
INTRODUCTION
Recent developments in sequencing cancer genomes have advanced our understanding of the complex molecular landscape of AML. Even after addressing the extensive network of cytogenetic abnormalities, molecular heterogeneity, epigenetic and gene expression profiles in AML, the prognostic stratification in AML mainly relies upon only cytogenetics and a handful of gene mutations. Subsequent to risk stratification and standard treatment, the 5 year survival is less than 60% in adult AML [1]. Therefore, there is a clinical need to develop assays that identify patients who have a suboptimal response to therapy and are at a high risk of relapse so that they can be treated with intensive post remission strategies such as allogeneic bone marrow transplantation. The detection of leukemic cells at a threshold below the morphologic limit of detection is called measurable residual disease (MRD). Studies over the last two decades have shown that the presence of MRD is an important prognostic factor that is highly predictive of outcome in a variety of haematological malignancies. Today, MRD detection is routinely performed for assessment of response to therapy as well as guiding post remission strategies in chronic myeloid leukemia and precursor B lineage acute lymphoblastic leukemia. MRD can be measured by sensitive real-time PCR for AMLs that harbour gene fusions (for e.g. RUNX1-RUNX1T1). However, for the majority of AML, immunophenotyping (FCM-MRD) remains as the most accepted method to detect MRD [2–4].
Acute myeloid leukemia (AML) with mutated NPM1 is a specific subtype of AML with a recurrent genetic abnormality and favourable outcome [5]. NPM1mut AML is one of the most common subsets of AML in adults and comprises around 35% overall adult AML cases and around 45% of normal karyotype AML [6, 7]. More than 50 types of insertion mutations have been detected in exon 12 of the NPM1 gene [8]. These mutations are specific to the blast compartment and are not present in mature myeloid and lymphoid cells, deeming NPM1 as reliable marker for MRD [9]. For MRD monitoring of NPM1mut AML, the use of molecular techniques has been more prevalent. Recent studies have shown that serial monitoring of NPM1 mutations from the blood or bone marrow during chemotherapy is highly predictive of relapse. Majority of these studies have been RNA based and have used mutation specific approaches for the detection of MRD [10–12]. A disadvantage of a mutation specific approach is that the NPM1 mutation must be characterized at diagnosis by sequencing and each type of NPM1 mutation must be validated as an independent MRD assay. This is further complicated by varying performance characteristics of mutation specific primers and fluorescent probe combinations.
DNA based next generation sequencing (NGS) is a scalable solution that has the potential for detection of MRD in AML [13–15]. As an NGS based assay can cover all occurring mutations in a specific genomic locus, it can potentially overcome the technical drawbacks of real time quantitative PCR (RQ-PCR) and provide a unique solution for detection of MRD. However, studies that have evaluated the clinical impact of NGS-MRD are largely lacking. In this study, we have validated an ultra-sensitive technique for NPM1mut AML that can detect NPM1 mutations at a frequency of 0.001%. We have then compared NGS-MRD with FCM-MRD, the gold standard for detection of MRD in AML as well as RQ-PCR for the commonly occurring type A NPM1 mutations. We demonstrate that NGS-MRD for NPM1mut AML is an independent prognostic factor significantly predictive of outcome in NPM1mut AML.
RESULTS
Patient outcome
The mean overall survival (OS) was 40.02 months (95%CI 33.73 to 46.32) for the entire cohort (median not reached). The mean relapse free survival (RFS) was 33.31 months (95%CI 26.85 to 39.77), median RFS was 19.43 months (95%CI 14.27 to 43.03) months (Supplementary Figure 1). The median follow-up was 23.5 months. Additional patient characteristics can be seen in Supplementary Table 1A and 1B.
Types of NPM1 mutations monitored & clinical relevance of NGS-MRD
Diagnostic DNA was available in 81 patients. These samples were sequenced to characterize the mutation, ensure the stability of NPM1 mutation over post-induction (PI) and first post consolidation (PC) time points and ascertain whether there was any switch in the mutation type in the course of treatment [16]. We did not detect a clonal switch in our cohort. Figure 1 shows the types of NPM1 mutations and their frequencies. Type A, B and D were the most common subtypes (83.95%, Supplementary Table 2). On comparing the prognosis of these common subtypes against the other uncommon subtypes no significant difference in OS and RFS could be observed. We also detected a novel 4 bp insertion, c.873_874insGCCA. Based on a one log reduction between PI and PC time points, out of 54 cases tested, 16 (29.63%) were NGS-MRD positive and 38 (70.37%) were NGS-MRD negative. Presence of NGS-MRD was significantly predictive of an inferior OS and RFS (Figure 2 and Table 1).
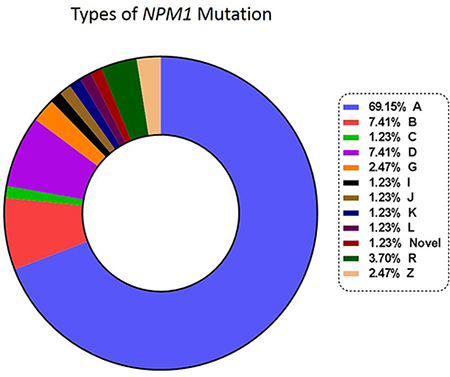
Figure 1: Types and frequencies of NPM1 mutations.
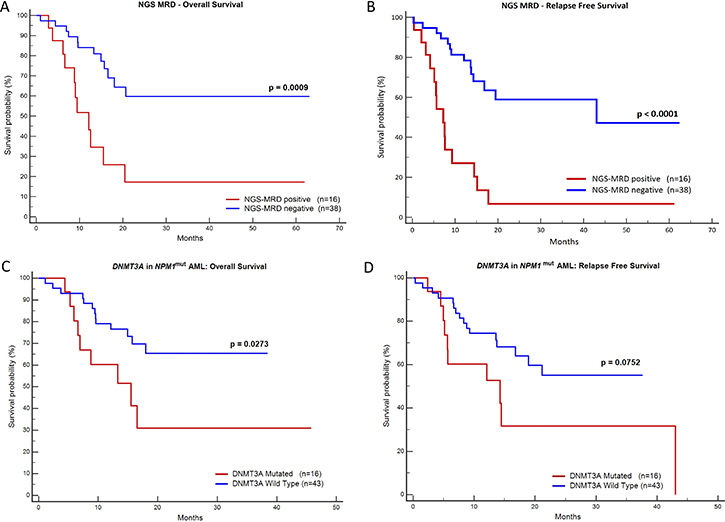
Figure 2: Kaplan-Meier graphs of NGS-MRD and presence of DNMT3A mutation. Plots (A) and (B) demonstrate that the presence of NGS MRD is highly predictive of inferior OS and RFS. The plots (C) & (D) show that presence of DNMT3A mutation is predictive of the inferior OS (C) and likely RFS (D).
Table 1: Prognostic significance of MRD in NPM1mut AML by univariate and multivariate Cox analysis
Univariate Cox analyses |
||||
|---|---|---|---|---|
Overall Survival (OS) |
Relapse Free Survival (RFS) |
|||
HR (95% CI) |
P |
HR (95% CI) |
P |
|
NGS MRD |
||||
MRD Negative |
1 |
0.002 |
1 |
0.0001 |
MRD Positive |
3.74 (1.64 to 8.54) |
4.5 (2.2 to 9.86) |
||
PI FCM-MRD |
||||
MRD Negative |
1 |
0.0081 |
1 |
0.012 |
MRD Positive |
2.6 (1.29 to 5.27) |
2.3 (1.2 to 4.26) |
||
PC FCM-MRD |
||||
MRD Negative |
1 |
0.26 |
1 |
0.5 |
MRD Positive |
1.84 (0.65 to 5.30) |
1.42 (0.51 to 3.92) |
||
FLT3-ITD |
||||
FLT3-ITD Negative |
1 |
0.83 |
1 |
0.96 |
FLT3-ITD Positive |
1.08 (0.53 to 2.23) |
1.02 (0.53 to 1.94) |
||
DNMT3A |
||||
DNMT3A Negative |
1 |
0.0330 |
1 |
0.08 |
DNMT3A Positive |
2.53 (1.08 to 5.93) |
2.08 (0.92 to 4.70) |
||
Multivariate Cox analyses |
||||
|---|---|---|---|---|
Overall Survival (OS) |
Relapse Free Survival (RFS) |
|||
HR (95% CI) |
P |
HR (95% CI) |
P |
|
NGS MRD |
3.64 (1.58 to 8.37) |
0.0025 |
4.8 (2.24 to 10.28) |
0.0001 |
PI FCM-MRD |
2.61 (1.14 to 6.00) |
0.0246 |
2.71 (1.27 to 5.8) |
0.0105 |
Abbreviations: HR, hazards ratio; CI, confidence interval; ITD, internal tandem duplication.
Comparison of MRD results obtained by NGS (VAF) and RQ-PCR (only for Type A mutant NPM1)
To compare NGS-MRD results using an orthogonal technique, we used a DNA based RQ-PCR based assay to detect MRD for Type-A NPM1 mutations at end of PI and PC. RQ-PCR was done in total 71 samples carrying Type-A NPM1 mutation at PI (44) and PC (27) time points. Values obtained from RQ-PCR were not significantly different from corresponding NGS variant allelic frequency (VAF) (p = 0.7134; Supplementary Figure 2). A significant concordance was seen between NGS-VAF and RQ-PCR data for MRD samples with minimal bias (r2 = 0.9375, p < 0.0001; Supplementary Figure 2). NGS-MRD was more sensitive as compared to RQ-PCR. Ten samples were positive by NGS-MRD (range 0.0016 to 0.018, median = 0.0045) and RQ-PCR negative. Similarly, only 3 samples were RQ-PCR positive but NGS-MRD negative.
Relevance of DNMT3A mutations in NPM1mut AML
A total of 59 NPM1mut AML samples with optimum DNA concentration at baseline were subjected for analysis of DNMT3A mutation. For baseline samples median coverage was 987x. Out of 59 patients tested, 16 (26.6%) were positive for DNMT3A mutation with VAF ranging from 3.5% to 50%. The arginine 882 hotspot was the most commonly affected amino acid in our cohort (43.75%). Presence of DNMT3A mutations in NPM1mut AML were predictive of inferior OS (p = 0.027) and possibly inferior RFS (p = 0.075; Figure 2). This agrees with other studies where DNMT3A mutations have been reported to have adverse prognosis in NPM1mutAML [10, 17, 18].
Dynamics of VAF changes in DNMT3A and comparison with NPM1-VAF
The details of DNMT3A mutations and the dynamics of these mutations at post therapeutic time points can be seen in Supplementary Table 3. In 7 out of 16 cases, DNMT3A mutations were persistent at the end of induction and 3 cases (out of 7 tested) at end of consolidation. The corresponding NPM1 VAF can be also seen in Supplementary Table 3 and Figure 3.
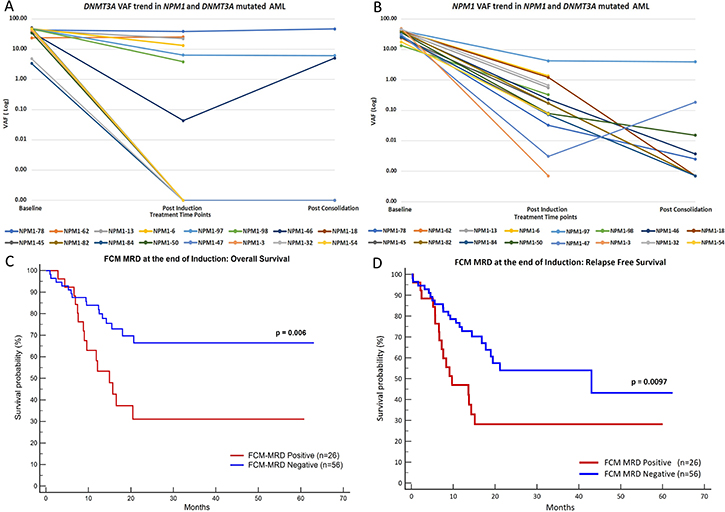
Figure 3: Plot (A) and (B) represent the trends of VAF during treatment for DNMT3A and NPM1 in dual mutated patients. Plots (C) and (D) show Kaplan-Meier graphs of FCM-MRD, depicting that FCM-MRD at the end of induction is predictive of inferior OS (C) and RFS (D).
Clinical relevance of FCM-MRD
At the end of induction, FCM-MRD was detected in 26 out of 82 (31.7%) patients at levels ranging from 0.02 to 6.32% (median: 0.7%). Similarly, at end of consolidation FCM-MRD was detected in 8 out of 40 (20%) patients at levels ranging from 0.01 to 1.8% (median: 0.05%). The presence of FCM-MRD at the end of induction was highly predictive of inferior OS as well as and RFS (Figure 3). However, FCM-MRD measured at the PC time point did not influence either OS or RFS as can be seen by results of univariate analysis in Table 1.
Comparison of MRD results obtained by NGS (VAF) and FCM-MRD values
A total 82 cases at the PI timepoint and 40 at PC timepoint were analyzed for MRD using FCM and NGS. A comparison of these results can be seen in Supplementary Figure 3. MRD values measured using these two modalities correlated poorly (r = 0.1685, p = 0.07).
Multivariate analysis
On a multivariate Cox proportional hazards regression analysis, the presence of NGS-MRD was an independent factor predictive of inferior OS and RFS. In fact, NGS-MRD and PI FCM-MRD were the only significant prognostic factors for relapse and death (Table 1). NGS-MRD had a higher hazard ratio as compared to FCM-MRD.
DISCUSSION
NPM1 gene mutations are the largest subset of molecular alterations seen in adult AML. It is therefore imperative that we develop effective strategies for monitoring MRD. Most of the molecular techniques that have been published for MRD detection in NPM1mut MRD are based on quantitative measurement of NPM1-mutant transcripts [10, 17–20] or DNA based RQ-PCR for the detection of mutant NPM1 alleles [2, 17, 21]. Recently, droplet digital PCR has also been used as tool to measure MRD in NPM1mut AML [12, 22]. The importance of sequential monitoring of NPM1 mutations has been described by a few cohorts [10, 20, 21]. These papers have established the utility of RNA based MRD detection in NPM1mut AML. In fact, Ivey and colleagues indicated that real time PCR based NPM1 MRD status was the only important predictor of relapse or death [10]. They conclusively showed that NPM1 mutations are a reliable marker for monitoring disease progression. There are very few studies that have evaluated the utility of molecular NPM1-MRD in comparison with immunophenotypic MRD. Here, we demonstrate that at the early time-points of therapy (PI) FCM-MRD is important in prediction of relapse. As seen in Supplementary Table 4, FCM-MRD positive patients had a shorter OS and RFS as compared to FCM-MRD negative patients.
DNA based NGS assays overcome many of the obstacles of real time PCR as they do not need patient specific sequence information prior to monitoring. In our dataset, even though majority of the mutations were of type A, B and D, we detected nine uncommon NPM1 mutation subtypes (Figure 1, Supplementary Table 2). These patients could have been missed by RQ-PCR assays that target common types of NPM1 mutations (type A, B and D). Our dataset shows the importance of a generalized approach to the detection of MRD in NPM1mut AML. The fact that we could identify and monitor rare NPM1 mutations using this technique validates our approach. There have been reports of switch of NPM1 mutations at relapse [16]. Although, we did not find any evidence of switch in the NPM1 mutation type either at post induction or post consolidation time points, an NGS based approach provides a definitive advantage over mutation specific testing in potentially detecting a NPM1 clonal switch. There have been contradictory observations in the literature regarding the prognosis of uncommon NPM1 mutation as against the common subtypes [23, 24]. In our cohort, we did not find significant difference between common and uncommon subtypes.
Recent data has shown that NGS based MRD assays are potentially more sensitive than flow cytometry and offer comparable sensitivity as RQ-PCR [13, 25–28]. Based on a 1-log change in VAF between PI and PC time points, patients were categorised into NGS-MRD negative and NGS-MRD positive. Failure to achieve a 1-log reduction at PC time point predicted shorter relapse free and overall survival. Thus, this criterion can be used prospectively in determination of MRD in NPM1mut AML patients. As seen in Supplementary Table 4, NGS-MRD positive patients had worse OS and RFS as compared to FCM-MRD positive patients.
In our cohort, significant number of patients who were FCM-MRD-negative had detectable MRD levels using deep sequencing. This resulted in a poor correlation between FCM-MRD and NGS-MRD VAF (Supplementary Figure 3). Unlike precursor B lineage acute lymphoblastic leukaemia which has a high frequency of leukemia associated immunophenotype (LAIP), AML MRD detection by flow cytometry is inherently complex. This is because FCM-MRD detection in AML is conceptually different from B or T-ALL because of lack of a common identifier for abnormal myeloid blasts [29, 30]. An inherent problem with FCM-MRD for AML is inability to identify leukemic populations below the threshold of 0.1% with a high level of confidence [31]. Furthermore, FCM-MRD detection in AML is difficult when the leukemia associated immunophenotype (LAIP) is expressed only by the subpopulation of leukemic cells, as most of the leukemic cells have similar immunophenotype compared to normal population. LAIP may change during the treatment, thereby making initial MRD markers irrelevant in subsequent time points [32–34]. It has been proven that deep sequencing based MRD has a higher sensitivity as compared to FCM-MRD in NPM1mut AML [13, 35]. In our study NGS MRD had a one log higher sensitivity in dilution experiments. Recently, Malmberg et al. demonstrated a consistent lower estimation of leukemic cell burden using FCM-MRD compared to targeted deep sequencing. MRD was even detected using targeted deep sequencing in cases where FCM-MRD was below the level of LOD [36]. These reasons may be an explanation for poor correlation between FCM and NGS based MRD observed in our study.
We saw agreement with previous studies that NPM1 mutations often co-occur with DNMT3A mutations and portend a poor prognosis [37–39]. Furthermore, we detected persistence of these DNMT3A mutations in 7 post treatment samples even when there was significant reduction in the NPM1 mutant levels. These are likely to be associated with age related clonal haematopoiesis [40, 41]. It has been reported that the presence of FLT3-ITD and high allelic ratio of FLT3-ITD is a poor prognostic factor in NPM1mut AML [4, 42]. However we could not confirm these findings in our patients.
We found a reasonable correlation between paired bone marrow and peripheral blood NPM1 mutant VAF in 19 samples (see Supplementary Methods for equivalence studies, Supplementary Figure 4). It has previously been demonstrated that monitoring of NPM1 mutant levels can be done using peripheral blood or plasma [10, 20, 43, 44]. A blood sample can be obtained easily as opposed to a painful bone marrow aspiration, however, there is a risk of missing very low level mutations in peripheral blood samples [45]. Such a recommendation has also been made by ELN MRD working party [46]. Nonetheless, to negate a bias from inclusion of blood samples, we evaluated the prognostic relevance of sequential monitoring of NPM1 NGS-MRD in only bone marrow samples (Supplementary Methods, Supplementary Figure 5). Thus, we can conclude that, patients can be sequentially monitored using bone marrow as well as blood during post treatment time points for NPM1 NGS MRD.
Additional somatic mutations that are known to occur in NPM1 mutated AML could not be monitored using this assay [10, 24]. As this assay focusses only on the NPM1 mutation for calculating AML MRD, even though rare; clonal evolution leading to relapse because of the non-NPM1 mutations could not be detected [44, 47]. Another potential limitation of the original assay [25] was the possibility of barcode cross contamination leading to a misattribution of the samples contaminating the library and thereby obtaining inaccurate results. To avoid this problem we incorporated a dual indexing strategy [48]. The latter is used routinely in sample multiplexing and allows detection of rare mutations in multiplexed samples with minimal errors.
To summarize, this is one of the few papers that has evaluated the clinical relevance of NGS MRD in NPM1mut AML. Shayegi et al. used the IonTorrent PGM chemistry to deep sequence NPM1 gene for mutations in a small subgroup of 10 patients using genomic DNA as a template [49]. They concluded that an RNA expression value of 1% NPM1 mutant/ABL corresponds to a value of 0.016% NPM1 mutant alleles on genomic DNA (per NPM1 wild-type alleles). However, we could not confirm the clinical relevance of using such a cut-off in our dataset at either PI or PC time points. The results of Shayegi and colleagues as well as our results need to be confirmed by larger cohorts for incorporation into treatment algorithms.
MATERIALS AND METHODS
Patients
A total of 83 NPM1mut AML were accrued into the study after informed consent from October 2012 to May 2017 (after exclusion of patients with induction deaths and refractory disease). Patients were diagnosed as per current WHO 2016 criteria. NPM1 mutations were detected initially using fragment length analysis [50] and subsequently characterized using the NGS assay (see below). These patients were treated with standard 3 + 7 induction followed by 3 cycles of high dose cytarabine (12–18g/m2) and followed up till January 2018. FCM-MRD was assessed from the bone marrow at the end of induction (PI) (BM, n = 82) and at the end of first consolidation (PC, n = 40). NGS-MRD was assessed from 82 PI samples (all BM) and 55 PC samples, of which 13 samples were sourced from the peripheral blood (PB). In total, there were 54 paired samples between PI and PC time points. (Supplementary Figure 6). FLT3-ITD was detected on the diagnostic sample by fragment length analysis [50].
Detection of MRD using NGS in NPM1mut AML
We adopted a strategy as described by Salipante et al. with minor modifications as detailed below [25]. Briefly the assay incorporated a one-step strategy wherein, Illumina adapter linked locus specific primers were designed for amplification of exon 12 of NPM1 (Integrated DNA Technologies (Coralville, IA, USA)). To prevent contamination from other sample indices, we used a dual indexing strategy with 10 bp sample specific index barcode in both forward and reverse primers. The primer sequences are as listed in Supplementary Table 5.
Assay setup
Assay setup was as follows: For a total reaction volume of 75 μl, 37.5 μl of NEBNext High-Fidelity 2X PCR Master Mix (New England BioLabs Inc., Massachusetts, USA), and 10 uM each forward and reverse primers were used to amplify 600ng of genomic DNA (approximately 1,00,000 genomic cell equivalents). PCR cycling conditions were as follows: Initial denaturation of 95°C for 15 minutes; then 35 cycles of denaturation at 94°C for 1 minute, annealing at 65°C for 1 minute, and extension at 72°C for 1 minute; followed by extension cycle of 72°C for 45 seconds. PCR products were size selected using Agencourt AMPure XP beads (Beckman Coulter Inc., California, USA) and quantified using Qubit dsDNA HS assay (Thermo Fisher Scientific, Massachusetts, USA). Samples were deep sequenced after pooling in equimolar concentration on an Illumina MiSeq (Illumina, San Diego, CA, USA) next generation sequencer using a 150 bp paired end V2 chemistry.
Data analysis
Runs were demultiplexed using the MiSeq onboard software. Paired-end reads were self-assembled using PANDAseq [51]. Self-assembled reads were mapped to the human genome (GrCh37) using bwa v0.7.12 [52]. Samtools v0.1.19 [53] was used to sort the bam file and create a mpileup file. Variant indels were called using VarScan v2.3.7 [54]. Finally, once the .vcf file was generated, it was annotated using an internal database and custom scripts. For additional redundancy, we also introduced a synthetic control in each sequencing dataset. This ‘bioinformatics control’ with 20 bp insertion served as mechanism to ensure that the pipeline worked as expected. To make a variant call, a minimum of two mutant reads had to be obtained.
NPM1 NGS-MRD assay validation
We increased the analytical sensitivity of the original NGS-MRD assay (as described by Salipante) to detect NPM1 mutations at a high sensitivity of 0.001%. A limit of detection experiment demonstrated that the assay could detect the NPM1 mutation at 1:100,000 frequency. To ensure that the assay did not make false NPM1 insertion calls we performed a limit of blank experiment on 30 normal DNA samples. The details pertaining to these experiments can be seen in Supplementary Methods.
Controls
To account for systemic drift, we assayed two (high and low level) precision controls (OCI-AML3 cell line diluted in normal DNA at 0.2 and 0.02% VAF respectively) in every run. The results of these experiments can be seen in Supplementary Methods. In addition, we also used a NA12878 control DNA as a negative MRD control.
Calculation and reporting of NGS-MRD
NPM1 variant allele frequencies obtained after ultradeep sequencing at PI and PC time points were used to calculate log reduction values. (Supplementary Methods) ROC curves of log reduction values were used to calculate the cut off value that provided the optimal classification of patients in terms of overall survival with the help of Youden’s index. We determined that a 1-log reduction represented the optimal sensitivity and specificity with the corresponding Youden’s index of 0.42. We used a 1-log reduction between PI and PC time points to classify patients as NGS-MRD positive (<1-log reduction) or negative (>1-log reduction). Cases in which PI and PC values were both zero were noted as NGS-MRD negative.
Assessment of DNMT3A mutations in NPM1mut AML
We designed 61 single molecule molecular inversion probes (smMIPS) to construct a library that spanned the entire DNMT3A coding region. These libraries were balanced to ensure similar capture efficiencies of targeted regions as described previously [55]. Sequencing was done on an Illumina MiSeq (Illumina, San Diego, CA, USA) using the V2-300 cycle chemistry. Data was analysed using a custom pipeline that incorporates adapter trimming using ea-utils [56], read self-assembly using PEAR [57], alignment to the human genome (build hg19) using bwa (v0.7.12) [52], pre-processing of aligned files using samtools (v. 0.1.19) [53] & GATK v3.8 [58]. Variant calling was done using Varscan (v. 2.3) [54], Mutect 2.0 [58], and Platypus v0.81 [59]. Using annovar [60] the variants were annotated with population frequency databases as well as the COSMIC (v.84) [61] database.
DNA based RQ-PCR assay for Type-A NPM1mut AML
DNA extracted from OCI-AML3 cell lines carrying Type-A NPM1 mutation were subjected to PCR to amplify exon 12 of NPM1. PCR products were purified and cloned into a pJET1.2/blunt cloning vector (ThermoFisher Scientific, Foster City, CA, USA) following the manufacturer’s instructions. Plasmids carrying mutant and wild type NPM1 were confirmed by sanger sequencing, linearized and used to generate standard curves in further RQ-PCR experiments. MRD detection for type-A NPM1 mutations using DNA as a template was done using a previously published protocol on a LightCycler 96 (Roche, Basel, Switzerland) [21].
Detection of MRD using FCM (FCM-MRD) in NPM1mut AML
Patients accrued from March 2012 to February 2015 (25 cases) had been acquired on an 8 colour BD FACSCanto II or two 10 colour BC Navios instruments using a three tube 8 colour MRD assay as seen in Supplementary Table 6. Subsequently diagnostic and follow up samples of 57 patients were acquired on two 10 colour BC Navios instruments using a two tube 10 colour MRD assay as seen in Supplementary Table 7. An identical panel was used for diagnostic sample, post induction and post consolidation time points. More than 500,000 events were acquired per tube with the 3-tube assay and 1.6 million events per tube obtained per tube with the 2 tubes, 10 colour assays. Kaluza software (v1.2) was used to analyse the. fcs/. lmd files. Boolean gating was used to focus on progenitors, monocytes while excluding lymphocytes and hematogones [62]. The focus of analysis was to study progenitor cells as they matured into monocytes and myeloid cells. MRD was calculated as a percentage of abnormal leukemic cells per total nucleated cells. Normal templates were periodically updated (once in 2 weeks). Additional experiments pertaining to the FCM-MRD assay (limit of dilution and example of approach to FCM-MRD detection) can be seen in Supplementary Methods.
Survival analysis
Overall survival (OS) was defined as time from start of induction therapy to time of last follow up or death. Relapse free survival (RFS) was calculated from date of achievement of remission to the date of relapse, if not, death from any cause. Patient who did not relapse and were alive were censored on the last follow up date [63]. Results of the FCM-MRD and NGS-MRD assays were analysed for their impact on OS and DFS using the Kaplan-Meier technique and compared using log-rank test. Cox proportional-hazards regression was used to calculate hazard ratios assessing the prognosis of different variables with univariate and multivariate analysis. MedCalc Statistical Software version 14.8.1 (MedCalc Software, Ostend, Belgium) and SPSS (IBM Corp. Released 2015. IBM SPSS Statistics for Windows, Version 23.0. Armonk, NY: IBM Corp.) were used to perform the statistical analysis.
CONCLUSIONS
We have evaluated the clinical utility of an ultrasensitive NGS assay for sequential monitoring of NPM1 mutation. This assay has a prognostic impact and can be used in the prospective monitoring of NPM1mut AMLs. It has advantages as compared to FCM-MRD in terms of a lower cost of testing, stability of DNA for shipped samples, higher sensitivity and easier interpretation. In addition, it is comparable to established RQ-PCR assay based MRD results with definite advantages. We feel that in view of the high sensitivity of the NGS assay, monitoring of NPM1 MRD from the peripheral blood is feasible.
Author contributions
NP and RK designed the study, performed the research, analysed the data and wrote the manuscript. RK, CK, GR, PB, SJ, SC, YB, SG, SHK performed flow and NGS experiments. SK aided in statistics, DS performed cytogenetics. SK, AG, SP, HJ, BB, HM, MS, NK aided in recruitment of patients. PGS and SG supervised the research.
ACKNOWLEDGMENTS
We are grateful for the training imparted to Dr Nikhil Patkar in the field genome sequencing technologies by Dr David Wu, Dr Stephen Salipante and Dr Brent Wood. We would like to acknowledge the mentorship of Nikhil Patkar by Dr David Wu, Dr Stephen Salipante and Dr Brent Wood in the Department of Laboratory Medicine, University of Washington, USA. We acknowledge Dr Wu for critically reviewing this manuscript and providing thoughtful insights.
CONFLICTS OF INTEREST
The authors have no commercial or other relationships that could contribute a conflict of interest.
FUNDING
This work was supported by the Wellcome Trust/DBT India Alliance Fellowship [grant number IA/CPHI/14/1/501485] awarded to Dr Nikhil Patkar. Initial part of the study (8 colour FCM) was funded by a Lady Tata Memorial Trust Grant awarded to Prof Navin Khattry.
REFERENCES
1. Howlader N, Noone A, Krapcho M, Miller D, Bishop K, Kosary C, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis D, Chen H, Feuer E, et al. SEER Cancer Statistics Review, 1975–2014. Natl Cancer Inst. 2017; 1992–2014. Available from https://seer.cancer.gov/csr/1975_2014/results_merged/sect_13_leukemia.pdf.
2. Dvorakova D, Racil Z, Jeziskova I, Palasek I, Protivankova M, Lengerova M, Razga F, Mayer J. Monitoring of minimal residual disease in acute myeloid leukemia with frequent and rare patient-specific NPM1 mutations. Am J Hematol. 2010; 85:926–29. https://doi.org/10.1002/ajh.21879.
3. Freeman SD, Virgo P, Couzens S, Grimwade D, Russell N, Hills RK, Burnett AK. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol. 2013; 31:4123–31. https://doi.org/10.1200/JCO.2013.49.1753.
4. Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, Linch DC, and Medical Research Council Adult Leukaemia Working Party. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008; 111:2776–84. https://doi.org/10.1182/blood-2007-08-109090.
5. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–405. https://doi.org/10.1182/blood-2016-03-643544.
6. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, et al, and GIMEMA Acute Leukemia Working Party. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005; 352:254–66. https://doi.org/10.1056/NEJMoa041974.
7. Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, Ehninger G. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006; 107:4011–20. https://doi.org/10.1182/blood-2005-08-3167.
8. Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, Haferlach T. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011; 117:1109–20. https://doi.org/10.1182/blood-2010-08-299990.
9. Getta BM, Devlin SM, Levine RL, Arcila ME, Mohanty AS, Zehir A, Tallman MS, Giralt SA, Roshal M. Multicolor Flow Cytometry and Multigene Next-Generation Sequencing Are Complementary and Highly Predictive for Relapse in Acute Myeloid Leukemia after Allogeneic Transplantation. Biol Blood Marrow Transplant. 2017; 23:1064–71. https://doi.org/10.1016/j.bbmt.2017.03.017.
10. Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, Patel Y, Bhudia N, Farah H, Mason J, Wall K, Akiki S, Griffiths M, et al, and UK National Cancer Research Institute AML Working Group. Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med. 2016; 374:422–33. https://doi.org/10.1056/NEJMoa1507471.
11. Krönke J, Schlenk RF, Jensen KO, Tschürtz F, Corbacioglu A, Gaidzik VI, Paschka P, Onken S, Eiwen K, Habdank M, Späth D, Lübbert M, Wattad M, et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol. 2011; 29:2709–16. https://doi.org/10.1200/JCO.2011.35.0371.
12. Mencia-Trinchant N, Hu Y, Alas MA, Ali F, Wouters BJ, Lee S, Ritchie EK, Desai P, Guzman ML, Roboz GJ, Hassane DC. Minimal Residual Disease Monitoring of Acute Myeloid Leukemia by Massively Multiplex Digital PCR in Patients with NPM1 Mutations. J Mol Diagn. 2017; 19:537–48. https://doi.org/10.1016/j.jmoldx.2017.03.005.
13. Waalkes A, Penewit K, Wood BL, Wu D, Salipante SJ. Ultrasensitive detection of acute myeloid leukemia minimal residual disease using single molecule molecular inversion probes. Haematologica. 2017; 102:1549–57. https://doi.org/10.3324/haematol.2017.169136.
14. Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, Erpelinck-Verschueren CA, Gradowska PL, Meijer R, Cloos J, Biemond BJ, Graux C, van Marwijk Kooy M, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med. 2018; 378:1189–99. https://doi.org/10.1056/NEJMoa1716863.
15. Young AL, Wong TN, Hughes AE, Heath SE, Ley TJ, Link DC, Druley TE. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia. 2015; 29:1608–11. https://doi.org/10.1038/leu.2015.17.
16. Webersinke G, Kranewitter W, Deutschbauer S, Zach O, Hasenschwandtner S, Wiesinger K, Erdel M, Marschon R, Böhm A, Tschurtschenthaler G. Switch of the mutation type of the NPM1 gene in acute myeloid leukemia (AML): relapse or secondary AML? Blood Cancer J. 2014; 4:e221. https://doi.org/10.1038/bcj.2014.42.
17. Gorello P, Cazzaniga G, Alberti F, Dell’Oro MG, Gottardi E, Specchia G, Roti G, Rosati R, Martelli MF, Diverio D, Lo Coco F, Biondi A, Saglio G, et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006; 20:1103–08. https://doi.org/10.1038/sj.leu.2404149.
18. Bacher U, Badbaran A, Fehse B, Zabelina T, Zander AR, Kröger N. Quantitative monitoring of NPM1 mutations provides a valid minimal residual disease parameter following allogeneic stem cell transplantation. Exp Hematol. 2009; 37:135–42. https://doi.org/10.1016/j.exphem.2008.09.014.
19. Schnittger S, Kern W, Tschulik C, Weiss T, Dicker F, Falini B, Haferlach C, Haferlach T. Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood. 2009; 114:2220–31. https://doi.org/10.1182/blood-2009-03-213389.
20. Balsat M, Renneville A, Thomas X, de Botton S, Caillot D, Marceau A, Lemasle E, Marolleau JP, Nibourel O, Berthon C, Raffoux E, Pigneux A, Rodriguez C, et al. Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with NPM1 mutation: A study by the acute leukemia French association group. J Clin Oncol. 2017; 35:185–93. https://doi.org/10.1200/JCO.2016.67.1875.
21. Chou WC, Tang JL, Wu SJ, Tsay W, Yao M, Huang SY, Huang KC, Chen CY, Huang CF, Tien HF. Clinical implications of minimal residual disease monitoring by quantitative polymerase chain reaction in acute myeloid leukemia patients bearing nucleophosmin (NPM1) mutations. Leukemia. 2007; 21:998–1004. https://doi.org/10.1038/sj.leu.2404637.
22. Bacher U, Dicker F, Haferlach C, Alpermann T, Rose D, Kern W, Haferlach T, Schnittger S. Quantification of rare NPM1 mutation subtypes by digital PCR. Br J Haematol. 2014; 167:710–14. https://doi.org/10.1111/bjh.13038.
23. Pastore F, Greif PA, Schneider S, Ksienzyk B, Mellert G, Zellmeier E, Braess J, Sauerland CM, Heinecke A, Krug U, Berdel WE, Buechner T, Woermann B, et al. The NPM1 mutation type has no impact on survival in cytogenetically normal AML. PLoS One. 2014; 9:e109759. https://doi.org/10.1371/journal.pone.0109759.
24. Alpermann T, Schnittger S, Eder C, Dicker F, Meggendorfer M, Kern W, Schmid C, Aul C, Staib P, Wendtner CM, Schmitz N, Haferlach C, Haferlach T. Molecular subtypes of NPM1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica. 2016; 101:e55–58. https://doi.org/10.3324/haematol.2015.133819.
25. Salipante SJ, Fromm JR, Shendure J, Wood BL, Wu D. Detection of minimal residual disease in NPM1-mutated acute myeloid leukemia by next-generation sequencing. Mod Pathol. 2014; 27:1438–46. https://doi.org/10.1038/modpathol.2014.57.
26. Wu D, Sherwood A, Fromm JR, Winter SS, Dunsmore KP, Loh ML, Greisman HA, Sabath DE, Wood BL, Robins H. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012; 4:134ra63. https://doi.org/10.1126/scitranslmed.3003656.
27. Wood B, Wu D, Crossley B, Dai Y, Williamson D, Gawad C, Borowitz MJ, Devidas M, Maloney KW, Larsen E, Winick N, Raetz E, Carroll WL, et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood. 2018; 131:1350–59. https://doi.org/10.1182/blood-2017-09-806521.
28. Faham M, Zheng J, Moorhead M, Carlton VE, Stow P, Coustan-Smith E, Pui CH, Campana D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012; 120:5173–80. https://doi.org/10.1182/blood-2012-07-444042.
29. Wood BL. Hematological Malignancies. Czader M, editor. Methods in Molecular Biology (Methods and Protocols). 2013; p.123–136. https://doi.org/10.1007/978-1-62703-357-2.
30. Soma L, Wood B. Minimal residual disease testing in acute leukemia. Int J Hematol Oncol. 2013; 2:467–85. https://doi.org/10.2217/ijh.13.57.
31. Walter RB, Buckley SA, Pagel JM, Wood BL, Storer BE, Sandmaier BM, Fang M, Gyurkocza B, Delaney C, Radich JP, Estey EH, Appelbaum FR. Significance of minimal residual disease before myeloablative allogeneic hematopoietic cell transplantation for AML in first and second complete remission. Blood. 2013; 122:1813–21. https://doi.org/10.1182/blood-2013-06-506725.
32. Feller N, van der Pol MA, van Stijn A, Weijers GW, Westra AH, Evertse BW, Ossenkoppele GJ, Schuurhuis GJ. MRD parameters using immunophenotypic detection methods are highly reliable in predicting survival in acute myeloid leukaemia. Leukemia. 2004; 18:1380–90. https://doi.org/10.1038/sj.leu.2403405.
33. Baer MR, Stewart CC, Dodge RK, Leget G, Sulé N, Mrózek K, Schiffer CA, Powell BL, Kolitz JE, Moore JO, Stone RM, Davey FR, Carroll AJ, et al. High frequency of immunophenotype changes in acute myeloid leukemia at relapse: implications for residual disease detection (Cancer and Leukemia Group B Study 8361). Blood. 2001; 97:3574–80. https://doi.org/10.1182/blood.V97.11.3574.
34. Kern W, Voskova D, Schoch C, Hiddemann W, Schnittger S, Haferlach T. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood. 2004; 104:3078–85. https://doi.org/10.1182/blood-2004-03-1036.
35. Delsing Malmberg E, Johansson Alm S, Nicklasson M, Lazarevic V, Ståhlman S, Samuelsson T, Lenhoff S, Asp J, Ehinger M, Palmqvist L, Brune M, Fogelstrand L. Minimal residual disease assessed with deep sequencing of NPM1 mutations predicts relapse after allogeneic stem cell transplant in AML. Leuk Lymphoma. 2018; 0:1–9. https://doi.org/10.1080/10428194.2018.1485910.
36. Malmberg EB, Ståhlman S, Rehammar A, Samuelsson T, Alm SJ, Kristiansson E, Abrahamsson J, Garelius H, Pettersson L, Ehinger M, Palmqvist L, Fogelstrand L. Patient-tailored analysis of minimal residual disease in acute myeloid leukemia using next-generation sequencing. Eur J Haematol. 2017; 98:26–37. https://doi.org/10.1111/ejh.12780.
37. Gale RE, Lamb K, Allen C, El-Sharkawi D, Stowe C, Jenkinson S, Tinsley S, Dickson G, Burnett AK, Hills RK, Linch DC. Simpson’s paradox and the impact of different DNMT3A mutations on outcome in younger adults with acute myeloid leukemia. J Clin Oncol. 2015; 33:2072–83. https://doi.org/10.1200/JCO.2014.59.2022.
38. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363:2424–33. https://doi.org/10.1056/NEJMoa1005143.
39. Peterlin P, Renneville A, Ben Abdelali R, Nibourel O, Thomas X, Pautas C, de Botton S, Raffoux E, Cayuela JM, Boissel N, Terré C, Celli-Lebras K, Castaigne S, et al. Impact of additional genetic alterations on the outcome of patients with NPM1-mutated cytogenetically normal acute myeloid leukemia. Haematologica. 2015; 100:e196–99. https://doi.org/10.3324/haematol.2014.115576.
40. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med. 2014; 371:2477–2487. https://doi.org/10.1056/NEJMoa1409405.
41. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371:2488–98. https://doi.org/10.1056/NEJMoa1408617.
42. Schnittger S, Bacher U, Kern W, Alpermann T, Haferlach C, Haferlach T. Prognostic impact of FLT3-ITD load in NPM1 mutated acute myeloid leukemia. Leukemia. 2011; 25:1297–304. https://doi.org/10.1038/leu.2011.97.
43. Ma W, Kantarjian H, Zhang X, Jilani I, Sheikholeslami MR, Donahue AC, Ravandi F, Estey E, O’Brien S, Keating M, Giles FJ, Albitar M. Detection of nucleophosmin gene mutations in plasma from patients with acute myeloid leukemia: clinical significance and implications. Cancer Biomark. 2009; 5:51–58. https://doi.org/10.3233/CBM-2009-0583.
44. Papadaki C, Dufour A, Seibl M, Schneider S, Bohlander SK, Zellmeier E, Mellert G, Hiddemann W, Spiekermann K. Monitoring minimal residual disease in acute myeloid leukaemia with NPM1 mutations by quantitative PCR: clonal evolution is a limiting factor. Br J Haematol. 2009; 144:517–23. https://doi.org/10.1111/j.1365-2141.2008.07488.x.
45. Stahl T, Badbaran A, Kröger N, Klyuchnikov E, Zabelina T, Zeschke S, Schafhausen P, Schultz W, Asenova S, Smirnova A, Wolschke C, Ayuk F, Zander AR, et al. Minimal residual disease diagnostics in patients with acute myeloid leukemia in the post-transplant period: comparison of peripheral blood and bone marrow analysis. Leuk Lymphoma. 2010; 51:1837–43. https://doi.org/10.3109/10428194.2010.508822.
46. Schuurhuis GJ, Heuser M, Freeman S, Béné MC, Buccisano F, Cloos J, Grimwade D, Haferlach T, Hills RK, Hourigan CS, Jorgensen JL, Kern W, Lacombe F, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018; 131:1275–91. https://doi.org/10.1182/blood-2017-09-801498.
47. Krönke J, Bullinger L, Teleanu V, Tschürtz F, Gaidzik VI, Kühn MW, Rücker FG, Holzmann K, Paschka P, Kapp-Schwörer S, Späth D, Kindler T, Schittenhelm M, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013; 122:100–08. https://doi.org/10.1182/blood-2013-01-479188.
48. Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012; 40:e3. https://doi.org/10.1093/nar/gkr771.
49. Shayegi N, Kramer M, Bornhäuser M, Schaich M, Schetelig J, Platzbecker U, Röllig C, Heiderich C, Landt O, Ehninger G, Thiede C, and Study Alliance Leukemia (SAL). The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood. 2013; 122:83–92. https://doi.org/10.1182/blood-2012-10-461749.
50. Huang Q, Chen W, Gaal KK, Slovak ML, Stein A, Weiss LM. A rapid, one step assay for simultaneous detection of FLT3/ITD and NPM1 mutations in AML with normal cytogenetics. Br J Haematol. 2008; 142:489–92. https://doi.org/10.1111/j.1365-2141.2008.07205.x.
51. Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012; 13:31. https://doi.org/10.1186/1471-2105-13-31.
52. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
53. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–79. https://doi.org/10.1093/bioinformatics/btp352.
54. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–76. https://doi.org/10.1101/gr.129684.111.
55. Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013; 23:843–54. https://doi.org/10.1101/gr.147686.112.
56. Aronesty E. Command-line tools for processing biological sequencing data. 2011. Available from https://github.com/ExpressionAnalysis/ea-utils.
57. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014; 30:614–20. https://doi.org/10.1093/bioinformatics/btt593.
58. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303. https://doi.org/10.1101/gr.107524.110.
59. Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Wilkie AO, McVean G, Lunter G, and WGS500 Consortium. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014; 46:912–18. https://doi.org/10.1038/ng.3036.
60. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. https://doi.org/10.1093/nar/gkq603.
61. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008; Chapter 10:11. https://doi.org/10.1002/0471142905.hg1011s57.
62. Loken MR, Alonzo TA, Pardo L, Gerbing RB, Raimondi SC, Hirsch BA, Ho PA, Franklin J, Cooper TM, Gamis AS, Meshinchi S. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood. 2012; 120:1581–88. https://doi.org/10.1182/blood-2012-02-408336.
63. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129:424–47. https://doi.org/10.1182/blood-2016-08-733196.