
INTRODUCTION
The main components of the conventional axis of the renin-angiotensin system (RAS) [such as angiotensinogen (AGT), renin, angiotensin-converting enzyme (ACE), angiotensin I (Ang I), angiotensin II (Ang II)], function as an intricate peptide signaling network through several receptors [including angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R)] [1]. AGT is produced and released into circulation by the liver, then AGT is hydrolyzed by renin, which is produced by the juxtaglomerular cells of the kidney to form Ang I [1]. Afterwards Ang I is hydrolyzed by ACE in the endothelial cells of the lungs, to produce the biologically active Ang II. Ang II interacts with two different receptors, AT1R and AT2R [1]. The alternative axis comprises ACE2, which is known as ACE-related carboxypeptidase or angiotensin-converting enzyme homolog (ACEH). ACE2 is mostly found in the vascular endothelial cells and renal tubular epithelium [2] ACE2 cleaves Ang II to Ang-(1-7), whereas ACE produces Ang-(1-7) by cleaving Ang-(1-9). Ang-(1-7) commonly acts via mitochondrial assembly receptor (Mas receptor or MasR) [3]. Another peptide alamandine is generated either by the cleavage of Ang A or Ang-(1-7), by ACE and decarboxylase (DC) respectively. Alamandine binds through MAS-related G protein couple receptor D (MRGD) [3]. Several other truncated bioactive peptides have been previously characterized including Angiotensin III [4], Angiotensin IV [5], and Ang A [6], as part of the RAS network (Figure 1).
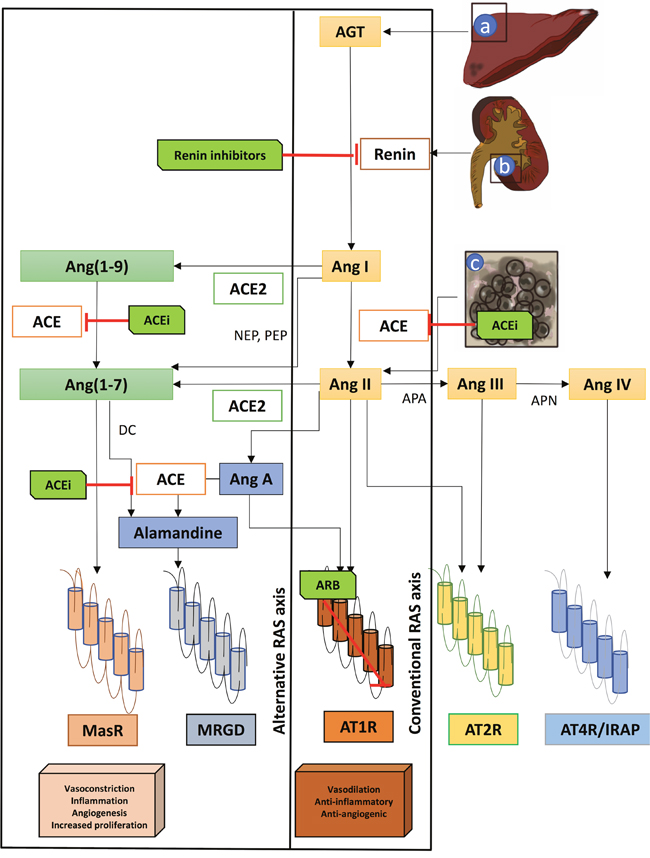
Figure 1: The pro-tumor and anti-tumor mediated effects of the conventional and alternative RAS axis. Angiotensinogen (AGT) is produced and released into circulation by the liver (a), then AGT is hydrolyzed by renin. Renin is produced by the juxtaglomerular cells of the kidney (b) to form Angiotensin I (Ang I). Afterwards Ang I is hydrolyzed by angiotensin-converting enzyme (ACE) in the endothelial cells of the lungs (c), to produce the biologically active angiotensin II (Ang II). Ang II interacts with two different receptors, angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R). ACE2 cleaves Ang II to Angiotensin (1-7) (Ang (1-7)), whereas ACE produces Ang (1-7) by cleaving Angiotensin (1-9) (Ang (1-9)). Ang (1-7) commonly acts via mitochondrial assembly receptor (Mas receptor or MasR). Alamandine is generated either by the cleavage of Ang A or Ang (1-7), by ACE and decarboxylase (DC) respectively. Alamandine binds through MAS-related G protein couple receptor D (MRGD). Angiotensin II is metabolized to Angiotensin III (Ang III) by aminopeptidase A (APA), whereas Ang III is metabolized to Angiotensin IV (Ang IV) by aminopeptidase N (APN). AT4R or Insulin-regulated membrane aminopeptidase (IRAP) is activated by Ang IV. NEP, neutral endopeptidase; PEP, prolyendopeptidase. The inhibition of the conventional axis by ACEi or ARB reduces inflammation, vasoconstriction, and angiogenesis means by which also inhibits tumor growth. Opposite effects have been associated with the activation of the alternative axis including vasodilatation, antinflammatory and anti-angiogenic effects.
In the mid-1970s it became possible for the first time to therapeutically target the components of Ang II/AT1R axis using the angiotensin-converting enzyme inhibitor, captopril [7], and followed by selective blockade of the AT1R with losartan [8]. Renin-angiotensin system inhibitors (RASi) includes two classes of pharmacological agents, angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARBs) [8]. A third class of RASi, a direct renin inhibitor such as aliskiren is also included in the list of RASi approved by the U.S. Food and Drug administration (FDA) [9].
RASi are widely used to treat heart failure [10, 11], myocardial infarction [12], and systemic hypertension [13] and have proved to be effective in preventing cardiovascular [14] and renal associated comorbidities [13]. Additionally RASi have shown potential anti-tumor effects that promise a significant impact in cancer therapy [15]. Several studies have yielded diverging findings on the role of RASi on the incidence of recurrence, metastasis and survival in cancer patients [16]. To comprehend the potential benefit of RASi in cancer patients consideration should be given to factors such as tumor heterogeneity, tumor stage, hormone receptor status, human epidermal growth factor receptor 2 (EGFR2) over-expression, and (neo)adjuvant treatment regimen [15]. A meta-analysis including 55 studies suggested that RASi may improve the survival of cancer patients depending on cancer type and class of RASi [16].
Differences between the mediated effects of the RAS conventional and alternative axis has been previously described. In general, the over-expression of RAS components within the Ang II/AT1R axis (such as ACE and AT1R) is associated with tumor growth and with more aggressive tumor features in several types of human cancer, including breast cancer, ovarian cancer and renal cancer [17–19], whereas Ang II/AT2R and Ang(1-7)/MasR showed opposite effects [20]. Nonetheless there are also conflicting evidence, which may suggest tumor-type specific differences [18]. Similarly, ACE2/Ang-(1-7)/MasR axis dysregulation has been shown to be up-regulated or down-regulated depending on the type of cancer [21–23] (Figure 1).
The role of the various RASi in modulating pathological processes involving cell inflammation [24, 25], fibrosis [26–28], and tumor growth [29–32], has been well-documented, but its influence on anti-tumor immunity is uncertain. The aim of this literature review is to analyze current evidence on the effects of RASi on anti-tumor immunity by reprogramming the tumor microenvironment (TME).
The pathological processes and cellular functions inside the TME which appear to be influenced by RASi include tumor angiogenesis, hypoxia and acidosis within the tumor stroma, inflammatory signaling pathways, oxidative stress, immune cell modulation and the role of kalli krein kinin system (KKS) (Figure 2).
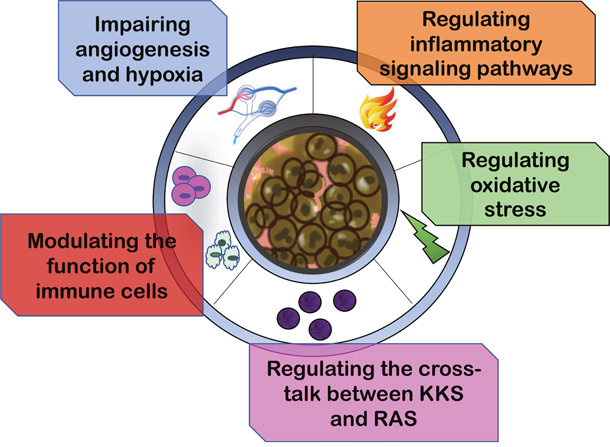
Figure 2: Renin-angiotensin inhibitors reprogram tumor immune microenvironment. The pathological processes and cellular functions inside the tumor microenvironment which appear to be influenced by RASi include tumor angiogenesis, hypoxia and acidosis within the tumor stroma, inflammatory signaling pathways, oxidative stress, immune cell modulation and the role of kalli krein kinin system (KKS).
Regulation of tumor angiogenesis using renin-angiotensin inhibitors impacts cancer progression
Conclusive evidence has shown that targeting Ang II/AT1R impairs neovascularization and vascular permeability and decreases microvessel density by reducing vascular endothelial growth factor (VEGF)-expression [19, 33–35]. Angiogenesis is initiated by the disruption of the endothelial cells (EC) monolayer, and their invasion into the surrounding stroma. The recruitment of pericytes along the basement membrane into the new vessels provides both, mechanical stability and molecular cross talk with EC, which induces VEGF production and survival signaling [36, 37]. The cellular infiltrating populations into the TME can originate either in the blood vessels, the stroma or the bone marrow [37]. For example, cancer-associated fibroblast (CAFs) can be derived from both, tumor stroma and from bone marrow precursors [37]. This cellular plasticity is mediated by the epithelial-mesenchymal transition (EMT) [37]. Many of the different cell types involved in tumor vasculature, and-/or are part of the TME such as pericytes [38, 39] and CAFs [40] express RAS components. Regulating angiogenesis using RASi may extend beyond the converging point of VEGF-response. ACE2 acts as potent counter-regulator against ACE and Ang II activity [20, 41, 42]. ACE2 overexpression or ACE2-Ang-(1-7)- MasR activation may suppress angiogenesis either by inhibiting the production of VEGFa in NSCLC [43], or VEGF receptors attenuation in nasopharyngeal carcinoma [44], respectively.
The use of RASi concomitant with anti-VEGF therapy was associated with better survival in metastatic renal cell carcinoma [45–48], metastatic CRC [49], advanced HCC [50] and glioblastoma [51]. Increased resistance has been reported to VEGF inhibition [52, 53]. The tumor vasculature interacts with the TME via several other mechanisms offering potential new therapeutic targets [54]. The regulation of TME by emerging anti-angiogenic therapies are aiming to counteract tumor progression and to achieve greater tumor destruction. Rather than acute vessel disrupting strategies, there is now a focus on anti-inflammatory regimens. For instance, cabozantinib inhibits the activity of VEGFR, c-MET, AXL and other tyrosine kinases, thereby leading not only to impair angiogenesis but also the disruption of other processes inside the TME [55].
Targeting Ang II/AT1R axis to impair hypoxia and acidosis within the tumor stroma
Local RAS works synergistically and independently of systemic RAS in a paracrine fashion [56]. Ang II mediates effects that reduce tumor perfusion and oxygenation, resulting in hypoxia and subsequent acidosis within the tumor stroma [57]. For example, local Ang II, predominantly exists in hypoxic regions of nasopharyngeal carcinoma and breast cancer cells, where it is autocrinely produced by chymase-dependant rather than ACE dependent mechanism [58]. In which case the action of ACEi will not be effective to inhibit tumor growth. Tumor hypoxia and acidosis trigger a cascade of up-regulation of transcription factors, growth factors and cytokines, including hypoxia inducible factor (HIF), VEGF and Transforming growth factor beta (TGF-β) that together promote immunosuppression within the TME [59, 60]. An immunosuppressive microenvironment includes impaired function of T cells [61], dendritic cells [62] the accumulation of M2-like macrophages [63] and MDSC, but also increased expression of immune check point molecules such as PD-L1 / PD-1 on tumour / stromal cells and the immune cells [64].
In the liver, the local RAS is up-regulated in response to tissue injury and hypoxia [31]. The main effector is Ang II, which normally maintains tissues homeostasis, in this scenario can stimulate the expression pro-angiogenic VEGF via AT1R signaling, leading to wound healing in tissue injury or to tumor neovascularization in hypoxic tumors [65]. ACEi mechanism of action is to reduce the production of Ang II, while ARBs selectively block the action of AT1R receptors inhibiting tumor-associated angiogenesis [16].
Renin-angiotensin inhibitors regulate inflammatory signaling pathways and oxidative stress in tumor biology
The TME is bound to the dynamic between malignant and non-transformed cells [66]. Non-malignant cells are reprogrammed to accomplish tumor-promoting functions during all stages of carcinogenesis [66]. Intercellular interactions modulate the chemical and physical properties of any tissue through a diverse pool of secreted cytokines, chemokines, growth factors, and inflammatory or matrix remodeling enzymes [67]. TGF-β suppresses the differentiation and function of T helper ( TH), CD8+ cells, Natural Killer (NK) cells, and tumor-associated neutrophils (TANs), tumor associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) [67]. Tumor supporting cytokines are released from tumor and stromal cells upon AT1R activation via Ang II including, TGF-β Interleukins (IL-1a, IL-1B, IL-6, IL-8) and MCP-1: (monocyte chemoattractant protein 1), macrophage colony-stimulating factor (M-CSF), cyclooxygenase 2(COX-2), C- reactive protein (CRP) [68]. Immunomodulatory cytokines may up-regulate immunosuppressive pathways, i.e. COX-2 via prostaglandin E2 synthesis, and impair dendritic cell (DC) function by reducing their migration [68, 69]. Ang II/AT1R signaling induces reactive oxygen species (ROS) generation and related proteins such as inducible nitric oxide synthase in the tumor cells and stroma cell [70]. Exposure to ROS in the TME can impair T cell function while enhancing T regs and TAMs, as has been previously reported in prostate cancer [69]. Treatment with the ARB candesartan diminishes ROS generation [71].
Renin-angiotensin inhibitors modulate the function of immune cells within the tumor microenvironment
There are three major immunosuppressive cell types within the tumor mass; MDSCs, TAMs and CAFs. MDSC include both myeloid progenitors cells and immature myeloid cells [72]. These cell have been characterized as a population of inhibitory immune cells that lack typical mature myeloid markers, in mouse and human cancers [72]. Murine and human MDSCs exert their suppressive effects through several mechanisms that inhibit CD8+T cell activation [73], Additionally they induce the development of T regs population, the polarization of macrophages to a TAM-like phenotype, the production of ROS, the upregulation of nitric oxide and depletion of nutrients via increased activity of L-arginase [73]. ACE is required for normal myelopoiesis, as an increased level of CD11(+) bGrl (+) cell-similar to MDSCs phenotype were detected after extramedullary myelopoiesis compensatory to bone marrow incompetence [74]. Likewise, ACE may have a role in the accumulation of MDSCs in the TME. However, the inhibitory function of MDSCs over CD8+T cells in the TME requires further investigation [74].
Similarly, TAMs are usually pro-tumorigenic and have shown that a polarized M2-like phenotype contributes to immunosuppression, whereas M1-like phenotype induces anti-tumor immunity [63]. TAMs tend to accumulate in hypoxic and/or necrotic areas of the tumor [75]. Studies with ACE KO and ACE 10/10 mouse model indicated that ACE plays key role in macrophages to regulate the production of pro-inflammatory cytokines such as IL-12, nitride, and TNF-α in response to lipopolysaccharide (LPS) or chitin (a polymer of N-acetylglucosamine). These induce M1 macrophages or activate M2, respectively [76]. Additionally, M1 induction through granulocyte-macrophage colony stimulating factor (GM-CSF) upregulate ACE expression in human monocytes [76, 77]. Recent findings remain inconclusive as to how RAS modulation establishes M2 macrophage induction and polarization. Some studies suggest that there is no difference between ACE KO, ACE 10/10 and WT model [74]. Others have found an increased or decreased M2 response by RAS blockade [78, 79]. Our laboratory has shown the immunomodulatory role of RAS in influencing TAMs. Blockade of ACE with captopril increased Kupffer cells infiltration in the tumor-bearing liver during an early stage of tumor progression in CRC liver metastasis [80].
Myofibroblasts also known as CAFs are derived from multiple precursors, such as myoepithelial cells, mesenchymal stem cells, smooth muscle cells and EC [81]. Their capability to secrete EGF family growth factors hepatocyte and fibroblast growth factors and insulin growth factor confer them the potential to induce malignant transformation of cells [82, 83]. CAF express AT1R [40], through which Ang II [40] stimulates their proliferation and induces the production of various cytokines including TGF-β [40]. TGF-β in addition to its immunosuppressive role as outlined above, can stimulate EMT in malignant cells contributing to an immunosuppressive TME and therapy resistance [84, 85]. Our laboratory found that RAS is implicated in the regulation of EMT in CRC via both AT1 and AT2 receptors by Ang II modulated migration [86]. Additionally, CAF tumor promoting effects have been associated with promoting neovascularization and recruitment of immune cells in the TME via NF-κβ signaling pathway [87]. A recent study concluded that local RAS activity can modulate the function MDSCs, TAMs and CAFs, and indeed is a potent inducer of immunosuppression in the TME [40]. Inhibition of local RAS augments the induction and infiltration of tumor antigen-specific T cells, reduces the T cell suppressive activity of tumor-infiltrating CD11b+ cells including MDSCs and enhances the T-cell stimulatory activity of CAFs [47].
In addition to CAFs, other types of immune cells also express RAS components. These include monocytes, neutrophils, dendritic cell and T cells, thus presenting pivotal targets for immune therapy [15]. For example, the stimulatory and coinhibitory interactions between lymphocytes infiltrating tumor stroma and tumor cells expressing receptor-ligands pairs, like immune check point inhibitors [cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed cell death-1 (PD-1), and its ligand (PD-L1)] correlates with T cell dysfunction [37]. The inhibition of AT1R enhances the induction of tumor antigen-specific CD8+ cytotoxic T lymphocytes via gp70-Tcells [40]. Additionally, a clinical study of non-metastatic pancreatic ductal adenocarcinoma (PDAC) patients reported significant overall survival in the chronic RASi users, while unbiased gene profiling of resected tumors from this group presented enriched gene signatures associated with antigen processing and presentation, and activity of T cells, and reduced gene signatures associated with tumour aggressiveness. These findings may suggest that RAS signaling could modulate the efficacy of immunotherapy on this type of cancer [88]. Altogether the synergistic use of RASi combined with immune check point inhibitors may improve anti-tumor efficacy by reprograming the TME towards an immune stimulatory milieu.
Targeting tumor non-cellular components using renin-angiotensin inhibitors may enhance antitumor immunity
RAS has been studied under the scope of chronic wound healing and fibrosis [89]. During stromagenesis activated fibroblasts, have a critical function in orchestrating ECM remodeling. The effects of tissue renin angiotensin (tRAS) on chronic wound healing can be divided into two opposite axes: (a) proinflammatory/pro-fibrotic and (b) anti-inflammatory/anti-fibrotic [89]. RAS signaling modulates ECM remodeling via AT1 by stimulation in dermal fibroblast, which promotes deposition of fibrotic ECM [90]. The contrary effect was observed via AT2 stimulation, which reduced collagen deposition counteracting AT1 signaling through engagement of tyrosine phosphatase SHP-1 [91]. In the case of tumor-associated stromagenesis, the pathophysiologic response fails to resolve. RAS activated CAFs secrete collagen and other ECM components which leads to a fibrotic reaction so-called tumor desmoplasia [92]. A tumor desmoplastic environment can be either a physical barrier to immune cell infiltration [93], or can provide the substratum to their interstitial migration [94]. Additionally, RAS activated CAFs and other stromal cells produce immunosuppressive cytokines/ growth factors especially TGF-β in the TME leading to chronic inflammation and effector T- cell disfunction, apoptosis and failure to infiltrate deep into the tumour [61]. RASi targeting of the tumor stroma resulted in collagen I synthesis inhibition by CAFs in a dose-dependent manner in different desmoplastic models including human breast, pancreatic, and skin tumors in mice [95]. A collateral effect of decreasing tumor desmoplasia is the decompression of tumor blood vessels and subsequent reduction of tumor hypoxia by increasing perfusion, which may also enhance antitumor immunity [96].
The cross-regulation of kalli-krein kinin system and renin-angiotensin system by angiotensin-converting enzyme has pleotropic effects in the tumor microenvironment
The KKS comprises several peptides that are produced and cleaved in various sites to finally release the vasoactive kinins [97]. Kinins are originated from their parental molecules, the kininogens [97]. Diverse enzymes known also as kininases such as ACE, neutral endopeptidase (NEP), carboxypeptidase N, carboxypeptidase M, cathepsin X and aminopeptidase metabolize the kinins, [98] (Figure 3). Bradykinin (BK) is the final bioactive peptide produced upon the activation of the KKS, which has pleiotropic functions depending on the pattern signaling triggered through its two different receptors [99]. The kinin receptor 1 (B1R) and the kinin receptor 2 (B2R) activate the same signaling pathway, while inducing a different duration and intensity of the signal modulated by calcium influx [100]. B1R and B2R can be expressed in the same cell type, such as EC, fibroblast and several tumor cells [101]. While B1R expression is induced upon inflammatory conditions and tissue injury, B2R is constituvely expressed [102]. The overall effect of BK through either B1R, B2R, or both is to promote vascular cell proliferation [103], influence barrier permeability [104] and possibly stimulate the release of pro-inflammatory cytokines [105]. The role of kinin receptors has been investigated in various types of cancer, including melanoma, renal, prostate, lung and breast and mesothelioma [106–110], among others. Recently our laboratory found that both human and mouse CRC cell lines showed a strong positive expression of B1R and B2R, and while the inhibition of both receptors delayed tumor growth, only B1R blockade reduced tumor load and increased tumor apoptosis [105]. Indistinctively, blockade of B1R or B2R diminished tumor vascularization in vivo, and impaired proliferation and migration of CRC cell in vitro [111]. The effect of RASi -ACEi- leads to accumulation of kinins and subsequent B1R activation [112], which may influence not only the cellular components part of the TME by activating macrophages and DC, but also can up-regulate cytokine production of IL-6 and IFN-γ [97]. ACEi activate B2R on DC, with increased production IL-12 [113]. Further studies are required to elucidate the therapeutic opportunities of modulating kinin receptors with ACEi and their influence on the TME.
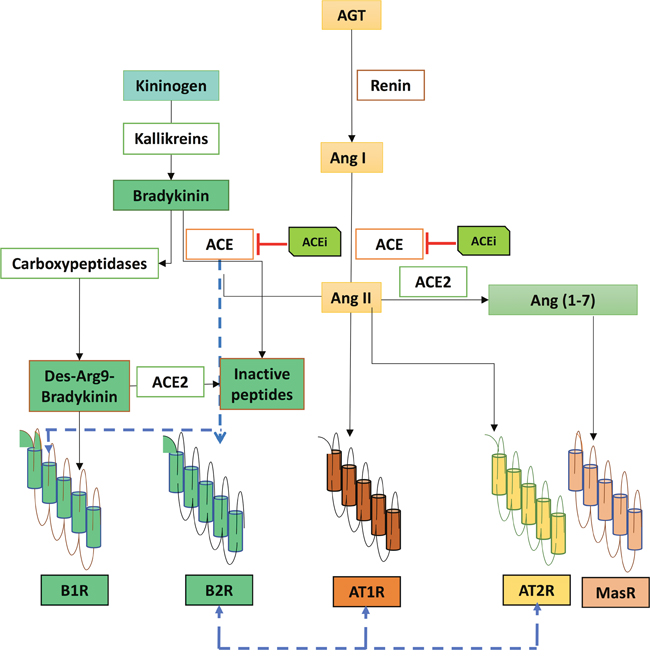
Figure 3: Renin-angiotensin system components intersecting with the kalli krein kinin system. Angiotensin-converting enzyme (ACE) is a major hub intersecting between the crosstalk of both systems, by regulating the levels of Angiotensin II (Ang II) and kinins. ACE metabolizes bradykinin (BK), and converts Angiotensin I (Ang I) into Ang II. Interactions between ACE and kinin receptor 1 (B1R) and/or kinin receptor 2 (B2R) have been reported, same as interactions between angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R) with B2R.
Perspectives and significance
Over the last decade a large number of clinical studies have shown the benefits of using RASi in patients at different stages in several types of cancer. The evidence of RASi impairing tumor growth beyond the function of modulating tumor vasculature is rapidly increasing. A major challenge in the field of cancer therapeutics is the increasing rate of resistance to chemotherapy [114], and immunotherapy [115]. A better understanding of the complex interaction between non-cellular components, tumor cells, tissue resident immune cells and infiltrating immune cells within TME is required to develop new cancer treatment strategies. An immunosuppressive TME affects the efficacy of immune checkpoint therapy, which is reflected in two different clinical scenarios, overall survival and adverse side effects. RASi are able to reprogram the TME, using mechanisms by which they impair hypoxia and acidosis within the tumor stroma, regulate inflammatory signaling pathways and oxidative stress, modulate the function of the non-cellular components and immune cells, and regulate the cross-talk between KKS and RAS. Targeting RAS conventional axis (Ang II/AT1R), KKS and enhancing RAS alternative axis (ACE2/Ang-(1-7)/MasR) seem to be promising strategies to effectively influence TME toward an immunostimulatory milieu, and subsequently improve immunotherapy outcome for a larger population of cancer patients. Further pre-clinical and clinical studies are necessary to propose that the concomitant use of RASi and immunotherapy could not only improve the overall survival of cancer patients, but also could decrease the immunotherapy related side effects. Lastly, additional studies may be required in order to conclude if the effects of RASi on the tumor stroma, not only would depend upon the degree of tumor desmoplasia, but also may depend on how they can influence tumor immune infiltration by either impairing or promoting their migration throughout the interstitial space.
ACKNOWLEDGMENTS
This work was supported by the Department of Science, Technology, and Innovation (COLCIENCIAS-Colombia) (Ph. D overseas 679 program) and University of Melbourne, Faculty of Medicine, Dentistry and Health Sciences (Scholarship 2018-2021).
CONFLICTS OF INTEREST
The authors of this manuscript have no conflicts of interest to declare.
REFERENCES
1. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003; 24:261–71.
2. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000; 87:E1–9.
3. Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013; 112:1104–11.
4. Moeller I, Allen AM, Chai SY, Zhuo J, Mendelsohn FA. Bioactive angiotensin peptides. J Hum Hypertens. 1998; 12:289–93.
5. Mustafa T, Lee JH, Chai SY, Albiston AL, McDowall SG, Mendelsohn FA. Bioactive angiotensin peptides: focus on angiotensin IV. J Renin Angiotensin Aldosterone Syst. 2001; 2:205–10.
6. Walmor De M. Beyond the conventional Renin Angiotensin System. Frontiers. 2014.
7. Erdös EG. Conversion of angiotensin I to angiotensin II. Am J Med. 1976; 60:749–59.
8. Bhardwaj G. How the antihypertensive losartan was discovered. Expert Opin Drug Discov. 2006; 1:609–18.
9. Müller DN, Luft FC. Direct renin inhibition with aliskiren in hypertension and target organ damage. Clin J Am Soc Nephrol. 2006; 1:221–28.
10. Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN, and SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991; 325:293–302.
11. CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987; 316:1429–35.
12. Okura H, Kataoka T, Yoshida K. Renin-angiotensin system inhibitors in patients with myocardial infarction and secondary mitral regurgitation. Heart. 2016; 102:694–700.
13. Daien V, Duny Y, Ribstein J, du Cailar G, Mimran A, Villain M, Daures JP, Fesler P. Treatment of hypertension with renin-angiotensin system inhibitors and renal dysfunction: a systematic review and meta-analysis. Am J Hypertens. 2012; 25:126–32.
14. Tardif JC, Grégoire J. Renin-angiotensin system inhibition and secondary cardiovascular prevention. JAMA. 2013; 310:1130–31.
15. Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: implications for immunotherapy. Sci Transl Med. 2017; 9:eaan5616.
16. Sun H, Li T, Zhuang R, Cai W, Zheng Y. Do renin-angiotensin system inhibitors influence the recurrence, metastasis, and survival in cancer patients?: evidence from a meta-analysis including 55 studies. Medicine (Baltimore). 2017; 96:e6394.
17. Arrieta O, Pineda-Olvera B, Guevara-Salazar P, Hernández-Pedro N, Morales-Espinosa D, Cerón-Lizarraga TL, González-De la Rosa CH, Rembao D, Segura-Pacheco B, Sotelo J. Expression of AT1 and AT2 angiotensin receptors in astrocytomas is associated with poor prognosis. Br J Cancer. 2008; 99:160–66.
18. Arrieta O, Villarreal-Garza C, Vizcaíno G, Pineda B, Hernández-Pedro N, Guevara-Salazar P, Wegman-Ostrosky T, Villanueva-Rodríguez G, Gamboa-Domínguez A. Association between AT1 and AT2 angiotensin II receptor expression with cell proliferation and angiogenesis in operable breast cancer. Tumour Biol. 2015; 36:5627–34.
19. Ino K, Shibata K, Kajiyama H, Yamamoto E, Nagasaka T, Nawa A, Nomura S, Kikkawa F. Angiotensin II type 1 receptor expression in ovarian cancer and its correlation with tumour angiogenesis and patient survival. Br J Cancer. 2006; 94:552–60.
20. Xu J, Fan J, Wu F, Huang Q, Guo M, Lv Z, Han J, Duan L, Hu G, Chen L, Liao T, Ma W, Tao X, Jin Y. The ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Pleiotropic Roles in Cancer. Front Physiol. 2017; 8:276.
21. Yu C, Tang W, Wang Y, Shen Q, Wang B, Cai C, Meng X, Zou F. Downregulation of ACE2/Ang-(1-7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016; 376:268–77.
22. Zheng S, Yang Y, Song R, Yang X, Liu H, Ma Q, Yang L, Meng R, Tao T, Wang S, He J. Ang-(1-7) promotes the migration and invasion of human renal cell carcinoma cells via Mas-mediated AKT signaling pathway. Biochem Biophys Res Commun. 2015; 460:333–40.
23. Zhao Y, Chen X, Cai L, Yang Y, Sui G, Fu S. Angiotensin II/angiotensin II type I receptor (AT1R) signaling promotes MCF-7 breast cancer cells survival via PI3-kinase/Akt pathway. J Cell Physiol. 2010; 225:168–73.
24. Husain K, Hernandez W, Ansari RA, Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J Biol Chem. 2015; 6:209–17.
25. Pacurari M, Kafoury R, Tchounwou PB, Ndebele K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflam. 2014; 2014:689360.
26. Kim G, Kim J, Lim YL, Kim MY, Baik SK. Renin-angiotensin system inhibitors and fibrosis in chronic liver disease: a systematic review. Hepatol Int. 2016; 10:819–28.
27. Saber S, Mahmoud AA, Helal NS, El-Ahwany E, Abdelghany RH. Renin-angiotensin system inhibition ameliorates CCl4-induced liver fibrosis in mice through the inactivation of nuclear transcription factor kappa B. Can J Physiol Pharmacol. 2018; 96:569–76.
28. Zhu Q, Li N, Li F, Zhou Z, Han Q, Lv Y, Sang J, Liu Z. Therapeutic effect of renin angiotensin system inhibitors on liver fibrosis. J Renin Angiotensin Aldosterone Syst. 2016; 17:1470320316628717.
29. Sugimoto M, Yamaoka Y, Shirai N, Furuta T. Role of renin-angiotensin system in gastric oncogenesis. J Gastroenterol Hepatol. 2012; 27:442–51.
30. Wegman-Ostrosky T, Soto-Reyes E, Vidal-Millán S, Sánchez-Corona J. The renin-angiotensin system meets the hallmarks of cancer. J Renin Angiotensin Aldosterone Syst. 2015; 16:227–33.
31. Neo JH, Ager EI, Angus PW, Zhu J, Herath CB, Christophi C. Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer. 2010; 10: 134.
32. Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008; 29:1675–84.
33. Carbajo-Lozoya J, Lutz S, Feng Y, Kroll J, Hammes HP, Wieland T. Angiotensin II modulates VEGF-driven angiogenesis by opposing effects of type 1 and type 2 receptor stimulation in the microvascular endothelium. Cell Signal. 2012; 24:1261–69.
34. Usui T, Sugisaki K, Iriyama A, Yokoo S, Yamagami S, Nagai N, Ishida S, Amano S. Inhibition of corneal neovascularization by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci. 2008; 49:4370–76.
35. Shirotake S, Miyajima A, Kosaka T, Tanaka N, Maeda T, Kikuchi E, Oya M. Angiotensin II type 1 receptor expression and microvessel density in human bladder cancer. Urology. 2011; 77:1009.e19–25.
36. Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol. 2011; 3:a005090.
37. Mittal K, Ebos J, Rini B. Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Oncol. 2014; 41:235–51.
38. Stefanska A, Kenyon C, Christian HC, Buckley C, Shaw I, Mullins JJ, Péault B. Human kidney pericytes produce renin. Kidney Int. 2016; 90:1251–61.
39. Berg AC, Chernavvsky-Sequeira C, Lindsey J, Gomez RA, Sequeira-Lopez ML. Pericytes synthesize renin. World J Nephrol. 2013; 2:11–16.
40. Nakamura K, Yaguchi T, Ohmura G, Kobayashi A, Kawamura N, Iwata T, Kiniwa Y, Okuyama R, Kawakami Y. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2018; 109:54–64.
41. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ Res. 2016; 118:1313–26.
42. Han CD, Ge WS. Up-Regulation of Angiotensin-Converting Enzyme (ACE) Enhances Cell Proliferation and Predicts Poor Prognosis in Laryngeal Cancer. Med Sci Monit. 2016; 22:4132–38.
43. Cheng Q, Zhou L, Zhou J, Wan H, Li Q, Feng Y. ACE2 overexpression inhibits acquired platinum resistance-induced tumor angiogenesis in NSCLC. Oncol Rep. 2016; 36:1403–10.
44. Pei N, Wan R, Chen X, Li A, Zhang Y, Li J, Du H, Chen B, Wei W, Qi Y, Zhang Y, Katovich MJ, Sumners C, et al. Angiotensin-(1-7) Decreases Cell Growth and Angiogenesis of Human Nasopharyngeal Carcinoma Xenografts. Mol Cancer Ther. 2016; 15:37–47.
45. Izzedine H, Derosa L, Le Teuff G, Albiges L, Escudier B. Hypertension and angiotensin system inhibitors: impact on outcome in sunitinib-treated patients for metastatic renal cell carcinoma. Ann Oncol. 2015; 26:1128–33.
46. Keizman D, Gottfried M, Ish-Shalom M, Maimon N, Peer A, Neumann A, Hammers H, Eisenberger MA, Sinibaldi V, Pili R, Hayat H, Kovel S, Sella A, et al. Active smoking may negatively affect response rate, progression-free survival, and overall survival of patients with metastatic renal cell carcinoma treated with sunitinib. Oncologist. 2014; 19:51–60.
47. Keizman D, Huang P, Eisenberger MA, Pili R, Kim JJ, Antonarakis ES, Hammers H, Carducci MA. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: a retrospective examination. Eur J Cancer. 2011; 47:1955–61.
48. McKay RR, Rodriguez GE, Lin X, Kaymakcalan MD, Hamnvik OP, Sabbisetti VS, Bhatt RS, Simantov R, Choueiri TK. Angiotensin system inhibitors and survival outcomes in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2015; 21:2471–79.
49. Osumi H, Matsusaka S, Wakatsuki T, Suenaga M, Shinozaki E, Mizunuma N. Angiotensin II type-1 receptor blockers enhance the effects of bevacizumab-based chemotherapy in metastatic colorectal cancer patients. Mol Clin Oncol. 2015; 3:1295–300.
50. Pinter M, Weinmann A, Wörns MA, Hucke F, Bota S, Marquardt JU, Duda DG, Jain RK, Galle PR, Trauner M, Peck-Radosavljevic M, Sieghart W. Use of inhibitors of the renin-angiotensin system is associated with longer survival in patients with hepatocellular carcinoma. United European Gastroenterol J. 2017; 5:987–96.
51. Levin VA, Chan J, Datta M, Yee JL, Jain RK. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J Neurooncol. 2017; 134:325–30.
52. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014; 17:471–94.
53. Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT, Sorensen AG. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009; 6:327–38.
54. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018; 9:115.
55. Tannir NM, Schwab G, Grünwald V. Cabozantinib: an Active Novel Multikinase Inhibitor in Renal Cell Carcinoma. Curr Oncol Rep. 2017; 19:14.
56. Campbell DJ. Clinical relevance of local Renin Angiotensin systems. Front Endocrinol (Lausanne). 2014; 5:113.
57. Thews O, Kelleher DK, Vaupel P. Disparate responses of tumour vessels to angiotensin II: tumour volume-dependent effects on perfusion and oxygenation. Br J Cancer. 2000; 83:225–31.
58. Xie G, Liu Y, Yao Q, Zheng R, Zhang L, Lin J, Guo Z, Du S, Ren C, Yuan Q, Yuan Y. Hypoxia-induced angiotensin II by the lactate-chymase-dependent mechanism mediates radioresistance of hypoxic tumor cells. Sci Rep. 2017; 7:42396. https://www.nature.com/articles/srep42396#supplementary-information.
59. Riemann A, Reime S, Thews O. Tumor Acidosis and Hypoxia Differently Modulate the Inflammatory Program: Measurements In Vitro and In Vivo. Neoplasia. 2017; 19:1033–42.
60. Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron. 2008; 1:53–68.
61. Anderson KG, Stromnes IM, Greenberg PD. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell. 2017; 31:311–25.
62. Shurin GV, Ma Y, Shurin MR. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron. 2013; 6:159–67.
63. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014; 6:1670–90.
64. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017; 8:561.
65. Walther T, Menrad A, Orzechowski HD, Siemeister G, Paul M, Schirner M. Differential regulation of in vivo angiogenesis by angiotensin II receptors. FASEB J. 2003; 17:2061–67.
66. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012; 125:5591–96.
67. Fearon DT. Immune-Suppressing Cellular Elements of the Tumor Microenvironment. Annual Review of Cancer Biology. 2017; 1:241–55.
68. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HM, Signori E, Honoki K, Georgakilas AG, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015 (Suppl ); 35:S185–98.
69. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015; 162:1257–70.
70. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004; 5:818–27.
71. Uemura H, Ishiguro H, Ishiguro Y, Hoshino K, Takahashi S, Kubota Y. Angiotensin II induces oxidative stress in prostate cancer. Mol Cancer Res. 2008; 6:250–58.
72. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016; 7:12150.
73. Pyzer AR, Cole L, Rosenblatt J, Avigan DE. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int J Cancer. 2016; 139:1915–26.
74. Okwan-Duodu D, Landry J, Shen XZ, Diaz R. Angiotensin-converting enzyme and the tumor microenvironment: mechanisms beyond angiogenesis. Am J Physiol Regul Integr Comp Physiol. 2013; 305:R205–15.
75. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004; 104:2224–34.
76. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010; 11:936–44.
77. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. 2009; 86:411–21.
78. Aki K, Shimizu A, Masuda Y, Kuwahara N, Arai T, Ishikawa A, Fujita E, Mii A, Natori Y, Fukunaga Y, Fukuda Y. ANG II receptor blockade enhances anti-inflammatory macrophages in anti-glomerular basement membrane glomerulonephritis. Am J Physiol Renal Physiol. 2010; 298:F870–82.
79. Kohlstedt K, Trouvain C, Namgaladze D, Fleming I. Adipocyte-derived lipids increase angiotensin-converting enzyme (ACE) expression and modulate macrophage phenotype. Basic Res Cardiol. 2011; 106:205–15.
80. Wen SW, Ager EI, Neo J, Christophi C. The renin angiotensin system regulates Kupffer cells in colorectal liver metastases. Cancer Biol Ther. 2013; 14:720–27.
81. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010; 15: 166-79.
82. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. 2017; 14:2611–20.
83. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel). 2015; 7:2443–58.
84. Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, Chouaib S. New insights into the role of EMT in tumor immune escape. Mol Oncol. 2017; 11:824–46.
85. Chae YK, Chang S, Ko T, Anker J, Agte S, Iams W, Choi WM, Lee K, Cruz M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci Rep. 2018; 8:2918.
86. Nguyen L, Ager EI, Neo J, Christophi C. Regulation of colorectal cancer cell epithelial to mesenchymal transition by the renin angiotensin system. J Gastroenterol Hepatol. 2016; 31:1773–82.
87. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010; 17:135–47.
88. Liu H, Naxerova K, Pinter M, Incio J, Lee H, Shigeta K, Ho WW, Crain JA, Jacobson A, Michelakos T, Dias-Santos D, Zanconato A, Hong TS, et al. Use of Angiotensin System Inhibitors Is Associated with Immune Activation and Longer Survival in Nonmetastatic Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017; 23:5959–69.
89. Bernasconi R, Nyström A. Balance and circumstance: the renin angiotensin system in wound healing and fibrosis. Cell Signal. 2018; 51:34–46.
90. Min LJ, Cui TX, Yahata Y, Yamasaki K, Shiuchi T, Liu HW, Chen R, Li JM, Okumura M, Jinno T, Wu L, Iwai M, Nahmias C, et al. Regulation of collagen synthesis in mouse skin fibroblasts by distinct angiotensin II receptor subtypes. Endocrinology. 2004; 145:253–60.
91. Cui T, Nakagami H, Iwai M, Takeda Y, Shiuchi T, Daviet L, Nahmias C, Horiuchi M. Pivotal role of tyrosine phosphatase SHP-1 in AT2 receptor-mediated apoptosis in rat fetal vascular smooth muscle cell. Cardiovasc Res. 2001; 49:863–71.
92. Schnittert J, Bansal R, Storm G, Prakash J. Integrins in wound healing, fibrosis and tumor stroma: high potential targets for therapeutics and drug delivery. Adv Drug Deliv Rev. 2018; 129:37–53.
93. Watt J, Kocher HM. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology. 2013; 2:e26788.
94. Mrass P, Kinjyo I, Ng LG, Reiner SL, Puré E, Weninger W. CD44 mediates successful interstitial navigation by killer T cells and enables efficient antitumor immunity. Immunity. 2008; 29:971–85.
95. Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011; 108:2909–14.
96. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013; 4:2516.
97. da Costa PL, Sirois P, Tannock IF, Chammas R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014; 345:27–38.
98. Blaes N, Girolami JP. Targeting the ‘Janus face’ of the B2-bradykinin receptor. Expert Opin Ther Targets. 2013; 17:1145–66.
99. Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005; 57:27–77.
100. Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci. 2005; 99:6–38.
101. Wu J, Akaike T, Hayashida K, Miyamoto Y, Nakagawa T, Miyakawa K, Müller-Esterl W, Maeda H. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int J Cancer. 2002; 98:29–35.
102. Hall JM. Bradykinin receptors. Gen Pharmacol. 1997; 28:1–6.
103. Ikeda Y, Hayashi I, Kamoshita E, Yamazaki A, Endo H, Ishihara K, Yamashina S, Tsutsumi Y, Matsubara H, Majima M. Host stromal bradykinin B2 receptor signaling facilitates tumor-associated angiogenesis and tumor growth. Cancer Res. 2004; 64:5178–85.
104. Ishihara K, Kamata M, Hayashi I, Yamashina S, Majima M. Roles of bradykinin in vascular permeability and angiogenesis in solid tumor. Int Immunopharmacol. 2002; 2:499–509.
105. Bras G, Bochenska O, Rapala-Kozik M, Guevara-Lora I, Faussner A, Kozik A. Extracellular aspartic protease SAP2 of Candida albicans yeast cleaves human kininogens and releases proinflammatory peptides, Met-Lys-bradykinin and des-Arg(9)-Met-Lys-bradykinin. Biol Chem. 2012; 393:829–39.
106. Maria AG, Dillenburg-Pilla P, Reis RI, Floriano EM, Tefé-Silva C, Ramos SG, Pesquero JB, Nahmias C, Costa-Neto CM. Host kinin B1 receptor plays a protective role against melanoma progression. Sci Rep. 2016; 6:22078. https://www.nature.com/articles/srep22078#supplementary-information.
107. Chee J, Singh J, Naran A, Misso NL, Thompson PJ, Bhoola KD. Novel expression of kallikreins, kallikrein-related peptidases and kinin receptors in human pleural mesothelioma. Biol Chem. 2007; 388:1235–42.
108. Dlamini Z, Bhoola KD. Upregulation of tissue kallikrein, kinin B1 receptor, and kinin B2 receptor in mast and giant cells infiltrating oesophageal squamous cell carcinoma. J Clin Pathol. 2005; 58:915–22.
109. Molina L, Matus CE, Astroza A, Pavicic F, Tapia E, Toledo C, Perez JA, Nualart F, Gonzalez CB, Burgos RA, Figueroa CD, Ehrenfeld P, Poblete MT. Stimulation of the bradykinin B(1) receptor induces the proliferation of estrogen-sensitive breast cancer cells and activates the ERK1/2 signaling pathway. Breast Cancer Res Treat. 2009; 118:499–510.
110. Moodley R, Snyman C, Odhav B, Bhoola KD. Visualisation of transforming growth factor-beta 1, tissue kallikrein, and kinin and transforming growth factor-beta receptors on human clear-cell renal carcinoma cells. Biol Chem. 2005; 386:375–82.
111. da Costa PL, Wynne D, Fifis T, Nguyen L, Perini M, Christophi C. The kallikrein-Kinin system modulates the progression of colorectal liver metastases in a mouse model. BMC Cancer. 2018; 18:382.
112. Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG. Activation of bradykinin B1 receptor by ACE inhibitors. Int Immunopharmacol. 2002; 2:1787–93.
113. Aliberti J, Viola JP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol. 2003; 170:5349–53.
114. Zuccala E. Chemotherapy: clocking up resistance. Nat Rev Cancer. 2016; 16:343.
115. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018; 118:9–16.