
INTRODUCTION
Heterozygous mutations in the isocitrate dehydrogenase 1 (IDH1) gene are found most frequently in glioma, predominantly resulting in the mutant enzyme IDH1R132H with histidine substitution at arginine 132 [1–3]. The biological function of IDH1R132H, however, remains controversial. The prevailing belief is that IDH1R132H is oncogenic owing to the gain of neomorphic activity that converts 2-oxoglutarate (aka α-ketoglutarate)—the product of wild-type IDH1—in an NADPH-dependent reduction to an “oncometabolite” D-2-hydroxyglutarate (D2-HG), which in turn inhibits a class of 2-oxoglutarate-dependent dioxygenases involved in epigenetic regulation, collagen synthesis, and cell signaling [4]. Supporting evidence for this theory includes 1) the association of IDH1 mutations with glioma evolution, glioma CpG island methylator phenotype, and proneural subtype; 2) the induction of methylator phenotype in normal human astrocyte by IDH1R132H transduction or D2-HG treatment; and 3) the association of IDH1 mutations with repressive histone methylation marks that contribute to a less differentiated or stem-like state [5]. Despite the circumstantial evidence, the exact mechanism by which IDH1R132H drives glioma initiation remains ill-defined, and, moreover, evidence from recent studies apparently challenges this belief.
Specifically, despite effective reduction of D2-HG by small-molecule inhibitors specific to mutant IDH1, treated glioma cells, unexpectedly, accelerated proliferation and shortened survival in an animal model [6]. Therapeutic sensitivity is important to improved survival of glioma patients with IDH1 mutations, but mutant IDH1 inhibitors desensitized tumors cells to irradiation and chemotherapy [7]. Apparently, these counterintuitive findings not only argue against therapeutic targeting of IDH1 mutations but also question the presumptive oncogenic activity of IDH1R132H. Consistently, previous studies showed that IDH1R132H transduction inhibited rather than stimulated tumor growth [8, 9], and gliomas with IDH1 mutations possessed attenuated oncogenic signaling in comparison with those without [8, 10–13]. These studies have led us to posit that IDH1 mutations are tumor-suppressive on the contrary; the biological consequence of IDH1 mutations in glioma is to ameliorate patient survival, at least in part, by inhibiting oncogenic signaling [13]. This concept is in accordance with the experimental demonstration of anti-tumor effects of D2-HG, which decreases the stability of MYC/CEBPA transcripts via N6-methyladenosine RNA modification and thereby inhibits tumor cell survival and proliferation [14]. We have reported recently that whereas heterozygous but not hemizygous IDH1R132H suppresses anchorage-independent growth of glioma cells, the surviving cells conversely selects against IDH1R132H heterozygosity [15]. Our findings not only support the concept of IDH1R132H being anti-oncogenic but also suggest the strong antagonism between tumor growth and heterozygous IDH1R132H expression in the experimental setting. This interpretation is consistent with the requirement of a wild-type IDH1 allele for D2-HG production [16, 17] and the frequent loss of either wild-type or mutant IDH1 allele in patient-derived xenograft, ex vivo neurosphere culture, and glioma recurrence and progression [11, 16, 18, 19], even though the underlying mechanism of copy number alteration remains unclear.
The concept that IDH1R132H heterozygosity is anti-oncogenic and incompatible with tumor growth seems at odds with the fact that greater than 70% of WHO grade II and grade III gliomas and secondary glioblastomas harbor IDH1 mutations [1–3]. Moreover, IDH1R132H-specific inhibitor and mutant IDH1 pan-inhibitor have been shown to be effective in animal studies [20, 21]. It is noteworthy, however, that the anti-oncogenic activity of heterozygous IDH1R132H can be circumvented by genetic and metabolic alterations, including the loss of IDH1R132H heterozygosity and use of reducing equivalent [15]. Furthermore, deletion or amplification of either mutant or wild-type IDH1 allele decreases D2-HG in glioma recurrence [19]. Moreover, glutamate—a neurotransmitter rich in the cerebral cortex—is sufficient to bypass the inhibitory effect of IDH1R132H on glioma progenitor proliferation [9]. These findings altogether indicate the delicate nature of heterozygous IDH1R132H, whose tumor-suppressive activity can be compromised by genetic alterations and tumor microenvironment.
RESULTS
IDH1R132H transduction suppresses subcutaneous tumor growth
We reported recently that heterozygous IDH1R132H is functionally anti-oncogenic, as evidenced by the antagonism between IDH1R132H heterozygosity and anchorage-independent growth; whereas heterozygous IDH1R132H suppressed neurosphere genesis, the surviving neurosphere selected against the expression of either IDH1R132H or IDH1 transgene and reduced D2-HG levels [15]. To ascertain the tumor-suppressive activity of IDH1R132H in vivo, we first established subcutaneous tumor growth of mouse astrocyte NA1 that had been transduced with luc–PDGFB, which expresses luciferase and platelet-derived growth factor B (PDGFB) upon P2A cleavage [15]. PDGFB has been used extensively for gliomagenesis in vivo [9, 22–26]. Accordingly, the transduced NA1 developed robust tumor growth with a volume-based doubling time of 9.4 days in contrast to NA1 transduced with luc*, which expresses the same transcript harboring a stop codon engineered at the P2A (Supplementary Figure 1).
Next, we sought to test whether IDH1R132H co-transduction inhibits tumor growth using YFP–IDH1R132H, which expresses nuclear yellow fluorescent protein (YFP) and IDH1R132H upon P2A cleavage [15]. As expected, YFP–IDH1R132H significantly inhibited cell proliferation, resulting in 20% increase of the mean doubling time to 28.8 hours compared with 24.0 hours of its control YFP–IDH1 (Supplementary Figure 2A). In keeping with this, YFP–IDH1R132H cells showed G2/M arrest compared with YFP–IDH1 cells (Supplementary Figure 2B). In agreement with its effect on neurosphere genesis [15], YFP–IDH1R132H markedly inhibited tumor growth, as indicated by bioluminescent imaging and confirmed by tumor weight analysis (Figure 1A–1C). The mean volume-based doubling time of YFP–IDH1R132H tumors increased 66% to 15.8 days from 9.5 for YFP–IDH1 tumors. Histological examination confirmed reduced cellularity, nuclear–cytoplasm ratio, and nuclear pleomorphism in YFP–IDH1R132H tumors compared with YFP–IDH1 tumors (Figure 1D). Furthermore, immunohistochemistry showed decreased Ki67 as well as PDGFB staining in YFP–IDH1R132H tumors. It is noteworthy, however, in contrast to the staining of HA-tagged wild-type IDH1 in the control, HA-tagged IDH1R132H was nearly undetectable in YFP–IDH1R132H tumors and sparsely stained with an anti-IDH1R132H antibody (Supplementary Figure 2C). Taken together, these results support the concept that IDH1R132H is not only tumor-suppressive but is also selected against in the surviving tumors, as reported previously in anchorage-independent growth [15].
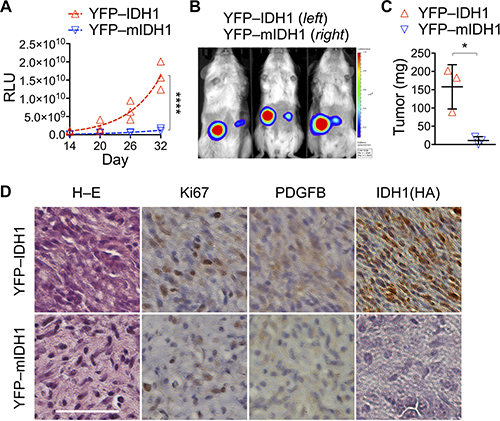
Figure 1: IDH1R132H transduction suppresses subcutaneous tumor growth. (A–C) Subcutaneous tumors derived from NA1 astrocytes that had been transduced with luc–PDGFB showing significant growth suppression by co-transduction with YFP–IDH1R132H (YFP-mIDH1) compared with YFP–IDH1 (A). Nonlinear regression curve fit was performed using exponential growth equation and two-way ANOVA for the analysis of statistical significance. RLU, relative luminescent units. ****p < 0.0001. Suppression of tumor growth was supported by bioluminescent imaging (B) and autopsied tumor weight (C). Unpaired t-tests were performed using two-tailed p value. *p < 0.05. (D) Hematoxylin–eosin (H–E) and immunohistochemistry staining revealed less malignant histologic features, decreased Ki67 and PDGFB expression, and weak HA-tagged IDH1R132H staining in YFP–IDH1R132H tumor compared with YFP–IDH1 tumor. Scale bar: 50 μm.
Antagonism between IDH1R132H transgene expression and tumor growth
To provide further evidence for the selection against IDH1R132H expression in tumor growth, we observed 76% reduction of YFP–IDH1R132H transcript levels accompanied by 35% reduction of PDGFB transcript levels compared with those in YFP–IDH1 tumors (Figure 2A, 2B), a finding in agreement with the selection against IDH1R132H transgene in neurosphere culture [15]. No reduction of IDH1R132H copy number, however, was observed at the genomic DNA level (Figure 2C), suggesting non-genetic event(s) for IDH1R132H downregulation.
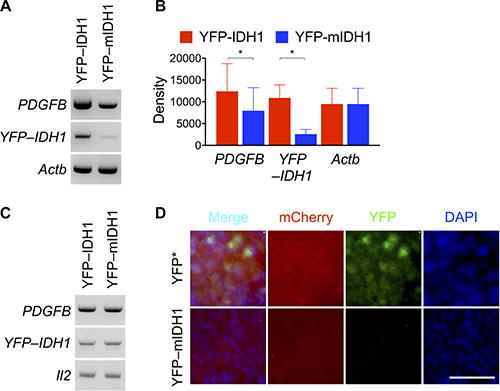
Figure 2: Markedly decreased YFP–IDH1R132H expression in subcutaneous tumor. (A–C) Reverse transcription–PCR showing marked decrease of YFP–IDH1R132H mRNA levels in the subcutaneous tumor along with modest decrease of PDGFB mRNA levels (A). Densitometry analysis supporting significant decreases of YFP–IDH1R132H (n = 3) and PDGFB (n = 5) mRNA levels (B). PCR amplification of genomic DNA showing no copy number alterations (C). (D) Fluorescent microscopy confirming loss of IDH1R132H expression and decreased PDGFB expression, as indicated by respective YFP and mCherry signals, in subcutaneous tumor derived from NA1 transduced with mCherry–PDGFB. Scale bar: 50 μm.
Next, we employed fluorescent microscopy to visualize the suppression of IDH1R132H transgene expression by examining tumor cells transduced with mCherry–PDGFB, which expresses the fluorescent mCherry and PDGFB upon P2A cleavage [15]. To that end, we transduced NA1 cells with mCherry–PDGFB and YFP–IDH1R132H or its control YFP*, which expresses only YFP protein from the same YFP–IDH1R132H transcript that harbors an engineered stop codon [15]. Of note, we opted for YFP* as a more appropriate control because the upregulation of wild-type IDH1 promotes aggressive growth of malignant glioma [27]. As expected, YFP–IDH1R132H co-transduction resulted in significant decreases in cell proliferation and subcutaneous tumor growth compared with YFP* co-transduction (Supplementary Figure 3). Fluorescent microscopy revealed few cells that were YFP+ in the surviving YFP–IDH1R132H tumor in contrast to widespread YFP+ cells in the control (Figure 2D), which is consistent with the previous finding in neurosphere culture [15]. We conclude therefore that although IDH1R132H transduction suppresses subcutaneous tumor growth, the surviving tumors, conversely, select against the transgene expression.
IDH1R132H tumors are histologically indistinguishable from IDH1-wildtype tumors in orthotopic models
Unexpectedly, IDH1R132H suppression of tumor growth could not be reproduced in orthotopic transplantation models, as shown by bioluminescent imaging and histological examination (Figure 3A, 3B). Indeed, YFP–IDH1R132H transduction failed to inhibit PDGFB-driven orthotopic tumor growth, resulting in similar bioluminescent readings in reference to the control. Histological examination showed similar tumor cell proliferation and invasion in both groups of mice (Figure 3C), an observation consistent with previous reports [25, 28, 29]. The lack of clear tumor suppression in the orthotopic model indicates a tissue-specific role of the cerebral cortex in the biological effect of IDH1R132H.
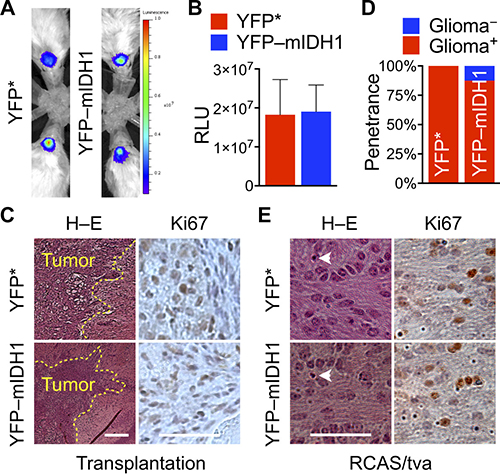
Figure 3: Loss of IDH1R132H suppression of tumor initiation and growth in orthotopic models. (A–C) YFP–IDH1R132H failed to suppress PDGFB-driven tumor growth in intracranial transplantation compared with YFP*, as indicated by bioluminescent imaging (A), the mean bioluminescent values (B, n = 4), and invasive histologic presentation and similar patterns of Ki67 staining (C). RCAS/tva mouse model showing similar glioma penetrance (D) and malignant features including mitotic indices (arrowhead), infiltration, and Ki67 staining (E) between mice subjected to RCAS/PDGFB and RCAS/YFP* infection and RCAS/PDGFB and RCAS/YFP–IDH1R132H infection (n = 8 for each group).
We next sought to corroborate the tissue-specific effect of IDH1R132H in a spontaneous glioma mouse model, which combines the replication-competent avian sarcoma-leukosis virus long-terminal repeat with splice acceptor (RCAS) for transgene delivery [30–32] with a transgenic line (Nes-tva) carrying Nestin promoter-driven expression of avian retroviral receptor tva [33]. PDGFB has been used as a potent inducer of gliomagenesis in Nes-tva mice [22, 24, 26, 34, 35]. Consistently, we observed equivalent glioma penetrance between mice co-transduced with PDGFB and YFP* (100% or 8/8) and those co-transduced with PDGFB and YFP–IDH1R132H (88% or 7/8) (Figure 3D). Our earlier investigation using PDGFB and IDH1 or IDH1R132H showed similar penetrance: 77% (7/9) or 75% (6/8). Furthermore, similar invasion of the cerebral cortex and Ki67 and Olig2 staining were observed in both tumor types (Figure 3E; Supplementary Figure 4), as shown previously [28, 29]. Taken together, these results suggest that the tumor-suppressive effect of IDH1R132H is functionally compromised by the cerebral cortex in the experimental setting.
IDH1R132H expression becomes permissible in glioma with Cdkn2a deletion
Although orthotopic transplantation exhibited similar tumor growth between YFP–IDH1R132H and YFP* control cells, strong nuclear staining of YFP was seen in YFP*, but not YFP–IDH1R132H, tumors by immunohistochemistry (Figure 4A). Additionally, IDH1R132H staining was scattered in YFP–IDH1R132H tumor cells (Supplementary Figure 5A). Likewise, in the RCAS/PDGFB glioma model, weak YFP staining was seen in YFP–IDH1R132H glioma cells in contrast to prevalent nuclear staining in YFP* glioma cells (Figure 4B; Supplementary Figure 5B). Furthermore, in RCAS/mCherry–PDGFB-induced gliomas, fluorescent YFP signal was visualized only in the YFP–IDH1, but not YFP–IDH1R132H, tumors despite a modest decrease of mCherry signal in the latter (Figure 4C). These results strongly indicate that IDH1R132H transgene expression is selected against during glial tumor growth irrespective of tumor size and microenvironment, supporting the notion of antagonism between IDH1R132H expression and tumor growth.
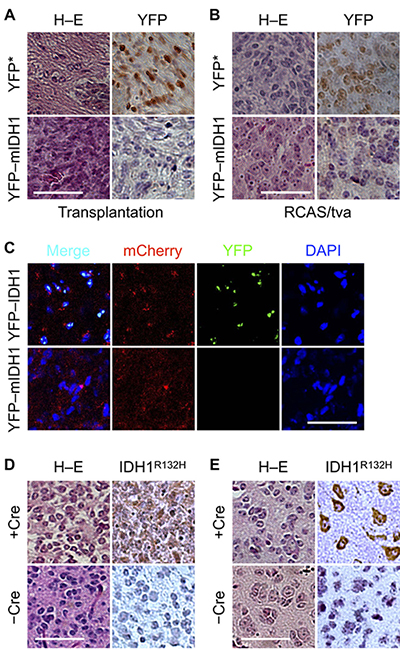
Figure 4: Dependence of IDH1R132H expression on Cdkn2a deletion in orthotopic tumor. Pronounced reduction of YFP staining in YFP–IDH1R132H intracranial transplantation (A) and RCAS/tva glioma (B) models compared with their YFP* controls. (C) Fluorescent microscopy confirming the loss of YFP–IDH1R132H, but not YFP–IDH1, expression in RCAS/tva glioma driven by mCherry–PDGFB. Cdkn2a deletion with RCAS/Cre (+Cre) in Nes-tva;Cdkn2afl/fl mice increased IDH1R132H expression in the cytoplasm of tumor cells (D) and those involved in perineuronal satellitosis (E). Scale bar: 50 μm.
Our findings are apparently at odds with the fact that IDH1R132H is detectable immunologically in human gliomas and tumor transplantations [10, 36–40]. In fact, IDH1R132H staining was strong and conspicuous in RCAS/PDGFA gliomas when combined with Cdkn2a knockout and Trp53 knockdown [28]. In light of frequent mutations in various tumor-suppressor genes associated with IDH-mutant glioma [41], we hypothesized that the inactivation of tumor-suppressor gene(s) renders glioma more permissible to IDH1R132H expression. To test this notion, we compared immunohistochemical IDH1R132H staining between tumors developed in Cdkn2a-intact and -deleted genetic background. Indeed, IDH1R132H gliomas derived from Cdkn2afl/fl mice showed much increased immunohistochemical staining of IDH1R132H with Cre co-transduction compared with those without (Figure 4D). Additionally, IDH1R132H staining was seen in glioma cells forming perineuronal satellitosis (Figure 4E), as reported previously [36]. Taken together, these results support the selection against IDH1R132H transgene in PDGFB-driven tumors and the dependence of IDH1R132H expression on inactivation of tumor-suppressor gene(s).
Expression of IDH1R132H and PDGFB from the same transcript obliterates gliomagenesis
Although recent studies indicated anti-tumor effects of IDH1R132H [14, 19], whether IDH1R132H suppresses gliomagenesis remains unclear. To provide evidence that IDH1R132H is tumor-suppressive, we engineered a RCAS vector that expresses IDH1R132H, P2A, and PDGFB from the same transcript. This tandem design of IDH1R132H–PDGFB not only ensures the expression of the two at 1:1 molar ratio within the same cells but also precludes selection against IDH1R132H expression especially in the cerebral cortex (Figure 4). Similarly, IDH1–PDGFB was constructed as control.
We confirmed equivalent transgene expression between NA1 cells transduced with IDH1–PDGFB and IDH1R132H–PDGFB at mRNA and protein levels (Supplementary Figure 6A, 6B). We observed the mean D2-HG levels at 3,583 nmol per mg protein in IDH1R132H–PDGFB cells (Figure 5A), a concentration similar to those in human IDH1R132H glioma cells [15] and fourfold greater than that in IDH1–PDGFB cells. IDH1R132H–PDGFB cells showed a remarkable decrease in proliferation compared with IDH1–PDGFB cells, resulting in 47% increase of doubling time to 41.9 hours from 28.5 (Figure 5B). Furthermore, we determined the ability of single cells to form neurosphere; consistent with the inhibitory effect of YFP–IDH1R132H when co-expressed with luc–PDGFB or mCherry–PDGFB from different transcripts [15], IDH1R132H–PDGFB cells had a fivefold reduction of neurosphere genesis from 3.5% in IDH1–PDGFB cells to 0.7, a level equivalent to the parental NA1 (Figure 5C; Supplementary Figure 6C). These results indicate that IDH1R132H–PDGFB is a functional platform demonstrating that a single-nucleotide difference in IDH1 is sufficient to confer suppression of anchorage-independent growth by overriding the oncogenic activity of PDGFB.
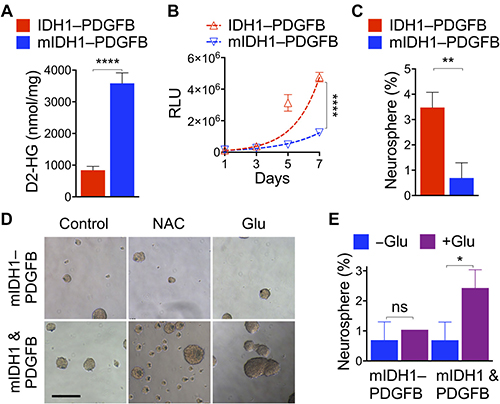
Figure 5: IDH1R132H overrides oncogenic PDGFB in tandem expression. (A) An extremely significant increase of D2-HG levels (n = 6) in NA1 transduced with IDH1R132H–PDGFB (mIDH1–PDGFB) in reference to the control. Significant decreases in NA1 proliferation (B, n = 6) and neurosphere genesis from single cells (C, n = 3) upon transduction with IDH1R132H–PDGFB compared with IDH1–PDGFB. (D) Treatment with 1 mM N-acetyl cysteine (NAC) or glutamate (Glu) failed to increase the size and number of neurosphere growth that had been transduced with IDH1R132H–PDGFB but did so in those co-transduced with YFP–IDH1R132H and mCherry–PDGFB (mIDH1 & PDGFB). (E) Quantitative analysis confirming the ineffectiveness of glutamate on neurosphere genesis from single mIDH1–PDGFB cells (n = 3). Unpaired t-tests were performed using two-tailed p value. ns, not significant, *p < 0.05; **p < 0.01; ****p < 0.001. Scale bar: 200 μm.
Previous studies have indicated the importance of glutamate anaplerosis in IDH-mutant glioma metabolism and growth [9, 42]. In particular, the addition of glutamate reversed IDH1R132H-mediated proliferative inhibition of neural progenitor cells co-transduced with PDGFB [9]. Likewise, we previously showed that the addition of reducing equivalent N-acetyl cysteine reversed inhibition of anchorage-independent growth by heterozygous IDH1R132H [15]. We sought to determine whether IDH1R132H–PDGFB cells would respond differently to the treatment of glutamate or N-acetyl cysteine in neurosphere genesis. Indeed, treatment with sodium glutamate or N-acetyl cysteine markedly increased size and number of PDGFB-driven neurospheres when YFP–IDH1R132H was expressed from different transcripts (Figure 5D, bottom); however, neither treatment had noticeable effect on those transduced with IDH1R132H–PDGFB (top). Furthermore, results from single-cell analysis confirmed that glutamate treatment had virtually no effect on IDH1R132H–PDGFB cells in contrast to a 3.5-fold increase in the co-transduced cells from 0.69% to 2.43 (Figure 5E). Therefore, these results not only further support the concept that IDH1R132H is intrinsically tumor-suppressive but also suggest a complete suppression of glioma development if IDH1R132H is co-expressed with PDGFB from the same transcript.
Given the overriding role of IDH1R132H against oncogenic PDGFB in anchorage-independent growth, we predicted that expression of IDH1R132H with PDGFB from the same transcript would prevent spontaneous glioma initiation and growth even in the glutamine-rich microenvironment. Indeed, none of the mice (13/13) developed glioma with RCAS/IDH1R132H–PDGFB in contrast to 93% incidence (14/15) in those with RCAS/IDH1–PDGFB (Figure 6A, 6B). In addition, immunohistochemistry showed widespread Olig2 staining in tumor cells but not in the cortex of IDH1R132H–PDGFB mice (Figure 6B). Furthermore, Kaplan–Meier analysis revealed that IDH1R132H was remarkably beneficial to the survival of IDH1R132H–PDGFB mice; none of them exhibited clear neurologic signs by the end of the 8-week period, whereas 73% IDH1–PDGFB mice had to be sacrificed, thereby significantly decreasing the median survival to 43 days (Figure 6C). Histological examination and Olig2 immunohistochemical staining confirmed the presence or absence of glioma lesions in all of the mice (data not shown). Thus, we conclude that IDH1R132H is intrinsically suppressive of glioma initiation as well as glioma growth.
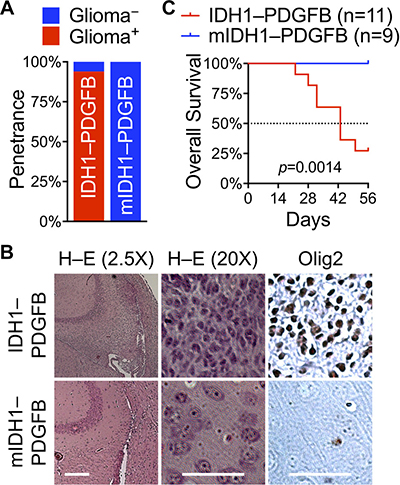
Figure 6: IDH1R132H obliterates PDGFB-driven gliomagenesis when expressed in tandem. (A) Ninety-three percent of glioma penetrance (Glioma+) with RCAS/IDH1–PDGFB (14/15) in contrast to zero percent of glioma penetrance (Glioma−) with RCAS/IDH1R132H–PDGFB (13/13) in Nes-tva mice. (B) Lack of glioma lesions in the brain of RCAS/IDH1R132H–PDGFB mice, as determined by histologic examination and Olig2 staining. Scale bar: short, 500 μm; long, 50 μm. (C) A Kaplan–Meier plot showing a striking survival difference between RCAS/IDH1–PDGFB mice and RCAS/IDH1R132H–PDGFB mice.
DISCUSSION
We present evidence in this study that the outcome of IDH1R132H transduction in glioma initiation and growth is context dependent even though IDH1R132H is intrinsically tumor-suppressive. Specifically, we demonstrate that when IDH1 and PDGFB are expressed from the same transcript, a single-nucleotide change of IDH1 at codon 132 determines the fate of gliomagenesis and overall survival of Nes-tva mice. Our results provide direct evidence that IDH1R132H is not only intrinsically tumor-suppressive but also resistant to functional compromise by the environmental glutamate or reducing power, which would otherwise attenuate the antagonism of IDH1R132H toward the oncogenic PDGFB when both are expressed from different transcripts. This finding offers an explanation for the distinct effects of IDH1R132H on tumor growth between the subcutaneous and the glutamate-rich cerebral environment in this study. Our observation that the addition of glutamate markedly decreased the inhibitory effect of IDH1R132H on neurosphere growth and genesis from single cells is in agreement with glutamate reversal of IDH1R132H inhibition of neural progenitor cell proliferation [9] and is consistent with the metabolic dependence of IDH1-mutant glioma on glutamate [43]. Although we did not provide evidence that there is sufficient glutamate in the cerebral cortex to feed glioma in our models, these results nevertheless support the concept that IDH1R132H-mediated tumor suppression can be compromised by the microenvironmental factors including glutamate and reducing equivalent as escape mechanisms of glioma progression [15]. The study may also account for the prevalence of IDH mutations in glioma and the nonexistence in most other cancer types [2, 3, 44, 45]. Furthermore, our results may provide a clue to the challenging issue of maintaining IDH1R132H heterozygosity in glioma cell culture [11, 18, 46].
In agreement with our initial concept that the biological consequence of IDH mutations is antagonistic toward oncogenic signaling [13], the conclusion of IDH1R132H being intrinsically tumor-suppressive is further supported by the observations that IDH1R132H is anti-tumor growth or incompatible with glioma progression [6, 8–10, 14, 15, 19, 28]. Moreover, genetic studies indicate that endogenous Idh1R132H/+ expression alone is non-tumorigenic in hematopoietic and neural tissues [29, 47–49]. Conditional expression of transgenic IDH2 mutation in knock-in mice caused cardiomyopathy and neurodegeneration instead [50]. Importantly, endogenous Idh1R132H/+ expression through Nes-CreERT2 resulted in 70% decrease in glioma penetrance induced by Trp53 deletion and extended mouse survival in reference to Idh1 wild-type expression [29]. Likewise, IDH1R132H gliomas driven by PDGFA transduction and Trp53 knockdown show significantly extended survival in comparison with IDH1 gliomas [28]. Therefore, the genetic evidence supports the conclusion that IDH1R132H is intrinsically tumor-suppressive.
In accordance with the incompatibility between IDH1R132H heterozygosity and anchorage-independent growth [15], we observed the strong antagonism between IDH1R132H expression and PDGFB-driven tumor growth. Interestingly, IDH1R132H transgene expression was markedly attenuated but more permissible at the expense of Cdkn2a deletion. We anticipate additional Trp53 knockdown would result in even greater IDH1R132H transgene expression, as shown previously [28]. The biological consequence of tumor-suppressor gene inactivation, however, is the erosion of IDH1R132H tumor-suppressive activity, as indicated by the complete loss of IDH1R132H survival benefit in Cdkn2a−/− mice in contrast to Cdkn2a+/+ and Cdkn2a−/+ mice [28]. In light of the association of IDH-mutant gliomas with mutations in tumor-suppressor genes including TP53, ATRX, CIC, NOTCH1, and FUBP1 [41], it stands to reason that IDH1R132H expression becomes permissible and therefore detectable in these gliomas of various grades [36]. The notion that inactivation of tumor-suppressor gene(s) permits IDH1R132H existence and expression in glioma may account for the continuous presence of IDH1R132H in recurrent gliomas [51, 52], even though some recurrent gliomas underwent genetic deletion of mutant IDH1 allele and copy number alterations [16, 19]. Whether IDH1R132H detected in recurrent glioma is fully functional remains to be investigated; the finding that IDH1R132H and D2-HG are nonessential at recurrence nevertheless has raised the question of targeting IDH1R132H for therapeutic intervention [19]. It is interesting to note that although the IDH1R132H-specific inhibitor AGI-5198 was shown initially to be effective in inhibiting glioma cell growth in subcutaneous xenograft [20], followup studies found lack of tumor regression in the same tumor model [53]. Despite the high potency in 2HG suppression among available mutant IDH1 inhibitors, their therapeutic efficacies in survival experiments vary from modest to harmful [6, 21]. Additionally, studies of gliomagenesis in cell culture models also indicate that 2-HG is nonessential to cell growth [54].
Given the association of PDGFRα with IDH-mutant glioma [39, 55, 56], the use of PDGFA as an oncogenic driver seems more relevant because PDGFA activates only PDGFRα [57]. Furthermore, since Trp53 deletion is sufficient to induce glioma [29], it will be interesting to test further whether IDH1R132H obliterates gliomagenesis driven by PDGFA transduction or Trp53 knockdown when expressed from the same transcript. It should be noted that this design, albeit artificial, has enabled us to tease out the intrinsic function of IDH1R132H, similar to what the genetic engineering of heterozygous IDH1R132H/+ in HCT116 colon cancer cells, for instance, has done to advance the understanding of glioma biology. Thus far, our tandem design arguably has the advantage of directly determining the antagonism between IDH1R132H and oncogenic activities in gliomagenesis.
In summary, this study has shown that IDH1R132H is intrinsically tumor-suppressive, and yet its anti-tumor activity can be compromised by internal factors, such as inactivation of tumor-suppressor gene, and external factors, such as glutamate. The context-dependent effects of IDH1R132H on tumor initiation and growth may have implications in glioma etiology, model development, and therapeutic targeting.
MATERIALS AND METHODS
RCAS vectors and retroviral generation
All RCAS vectors including RCAS/luc–PDGFB, RCAS/mCherry–PDGFB, RCAS/YFP–IDH1, RCAS/YFP–IDH1R132H, and RCAS/YFP* were created as described previously [15]. RCAS/IDH1–PDGFB and RCAS/IDH1R132H–PDGFB vectors were constructed using the Gibson assembly (New England Biolabs) as described previously [15]. RCAS/luc* was derived from RCAS/luc–PDGFB with the introduction of a stop codon at P2A. Recombinant retroviruses were generated essentially as described previously [15, 26].
Cell culture, retroviral infection, and neurosphere culture
Immortalized mouse astrocyte cell line NA1 was prepared and subjected to retroviral infection as described previously [15]. Likewise, resultant cells with fluorescent signals were enriched by flow cytometry, and the IDH1R132H status was verified by DNA sequencing. Neurosphere growth was compared qualitatively by seeding 10,000 cells in a 48-well plate with 500 μL of neural stem cell medium [Neurobasal media supplemented with B-27, 10 ng/mL bFGF, and 20 ng/mL EGF (Invitrogen)]. An additional 100 μL of neural stem cell medium was added after 4 days. Micrographs were acquired 1 week following seeding. To determine the ability to form neurosphere from single cells, we seeded cells of interest at 1 or 5 cells per well in a 96-well plate in triplicate and replenished fresh medium every 2–3 days. Sodium glutamate or N-acetyl cysteine was added at 1 mM and refreshed every 2–3 days. Spheres over 50 μm in diameter were counted after 14 days.
D2-HG analysis
D-2-Hydroxyglutarate Colorimetric Assay Kit (BioVision) was used to measure the intracellular level of D2-HG, as per manufacturer recommendations. Briefly, cell lysates from 1 × 105 cells were split into three parts to determine the absorbance of the sample, 5 nmol D2-HG spiked sample, and sample background. The protein concentration of cell lysate was determined using the BCA Protein Assay Kit (Thermo Scientific). D2-HG was determined in triplicate according to the manufacturer’s protocol and expressed as nmol/mg protein.
Cell proliferation and cell-cycle analysis
Cells expressing luciferase were seeded in a 96-well plate at 100 cells per well in triplicate. Cell proliferation was determined through luciferase activity every 24 hours for 6 consecutive days with a luciferase assay kit (Biotium) or cell viability kit (Promega) and a microplate reader (Turner BioSystems). Relative luciferase units were normalized with background subtraction. Nonlinear regression curve fit was performed using exponential growth equation, and two-way ANOVA was used for the analysis of statistical significance (GraphPad Prism 7.0). Cell-cycle profiling was performed in quadruplicate by labeling the cells with 4′,6-diamidino-2-phenylindole (DAPI) to a final concentration of 10 μg/mL. Cells were then analyzed by flow cytometry (BD FACSCanto, BD Biosciences) with BD FACSDiva Software (BD Biosciences).
Gene expression
Gene expression analysis at genomic DNA, RNA, and protein levels was performed essentially as described before [15]. Amplicon intensities were quantified using an open-source image analysis platform (Fiji) and normalized to Actb expression. Dilutions of primary antibodies for Western blotting are as follows: 1:1000 anti-PDGFB (Santa Cruz Biotechnology), 1:5000 anti-β-actin (Sigma), and 1:500 anti-HA (Abcam).
Mouse models and bioluminescent imaging
All experiments and procedures involving live mice were approved by the University of Utah Institutional Animal Care and Use Committee. Transplantation of transduced cells into the subcutaneous and intracranial sites and bioluminescent imaging of tumor volume were performed essentially as described previously [58]. Alternatively, subcutaneous tumor growth was measured with an electronic caliper, once a week for 6 weeks. The tumor volume was calculated using the formula (length × width2) / 2.
Spontaneous gliomas were generated in Nes-tva or Nes-tva;Cdkn2a mice as described previously [15, 26]. Briefly, 1–2-day-old newborns were subjected to intracranial injection of DF-1 producer cells expressing genes of interest. The cell number per injection was 3 × 104 PDGFB mixed with equal numbers of IDH1 or IDH1R132H or 3 × 104 PDGFB or mCherry–PDGFB mixed with 1 × 105 YFP* or YFP–IDH1R132H. For Cdkn2a deletion, additional 1 × 105 DF-1 cells expressing Cre recombinase was included. These mice were terminated by the end of 5 weeks post injection or earlier. The cell number for IDH1–PDGFB or IDH1R132H–PDGFB was 5 × 105. For survival analysis, these mice were sacrificed by the end of 8 weeks or earlier if any of the following symptoms was found: severe lethargy, pronounced hydrocephalus, and severe cachexia.
Autopsied brains were embedded in paraffin after formalin fixation, sectioned at 3 μm, and stained with hematoxylin and eosin for histological analysis. For fluorescent microscopy, samples were flash-frozen in liquid nitrogen, embedded in OCT compound, and sectioned at 30 μM thickness with a cryostat microtome (Leica CM1950). Sections were mounted with 30% glycerin containing 10 μg/mL DAPI prior to imaging with a Nikon A1R confocal microscope and NIS-elements confocal software (Nikon Instruments). The image was converted with an open-source image analysis platform (Fiji).
Immunohistochemistry
Immunohistochemistry was performed in 3-μm sections of formalin-fixed and paraffin-embedded tissues. Primary antibodies and their corresponding dilutions are as follows: 1:200 anti-Ki67 (EMD Millipore), 1:100 anti-PDGFB (Santa Cruz Biotechnology), 1:100 anti-GFP (Novus Biologicals), 1:500 anti-HA (Abcam), 1:200 anti-IDH1R132H (HistoBioTec DIA-H09) or 1:200 anti-IDH1R132H (EMD Millipore), and 1:2000 anti-Olig2 (EMD Millipore). Secondary antibodies used were anti-mouse Fab antibody (Dako) at 1:100 dilution, or ready-to-use kits ImmPRESS™ HRP Anti-Rabbit IgG Polymer Detection Kit (Vector Laboratories), ImmPRESS™ HRP Anti-Goat IgG Polymer Detection Kit (Vector Laboratories), and Mouse on Mouse Elite Peroxidase Kit (Vector Laboratories) followed by DAB (3-3’ diaminobenzidine) as the chromogen and counterstained with hematoxylin.
Abbreviations
D2-HG: D-2-hydroxyglutarate; IDH1: Isocitrate dehydrogenase 1; IDH1R132H: IDH1 with histidine substitution at arginine 132; luc: Luciferase; NADPH: Nicotinamide adenine dinucleotide phosphate; PDGFB: Platelet-derived growth factor B; RCAS: Replication-competent avian sarcoma-leukosis virus long-terminal repeat with splice acceptor; YFP: Yellow fluorescent protein; WHO: World Health Organization.
Author contributions
P.D.B.T. and L.E.H. conceived and designed the study and wrote the manuscript. P.D.B.T., B.X., Y.C., S.A., S.T., and D.G. performed experiments. All authors reviewed and commented on the manuscript.
ACKNOWLEDGMENTS
Authors wish to thank Kristin Kraus for editorial assistance and Shauna Berg, Sean Lyne, and Tallis Blossom for technical assistance. Authors also acknowledge technical support from the Preclinical Research Resource and Research Histology Core at the Huntsman Cancer Institute and Fluorescence Microscopy Core at the University of Utah. The cores were supported by the award P30CA042014 from the National Cancer Institute and the NCRR Shared Equipment Grant 1S10RR024761-01).
CONFLICTS OF INTEREST
The authors declare no competing interest.
FUNDING
B.X. and Y.C. were supported by Jiangxi Province Foreign Science and Technology Cooperation Plan (20151BDH80009). L.E.H. was supported by the Department of Neurosurgery at the University of Utah and awards from the Huntsman Cancer Foundation.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807–12. https://doi.org/10.1126/science.1164382.
2. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008; 116:597–602. https://doi.org/10.1007/s00401-008-0455-2.
3. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–73. https://doi.org/10.1056/NEJMoa0808710.
4. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013; 3:730–41. https://doi.org/10.1158/2159-8290.CD-13-0083.
5. Miller JJ, Shih HA, Andronesi OC, Cahill DP. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer. 2017; 123:4535–46. https://doi.org/10.1002/cncr.31039.
6. Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, He T, Shi X, Gerszten RE, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015; 28:773–84. https://doi.org/10.1016/j.ccell.2015.11.006.
7. Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018; 37:1949–60. https://doi.org/10.1038/s41388-017-0077-z.
8. Bralten LBC, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM, Jansen EEW, Struys EA, Jakobs C, Salomons GS, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011; 69:455–63. https://doi.org/10.1002/ana.22390.
9. Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, Forrest WF, Kwong M, Hedehus M, Hatzivassiliou G, Friedman LS, Phillips HS. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci USA. 2014; 111:14217–22. https://doi.org/10.1073/pnas.1409653111.
10. Birner P, Pusch S, Christov C, Mihaylova S, Toumangelova-Uzeir K, Natchev S, Schoppmann SF, Tchorbanov A, Streubel B, Tuettenberg J, Guentchev M. Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer. 2014; 120:2440–7. https://doi.org/10.1002/cncr.28732.
11. Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, Pieper RO, Ronen SM, Weiss S, Luchman HA, Cairncross JG. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014; 16:686–95. https://doi.org/10.1093/neuonc/not243.
12. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, et al, and TCGA Research Network. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016; 164:550–63. https://doi.org/10.1016/j.cell.2015.12.028.
13. Huang LE, Cohen AL, Colman H, Jensen RL, Fults DW, Couldwell WT. IGFBP2 expression predicts IDH-mutant glioma patient survival. Oncotarget. 2017; 8:191–202. https://doi.org/10.18632/oncotarget.13329.
14. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, Yu M, Skibbe J, Dai Q, et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell. 2018; 172:90–105.e23. https://doi.org/10.1016/j.cell.2017.11.031.
15. Tiburcio PDB, Xiao B, Berg S, Asper S, Lyne S, Zhang Y, Zhu X, Yan H, Huang LE. Functional requirement of a wild-type allele for mutant IDH1 to suppress anchorage-independent growth through redox homeostasis. Acta Neuropathol. 2018; 135:285–98. https://doi.org/10.1007/s00401-017-1800-0.
16. Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA, Yang R, Lopez GY, He Y, McLendon RE, Bigner DD, Yan H. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013; 73:496–501. https://doi.org/10.1158/0008-5472.CAN-12-2852.
17. Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK, Thompson CB. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. 2013; 288:3804–15. https://doi.org/10.1074/jbc.M112.435495.
18. Borodovsky A, Salmasi V, Turcan S, Fabius AWM, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, Riggins GJ. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013; 4:1737–47. https://doi.org/10.18632/oncotarget.1408.
19. Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, Smirnov IV, Marshall R, Souza CF, Shen Y, Viswanath P, Noushmehr H, Ronen SM, et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutantIDH1. Proc Natl Acad Sci USA. 2017; 114:10743–8. https://doi.org/10.1073/pnas.1708914114.
20. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013; 340:626–30. https://doi.org/10.1126/science.1236062.
21. Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, Capper D, Sahm F, Eisel J, Beck AC, Jugold M, Eichwald V, Kaulfuss S, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017; 133:629–44. https://doi.org/10.1007/s00401-017-1677-y.
22. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001; 15:1913–25. https://doi.org/10.1101/gad.903001.
23. Tchougounova E, Kastemar M, Bråsäter D, Holland EC, Westermark B, Uhrbom L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene. 2007; 26:6289–96. https://doi.org/10.1038/sj.onc.1210455.
24. Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009; 28:2266–75. https://doi.org/10.1038/onc.2009.76.
25. Waitkus MS, Pirozzi CJ, Moure CJ, Diplas BH, Hansen LJ, Carpenter AB, Yang R, Wang Z, Ingram BO, Karoly ED, Mohney RP, Spasojevic I, McLendon RE, et al. Adaptive Evolution of the GDH2 Allosteric Domain Promotes Gliomagenesis by Resolving IDH1R132H-Induced Metabolic Liabilities. Cancer Res. 2018; 78:36–50. https://doi.org/10.1158/0008-5472.CAN-17-1352.
26. Tiburcio PD, Lyne SB, Eric Huang L. In Vivo Manipulation of HIF-1α Expression During Glioma Genesis. Methods Mol Biol. 2018; 1742:227–35. https://doi.org/10.1007/978-1-4939-7665-2_20.
27. Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, Kachman M, May JL, Bartom E, Hua Y, Mishra RK, Schiltz GE, Dubrovskyi O, et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Reports. 2017; 19:1858–73. https://doi.org/10.1016/j.celrep.2017.05.014.
28. Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, Hockenbery D, Houghton AM, Holland EC. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017; 31:774–86. https://doi.org/10.1101/gad.294991.116.
29. Pirozzi CJ, Carpenter AB, Waitkus MS, Wang CY, Zhu H, Hansen LJ, Chen LH, Greer PK, Feng J, Wang Y, Bock CB, Fan P, Spasojevic I, et al. Mutant IDH1 Disrupts the Mouse Subventricular Zone and Alters Brain Tumor Progression. Mol Cancer Res. 2017; 15:507–20. https://doi.org/10.1158/1541-7786.MCR-16-0485.
30. Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987; 61:3004–12.
31. Boerkoel CF, Federspiel MJ, Salter DW, Payne W, Crittenden LB, Kung HJ, Hughes SH. A new defective retroviral vector system based on the Bryan strain of Rous sarcoma virus. Virology. 1993; 195:669–79. https://doi.org/10.1006/viro.1993.1418.
32. Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, Williams BO, Varmus HE. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene. 1999; 18:5253–60. https://doi.org/10.1038/sj.onc.1203087.
33. Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci USA. 1998; 95:1218–23.
34. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009; 2:89–95.
35. Moore LM, Holmes KM, Smith SM, Wu Y, Tchougounova E, Uhrbom L, Sawaya R, Bruner JM, Fuller GN, Zhang W. IGFBP2 is a candidate biomarker for Ink4a-Arf status and a therapeutic target for high-grade gliomas. Proc Natl Acad Sci USA. 2009; 106:16675–9. https://doi.org/10.1073/pnas.0900807106.
36. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jäger D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010; 20:245–54. https://doi.org/10.1111/j.1750-3639.2009.00352.x.
37. Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, Nguyen SA, Chan JA, Weljie AM, Cairncross JG, Weiss S. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012; 14:184–91. https://doi.org/10.1093/neuonc/nor207.
38. Navis AC, Niclou SP, Fack F, Stieber D, van Lith S, Verrijp K, Wright A, Stauber J, Tops B, Otte-Holler I, Wevers RA, van Rooij A, Pusch S, et al. Increased mitochondrial activity in a novel IDH1-R132H mutant human oligodendroglioma xenograft model: in situ detection of 2-HG and α-KG. Acta Neuropathol Commun. 2013; 1:18. https://doi.org/10.1186/2051-5960-1-18.
39. Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, Dias-Santagata D, Ellisen LW, Borger DR, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014; 20:2898–909. https://doi.org/10.1158/1078-0432.CCR-13-3052.
40. Pellegatta S, Valletta L, Corbetta C, Patanè M, Zucca I, Riccardi Sirtori F, Bruzzone MG, Fogliatto G, Isacchi A, Pollo B, Finocchiaro G. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun. 2015; 3:4. https://doi.org/10.1186/s40478-014-0180-0.
41. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, et al, and Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015; 372:2481–98.
42. Khurshed M, Molenaar RJ, Lenting K, Leenders WP, van Noorden CJF. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget. 2017; 8:49165–77. https://doi.org/10.18632/oncotarget.17106.
43. Lenting K, Khurshed M, Peeters TH, van den Heuvel CNAM, van Lith SAM, de Bitter T, Hendriks W, Span PN, Molenaar RJ, Botman D, Verrijp K, Heerschap A, Ter Laan M, et al. Isocitrate dehydrogenase 1-mutated human gliomas depend on lactate and glutamate to alleviate metabolic stress. FASEB J. 2018 Jul 12 [Epub ahead of print].
44. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009; 30:7–11. https://doi.org/10.1002/humu.20937.
45. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009; 125:353–5. https://doi.org/10.1002/ijc.24379.
46. Piaskowski S, Bienkowski M, Stoczynska-Fidelus E, Stawski R, Sieruta M, Szybka M, Papierz W, Wolanczyk M, Jaskolski DJ, Liberski PP, Rieske P. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br J Cancer. 2011; 104:968–70. https://doi.org/10.1038/bjc.2011.27.
47. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, Harris IS, Holmes R, Wakeham A, Haight J, You-Ten A, Li WY, Schalm S, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012; 488:656–9. https://doi.org/10.1038/nature11323.
48. Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, Cairns RA, McCracken S, Wakeham A, Haight J, Ten AY, Snow B, Ueda T, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012; 26:2038–49. https://doi.org/10.1101/gad.198200.112.
49. Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L, Pfeffer I, Sibson N, Goldin R, et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell. 2016; 30:578–94. https://doi.org/10.1016/j.ccell.2016.08.017.
50. Akbay EA, Moslehi J, Christensen CL, Saha S, Tchaicha JH, Ramkissoon SH, Stewart KM, Carretero J, Kikuchi E, Zhang H, Cohoon TJ, Murray S, Liu W, et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014; 28:479–90. https://doi.org/10.1101/gad.231233.113.
51. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014; 343:189–93. https://doi.org/10.1126/science.1239947.
52. Bai H, Harmancı AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, Krischek B, Özduman K, Omay SB, Sorensen EA, Turcan Ş, Bakırcığlu M, Carrión-Grant G, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016; 48:59–66. https://doi.org/10.1038/ng.3457.
53. Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, Viale A, Riggins GJ, Chan TA. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013; 4:1729–36. https://doi.org/10.18632/oncotarget.1412.
54. Johannessen TA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R, Pieper RO. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol Cancer Res. 2016; 14:976–83. https://doi.org/10.1158/1541-7786.MCR-16-0141.
55. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al, and Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98–110. https://doi.org/10.1016/j.ccr.2009.12.020.
56. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2015; 529:110–4. https://doi.org/10.1038/nature16490.
57. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008; 22:1276–312. https://doi.org/10.1101/gad.1653708.
58. Choi H, Gillespie DL, Berg S, Rice C, Couldwell S, Gu J, Colman H, Jensen RL, Huang LE. Intermittent induction of HIF-1α produces lasting effects on malignant progression independent of its continued expression. PLoS One. 2015; 10:e0125125. https://doi.org/10.1371/journal.pone.0125125.