
There is no novelty in stating that conventional cancer therapeutics are highly mutagenic, unselective and toxic. Hair loss, neutropenia, vomiting, and immune suppression are just some of the immediate side effects. In addition, genomic damage induced by these agents can result in second tumours later in life. Cancer cells differ from normal cells in a number of hallmarks that define the tumour phenotype [1]. These traits, which involve genetic and epigenetic changes in tumour suppressors and oncogenes, constitute a double-edged sword for cancer cells. They provide a proliferative advantage but, importantly from a treatment prospective, also mean an increased dependence on certain cellular functions as well as an increased vulnerability to particular insults. The goal of cancer treatment is to exploit this differential sensitivity to selectively kill tumour cells whilst sparing normal tissues. Classically, the search for new anticancer therapies has focused on the discovery of agents that annihilate tumour cells at least as effectively as existing chemotherapeutics whilst having weaker or negligible effects on healthy tissues. Alternatively, so-called cyclotherapy strategies aim at improving the therapeutic window by selectively shielding normal cells from conventional anticancer drugs [2,3]. The majority of such drugs target dividing cells. Hence, if we could transiently and selectively pause proliferation in normal tissues, subsequent treatment with S- or M-phase poisons should eradicate cycling tumour cells only.
Nutlin-like compounds are well-established non-genotoxic activators of the p53 pathway [4] currently undergoing clinical trials for treating patients with cancers retaining wild-type p53. In addition, nutlin-3 has been proposed as a potential chemoprotective agent for patients bearing tumours with mutant p53. The rationale behind this use is that nutlin-3 has been shown to have a mild, reversible cytostatic effect on a variety of normal cells [5,6]. We believe it is important to highlight these observations, as a widely spread view is that activating p53 in vivo would trigger severe cytotoxic responses in normal tissues. Experimental data supporting this notion comes from animal studies where the expression of p53’s major negative regulator, mdm2, was suppressed [7]. However, such undesired side effects do not occur when p53 is activated in a buffered manner using nutlin-3, an agent that binds directly to mdm2 [4], partially impairs mdm2’s down-regulating activity on p53 [8], and stabilises mdm2 [9]. Indeed, nutlin-3 has been shown to protect normal cells from mitotic poisons such as the aurora kinase inhibitor VX680 [10], taxanes [5,6], and polo-like kinase inhibitors [11]. Most interestingly, nutlin-3 protects mice from PLK1 (polo-like kinase 1) inhibitor–induced neutropenia without abating the anticancer potency of the mitotic poison [11]. In their recent article, Apontes et al., (2011) provide additional evidence supporting the potential suitability of nutlin-3 for cyclotherapy purposes. Indeed, nutlin-3 also protects normal cells from nocodazole, a compound that −like the vinca alkaloids used in the clinic− inhibits tubulin polymerisation.
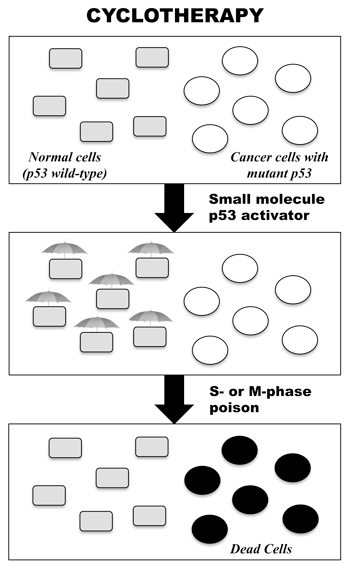
Figure 1: Cartoon illustrating the concept of p53-based cyclotherapy. The p53 tumour suppressor is mutated in about 50% of human solid tumours in adults. Hence, administering a small-molecule p53 activator to patients bearing p53-mutant tumours would selectively activate p53 in normal tissues, inducing a mild reversible cell-cycle arrest. Subsequent treatment with conventional anticancer drugs should then kill proliferating cancer cells, whilst leaving normal cells untouched.
The results summarised above suggest that nutlin-3 could constitute an ideal agent for chemoprotection purposes in patients with p53-deficient tumours. However, the clinical use of this compound has not yet been approved. In a recent paper, we tested the possibility of using low doses of actinomycin-D, an approved drug that at nanomolar concentrations can increase p53 activity in a similar manner to nutlin-3 [12]. Yet, there are some important differences between the p53 responses to nutlin-3 and LDActD that can be understood on the basis of their different from mechanisms of action [9]. Like nutlin-3, LDActD has a primarily cytostatic effect on p53 wild-type normal cells and protects them from subsequent treatment with VX680 [13]. Unfortunately, LDactD pretreament also protects p53-deficient tumour cells against VX680 to a small extent. Our hope is that LDActD might perform better in a p53-based cyclotherapy setting when combined with S-phase poisons than with mitotic poisons. In this line, nutlin-3 has been shown to prevent the mutagenic and cytotoxic effects of gemcitabine and cytosine arabinoside in normal cells without diminishing the killing effect of these compounds on p53-mutant tumour cells [14].
In the article featured here [6], aside from nutlin-3, Blagosklonny’s group evaluate other clinically-approved drugs as chemoprotective agents against the tubulin poisons nocodazole and paclitaxel. The two well-known drugs chosen for this purpose are rapamycin, a drug with striking anti-ageing properties, clinically used to prevent transplant organ rejection (in chronic administration with traditional immunosuppressants) [15], and the anti-diabetic drug metformin. The authors show that pretreatment with these drugs succeeds in protecting all normal cell types tested against these mitotic poisons, whilst having little impact on the vulnerability of a cancer cell line with mutant p53. The chemoprotective effects might be related to the interesting observations that glucose starvation selectively protects primary cells against cyclophosphamide in cell culture and fasting reduces the side effects of chemotherapy in patients [16,17]. Furthermore, evidence suggests that lack of p53 may sensitise cancer cells to metabolic stresses such as nutrient deprivation [18]. The study by Apontes et al. also addresses whether combining two chemoprotective agents could lead to an even greater increase in the therapeutic window of mitotic poisons. In this regard, nutlin-3 plus rapamycin and rapamycin plus metformin gave the most promising results.
Like in the case of the nutlins, the efficacy of metformin and rapamycin is currently being assessed in clinical trials for cancer [19,20]. Independently of whether these tests are successful, the encouraging data presented by Apontes and co-workers suggests that there might be other avenues for these compounds in cancer therapy.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74.
2. Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell Cycle 2002; 1:375-82.
3. Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res 2001; 61:4301-5.
4. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8.
5. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res 2005; 65:1918-24.
6. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture Oncotarget 2011; 2:222-33.
7. Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10:501-14.
8. Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell 2006; 23:251-63.
9. Van Leeuwen IMM, Higgins M, Campbell J, Brown CJ, McCarthy AR, Pirrie L, et al. Mechanism-specific signatures for small-molecule p53 activators. Cell Cycle 2011; 10:in press.
10. Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ 2010; 17:1486-500.
11. Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr., Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964-9.
12. Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 2009; 8:2810-8.
13. Rao B, Van Leeuwen IMM, Higgins M, Campbell J, Thompson AM, Lane DP, et al. Evaluation of an actinomycin-D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1.
14. Kranz D, Dobbelstein M. Nongenotoxic p53 activation protects cells against S-phase-specific chemotherapy. Cancer Res 2006; 66:10274-80.
15. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Discov Today 2007; 12:218-24.
16. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A 2008; 105:8215-20.
17. Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle 2010; 9:4474-6.
18. Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med 2011; 89:237-45.
19. Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol 2010; 7:209-19.
20. Jalving M, Gietema JA, Lefrandt JD, de Jong S, Reyners AK, Gans RO, et al. Metformin: taking away the candy for cancer? Eur J Cancer 2010; 46:2369-80.