
INTRODUCTION
Cetuximab, a chimeric human-mouse monoclonal antibody to the epidermal growth factor receptor (EGFR), has shown clinical efficacy in individuals with metastatic colorectal cancer (mCRC). However, a subset of mCRC patients fails to show an initial response (de novo resistance) to this agent, whereas others develop resistance after an initial response (acquired resistance). Well-established causes of de novo resistance to cetuximab include activating mutations in codon 12 or 13 of KRAS and in BRAF [1–4]. Various mechanisms responsible for acquired resistance to cetuximab in colorectal cancer have also been identified [5–7]. We previously established cetuximab-resistant cancer cells by exposing parental cells to increasing concentrations of cetuximab [8]. Analysis of these cells revealed that cell-derived heregulin confers cetuximab resistance through bypass signaling via HER2 (also known as ERBB2) and HER3 (also known as ERBB3). Heregulin is a ligand for HER3 and stabilizes the HER2-HER3 heterodimer [9]. We also found that high initial levels of serum heregulin protein and tumor heregulin mRNA were significantly associated with a poor clinical outcome in mCRC patients treated with cetuximab [8]. Furthermore, in patients who initially achieved a partial response to cetuximab-based therapy, the serum concentration of heregulin after the development of clinical cetuximab resistance was significantly higher than that before treatment [8]. These preclinical and clinical data indicate that increased levels of heregulin are associated with both de novo and acquired resistance to cetuximab.
Patritumab (U3-1287) is a first-in-class, fully human monoclonal antibody directed to the extracellular domain (ECD) of HER3 that is currently in clinical development, as are other HER3-targeted antibodies such as MM-121 and LJM716 (MM-121 prevents ligand binding, whereas LJM716 specifically binds to an epitope formed by ECD domains II and IV in the closed conformation of HER3 [10]). Patritumab has been shown both to inhibit ligand-induced HER3 phosphorylation and to suppress the growth of pancreatic, non–small cell lung cancer, and colorectal cancer xenograft tumors [11, 12]. To identify strategies or agents capable of overcoming resistance to cetuximab induced by heregulin, we have now established sublines of the cetuximab-sensitive human colorectal cancer cell line DiFi that stably express heregulin derived from transfected cDNA. With the use of these cells, we investigated the effects of patritumab on cetuximab resistance mediated by cell-derived heregulin both in vitro and in vivo.
RESULTS
DiFi cells stably overexpressing heregulin show resistance to cetuximab
The human colorectal cancer cell line DiFi, which harbors wild-type alleles of KRAS, BRAF, and PI3K, is highly sensitive to cetuximab [13]. To investigate whether cell-derived heregulin might induce cetuximab resistance in DiFi cells, we established DiFi sublines that stably overexpress this protein (DiFi-HRG4, DiFi-HRG5, and DiFi-HRG6) or that stably harbor the corresponding empty vector (DiFi- Mock1) as a result of retroviral infection. Heregulin is a soluble growth factor that is synthesized as a transmembrane precursor molecule of 105 kDa. Cell surface proteases catalyze cleavage of the extracellular domain of this precursor, which is then released and functions as a ligand for HER3. Immunoblot analysis revealed the presence of the transmembrane form of heregulin in DiFi-HRG cells (with its abundance being greatest in DiFi-HRG4 cells), whereas no such band was detected in DiFi-Mock1 cells or the parental DiFi cells (Fig. 1A). Analysis of conditioned medium from these cell lines with an enzyme-linked immunosorbent assay (ELISA) also revealed the presence of substantial amounts of heregulin in the medium from all DiFi-HRG cell lines but not in that from DiFi-Mock1 or the parental cells (Fig. 1B). To assess the effect of cetuximab on cell growth, we exposed DiFi-HRG and DiFi-Mock1 cells to various concentrations of the drug for 5 days and then measured cell viability. All DiFi-HRG cell lines showed a reduced sensitivity to cetuximab compared with DiFi-Mock1 cells, with median inhibitory concentration (IC50) values of > 100 μg/mL for the former cell lines and ~0.1 μg/mL for the latter (Fig. 1C). The DiFi-HRG cell lines also showed resistance to panitumumab, another antibody to EGFR (data not shown). These data thus suggested that DiFi-HRG cells are resistant to EGFR-targeted antibodies.
Heregulin maintains HER3 and AKT phosphorylation and survivin expression in the presence of cetuximab in DiFi-HRG cell lines
To investigate possible differences in signal transduction among the DiFi isogenic lines, we examined the effects of cetuximab (10 μg/mL) on EGFR, HER2, HER3, AKT, and extracellular signal–regulated kinase (ERK) phosphorylation (Fig. 2A). Immunoblot analysis revealed that cetuximab markedly inhibited the phosphorylation of all of these proteins in DiFi-Mock1 cells. In contrast, whereas cetuximab substantially reduced the level of EGFR and ERK phosphorylation in DiFi-HRG cells, it had little effect on the phosphorylation of HER2, HER3, or AKT. We next examined the effects of cetuximab on expression of the apoptosis-related proteins BIM (a proapoptotic BH3-only protein) and survivin (a member of the inhibitor of apoptosis, or IAP, family). We previously showed that inhibition of the MEK-ERK signaling pathway induces BIM expression, and that inhibition of the PI3K-AKT pathway suppresses survivin expression, with both of these effects being independently required for tyrosine kinase inhibitor (TKI)–induced apoptosis in lung cancer cells positive for EGFR mutation [14], breast cancer cells positive for HER2 amplification [15], and gastric cancer cells positive for MET amplification [16]. Consistent with these observations, we found that cetuximab induced both up-regulation of BIM and down-regulation of survivin in DiFi-Mock1 cells, resulting in generation of the cleaved form of poly(ADP-ribose) polymerase (PARP), a characteristic of apoptosis (Fig. 2B). In contrast, in DiFi-HRG cell lines, whereas cetuximab induced BIM expression, it had little effect on the abundance of survivin or PARP cleavage (Fig. 2B), suggesting that sustained AKT signaling and survivin expression confer resistance to cetuximab in these cell lines.
The HER3 neutralizing antibody patritumab abrogates cetuximab resistance induced by heregulin
To investigate further the role of HER3 and heregulin in the resistance of DiFi-HRG cell lines to cetuximab, we exposed DiFi-HRG4 cells to cetuximab, the fully human HER3-targeted monoclonal antibody patritumab, or the combination of both agents. We found that neither antibody alone substantially affected cell proliferation, whereas the combination of both agents induced marked inhibition of cell growth (Fig. 3A). We next examined the effects of these antibodies on apoptosis in DiFi-Mock1 and DiFi-HRG4 cells. An annexin V binding assay revealed that cetuximab alone induced a substantial level of apoptosis in DiFi-Mock1 cells but not in DiFi-HRG4 cells (Fig. 3B, C), suggesting that the operation of a heregulin autocrine loop in these latter cells inhibits cetuximab-induced apoptosis. However, exposure of DiFi-HRG4 cells to the combination of patritumab (10 μg/mL) and cetuximab (10 μg/mL) resulted in a marked increase in the proportion of apoptotic cells (Fig. 3B, C), suggesting that patritumab sensitizes DiFi-HRG cells to cetuximab such that the extent of apoptosis induced by both antibodies in these cells is similar to that induced by cetuximab alone in DiFi-Mock1 cells.
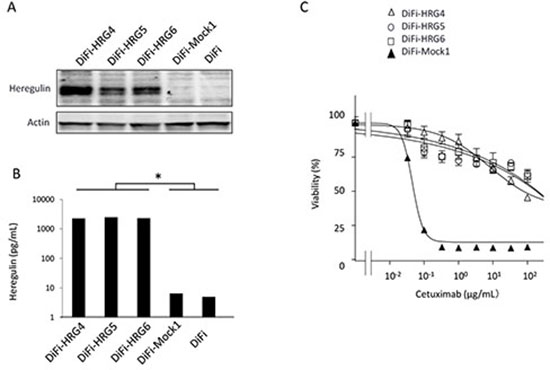
Figure 1: Characterization of DiFi isogenic cell lines. (A) DiFi isogenic cell lines (DiFi, DiFi-Mock1, DiFi-HRG4, DiFi-HRG5, and DiFi-HRG6) were cultured overnight in medium containing 10% serum and then incubated for 24 h in serum-free medium, after which the cells were lysed and subjected to immunoblot analysis with antibodies to heregulin and to β-actin (loading control). (B) Culture supernatants from cells cultured as described in Materials and Methods were assayed for heregulin with an ELISA. Data are means ± SE from three independent experiments. *P < 0.05 (Student's t test) for comparison of each DiFi-HRG line with DiFi-Mock1 or DiFi cells. (C) Cells were treated with cetuximab at the indicated concentrations for 5 days, after which cell viability was assessed. Data are means ± SE from three independent experiments.
We also examined the effects of patritumab alone or in combination with cetuximab on intracellular signaling. Immunoblot analysis showed that patritumab alone had little effect on such signaling in DiFi-Mock1 cells. In contrast, patritumab alone markedly inhibited the phosphorylation of HER3 and AKT, without affecting that of ERK, in DiFi-HRG4 cells (Fig. 3D). The combination of patritumab and cetuximab markedly attenuated the phosphorylation of EGFR, HER2, HER3, AKT, and ERK in DiFi-HRG4 cells (Fig. 3D). It also induced the cleavage of PARP in these cells to an extent similar to that observed in DiFi-Mock1 cells treated with cetuximab alone, and this effect was accompanied by both up-regulation of BIM and down-regulation of survivin expression (Fig. 3E). These results thus indicated that cetuximab resistance induced by heregulin is abrogated by patritumab through attenuation of AKT-suvivin signaling in DiFi-HRG4 cells.
Cell-derived heregulin induces cetuximab resistance and patritumab restores cetuximab sensitivity in tumor xenografts in vivo
To examine whether cell-derived heregulin induces cetuximab resistance as well as the efficacy of combined treatment with patritumab and cetuximab in vivo, we injected nude mice with DiFi-Mock1 or DiFi-HRG4 cells to allow the formation of tumor xenografts. Whereas cetuximab alone markedly inhibited the growth of DiFi-Mock1 xenografts (Fig. 4A), DiFi-HRG4 xenografts were resistant to this drug (Fig. 4B). Patritumab alone had little effect on the growth of tumors formed by either cell line. However, the combination of cetuximab and patritumab induced substantial regression of DiFi-HRG4 xenografts (Fig. 4B). These results thus suggested that heregulin produced by colorectal cancer tumors harboring wild-type KRAS induces cetuximab resistance, and that combination therapy with cetuximab and patritumab overcomes such resistance in vivo.
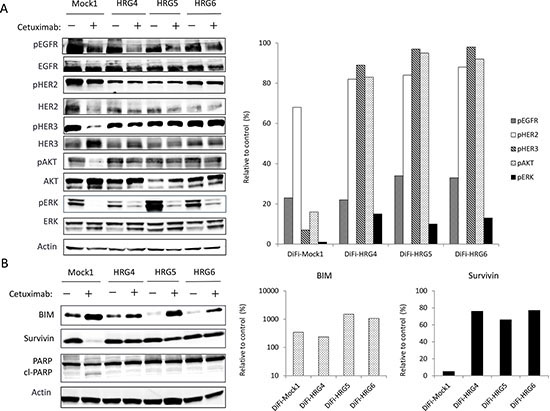
Figure 2: Effects of cetuximab on intracellular signaling and the expression of apoptosis-related proteins in DiFi isogenic cell lines. DiFi-Mock1, DiFi-HRG4, DiFi-HRG5, or DiFi-HRG6 cells were cultured overnight in medium containing 10% serum and then incubated for 6 h (A) or 24 h (B) in serum-free medium with or without cetuximab (10 μg/mL), after which cell lysates were prepared and subjected to immunoblot analysis with antibodies to phosphorylated (p) or total forms of the indicated proteins (left panels). A band corresponding to the cleaved (cl) form of PARP is indicated. The intensity of the bands corresponding to phosphorylated forms of EGFR, HER2, HER3, AKT, and ERK (A) or to BIM and survivin (B) was normalized by that of the corresponding total proteins or β-actin, respectively, and then expressed relative to the corresponding value for control cells not exposed to cetuximab (right panels).
DISCUSSION
Resistance to cetuximab is a major problem in the treatment of colorectal cancer. Although various mechanisms of cetuximab resistance have been identified [1–7, 17–20], the optimal treatment strategies for mCRC patients who show resistance to this drug remain unclear. We previously showed that tumor-derived heregulin mediates cetuximab resistance in preclinical models [8]. High levels of heregulin were also associated with a poor clinical outcome in mCRC patients treated with cetuximab-based regimens [8]. Moreover, increased heregulin levels were observed in such patients after the development of clinical resistance to cetuximab-based therapy [8]. Effective treatment options to overcome cetuximab resistance mediated by heregulin are thus urgently required.
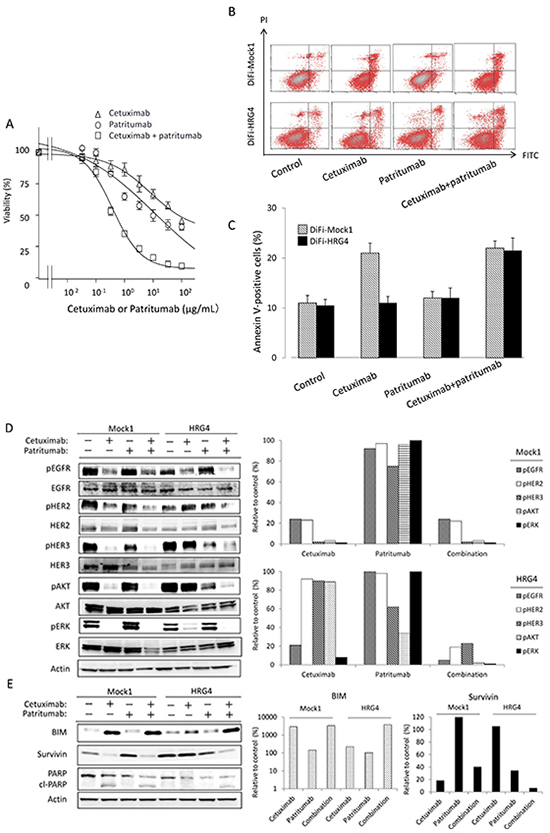
Figure 3: Effect of patritumab on heregulin-mediated cetuximab resistance in DiFi-HRG cells in vitro. (A) DiFi-HRG4 cells were incubated for 5 days with cetuximab alone, patritumab alone, or the combination of both drugs at the indicated concentrations, after which cell viability was assessed. Data are means ± SE from three independent experiments. (B, C) DiFi-Mock1 or DiFi-HRG4 cells were cultured overnight in medium containing 10% serum and then incubated for 48 h in the absence or presence of cetuximab alone (10 μg/mL), patritumab alone (10 μg/mL), or the combination of both drugs in serum-free medium, after which the number of apoptotic cells was determined by staining with propidium iodide (PI) and fluorescein isothiocyanate (FITC)–labeled annexin V followed by flow cytometry. Representative flow cytometric profiles are shown in (B), and quantitative data (means ± SE of three independent experiments) are shown in (C). (D, E) DiFi-Mock1 or DiFi-HRG4 cells were cultured overnight in medium containing 10% serum and then incubated for 6 h (D) or 48 h (E) in the absence or presence of cetuximab alone (10 μg/mL), patritumab alone (10 μg/mL), or the combination of both drugs in serum-free medium, after which cell lysates were prepared and subjected to immunoblot analysis with antibodies to the indicated proteins (left panels). The intensity of the bands corresponding to phosphorylated forms of EGFR, HER2, HER3, AKT, and ERK (D) or to BIM and survivin (E) was normalized by that of the corresponding total proteins or β-actin, respectively, and then expressed relative to the corresponding value for control cells not exposed to drug (right panels).
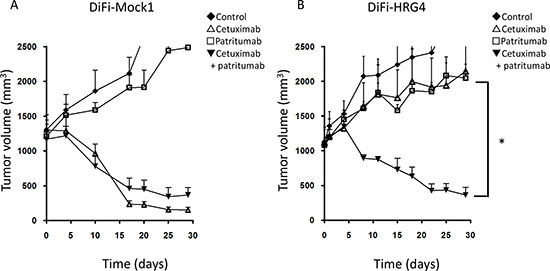
Figure 4: Effects of cetuximab, patritumab, and the combination of both drugs on the growth of DiFi-HRG tumor xenografts in vivo. Nude mice with tumor xenografts established by subcutaneous injection of DiFi-Mock1 (A) or DiFi-HRG4 (B) cells were treated for 4 weeks with vehicle (control), cetuximab (1.0 mg/body), patritumab (1.0 mg/body), or both drugs, as described in Materials and Methods. Tumor volume was determined at the indicated times after the onset of treatment. Data are means ± SE from six mice per group. *P < 0.05 for comparison of the combination of both drugs with cetuximab alone or patritumab alone (Student's t test).
We have here established heregulin-overexpressing sublines of DiFi cells (DiFi-HRG cells) and shown that these cells are resistant to cetuximab both in vitro and in vivo. Whereas the amount of the transmembrane form of heregulin was substantially higher in DiFi-HRG4 cells than in DiFi-HRG5 or DiFi-HRG6 cells (Fig. 1A), these three cell lines appeared to release similar amounts of the soluble form of heregulin into the culture medium (Fig. 1B). This difference in the relative abundance of the transmembrane and soluble forms of heregulin among DiFi-HRG cell lines might reflect a difference in the activity of cell surface proteases among the cell lines. Alternatively, the production of the transmembrane form of heregulin in DiFi-HRG4 cells might exceed the capacity of such proteases. To investigate the mechanism responsible for cetuximab resistance in DiFi-HRG cells, we examined differences in signal transduction between these cells and DiFi-Mock1 cells. In DiFi-Mock1 cells, cetuximab inhibited the phosphorylation of EGFR, HER2, HER3, AKT, and ERK as well as up-regulated BIM expression and down-regulated survivin expression, resulting in the induction of apoptosis. By contrast, in DiFi-HRG cell lines, whereas cetuximab inhibited EGFR and ERK phosphorylation, leading to BIM induction, it did not affect HER2, HER3, or AKT phosphorylation or survivin expression. Given that down-regulation both of AKT signaling and of the expression of its downstream target survivin is required for apoptosis induced by inhibition of receptor tyrosine kinases [14–16], our data suggest that sustained AKT-survivin signaling in the presence of cetuximab is responsible for the resistance of DiFi-HRG cell lines to this drug. To investigate further the relation between AKT signaling and the operation of a heregulin autocrine loop, we examined the effects of patritumab, a neutralizing monoclonal antibody to HER3, in DiFi-HRG4 cells. We found that exposure of these cells to patritumab in combination with cetuximab resulted in inhibition of EGFR, HER2, HER3, AKT, and ERK phosphorylation as well as in both up-regulation of BIM expression and down-regulation of survivin expression, leading to the induction of apoptosis. These results indicate that AKT signaling is triggered by heregulin binding to HER3.
Given that HER3 (a kinase-dead receptor) manifests impaired kinase activity [21], it requires dimerization with other HER family members to activate signaling after ligand binding [22, 23]. HER2 has been implicated as a dimerization partner of HER3, and heregulin stabilizes the HER2-HER3 heterodimer [9]. In this context, we examined the effects of lapatinib, a TKI for EGFR and HER2, in DiFi-HRG4 cells. Inhibition of EGFR and HER2 by lapatinib resulted in down-regulation of HER3, AKT, and ERK phosphorylation as well as in the induction of BIM and suppression of survivin expression in these cells, thereby triggering apoptosis (Supplementary Fig. S1A, S1B). These findings suggest that, in DiFi-HRG cells, HER3 is trans-phosphorylated by HER2 as a result of heregulin-induced HER2-HER3 heterodimerization, which in turn leads to the activation of AKT signaling [24] (Fig. 5A, 5B, 5C). The heterodimerization partner of HER3 is thus likely switched from EGFR to HER2 as a result of the overexpression of heregulin in DiFi-HRG cells. We also found that patritumab alone inhibited the phosphorylation of HER3 and AKT as well as down-regulated survivin expression in DiFi-HRG4 cells but not in DiFi-Mock1 cells (Fig. 3D, 3E). These results suggest that patritumab may prevent ligand-dependent HER3 phosphorylation by blocking HER2-HER3 heterodimerization, whereas it has little effect on ligand-independent HER3 phosphorylation.
In our DiFi-HRG xenograft model, we showed that combination therapy with patritumab and cetuximab inhibited tumor growth to the same extent as did cetuximab alone in the DiFi-Mock1 xenograft model. Antagonism of the heregulin-HER3 interaction by patritumab thus represents an effective strategy to abrogate cetuximab resistance induced by heregulin derived from tumor cells. Given that elevated circulating levels of heregulin are associated with both de novo and acquired cetuximab resistance in mCRC patients [8], our model systems based on stable overexpression of heregulin are clinically relevant and should prove useful for establishing strategies to overcome cetuximab resistance mediated by the heregulin autocrine loop. Indeed, a recent phase I/II study with refractory colorectal cancer patients revealed potential antitumor activity of the combination of cetuximab and pertuzumab, a HER2-targeted antibody that blocks ligand-dependent HER2-HER3 heterodimerization. However, this drug combination was not tolerable as a result of overlapping toxicities [25]. Given that the toxicity profile of patritumab differs from that of pertuzumab [26], the combination of cetuximab and patritumab warrants evaluation in the clinical setting. We also found that the IC50 value for the antiproliferative effect of the combination of cetuximab and patritumab in DiFi-HRG4 cells was ~10 times as high as that for cetuximab alone in DiFi-Mock1 cells (Fig. 1C, Fig. 3A). The discrepancy between these in vitro data and our in vivo findings may suggest that antibody-dependent cellular cytotoxicity [27] involving NK cell activation plays a role in tumor growth inhibition by the combination of both agents. Moreover, combination therapy with these two IgG1 antibodies may result in an enhanced antitumor activity mediated by cytotoxic T lymphocytes [28] in the clinical setting. Given the recent evidence implicating the importance of interactions between therapeutic antibodies and the immune system in the efficacy of antibody treatment [20], further investigation of such mechanisms is warranted.
In conclusion, we have shown that consecutive activation of HER2-HER3 and AKT by heregulin in an autocrine-dependent manner confers resistance to cetuximab, and that patritumab restores cetuximab sensitivity in tumors with heregulin-induced cetuximab resistance. Further studies of combination therapy with patritumab and cetuximab are thus warranted in mCRC patients with heregulin-induced resistance to EGFR-targeted antibodies.
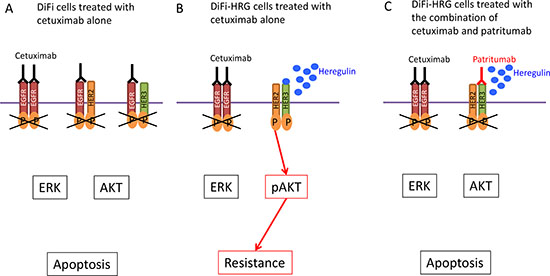
Figure 5: Model for intracellular signaling underlying the induction of apoptosis by cetuximab and patritumab in colorectal cancer cells. (A) DiFi colorectal cancer cells treated with cetuximab alone. (B) DiFi cells that stably overexpress heregulin (DiFi-HRG cells) are resistant to cetuximab as a result of HER2-HER3 heterodimerization and AKT activation induced by heregulin. (C) Patritumab abrogates cetuximab resistance mediated by the heregulin autocrine loop in DiFi-HRG cells.
MATERIALS AND METHODS
Cells and reagents
The DiFi human colorectal cancer cell line was kindly provided by P. A. Janne (Dana Farber Cancer Institute). DiFi cells were maintained under a humidified atmosphere of 5% CO2 in air at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose and supplemented with Ham’s F-12 and 10% fetal bovine serum (FBS). Cetuximab was obtained from Merck Serono, and patritumab was kindly provided by Daiichi-Sankyo (Tokyo, Japan). Recombinant human heregulin (NRG1-β1/HRG1-β1 extracellular domain) was obtained from R&D Systems.
Establishment of cells stably overexpressing heregulin
A full-length cDNA encoding human heregulin (NRG1, GenBank accession no. NM_013956) was obtained from Origene (Rockville, MD). The amplification product was verified by sequencing after its cloning into the pCR-Blunt II-TOPO vector (Invitrogen). The heregulin cDNA was then excised from pCR-Blunt II-TOPO and transferred to the pQCXIH retroviral vector (Clontech), and retroviruses encoding heregulin were produced and used to infect DiFi cells as described [29]. Cells stably expressing heregulin were then isolated by selection with hygromycin (Invivogen) at 500 μg/mL.
Cell growth inhibition assay
Cells were transferred to 96-well flat-bottomed plates and cultured for 24 h before exposure to various concentrations of cetuximab or patritumab in medium containing 1% FBS for 120 h. Cell Counting Kit-8 solution (Dojindo, Kumamoto, Japan) was then added to each well, and the cells were incubated for 3 h at 37°C before measurement of absorbance at 490 nm with a Multiskan Spectrum instrument (Thermo Labsystems). Absorbance values were expressed as a percentage of that for nontreated cells, and the IC50 of cetuximab for inhibition of cell growth was determined.
Heregulin ELISA
The concentration of heregulin in cell culture supernatants was measured with the use of a sandwich ELISA (NRG1-β1 DuoSet, R&D Systems) as previously described [30]. Cells were seeded in six-well plates at a density of 0.5 × 106 cells per well in DMEM supplemented with 10% FBS. After the cells had achieved confluence, the medium was replaced with 5 ml of DMEM supplemented with 0.1% FBS, the cells were incubated for 48 h, and the culture supernatants were collected for assay of heregulin.
Immunoblot analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and then lysed with 1 × Cell Lysis Buffer (Cell Signaling Technology) consisting of 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA (disodium salt), 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, leupeptin (1 μg/mL), and 1 mmol/L phenylmethylsulfonyl fluoride. The protein concentration of the lysates was determined with a bicinchoninic acid assay kit (Thermo Fisher Scientific), and equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis on a 7.5% gel (Bio-Rad). The separated proteins were transferred to a nitrocellulose membrane, which was then incubated with Blocking One solution (Nacalai Tesque) for 20 min at room temperature before incubation overnight at 4°C with primary antibodies. Antibodies to heregulin (NRG1-β1), to phosphorylated EGFR (phospho-Tyr1068), to phosphorylated HER2 (phospho-Tyr1248), to phosphorylated HER3 (phospho-Tyr1289), to phosphorylated or total forms of AKT, to phosphorylated ERK, to PARP, and to BIM were obtained from Cell Signaling Technology; those to total HER3, to total ERK, and to survivin were from Santa Cruz Biotechnology; those to total EGFR were from Zymed/Invitrogen; those to total HER2 were from Millipore; and those to β-actin were from Sigma. The membrane was then washed with PBS containing 0.05% Tween 20 before incubation for 1 h at room temperature with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare). Immune complexes were finally detected with ECL detection reagents (GE Healthcare).
Annexin V binding assay
The binding of annexin V to cells was measured with the use of an Annexin-V-FLUOS Staining Kit (Roche). Cells were harvested by exposure to trypsin-EDTA, washed with PBS, and centrifuged at 200 × g for 5 min. The cell pellets were resuspended in 100 μL of Annexin-V-FLUOS labeling solution, incubated for 10 to 15 min at 15° to 25°C, and then analyzed for fluorescence with a flow cytometer (FACSCalibur) and Cell Quest software (Becton Dickinson).
Tumor growth inhibition assay in vivo
All animal experiments were performed in accordance with the Recommendations for Handling of Laboratory Animals for Biomedical Research compiled by the Committee on Safety and Ethical Handling Regulations for Laboratory Animal Experiments, Kinki University. The ethical procedures followed conformed to the UKCCCR guidelines for the welfare and use of animals in cancer research [31]. The study was also reviewed and approved by the Animal Ethics Committee of Kinki University (approval no. KAME-22-018). Cells were injected subcutaneously into the axilla of 5- to 6-week-old female athymic nude mice (BALB/c nu/nu; CLEA Japan), and treatment was initiated when tumors in each group of six mice achieved an average volume of 1000 to 1200 mm3. Treatment groups consisted of vehicle control, cetuximab (1.0 mg/body), patritumab (1.0 mg/body), and the combination of both agents. Cetuximab and patritumab were administered by intraperitoneal injection twice a week for 4 weeks, with control animals receiving a 0.5% (w/v) aqueous solution of hydroxyl propylmethyl cellulose as vehicle. Tumor volume was determined from caliper measurements of tumor length (L) and width (W) according to the formula LW2/2. Both tumor size and body weight were measured twice weekly. For ethical reasons, animals were removed from the study if the tumor volume exceeded 2500 mm3.
Statistical analysis
Quantitative data are presented as means ± SE unless indicated otherwise. The significance of differences was evaluated with the unpaired two-tailed Student's t test. A P value of < 0.05 was considered statistically significant.
Conflict of interest statement
The authors declare no conflict of interest.
REFERENCES
1. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009; 360:1408–1417.
2. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009; 27:663–671.
3. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009; 360:563–572.
4. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008; 26:5705–5712.
5. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Serrano S, Himmelbauer H, Bellmunt J, Rovira A, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012; 18:221–223.
6. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532–536.
7. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, Lauricella C, Zanon C, Perera T, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013; 3:658–673.
8. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011; 3:99ra86.
9. Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995; 14:4267–4275.
10. Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014; 20:1410–1416.
11. Freeman D, Ogbagabreiel S, Rothe M, Radinsky R, Treder M. 2008; Fully human Anti-HER3 monoclonal antibodies (mAbs) have unique in vitro and in vivo functional and antitumor activities versus other HER family inhibitors (abstract). Proceedings of the Annual Meeting of the American Association for Cancer Research; San Diego, CA: AACR.
12. Treder M, Hartmann S, Ogbagabrei S, Borges E, Green L, Kang Jea (2008); Fully human Anti-HER3 monoclonal antibodies (mAbs) inhibit oncogenic signaling and tumor cell growth in vitro and vivo. Proceedings of the Annual Meeting of the American Association for Cancer Research; San Diego, CA: AACR
13. McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, Rothenberg SM, Supko JG, Sordella R, Ulkus LE, Iafrate AJ, Maheswaran S, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007; 104:19936–19941.
14. Okamoto K, Okamoto I, Okamoto W, Tanaka K, Takezawa K, Kuwata K, Yamaguchi H, Nishio K, Nakagawa K. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010; 70:10402–10410.
15. Tanizaki J, Okamoto I, Fumita S, Okamoto W, Nishio K, Nakagawa K. Roles of BIM induction and survivin downregulation in lapatinib-induced apoptosis in breast cancer cells with HER2 amplification. Oncogene. 2011; 30:4097–4106.
16. Okamoto W, Okamoto I, Arao T, Kuwata K, Hatashita E, Yamaguchi H, Sakai K, Yanagihara K, Nishio K, Nakagawa K. Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for MET amplification. Mol Cancer Ther. 2012; 11:1557–1564.
17. Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009; 101:1308–1324.
18. De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011; 12:594–603.
19. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010; 28:1254–1261.
20. Troiani T, Zappavigna S, Martinelli E, Addeo SR, Stiuso P, Ciardiello F, Caraglia M. Optimizing treatment of metastatic colorectal cancer patients with anti-EGFR antibodies: overcoming the mechanisms of cancer cell resistance. Expert Opin Biol Ther. 2013; 13:241–255.
21. Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL, 3rd. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994; 91:8132–8136.
22. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003; 100:8933–8938.
23. Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003; 144:1032–1044.
24. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996; 16:5276–5287.
25. Rubinson DA, Hochster HS, Ryan DP, Wolpin BM, McCleary NJ, Abrams TA, Chan JA, Iqbal S, Lenz HJ, Lim D, Rose J, Bekaii-Saab T, Chen HX, Fuchs CS, Ng K. Multi-drug inhibition of the HER pathway in metastatic colorectal cancer: results of a phase I study of pertuzumab plus cetuximab in cetuximab-refractory patients. Invest New Drugs. 2014; 32:113–122.
26. LoRusso P, Janne PA, Oliveira M, Rizvi N, Malburg L, Keedy V, Yee L, Copigneaux C, Hettmann T, Wu CY, Ang A, Halim AB, Beckman RA, Beaupre D, Berlin J. Phase I Study of U3-1287, a Fully Human Anti-HER3 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2013; 19:3078–3087.
27. Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007; 98:1275–1280.
28. Correale P, Botta C, Cusi MG, Del Vecchio MT, De Santi MM, Gori Savellini G, Bestoso E, Apollinari S, Mannucci S, Marra M, Abbruzzese A, Aquino A, Turriziani M, Bonmassar L, Caraglia M, Tagliaferri P. Cetuximab +/- chemotherapy enhances dendritic cell-mediated phagocytosis of colon cancer cells and ignites a highly efficient colon cancer antigen-specific cytotoxic T-cell response in vitro. Int J Cancer. 2012; 130:1577–1589.
29. Tanaka K, Arao T, Maegawa M, Matsumoto K, Kaneda H, Kudo K, Fujita Y, Yokote H, Yanagihara K, Yamada Y, Okamoto I, Nakagawa K, Nishio K. SRPX2 is overexpressed in gastric cancer and promotes cellular migration and adhesion. Int J Cancer. 2009; 124:1072–1080.
30. Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, Rogers AM, Johnson BE, Janne PA. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res. 2008; 14:6963–6973.
31. Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010; 102:1555–1577.