
INTRODUCTION
In most metastatic carcinomas, the lymph nodes are the first distant organs to be affected. [1]. Approximately half of the 246,000 annual U.S. cases of breast cancer involve women with nodal metastasis (NM) upon diagnosis. Of those without NM at diagnosis, another half will develop distant recurrence and/or relapse [2]. The presence of NM is also a concern in therapeutic and surgical decision making [3]. Yet, in the understanding of metastatic behavior, many of the molecular and genomic changes that occur during the transmission of primary tumor cells to distant sites of propagation remain undiscovered. Metastasis-focused research generally pairs primary-to-distant tumor samples or conflates metastasis, relapse, and death as one outcome in time-to-event studies. We propose that using NM as an alternative endpoint for tumor propagation offers both a point of observation in the metastatic process and a novel method for discovering the genetic underpinnings of NM.
The genomic characteristics of a primary tumor hold structural and functional clues to the behavior of tumor-originating cells in distant organs. There is extensive literature of use of gene expression for the profiling of primary breast tumors in the prediction of metastasis and outcomes [4–12]. Paired primary-to-distant research suggests that the capacity for metastasis is established early in primary tumor growth [13–16] and that distant metastases (nodal and otherwise) show molecular similarities to their primary tumors in both copy number variation (CNV) and mRNA transcription [15, 17–20]. The primary tumors of relapsed NM-negative patients have a higher total CNV burden as compared to relapse-free, NM-negative patients [21]. In NM-free patients, regions of CNV gains associated with a poor prognosis are chromosomes 8 (8p11-12), 11 (11q13-14), and 20 (20q13 33). In NM-positive primary tumors, survival-relevant regions of CNV loss are at 4p, 8p, 9p, 11q, 16q, 17p, and 18p, and areas of gains are at 1q, 8q, 16p, 17q, 19p, and 20q [21–27]. Yet, the findings are limited either by small sample size or by unknown reproducibility in other populations. Our study identifies features in both CNV and transcription platforms, validates findings in a second large dataset, and examines how the two measures interact in ways that are meaningful to NM.
This study presents a validated, gene-level landscape of genomic and intergenic regions associated to NM. We characterized the genomic regions at two levels, cancer-specific CNVs and mRNA expression. Later, we characterized CNV driven changes in mRNA unique to patients with NM. The molecular and genomic features of NM-positive samples were compared to control tumor samples which were MN-negative. To reduce false positives we described this landscape using concordant CNV to mRNA and validated analysis in two large breast cancer cohorts considering potential confounders.
RESULTS
Clinical associations to NM
Clinical covariates associations to NM showed a significant relationship between tumor size and NM. In METABRIC, half of the patients without NM had a tumor size ≤20 mm. In TCGA samples, the TNBC molecular subtype was also associated with a decreased instance of NM. Similarly, in the TCGA data, a higher proportion of women with NM were pre-menopausal and slightly younger than their non-NM counterparts. Table 1 shows the association of all covariates with NM in both METABRIC and TCGA.
Table 1: Association of nodal metastasis and patient features by data source
METABRIC |
TCGA |
|||||
|---|---|---|---|---|---|---|
Nodal metastasis present |
No |
Yes |
p-value |
No |
Yes |
p-value |
(N = 389) |
(N = 383) |
(N = 293) |
(N = 357) |
|||
Race |
0.021 |
0.678 |
||||
- Asian |
1 (0.3%) |
2 (0.5%) |
20 (6.8%) |
26 (7.3%) |
||
- Black |
0 (0.0%) |
0 (0.0%) |
40 (13.7%) |
45 (12.6%) |
||
- missing |
225 (57.8%) |
179 (46.7%) |
22 (7.5%) |
36 (10.1%) |
||
- Other |
3 (0.8%) |
4 (1.0%) |
0 (0.0%) |
1 (0.3%) |
||
- White |
160 (41.1%) |
198 (51.7%) |
211 (72.0%) |
249 (69.7%) |
||
Age at diagnosis (± SD) |
59.1 ± 12.2 |
60.8 ± 14.4 |
0.088 |
58.0 ± 12.4 |
56.1 ± 13.1 |
|
Receptor subtype |
0.572 |
0.043 |
||||
- missing |
0 (0.0%) |
0 (0.0%) |
29 (9.9%) |
23 (6.4%) |
||
- HER2 |
28 (7.2%) |
34 (8.9%) |
9 (3.1%) |
21 (5.9%) |
||
- Luminal |
285 (73.3%) |
282 (73.6%) |
212 (72.4%) |
276 (77.3%) |
||
- TNBC |
76 (19.5%) |
67 (17.5%) |
43 (14.7%) |
37 (10.4%) |
||
ER status |
0.708 |
0.102 |
||||
- missing |
0 (0.0%) |
0 (0.0%) |
17 (5.8%) |
16 (4.5%) |
||
- negative |
104 (26.7%) |
108 (28.2%) |
88 (30.0%) |
84 (23.5%) |
||
- positive |
285 (73.3%) |
275 (71.8%) |
188 (64.2%) |
257 (72.0%) |
||
PR status |
0.171 |
0.324 |
||||
- missing |
0 (0.0%) |
0 (0.0%) |
19 (6.5%) |
17 (4.8%) |
||
- negative |
184 (47.3%) |
200 (52.2%) |
110 (37.5%) |
121 (33.9%) |
||
- positive |
205 (52.7%) |
183 (47.8%) |
164 (56.0%) |
219 (61.3%) |
||
HER2 status |
0.138 |
0.591 |
||||
- missing |
0 (0.0%) |
0 (0.0%) |
96 (32.8%) |
114 (31.9%) |
||
- negative |
336 (86.4%) |
316 (82.5%) |
154 (52.6%) |
180 (50.4%) |
||
- positive |
53 (13.6%) |
67 (17.5%) |
43 (14.7%) |
63 (17.6%) |
||
Menopause status |
0.579 |
0.027 |
||||
- missing |
0 (0.0%) |
1 (0.3%) |
18 (6.1%) |
39 (10.9%) |
||
- post |
293 (75.3%) |
291 (76.0%) |
213 (72.7%) |
228 (63.9%) |
||
- pre |
96 (24.7%) |
91 (23.8%) |
62 (21.2%) |
90 (25.2%) |
||
Tumor size |
<0.0001 |
<0.0001 |
||||
- T1 (<20 mm) |
230 (59.1%) |
126 (32.9%) |
115 (39.3%) |
72 (20.2%) |
||
- T2 (>20 <50mm) |
157 (40.4%) |
234 (61.1%) |
160 (54.7%) |
229 (64.1%) |
||
- T3&4 (>50 mm) |
2 (0.5%) |
23 (6.0%) |
17 (5.9%) |
56 (15.7%) |
||
AJCC Stage |
<0.0001 |
<0.0001 |
||||
- I |
251 (64.5%) |
3 (0.8%) |
115 (39.2%) |
8 (2.2%) |
||
- II |
132 (33.9%) |
311 (81.2%) |
173 (59.0%) |
203 (56.9%) |
||
- III&IV |
6 (1.5%) |
69 (18.0%) |
3 (1.0%) |
129 (36.1%) |
||
- missing |
0 (0.0%) |
0 (0.0%) |
2 (0.7%) |
17 (4.8%) |
||
Abbreviations: HER2, human epidermal growth factor receptor 2; TNBC, triple negative/basal breast subtype; ER, estrogen receptor; PR, progesterone receptor.
*Tumor size is AJCC TNM staging.
Replicated CNV regions of interest (step 1: genomic landscape for NM)
Among all participants, 450 CNV genes of interest were associated with NM (as determined with a nominal p-value α ≤ 0.05) and replicated in both METABRIC and TCGA CNV losses in 314 genes had an association with NM. Specific regions of interest (regions having a density of significantly associated CNVs) were found in large areas of chromosomes 1 (1p32-1p36, 1q21-1q24, and 1q42), 3 (3q11, 3q22-3q26), 9 (9p24), 18 (18q11-18q12), and 19 (19p13 and 19q12). Genes in 1p34 with lowest odds of NM were AKIRIN1 (ORMETABRIC = 0.27, 95% CI 0.10–0.70, ORTCGA = 0.27, 95% CI 0.1–0.59), NDUFS5 (ORMETABRIC = 0.18, 95% CI 0.06–0.53, ORTCGA = 0.28, 95% CI 0.13–0.62), and RRAGC (ORMETABRIC = 0.18, 95% CI 0.05–0.68, ORTCGA = 0.29, 95% CI 0.13–0.62), (odds ratios are presented in Supplementary Table 1). In 136 genes, per-gene copy gains in CNV measures were associated with NM. There were regions of interest in chromosomes 5 (5q33-5q35), 12 (12q21 & 12q23), 14 (14q11-14q13, 14q21-14q23), and 15 (15q13-15q14, 15q21). Genes with the highest odds of NM were located on chromosome 5 (5q33.1d; ZNF300, CCDC69, SLC36A1, and SLC36A2) and 14 (14q 11: in gene SLC7A8; 14q13: gene AKAP6; and 14q2: gene CLEC14A). See Figure 1 and Supplementary Tables 1 and 2. After FDR adjustment for multiple testing, no single gene-level CNV had a significant and replicated association with NM.
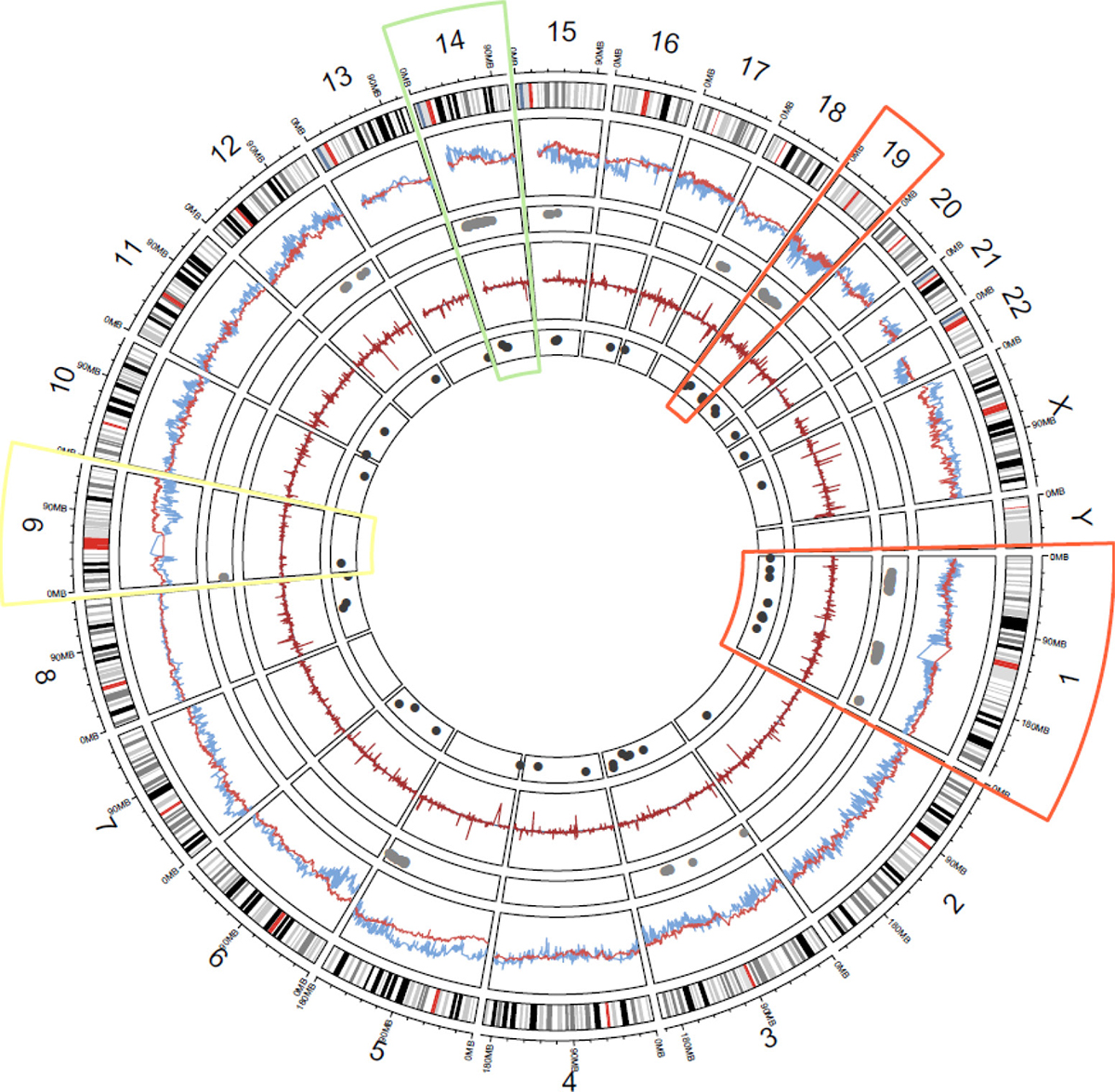
Figure 1: Genome-wide landscape of NM-associated CNV and mRNA: Layers, starting from outermost are (1) chromosome number and mega base scale; (2) ideogram of each chromosome (centromeres in red); (3) genome-wide model coefficient estimates of odds of NM for each gene (METABRIC = blue, TCGA = red); (4) replicated CNV genes associated with NM (grey dots); (5) genome-wide log-fold change between case and control mRNA (METABRIC = blue, TCGA = red); (6) replicated mRNA genes associated with NM (dark grey dots). Highlighted chromosomes bring attention to omic regions of interest (ROIs): orange = consistent CNV/RNA losses in ROIs (chromosomes 1 & 19), white = inconsistent CNV/mRNA measures in replicated ROIs (chromosome 9), green = consistent CNV/mRNA gains in replicated ROIs (chromosome 14).
Replicated significant mRNA regions of interest (step 1)
In both TCGA and METABRIC data, 48 genes overlapped with a concordant direction of association to NM. Regions of interest with replicated mRNA losses were found in 23 genes located at chromosomes 1 (1p32-1p36, 1q21, and 1q25), 3 (3q11, 3q22-3q24), 4 (4p32), 5 (5p15), 6 (6q22-6q23), 8 (8q24), 10 (10q23), 11 (11q13), 13 (13q33), 16 (16p11), 19 (19p13 and 19q13), 20 (20q11), and 22 (22q12). The largest decreases in mRNA log-fold change values in both data sets were found in CP (3q24), HS3ST5 (6q22), BAI1 (8q24), and CYP2C8 (10q23). Statistically significant gene-effect changes associated with cases of NM were found for 25 genes across datasets. Regions of interest were in chromosomes 1 (1p13, 1q23), 2 (2q31), 3 (3q26), 4 (4p14), 6 (6q24), 8 (8q21), 9 (9p21), 11 (11p15), 12 (12q24), 14 (14q12-14q13, 14q21), 15 (15q15, 15q21), 16 (16q22), 17 (17p13), 19 (19p13, 19q13), 20 (20p11), 21 (21q22), and X (Xp11). RNA transcripts with the highest log-fold change across datasets were DLX1 (2q31), TMEM156 (4p14), NOVA1 (14q12), and SHC4 (15q21). The lowest nominal p-value for log fold change was for NOVA1, 0.015 in METABRIC and 0.002 in TCGA. See Supplementary Tables 3 and 4 for details. While coding regions were not significant after FDR correction and validated across data sets, non-coding analysis in TCGA data found three regions significant at FDR 0.1; 6q24.1-6q23.3, 11q13.1, and 15q15.3 - 15q21.1.
Confirmed links between copy number and transcription (step 2: CNV to mRNA analysis)
A validated association between CNV and RNA was discovered in CTAGE5. For this gene, CNV copy gains and increased RNA were associated with NM-positive patients (Figure 2). CNV-driven loss of expression was found in the CRELD1 gene (Supplementary Table 5, Supplementary Figure 4). CNV-driven mRNA gains were present in chromosomes 8, 11, 17, and 20. CNV-based upregulation of several genes in chromosome 8 in region 8q23-24: PSCA, SLC30A8, and ZFPM2 were evident in approximately 6% of all METABRIC NM-positive patients and in 41% of all TCGA cases (Supplementary Table 6 and Supplementary Figure 5). In cases of NM, multiple genes in the chromosome 17q12-q21 region (CDC6, PSMD3, and STARD3 among them) showed a significant relationship of concordant CNV to RNA changes (Supplementary Figures 6–8). In both METABRIC and TCGA data, 20–25% of all women with NM had CNV copy gains or losses that were associated with same-direction RNA changes for this region. A similar result was found for EIF4EBP1 (8p12) and NDUFC2 (11q14) (Supplementary Table 7 and Supplementary Figures 9, 10).
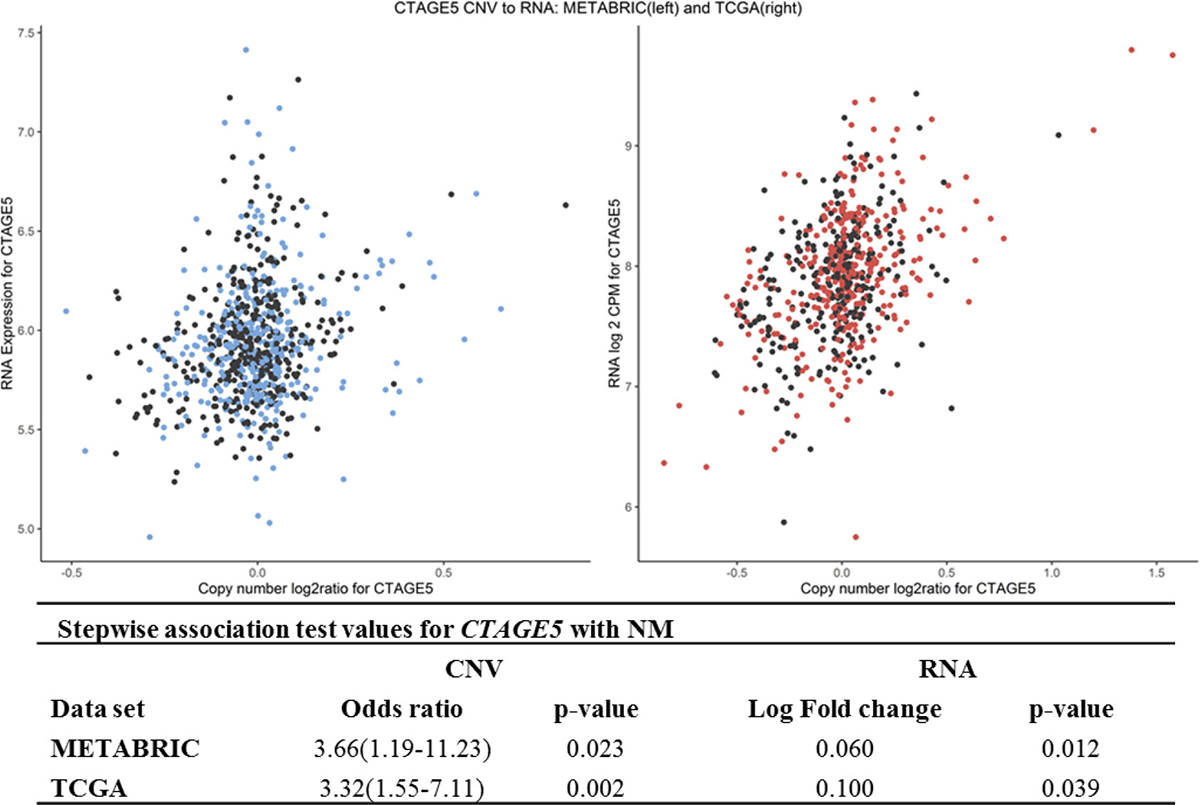
Figure 2: CNV to mRNA relationship in CTAGE5 across both datasets, including adjusted odds of NM and log-fold change between NM-positive and NM-negative samples. Black points are NM-negative; red and blue points are NM-positive.
Additional validation measures
Using only TCGA data and the results list from CNV-to-mRNA association in step 2, we performed a preliminary validation of CNV-driven results with two additional omics of protein and methylation. In general, genes with increased CNV-to-mRNA had an inverse relationship with CNV-to-methylation levels (HM450), and a positive correlation with CNV-to-protein levels. Additional omic information was available for four genes; CRELD1, EIF4EBP1, PSMD3, and STARD3. Only CRELD1 and EIF4EBP1 had significant changes in both protein and methylation which were unique to NM status. For both CRELD1 and EIF4EBP1, NM positive women had CNV-correlated protein increases (Pearson p-value of < 0.001 for both). In the same genes, methylation was inversely correlated to CNV (Pearson p-value of 0.02 and 0.02 respectively) (See Supplementary Table 8). In the top results from validated CNV-driven mRNA changes, we found the choline metabolism pathway in KEGG cell signaling (hsa05231), to be enriched in NM positive patients.
DISCUSSION
This investigation utilized TCGA and METABRIC data to identify and replicate genomic and transcriptional features in association with NM in ductal breast tumors. While we proposed NM as a proxy for metastatic behavior in general, the main objective of this paper is the creation of a descriptive landscape of omics in primary tumors with metastatic behavior. NM however is limited when considered as the outcome. While the overall objective of treatment is to achieve metastasis-free survival, rather than preventing nodal metastasis, NM has several advantages for the analysis, e.g., NM is cross-sectional and does not need follow up. Identified genes or other non-coding genomic regions will contribute to the understanding of the metastatic process. The genome-wide CNV association study revealed more than 400 gene-level areas of amplification and deletion that were replicated in both sets of data. Similar mRNA association testing gave a set of 48 gene-level transcripts consistently associated with NM status. Both sets of results were obtained without correction for multiple testing in order to be lax and reduce the number of false negatives. Genes were prioritized for discussion using the top results from statistical analysis in step 1(integrated CNV and mRNA landscape); significant genes (nominal p-value α ≤ 0.05) were then tested in step 2, CNV-to-mRNA genome-wide association testing. From these results, per-gene gains in CNV measurement had translational effects in the 8q12, 11q13-14, 14q, 20q1, and 17q14-24 regions. There were transcriptional corollaries across areas of CNV copy loss in chromosomes 1p and 1q and in 19p13. In chromosome 3 (3p25), there was a slight CNV-driven loss of transcription linked to CRELD1.
Chromosome 14 was of interest in regard to replicated CNV gains in the CTAGE5 gene (14q21). This gene is a member of the CTAGE family, and a variety of tumors, including those of the breast, express CTAGE5 exclusively [28]. The protein for this gene is involved in collagen VII transport in the endoplasmic reticulum [29]. The relationship between collagen density and tumorigenesis in mouse primary tumors and lymph nodes has been examined [30–32]. Although most research has focused on collagen I, collagen VII has been associated with in situ tumors in some cases of breast cancer [33]. An additional quality of CTAGE 5 is tumor-specific splicing [34], with an example of gene fusion in prostate cancer [35]. CTAGE5, as an antigen specific to tumors, has promise as a therapeutic target.
The gene for prostate stem cell antigen (PSCA) on chromosome 8 (8q24) had significantly associated CNV-to-mRNA expression only in cases of NM. For various tumor types, there was increased expression of this gene. For pancreatic and bladder cancers, both expression and copy number increase with metastases [36, 37]. For Asian populations, mutations in PSCA are linked to an increase in breast cancer risk, with an increased risk of NM. [38, 39] The genes NDUFC2 and EIF4EBP1 in 11q14 and 8q12 are amplified driver genes in several cancers [40–42], yet their link to NM in breast cancer is novel (Supplementary Figures 9, 10).
CNV-based genome-wide analysis of non-coding regions revealed three significant results after FDR correction. It is difficult to differentiate between protein coding and non-coding effects in these regions. In many patients the CNVs are large and involve both coding and non-coding regions. Loss of heterozygosity (LOH) at RAD51 15q14-15 loci and 6q23-24 at SASH1 loci in breast cancer has been linked to poor outcomes [43–45]. Furthermore, previous breast cancer research suggests that the consequences of CNV change on noncoding RNA seems to be less frequent than in protein coding and non-coding regions [46]. CNV-driven mRNA genes associated with NM were found to be enriched in the choline kinase pathway. Choline kinase has been observed as overexpressed in approximately 40% of breast tumors [47], and has been evaluated as a promising imaging tracer for breast tumors diagnosis [48, 49]. In Prostate cancer, choline PET/CT has been used successfully to detect recurrence and lymph node staging [50], and recent research in breast cancer has suggested a similar choline-based diagnosis strategy as promising [51, 52]. However, the relationship between choline and NM, as well as its association with CNV, presents novel topics of further research.
Relative to copy number aberrations and their mRNA consequences, we found more validation of per-gene CNV regions associated with NM than for the per-gene mRNA approach or CNV-driven mRNA analysis. This can be expected from a per-gene CNV approach, since mapping multi-gene segment units of copy number to an individual gene gives a distortion in interpreting measures of frequency. Empirically, approximates of per-gene transcription as correlated to CNV in all cancers suggest that ~60% [53] of the associated mRNA should be based in CNV. Breast cancer-specific studies of the CNV effect upon expression suggest ~12% concordance [54]. Our results show ~10% of CNV changes altered mRNA in a meaningful way for NM. Rather than predicting tumor versus normal, we examined NM within the situation of cancer. Therefore, expected proportions may not apply. It is important to note that the two steps of the analysis will not necessarily yield overlapping results, as they are geared towards different measures; The first step constructs a landscape of gene-level odds of NM for both CNV and mRNA measures while the second step identifies CNV-driven changes in mRNA unique to patients with NM. The strength of the second stage of the analysis (step 2) is built around the ability to validate gene-level, CNV-associated changes in mRNA. Differing approaches to CNV calls between sets of data may limit the effectiveness of our validation approach. A low rate of validation is expected; residual influences on validity, such as measurement errors, selection bias, and target population, would lead to spurious findings unique to both METABRIC and TCGA. Under the null hypothesis (i.e., if the gene has no effect on nodal metastasis), there will be at least 5% of false positives just from chance; however, the same false positive has a chance to be significant again only in the 0.25% of the tests. Finally, the limitation of lymph node metastasis as a proxy for overall metastatic behavior should be considered. Many cancers have alternate routes to distant metastasis, other than lymph nodes.
Our analysis found examples of CNV-driven relationships to mRNA that were unique to single sets of data, but they were not reproduced. In TCGA, the 15q21.1a region of chromosome 15 had the strongest statistical association with outcome. CNV unit increases in the gene SEMA6D had high odds of NM combined with a significant mRNA fold change in NM patients. Yet the closest significant regions of interest in METABRIC data were more than 1,000 kpb away from this result, and there was no clear link between CNV and mRNA. Large regions of CNV loss in chromosome 1 were validated across both sets of data, yet had few transcriptional correlates. Traditionally, joint analyses of copy number and expression data are used to guide internal validity through the discovery of CNV-driven mRNA effects [55–58].
MATERIALS AND METHODS
Patients and samples
Cancer genome data were obtained from two independent cohorts. The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) data (N = 772) were acquired from Synapse DREAM7 Breast Cancer Prognosis Challenge, already processed according to the METABRIC source paper [56]. A second set of data (N = 650) came from The Cancer Genome Atlas (TCGA) Data Portal [59]. TCGA level 3 data are also open-access and pre-processed. Since lobular breast cancer differs from ductal cancer in biological characteristics, indolence, and metastatic behavior, samples were exclusively from invasive ductal carcinomas [60, 61]. All patients were female, with no history of a prior malignancy or of neoadjuvant treatment. The response variable of NM was defined for both datasets using TNM pathologic staging [62] for lymph nodes (N). All TNM N values of 0 were considered controls (NM 0); N values greater than 0 were considered cases. Patient demographics and characteristics are described in Table 2 and Supplementary Figure 1.
Table 2: Patient features by data source
Features |
METABRIC |
TCGA |
|---|---|---|
(N = 772) |
(N = 650) |
|
Nodal metastasis present |
||
- No |
389 (50.4%) |
293 (45.1%) |
- Yes |
383 (49.6%) |
357 (54.9%) |
Race |
||
- Asian |
3 (0.4%) |
46 (7.1%) |
- Black |
0 (0.0%) |
85 (13.1%) |
- missing |
404 (52.3%) |
58 (8.9%) |
- Other |
7 (0.9%) |
1 (0.2%) |
- White |
358 (46.4%) |
460 (70.8%) |
Age at diagnosis (± SD) |
59.9 ± 13.4 |
56.9 ± 12.8 |
Receptor subtype |
||
- missing |
0 (0.0%) |
52 (8.0%) |
- HER2 |
62 (8.0%) |
30 (4.6%) |
- Luminal |
567 (73.4%) |
488 (75.1%) |
- TNBC |
143 (18.5%) |
80 (12.3%) |
ER status |
||
- missing |
0 (0.0%) |
33 (5.1%) |
- negative |
212 (27.5%) |
172 (26.5%) |
- positive |
560 (72.5%) |
445 (68.5%) |
PR status |
||
- missing |
0 (0.0%) |
36 (5.5%) |
- negative |
384 (49.7%) |
231 (35.5%) |
- positive |
388 (50.3%) |
383 (58.9%) |
HER2 status |
||
- missing |
0 (0.0%) |
210 (32.3%) |
- negative |
652 (84.5%) |
334 (51.4%) |
- positive |
120 (15.5%) |
106 (16.3%) |
Menopause status |
||
- missing |
1 (0.1%) |
57 (8.8%) |
- post |
584 (75.6%) |
441 (67.8%) |
- pre |
187 (24.2%) |
152 (23.4%) |
Tumor size* |
||
- T1 (<20 mm) |
356 (46.1%) |
187 (28.9%) |
- T2 (>20 <50 mm) |
391 (50.6%) |
389 (59.9%) |
- T3&4 (>50 mm) |
25 (3.2%) |
73 (11.2%) |
AJCC Stage |
||
- I |
254 (32.9%) |
123 (18.9%) |
- II |
443 (57.4%) |
376 (57.8%) |
- III&IV |
75 (9.7%) |
132 (20.3%) |
- missing |
0 (0.0%) |
19 (2.9%) |
Abbreviations: HER2, human epidermal growth factor receptor 2; TNBC, triple negative/basal breast subtype; ER, estrogen receptor; PR, progesterone receptor.
*Tumor size is AJCC TNM staging.
Copy number and transcriptome data
METABRIC and TCGA used Affymetrix Genome-Wide SNP array 6.0 to derive somatic copy number variations (CNVs). METABRIC preprocessing identified somatic CNV segments in tumors using the HMM-Dosage method [63]. A similar patient-by-gene matrix was created with TCGA data using normalized circular binary segmentation [64] files for each patient. A mean log2 ratio per segment was assigned to each genic and intergenic region within the segment according to METABRIC annotation. METABRIC used the Illumina HT-12v3 platform in gene expression analysis. Pre-processing included spatial artifact correction, summarization, and normalization of log2 intensities with bead-array and BASH R packages [65, 66]. In TCGA, normalized mRNA expression counts were derived from the TCGA Level 3 RNAseqV2 expression data. Illumina HiSeq 2000 was used to create the TCGA transcriptional data.
Association studies and omic integration
This study was performed in two steps (see workflow in Supplementary Figure 2). In the first step of the analysis, we created genome-wide landscapes of NM associated CNV and mRNA in both TCGA and METABRIC, and then integrated both CNV and mRNA results. This analysis was done for coding regions in TCGA and METABRIC and non-coding regions in TCGA. Non-coding analysis was done only in TCGA, since data was not available in METABRIC. Genome-wide, covariate-adjusted association tests were done at a gene level unit of analysis, separately evaluating the association between the levels first of CNV, and then mRNA, with the response variable yes/no NM. In total, association results were produced for five separate sets (see Supplementary Figure 3): TCGA protein coding CNV, TCGA mRNA, METABRIC protein coding CNV, METABRIC mRNA, and TCGA non-coding CNV. Equations for genome wide tests are:
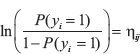
Where yi is a dummy variable representing subject’s i-th NM status (yes = 1, no = 0), and ηij is a linear predictor of the form:
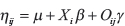
Where μ is a common intercept, Xi is the i-th row of the incidence matrix X representing different sets of covariates for each data set (METABRIC and TCGA), β is the vector of corresponding effects; Oij is the intensity of the j-th feature of the omic O in the i-th subject, and γ the corresponding effect. For METABRIC data, the columns of X consisted of grade, tumor size, age at the moment of diagnosis, and race. For TCGA data, they consisted of molecular subtype, tumor size, age at diagnosis, and race.
In the second step of the analysis we regressed a CNV to mRNA on a gene-by-gene basis [55, 58]. The analysis was conducted to examine the consequence of per-gene CNV gain/loss upon mRNA within the same sample. To identify the modifying effect of NM upon CNV related changes in expression, both datasets were stratified by NM status, and the following tests were performed with the iGC Bioconductor package [67]. Gene expression driven by CNV was identified first by grouping all per-gene CNVs as copy gains (log2 ratio ≥ 0.4), copy losses (log2 ratio ≤ −0.4), and between-threshold values as diploid/neutral (log2ratio null = 0). Thresholds for log2 ratio values were chosen at higher amplitudes than greater than or less than |0.1| of earlier approaches [68, 69] in order to better show gene-level, rather than chromosome-level, or arm-level, CNV events [70]. The variations in gene expression between CNV-gain genes and diploid normals and CNV-loss genes and diploid normals were tested with an unequal variance Student’s t-test. Filtering of results was based on the false discovery rate (FDR) adjusted p-value (α = 0.1) and consistent direction of CNV-to-RNA association. A relaxed p value threshold was selected to avoid losing genes that could be false negatives in a stringent testing by the cost of accepting more false positives. CNV-driven gene transcripts unique to NM status were found for both METABRIC and TCGA. Finally, significant genes in both datasets were then identified within each NM group.
Three additional measures of validation were used to supplement our findings. We performed an enrichment analysis on all significant CNV-driven mRNA genes in Enrichr [71]. Using only TCGA data, we checked for CNV-driven changes in the added omic measures of protein and methylation. In order to account for any non-coding CNVs of importance to our outcome, we also examined the association of non-coding regions of CNV to NM status. However this was done only in TCGA, since non-coding data is not available in METABRIC (see Statistical analysis section in Supplementary Materials).
CONCLUSIONS
In sum, we have identified, in invasive ductal beast carcinomas, CNV-based regions of interest that are associated with NM. Genes in regions 14q21, 8p12, 8q24, 11q14, and various locations on chromosome 1 and 17 may be associated with the development of NM, since the chromosome copy loss/gain happened after the development of NM, and the associated expressions of these genes were different by NM status, suggesting either a role in or a consequence of development of metastases.
Abbreviations
nM: nodal metastasis; CNV: copy number variation; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; TCGA: The Cancer Genome Atlas; kbp: kilobase pair.
Author contributions
MB and AV conceived the study. XC, SS, AG, AG, AV and MB participated in statistical approach. MB collected, processed and analyzed the data. MB, SS and AV wrote the manuscript. MB, UM, SS, and AV interpreted the data. All authors have reviewed and approved the manuscript.
ACKNOWLEDGMENTS
We thank Kyle Grimes for his edition work on this manuscript. We also thank The Cancer Genome Atlas network for data access (http://cancergenome.nih.gov/). METABRIC data were accessed through Synapse (http://synapse.sagebase.org), we thank them for making the data available.
Ethics statement
This study was approved by the UAB Institutional Review Board for Human Use, and performed in accordance with the ethical guidelines of the Declaration of Helsinki. Animals were not used in this study.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the following: METABRIC data can be downloaded from Synapse storage (https://www.synapse.org/#!Synapse:syn1688369/wiki/27311), and TCGA data is available via GDC data portal (https://portal.gdc.cancer.gov/projects/TCGA-BRCA).
CONFLICTS OF INTEREST
Dr. Manne is a member of the editorial board of this journal.
FUNDING
The Cancer Research Experiences for Students (CaRES) Program at UAB; 5R25CA76023 (Behring) and U54-CA118948 (Dr. U. Manne), funded by the National Cancer Institute; and a UAB Comprehensive Cancer Center Young Investigator Grant (Dr. Vazquez). Funding entities had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
REFERENCES
1. Sleeman J, Schmid A, Thiele W. Tumor lymphatics. Semin Cancer Biol. 2009; 19:285–97. https://doi.org/10.1016/j.semcancer.2009.05.005.
2. Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ. CK (eds). SEER Cancer Statistics Review, 1975–2011. Natl. Cancer Inst. 2013. Available from: http://seer.cancer.gov/csr/1975_2011/.
3. Hellman S, Harris JR. The appropriate breast cancer paradigm. Cancer Res. 1987; 47:339–42.
4. Ahr A, Karn T, Solbach C, Seiter T, Strebhardt K, Holtrich U, Kaufmann M. Identification of high risk breast-cancer patients by gene expression profiling. Lancet. 2002; 359:131–2. https://doi.org/10.1016/S0140-6736(02)07337-3.
5. Ahr A, Holtrich U, Solbach C, Scharl A, Strebhardt K, Karn T, Kaufmann M. Molecular classification of breast cancer patients by gene expression profiling. J Pathol. 2001; 195:312–20. https://doi.org/10.1002/path.955.
6. Hu Z, Fan C, Livasy C, He X, Oh DS, Ewend MG, Carey LA, Subramanian S, West R, Ikpatt F, Olopade OI, van de Rijn M, Perou CM. A compact VEGF signature associated with distant metastases and poor outcomes. BMC Med. 2009; 7:9. https://doi.org/10.1186/1741-7015-7-9.
7. West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, Zuzan H, Olson JA Jr, Marks JR, Nevins JR. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci U S A. 2001; 98:11462–67. https://doi.org/10.1073/pnas.201162998.
8. Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003; 100:10393–98. https://doi.org/10.1073/pnas.1732912100.
9. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747–52. https://doi.org/10.1038/35021093.
10. Vazquez AI, Veturi Y, Behring M, Shrestha S, Kirst M, Resende MF Jr, de Los Campos G. Increased proportion of variance explained and prediction accuracy of survival of breast cancer patients with use of whole-genome multiomic profiles. Genetics. 2016; 203:1425–38. https://doi.org/10.1534/genetics.115.185181.
11. Vazquez A, Wiener H, Shrestha S, Tiwari HK, de los Campos G. Integration of Multi-Layer Omic Data for Prediction of Disease Risk in Humans. 2014. Available from: https://doi.org/10.13140/2.1.4769.9200.
12. González-Reymúndez A, de Los Campos G, Gutiérrez L, Lunt SY, Vazquez AI. Prediction of years of life after diagnosis of breast cancer using omics and omic-by-treatment interactions. Eur J Hum Genet. 2017; 25:538–44. https://doi.org/10.1038/ejhg.2017.12.
13. Hunter KW, Alsarraj J. Gene expression profiles and breast cancer metastasis: a genetic perspective. Clin Exp Metastasis. 2009; 26:497–503. https://doi.org/10.1007/s10585-009-9249-8.
14. Lukes L, Crawford NP, Walker R, Hunter KW. The origins of breast cancer prognostic gene expression profiles. Cancer Res. 2009; 69:310–18. https://doi.org/10.1158/0008-5472.CAN-08-3520.
15. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003; 33:49–54.
16. Hoon DS, Korn EL, Cochran AJ. Variations in functional immunocompetence of individual tumor-draining lymph nodes in humans. Cancer Res. 1987; 47:1740–44.
17. Popławski AB, Jankowski M, Erickson SW, Díaz de Ståhl T, Partridge EC, Crasto C, Guo J, Gibson J, Menzel U, Bruder CE, Kaczmarczyk A, Benetkiewicz M, Andersson R, et al. Frequent genetic differences between matched primary and metastatic breast cancer provide an approach to identification of biomarkers for disease progression. Eur J Hum Genet. 2010; 18:560–68. https://doi.org/10.1038/ejhg.2009.230.
18. Wang C, Iakovlev VV, Wong V, Leung S, Warren K, Iakovleva G, Arneson NC, Pintilie M, Miller N, Youngson B, McCready DR, Done SJ. Genomic alterations in primary breast cancers compared with their sentinel and more distal lymph node metastases: an aCGH study. Genes Chromosomes Cancer. 2009; 48:1091–101. https://doi.org/10.1002/gcc.20711.
19. Suzuki M, Tarin D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: clinical implications. Mol Oncol. 2007; 1:172–80. https://doi.org/10.1016/j.molonc.2007.03.005.
20. Feng Y, Sun B, Li X, Zhang L, Niu Y, Xiao C, Ning L, Fang Z, Wang Y, Zhang L, Cheng J, Zhang W, Hao X. Differentially expressed genes between primary cancer and paired lymph node metastases predict clinical outcome of node-positive breast cancer patients. Breast Cancer Res Treat. 2007; 103:319–29. https://doi.org/10.1007/s10549-006-9385-7.
21. Isola JJ, Kallioniemi OP, Chu LW, Fuqua SA, Hilsenbeck SG, Osborne CK, Waldman FM. Genetic aberrations detected by comparative genomic hybridization predict outcome in node-negative breast cancer. Am J Pathol. 1995; 147:905–11.
22. Climent J, Martinez-Climent JA, Blesa D, Garcia-Barchino MJ, Saez R, Sánchez-Izquierdo D, Azagra P, Lluch A, Garcia-Conde J. Genomic loss of 18p predicts an adverse clinical outcome in patients with high-risk breast cancer. Clin Cancer Res. 2002; 8:3863–69.
23. Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat. 2007; 102:143–55. https://doi.org/10.1007/s10549-006-9317-6.
24. Nishizaki T, DeVries S, Chew K, Goodson WH 3rd, Ljung BM, Thor A, Waldman FM. Genetic alterations in primary breast cancers and their metastases: direct comparison using modified comparative genomic hybridization. Genes Chromosomes Cancer. 1997; 19:267–72. https://doi.org/10.1002/(SICI)1098-2264(199708)19:4<267::AID-GCC9>3.0.CO;2-V.
25. Teixeira MR, Pandis N, Heim S. Cytogenetic clues to breast carcinogenesis. Genes Chromosomes Cancer. 2002; 33:1–16. https://doi.org/10.1002/gcc.1206.
26. Kuukasjärvi T, Karhu R, Tanner M, Kähkönen M, Schäffer A, Nupponen N, Pennanen S, Kallioniemi A, Kallioniemi OP, Isola J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997; 57:1597–604.
27. Friedrich K, Weber T, Scheithauer J, Meyer W, Haroske G, Kunze KD, Baretton G. Chromosomal genotype in breast cancer progression: comparison of primary and secondary manifestations. Cell Oncol. 2008; 30:39–50.
28. Comtesse N, Niedermayer I, Glass B, Heckel D, Maldener E, Nastainczyk W, Feiden W, Meese E. MGEA6 is tumor-specific overexpressed and frequently recognized by patient-serum antibodies. Oncogene. 2002; 21:239–47. https://doi.org/10.1038/sj.onc.1205005.
29. Tanabe T, Maeda M, Saito K, Katada T. Dual function of cTAGE5 in collagen export from the endoplasmic reticulum. Mol Biol Cell. 2016; 27:2008–13. https://doi.org/10.1091/mbc.E16-03-0180.
30. Rizwan A, Bulte C, Kalaichelvan A, Cheng M, Krishnamachary B, Bhujwalla ZM, Jiang L, Glunde K. Metastatic breast cancer cells in lymph nodes increase nodal collagen density. Sci Rep. 2015; 5:10002. https://doi.org/10.1038/srep10002.
31. Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008; 6:11. https://doi.org/10.1186/1741-7015-6-11.
32. Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006; 4:38. https://doi.org/10.1186/1741-7015-4-38.
33. Wetzels RH, Holland R, van Haelst UJ, Lane EB, Leigh IM, Ramaekers FC. Detection of basement membrane components and basal cell keratin 14 in noninvasive and invasive carcinomas of the breast. Am J Pathol. 1989; 134:571–79.
34. Usener D, Schadendorf D, Koch J, Dübel S, Eichmüller S. cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J Invest Dermatol. 2003; 121:198–206. https://doi.org/10.1046/j.1523-1747.2003.12318.x.
35. Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X, Cui Z, Zhang J, Yi K, Xu W, Chen C, Wang F, Guo X, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012; 22:806–21. https://doi.org/10.1038/cr.2012.30.
36. Grubbs EG, Abdel-Wahab Z, Tyler DS, Pruitt SK. Utilizing quantitative polymerase chain reaction to evaluate prostate stem cell antigen as a tumor marker in pancreatic cancer. Ann Surg Oncol. 2006; 13:1645–54. https://doi.org/10.1245/s10434-006-9029-5.
37. Amara N, Palapattu GS, Schrage M, Gu Z, Thomas GV, Dorey F, Said J, Reiter RE. Prostate stem cell antigen is overexpressed in human transitional cell carcinoma. Cancer Res. 2001; 61:4660–65.
38. Wang M, Wang X, Fu SW, Liu X, Jin T, Kang H, Ma X, Lin S, Guan H, Zhang S, Liu K, Dai C, Zhu Y, Dai Z. Single-nucleotide polymorphisms in PSCA and the risk of breast cancer in a Chinese population. Oncotarget. 2016; 7:27665–75. https://doi.org/10.18632/oncotarget.8491.
39. Kim SY, Yoo JY, Shin A, Kim Y, Lee ES, Lee YS. Prostate stem cell antigen single nucleotide polymorphisms influence risk of estrogen receptor negative breast cancer in Korean females. Asian Pac J Cancer Prev. 2012; 13:41–48. https://doi.org/10.7314/APJCP.2012.13.1.041.
40. Karlsson E, Ahnstrom Waltersson M, Bostner J, Perez-Tenorio G, Olsson B, Fornander T, Stal O. Comprehensive Genomic and Transcriptomic Analysis of the 11q13 Amplicon in Breast Cancer. Cancer Res. 2009 (Suppl 24); 69. https://doi.org/10.1158/0008-5472.SABCS-09-5166.
41. Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013; 123:1068–81. https://doi.org/10.1172/JCI64264.
42. Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014; 9:e98293. https://doi.org/10.1371/journal.pone.0098293. Erratum in: PLoS One. 2014; 9:e107646.
43. Zeller C, Hinzmann B, Seitz S, Prokoph H, Burkhard-Goettges E, Fischer J, Jandrig B, Schwarz LE, Rosenthal A, Scherneck S. SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene. 2003; 22:2972–83. https://doi.org/10.1038/sj.onc.1206474.
44. Wick W, Petersen I, Schmutzler RK, Wolfarth B, Lenartz D, Bierhoff E, Hümmerich J, Müller DJ, Stangl AP, Schramm J, Wiestler OD, von Deimling A. Evidence for a novel tumor suppressor gene on chromosome 15 associated with progression to a metastatic stage in breast cancer. Oncogene. 1996; 12:973–78.
45. Schmutte C, Tombline G, Rhiem K, Sadoff MM, Schmutzler R, von Deimling A, Fishel R. Characterization of the human Rad51 genomic locus and examination of tumors with 15q14-15 loss of heterozygosity (LOH). Cancer Res. 1999; 59:4564–69. https://doi.org/10.1016/0022-2836(87)90689-9.
46. Jiang Z, Zhou Y, Devarajan K, Slater CM, Daly MB, Chen X. Identifying putative breast cancer-associated long intergenic non-coding RNA loci by high density SNP array analysis. Front Genet. 2012; 3:299. https://doi.org/10.3389/fgene.2012.00299.
47. Ramírez de Molina A, Gutiérrez R, Ramos MA, Silva JM, Silva J, Bonilla F, Sánchez JJ, Lacal JC. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene. 2002; 21:4317–22. https://doi.org/10.1038/sj.onc.1205556.
48. Contractor KB, Kenny LM, Stebbing J, Al-Nahhas A, Palmieri C, Sinnett D, Lewis JS, Hogben K, Osman S, Shousha S, Lowdell C, Coombes RC, Aboagye EO. [11C]choline positron emission tomography in estrogen receptor-positive breast cancer. Clin Cancer Res. 2009; 15:5503–10. https://doi.org/10.1158/1078-0432.CCR-09-0666.
49. Peñuelas I, Domínguez-Prado I, García-Velloso MJ, Martí-Climent JM, Rodríguez-Fraile M, Caicedo C, Sánchez-Martínez M, Richter JA. PET tracers for clinical imaging of breast cancer. J Oncol. 2012; 2012:710561. https://doi.org/10.1155/2012/710561.
50. Skanjeti A, Pelosi E. Lymph Node Staging with Choline PET/CT in Patients with Prostate Cancer: A Review. ISRN Oncol. 2011; 2011:219064. https://doi.org/10.5402/2011/219064.
51. Mao X, He J, Li T, Lu Z, Sun J, Meng Y, Abliz Z, Chen J. Application of imaging mass spectrometry for the molecular diagnosis of human breast tumors. Sci Rep. 2016; 6:21043. https://doi.org/10.1038/srep21043.
52. Hosokawa Y, Masaki N, Takei S, Horikawa M, Matsushita S, Sugiyama E, Ogura H, Shiiya N, Setou M. Recurrent triple-negative breast cancer (TNBC) tissues contain a higher amount of phosphatidylcholine (32:1) than non-recurrent TNBC tissues. PLoS One. 2017; 12:e0183724. https://doi.org/10.1371/journal.pone.0183724.
53. Gu W, Choi H, Ghosh D. Global associations between copy number and transcript mRNA microarray data: an empirical study. Cancer Inform. 2008; 6:17–23. https://doi.org/10.4137/CIN.S342.
54. Bergamaschi A, Kim YH, Wang P, Sørlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Børresen-Dale AL, Pollack JR. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006; 45:1033–40. https://doi.org/10.1002/gcc.20366.
55. Parris TZ, Danielsson A, Nemes S, Kovács A, Delle U, Fallenius G, Möllerström E, Karlsson P, Helou K. Clinical implications of gene dosage and gene expression patterns in diploid breast carcinoma. Clin Cancer Res. 2010; 16:3860–74. https://doi.org/10.1158/1078-0432.CCR-10-0889.
56. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, et al, and METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–52. https://doi.org/10.1038/nature10983.
57. Lipson D, Ben-Dor A, Dehan E, Yakhini Z. Joint analysis of DNA copy numbers and gene expression levels. Algorithms in Bioinformatics. 2004; 3240:135–46.
58. Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A. 2002; 99:12963–68. https://doi.org/10.1073/pnas.162471999.
59. TCGA. The Cancer Genome Atlas - Data Portal. Available from: https://tcga-data.nci.nih.gov/docs/publications/tcga/.
60. Guiu S, Wolfer A, Jacot W, Fumoleau P, Romieu G, Bonnetain F, Fiche M. Invasive lobular breast cancer and its variants: how special are they for systemic therapy decisions? Crit Rev Oncol Hematol. 2014; 92:235–57. https://doi.org/10.1016/j.critrevonc.2014.07.003.
61. Iorfida M, Maiorano E, Orvieto E, Maisonneuve P, Bottiglieri L, Rotmensz N, Montagna E, Dellapasqua S, Veronesi P, Galimberti V, Luini A, Goldhirsch A, Colleoni M, Viale G. Invasive lobular breast cancer: subtypes and outcome. Breast Cancer Res Treat. 2012; 133:713–23. https://doi.org/10.1007/s10549-012-2002-z.
62. NCI. PDQ® Breast Cancer Treatment. National Cancer Institute. 2013. Available from: https://www.cancer.gov/cancertopics/pdq/treatment/breast/healthprofessional.
63. Ha G, Shah S. Distinguishing somatic and germline copy number events in cancer patient DNA hybridized to whole-genome SNP genotyping arrays. Methods Mol Biol. 2013; 973:355–72. https://doi.org/10.1007/978-1-62703-281-0_22.
64. Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004; 5:557–72. https://doi.org/10.1093/biostatistics/kxh008.
65. Cairns JM, Dunning MJ, Ritchie ME, Russell R, Lynch AG. BASH: a tool for managing BeadArray spatial artefacts. Bioinformatics. 2008; 24:2921–22. https://doi.org/10.1093/bioinformatics/btn557.
66. Dunning MJ, Smith ML, Ritchie ME, Tavaré S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007; 23:2183–84. https://doi.org/10.1093/bioinformatics/btm311.
67. Lai YP, Wang LB, Wang WA, Lai LC, Tsai MH, Lu TP, Chuang EY. iGC-an integrated analysis package of gene expression and copy number alteration. BMC Bioinformatics. 2017; 18:35. https://doi.org/10.1186/s12859-016-1438-2.
68. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905. https://doi.org/10.1038/nature08822.
69. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007; 450:893–98. https://doi.org/10.1038/nature06358.
70. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011; 12:R41. https://doi.org/10.1186/gb-2011-12-4-r41.
71. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 14:128. https://doi.org/10.1186/1471-2105-14-128.