeneration and Hypertrophy. Stem Cells. 2017; 35:725–38. https://doi.org/10.1002/stem.2495.
215. Sahoo S, Meijles DN, Al Ghouleh I, Tandon M, Cifuentes-Pagano E, Sembrat J, Rojas M, Goncharova E, Pagano PJ. MEF2C-MYOCD and Leiomodin1 Suppression by miRNA-214 Promotes Smooth Muscle Cell Phenotype Switching in Pulmonary Arterial Hypertension. PLoS One. 2016; 11:e0153780. https://doi.org/10.1371/journal.pone.0153780.
216. Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 2014; 6:336–45. https://doi.org/10.1016/j.celrep.2013.12.025.
217. Al Madhoun AS, Voronova A, Ryan T, Zakariyah A, McIntire C, Gibson L, Shelton M, Ruel M, Skerjanc IS. Testosterone enhances cardiomyogenesis in stem cells and recruits the androgen receptor to the MEF2C and HCN4 genes. J Mol Cell Cardiol. 2013; 60:164–71. https://doi.org/10.1016/j.yjmcc.2013.04.003.
218. Nakatani T, Partridge NC. MEF2C Interacts With c-FOS in PTH-Stimulated Mmp13 Gene Expression in Osteoblastic Cells. Endocrinology. 2017; 158:3778–91. https://doi.org/10.1210/en.2017-00159.
219. Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, Nagano K, Baron R, Brooks D, Bouxsein M, Pajevic PD, Kronenberg HM. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J Bone Miner Res. 2015; 30:400–11. https://doi.org/10.1002/jbmr.2381.
220. Cortés R, Rivera M, Roselló-Lletí E, Martínez-Dolz L, Almenar L, Azorín I, Lago F, González-Juanatey JR, Portolés M. Differences in MEF2 and NFAT transcriptional pathways according to human heart failure aetiology. PLoS One. 2012; 7:e30915. https://doi.org/10.1371/journal.pone.0030915.
221. Wang E, Sun S, Qiao B, Duan W, Huang G, An Y, Xu S, Zheng Y, Su Z, Gu X, Jin L, Wang H. Identification of functional mutations in GATA4 in patients with congenital heart disease. PLoS One. 2013; 8:e62138. https://doi.org/10.1371/journal.pone.0062138.
222. Blanchard FJ, Collins B, Cyran SA, Hancock DH, Taylor MV, Blau J. The transcription factor Mef2 is required for normal circadian behavior in Drosophila. J Neurosci. 2010; 30:5855–65. https://doi.org/10.1523/JNEUROSCI.2688-09.2010.
223. Jiang Y, Liu H, Liu WJ, Tong HB, Chen CJ, Lin FG, Zhuo YH, Qian XZ, Wang ZB, Wang Y, Zhang P, Jia HL. Endothelial Aquaporin-1 (AQP1) Expression Is Regulated by Transcription Factor Mef2c. Mol Cells. 2016; 39:292–98. https://doi.org/10.14348/molcells.2016.2223.
224. Debnath I, Roundy KM, Pioli PD, Weis JJ, Weis JH. Bone marrow-induced Mef2c deficiency delays B-cell development and alters the expression of key B-cell regulatory proteins. Int Immunol. 2013; 25:99–115. https://doi.org/10.1093/intimm/dxs088.
225. Gekas C, Rhodes KE, Gereige LM, Helgadottir H, Ferrari R, Kurdistani SK, Montecino-Rodriguez E, Bassel-Duby R, Olson E, Krivtsov AV, Armstrong S, Orkin SH, Pellegrini M, Mikkola HK. Mef2C is a lineage-restricted target of Scl/Tal1 and regulates megakaryopoiesis and B-cell homeostasis. Blood. 2009; 113:3461–71. https://doi.org/10.1182/blood-2008-07-167577.
226. Fu W, Wei J, Gu J. MEF2C mediates the activation induced cell death (AICD) of macrophages. Cell Res. 2006; 16:559–65. https://doi.org/10.1038/sj.cr.7310073.
227. Wei X, Sun W, Fan R, Hahn J, Joetham A, Li G, Webb S, Garrington T, Dakhama A, Lucas J, Johnson GL, Gelfand EW. MEF2C regulates c-Jun but not TNF-alpha gene expression in stimulated mast cells. Eur J Immunol. 2003; 33:2903–09. https://doi.org/10.1002/eji.200324127.
228. Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, Kertser A, Baruch K, Rosenzweig N, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017; 8:717. https://doi.org/10.1038/s41467-017-00769-0.
229. Swanson BJ, Jäck HM, Lyons GE. Characterization of myocyte enhancer factor 2 (MEF2) expression in B and T cells: MEF2C is a B cell-restricted transcription factor in lymphocytes. Mol Immunol. 1998; 35:445–58. https://doi.org/10.1016/S0161-5890(98)00058-3.
230. Kawashima-Goto S, Imamura T, Tomoyasu C, Yano M, Yoshida H, Fujiki A, Tamura S, Osone S, Ishida H, Morimoto A, Kuroda H, Hosoi H. BCL2 Inhibitor (ABT-737): A Restorer of Prednisolone Sensitivity in Early T-Cell Precursor-Acute Lymphoblastic Leukemia with High MEF2C Expression? PLoS One. 2015; 10:e0132926. https://doi.org/10.1371/journal.pone.0132926.
231. Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, van Vlierberghe P, Ferrando AA, Cayuela JM, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011; 19:484–97. https://doi.org/10.1016/j.ccr.2011.02.008.
232. Agatheeswaran S, Chakraborty S. MEF2C and CEBPA: possible co-regulators in chronic myeloid leukemia disease progression. Int J Biochem Cell Biol. 2016; 77:165–70. https://doi.org/10.1016/j.biocel.2016.06.007.
233. Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, Tan TC, Clarke SJ, Robertson GR, Polly P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY). 2012; 4:133–43. https://doi.org/10.18632/aging.100436.
234. Xu Z, Yoshida T, Wu L, Maiti D, Cebotaru L, Duh EJ. Transcription factor MEF2C suppresses endothelial cell inflammation via regulation of NF-κB and KLF2. J Cell Physiol. 2015; 230:1310–20. https://doi.org/10.1002/jcp.24870.
235. Agatheeswaran S, Singh S, Biswas S, Biswas G, Chandra Pattnayak N, Chakraborty S. BCR-ABL mediated repression of miR-223 results in the activation of MEF2C and PTBP2 in chronic myeloid leukemia. Leukemia. 2013; 27:1578–80. https://doi.org/10.1038/leu.2012.339.
236. Neumann M, Heesch S, Gökbuget N, Schwartz S, Schlee C, Benlasfer O, Farhadi-Sartangi N, Thibaut J, Burmeister T, Hoelzer D, Hofmann WK, Thiel E, Baldus CD. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012; 2:e55. https://doi.org/10.1038/bcj.2011.49.
237. Zhang M, Zhu B, Davie J. Alternative splicing of MEF2C pre-mRNA controls its activity in normal myogenesis and promotes tumorigenicity in rhabdomyosarcoma cells. J Biol Chem. 2015; 290:310–24. https://doi.org/10.1074/jbc.M114.606277.
238. Huo Y, Zhao Q, Wang C, Zhao F, Du Y, Sun W. The involvement of myocyte enhancer factor 2D in regulating tumor biology of cardiac myxoma. Tumour Biol. 2016; 37:5405–11. https://doi.org/10.1007/s13277-015-4386-y.
239. Cao Q, Dong P, Wang Y, Zhang J, Shi X, Wang Y. miR-218 suppresses cardiac myxoma proliferation by targeting myocyte enhancer factor 2D. Oncol Rep. 2015; 33:2606–12. https://doi.org/10.3892/or.2015.3861.
240. Wang P, Wang L, Chen L, Sun X. Dual-specificity tyrosine-phosphorylation regulated kinase 1A Gene Transcription is regulated by Myocyte Enhancer Factor 2D. Sci Rep. 2017; 7:7240. https://doi.org/10.1038/s41598-017-07655-1.
241. Zeng W, Zhang W, Lu F, Gao L, Gao G. Resveratrol attenuates MPP+-induced mitochondrial dysfunction and cell apoptosis via AKT/GSK-3β pathway in SN4741 cells. Neurosci Lett. 2017; 637:50–56. https://doi.org/10.1016/j.neulet.2016.11.054.
242. Mount MP, Zhang Y, Amini M, Callaghan S, Kulczycki J, Mao Z, Slack RS, Anisman H, Park DS. Perturbation of transcription factor Nur77 expression mediated by myocyte enhancer factor 2D (MEF2D) regulates dopaminergic neuron loss in response to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). J Biol Chem. 2013; 288:14362–71. https://doi.org/10.1074/jbc.M112.439216.
243. Sala G, Arosio A, Stefanoni G, Melchionda L, Riva C, Marinig D, Brighina L, Ferrarese C. Rotenone upregulates alpha-synuclein and myocyte enhancer factor 2D independently from lysosomal degradation inhibition. Biomed Res Int. 2013; 2013:846725. https://doi.org/10.1155/2013/846725.
244. Chu Y, Mickiewicz AL, Kordower JH. α-synuclein aggregation reduces nigral myocyte enhancer factor-2D in idiopathic and experimental Parkinson’s disease. Neurobiol Dis. 2011; 41:71–82. https://doi.org/10.1016/j.nbd.2010.08.022.
245. Yang S, Gao L, Lu F, Wang B, Gao F, Zhu G, Cai Z, Lai J, Yang Q. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J Neuroinflammation. 2015; 12:33. https://doi.org/10.1186/s12974-015-0258-z.
246. Zhao Y, Li Y, Ma Y, Wang S, Cheng J, Yang T, Sun Z, Kuang Y, Huang H, Fan K, Gu J. Myocyte enhancer factor 2D promotes tumorigenicity in malignant glioma cells. Tumour Biol. 2016; 37:601–10. https://doi.org/10.1007/s13277-015-3791-6.
247. Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, McCastlain K, Reshmi SC, Payne-Turner D, Iacobucci I, Shao Y, Chen IM, Valentine M, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016; 7:13331. https://doi.org/10.1038/ncomms13331.
248. Zhang M, Mao D, Zhang W. The pathogenic role of MEF2D-SS18 fusion gene in B-cell acute lymphoblastic leukemia. Biochem Biophys Res Commun. 2018; 496:1331–36. https://doi.org/10.1016/j.bbrc.2018.02.013.
249. Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019; 32:147–153. https://doi.org/10.1038/s41379-018-0118-3.
250. Zhu HX, Shi L, Zhang Y, Zhu YC, Bai CX, Wang XD, Zhou JB. Myocyte enhancer factor 2D provides a cross-talk between chronic inflammation and lung cancer. J Transl Med. 2017; 15:65. https://doi.org/10.1186/s12967-017-1168-x.
251. Luan N, Wang Y, Liu X. Absent expression of miR-30a promotes the growth of lung cancer cells by targeting MEF2D. Oncol Lett. 2018; 16:1173–79. https://doi.org/10.3892/ol.2018.8719.
252. Du L, Chen T, Zhao K, Yang D. miR-30a suppresses osteosarcoma proliferation and metastasis by downregulating MEF2D expression. Onco Targets Ther. 2018; 11:2195–202. https://doi.org/10.2147/OTT.S102430.
253. Zhou Z, Lin Z, He Y, Pang X, Wang Y, Ponnusamy M, Ao X, Shan P, Tariq MA, Li P, Wang J. The Long Noncoding RNA D63785 Regulates Chemotherapy Sensitivity in Human Gastric Cancer by Targeting miR-422a. Mol Ther Nucleic Acids. 2018; 12:405–19. https://doi.org/10.1016/j.omtn.2018.05.024.
254. McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002; 27:40–7. https://doi.org/10.1016/S0968-0004(01)02031-X. Erratum in: Trends Biochem Sci 2002 May;57(5):271.
255. Clocchiatti A, Florean C, Brancolini C. Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med. 2011; 15:1833–46. https://doi.org/10.1111/j.1582-4934.2011.01321.x.
256. Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012; 90:85–94. https://doi.org/10.1038/icb.2011.100.
257. Roche J, Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016; 121:451–83. https://doi.org/10.1016/j.ejmech.2016.05.047.
258. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011; 3:166–79.
259. Sawas A, Radeski D, O’Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol. 2015; 6:202–8. https://doi.org/10.1177/2040620715592567.
260. Chowdhury S, Howell GM, Teggart CA, Chowdhury A, Person JJ, Bowers DM, Brattain MG. Histone deacetylase inhibitor belinostat represses survivin expression through reactivation of transforming growth factor beta (TGFbeta) receptor II leading to cancer cell death. J Biol Chem. 2011; 286:30937–48. https://doi.org/10.1074/jbc.M110.212035.
261. Tandon N, Ramakrishnan V, Kumar SK. Clinical use and applications of histone deacetylase inhibitors in multiple myeloma. Clin Pharmacol. 2016; 8:35–44. https://doi.org/10.2147/CPAA.S94021.
262. Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the Treatment of Multiple Myeloma. Clin Cancer Res. 2015; 21:4767–73. https://doi.org/10.1158/1078-0432.CCR-15-0530.
263. Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001; 20:6969–78. https://doi.org/10.1093/emboj/20.24.6969.
264. Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003; 22:3411–20. https://doi.org/10.1093/emboj/cdg315.
265. Zimmerman B, Sargeant A, Landes K, Fernandez SA, Chen CS, Lairmore MD. Efficacy of novel histone deacetylase inhibitor, AR42, in a mouse model of, human T-lymphotropic virus type 1 adult T cell lymphoma. Leuk Res. 2011; 35:1491–97. https://doi.org/10.1016/j.leukres.2011.07.015.
266. Sargeant AM, Rengel RC, Kulp SK, Klein RD, Clinton SK, Wang YC, Chen CS. OSU-HDAC42, a histone deacetylase inhibitor, blocks prostate tumor progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008; 68:3999–4009. https://doi.org/10.1158/0008-5472.CAN-08-0203.
267. Mukhi N, Sidhu G, Gonsky J, Shapira I. MP3: ROLE OF HISTONE DEACTYLASE (HDAC) INHIBITORS IN ADULT T-CELL LYMPHOMA/LEUKEMIA (ATL). Journal of Investigative Medicine. 2016; 64:805. https://doi.org/10.1136/jim-2016-000080.15.
268. Rossi L, Battistelli C, de Turris V, Noce V, Zwergel C, Valente S, Moioli A, Manzione A, Palladino M, Bordoni V, Domenici A, Menè P, Mai A, et al. HDAC1 inhibition by MS-275 in mesothelial cells limits cellular invasion and promotes MMT reversal. Sci Rep. 2018; 8:8492. https://doi.org/10.1038/s41598-018-26319-2.
269. Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med. 2011; 17:333–52. https://doi.org/10.2119/molmed.2011.00116.
270. Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. 2012; 119:2443–51. https://doi.org/10.1182/blood-2011-10-292003.
271. Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013; 21:277–85. https://doi.org/10.1016/j.tim.2013.02.005.
272. Zaikos TD, Painter MM, Sebastian Kettinger NT, Terry VH, Collins KL. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J Virol. 2018; 92:e02110-17. https://doi.org/10.1128/JVI.02110-17.
273. Engel JA, Jones AJ, Avery VM, Sumanadasa SD, Ng SS, Fairlie DP, Skinner-Adams T, Andrews KT. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int J Parasitol Drugs Drug Resist. 2015; 5:117–26. https://doi.org/10.1016/j.ijpddr.2015.05.004. Erratum in: Erratum to “Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites” [Int. J. Parasitol. Drugs Drug Res. 5 (2015) 117-126]. [Int J Parasitol Drugs Drug Resist. 2016].
274. Hesham HM, Lasheen DS, Abouzid KA. Chimeric HDAC inhibitors: comprehensive review on the HDAC-based strategies developed to combat cancer. Med Res Rev. 2018; 38:2058–109. https://doi.org/10.1002/med.21505.
275. Di Giorgio E, Gagliostro E, Brancolini C. Selective class IIa HDAC inhibitors: myth or reality. Cell Mol Life Sci. 2015; 72:73–86. https://doi.org/10.1007/s00018-014-1727-8.
276. Venza I, Visalli M, Oteri R, Cucinotta M, Teti D, Venza M. Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment Cell Melanoma Res. 2013; 26:193–204. https://doi.org/10.1111/pcmr.12049.
277. Panella S, Marcocci ME, Celestino I, Valente S, Zwergel C, Li Puma DD, Nencioni L, Mai A, Palamara AT, Simonetti G. MC1568 inhibits HDAC6/8 activity and influenza A virus replication in lung epithelial cells: role of Hsp90 acetylation. Future Med Chem. 2016; 8:2017–31. https://doi.org/10.4155/fmc-2016-0073.
278. Fleming CL, Ashton TD, Gaur V, McGee SL, Pfeffer FM. Improved synthesis and structural reassignment of MC1568: a class IIa selective HDAC inhibitor. J Med Chem. 2014; 57:1132–35. https://doi.org/10.1021/jm401945k.
279. Nebbioso A, Dell’Aversana C, Bugge A, Sarno R, Valente S, Rotili D, Manzo F, Teti D, Mandrup S, Ciana P, Maggi A, Mai A, Gronemeyer H, Altucci L. HDACs class II-selective inhibition alters nuclear receptor-dependent differentiation. J Mol Endocrinol. 2010; 45:219–28. https://doi.org/10.1677/JME-10-0043.
280. Ohguchi H, Hideshima T, Anderson KC. The biological significance of histone modifiers in multiple myeloma: clinical applications. Blood Cancer J. 2018; 8:83. https://doi.org/10.1038/s41408-018-0119-y.
281. Bayat S, Shekari Khaniani M, Choupani J, Alivand MR, Mansoori Derakhshan S. HDACis (class I), cancer stem cell, and phytochemicals: cancer therapy and prevention implications. Biomed Pharmacother. 2018; 97:1445–53. https://doi.org/10.1016/j.biopha.2017.11.065.