
INTRODUCTION
With 239,000 new cases per year, ovarian cancer is the seventh most frequent cancer in women in the world [1]. The highest age-adjusted incidence rates are seen in developed countries. In the United States alone, approximately 14,000 women per year die of ovarian cancer. Most patients are diagnosed at an advanced stage of disease [1]. Patients are initially treated with surgical debulking followed by platinum-based chemotherapy [2–4]. Approximately 75% of patients respond to primary treatment but quickly develop recurrent disease [5–9]. There is a high medical need particularly in recurrent disease and new active treatment modalities beyond chemotherapy are required [2].
Recently, targeted agents have been added as treatment options for ovarian cancer. The vascular endothelial growth factor (VEGF) inhibitor bevacizumab has been approved by the European Medicines Agency (EMA) and the U.S. Food & Drug Administration (FDA) for the treatment of advanced ovarian cancer patients in combination with chemotherapy [10], and the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib has been approved for BRCA1/BRCA2-mutated high-grade serous ovarian cancer [11, 12].
Mesothelin represents another candidate target for the treatment of ovarian cancer [13–17]. Mesothelin is highly expressed on the surface of tumor cells in various cancers, including ovarian cancer, whereas in normal tissue mesothelin shows limited expression. Mesothelin is frequently co-expressed with and binds to CA125, a well-established ovarian cancer biomarker, and may be involved in the peritoneal spread of ovarian cancer [13, 18, 19]. In epithelial ovarian cancer, high mesothelin expression has been shown to correlate with chemoresistance and poor prognosis [20]. Antibody-based approaches to target mesothelin include the chimeric IgG1 antibody amatuximab, which blocks mesothelin/CA125 interaction [21, 22]. Using 111In-labeled amatuximab and single-photon emission computed tomography-computed tomography (SPECT-CT), tumor-specific amatuximab uptake has been demonstrated in mesothelioma and pancreatic cancer patients [23]. Amatuximab was well tolerated in phase 1 clinical studies with stable disease as the best response as monotherapy [24, 25]. The amatuximab-derivative SS1P resulted in pleuritic chest pains as dose-limiting toxicity in phase 1 clinical trials, and only minor tumor responses and stable disease were achieved [26, 27]. Antitumor activity however was limited by neutralizing antibodies [26].
Antibody drug-conjugates (ADCs) consisting of a cytotoxic payload conjugated to an antibody binding to a tumor antigen have demonstrated efficacy in solid tumors [28–30]. The ADC anetumab ravtansine, a fully human anti-mesothelin antibody (MF-T) coupled via a reducible disulfide linker to a microtubule-targeting toxophore DM4, binds to mesothelin with high affinity and delivers the microtubule inhibitor DM4 to mesothelin-positive tumor cells [31]. Anetumab ravtansine has demonstrated potent antitumor activity and good tolerability as single agent in preclinical models including mesothelioma, pancreatic cancer and ovarian cancer [31].
Recent late-stage clinical trials revealed that addition of a third chemotherapy to the standard of care treatment of epithelial ovarian cancer, i.e. carboplatin and paclitaxel, shows increased toxicities without benefit in survival or tumor control [32, 33]. Therefore, targeted agents for the treatment of ovarian cancer either in monotherapy or in combination with chemotherapy should be explored as they may result in increased treatment benefit with more favorable tolerability [34]. Herein, the therapeutic potential of anetumab ravtansine (BAY-94-9343) in ovarian cancer was investigated in monotherapy and in combination with standard of care chemotherapy and targeted agents.
RESULTS
Mesothelin is internalized via the endosomal pathway and targeted to lysosomes for degradation
To study the internalization of anetumab ravtansine and its localization in cancer cells, the targeting antibody moiety of anetumab ravtansine (anetumab, MF-T) was coupled to a pH-sensitive fluorescent dye and incubated with HT29 human colon adenocarcinoma cells stably transfected with mesothelin (HT29-MSLN). The MF-T co-localized with the endocytosis marker clathrin at the sites of cell-to-cell contacts (Figure 1A). Co-localization with the lysosomal marker lysosome-associated membrane glycoprotein 1 (LAMP-1) was seen in cytoplasmic vesicles (Figure 1B). The MF-T-induced reduction of surface mesothelin expression was confirmed in endogenously mesothelin-positive NCI-H322 human lung cancer and OVCAR-3 human ovarian cancer cells by flow cytometry (Figure 1C).
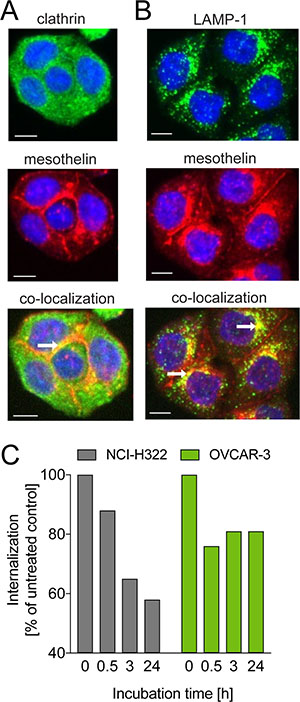
Figure 1: Internalization of the mesothelin-targeted antibody MF-T in cancer cells. Co-staining of mesothelin (red) with (A) clathrin (green) or (B) LAMP-1 (green) in HT29-MSLN colorectal adenocarcinoma cells transfected with human mesothelin as detected by fluorescence microscopy. Co-localization is indicated by yellow fluorescence pointed out by the white arrows. (C) Amount of mesothelin on cell surface in NCI-H322 and OVCAR-3 cells treated with the mesothelin-targeted antibody MF-T (anetumab). The data represent a mean of triplicates. All scale bars indicate 10 μm.
Anetumab ravtansine induces mesothelin degradation and re-synthesis of surface mesothelin
To study the mechanism of action of anetumab ravtansine, OVCAR-3 human ovarian cancer cells endogenously expressing mesothelin were incubated with 100 nM anetumab ravtansine for 4, 16, 24 or 48 h and the expression level of mesothelin was detected by Western blot. The OVCAR-3 cells treated with anetumab ravtansine showed decreased mesothelin expression compared to untreated cells, with the highest difference observed at 24 h (Figure 2A, 2B). A higher molecular weight precursor was induced in response to anetumab ravtansine treatment, indicating re-synthesis of mesothelin. The possible degradation and subsequent re-expression of mesothelin on the cell surface was further investigated by incubating OVCAR-3 cells with an excess amount of MF-T for 4, 24 or 48 h (Figure 2D). The cells were stained for mesothelin using the K1 antibody, which recognizes a different epitope than MF-T. At 4 h, strong mesothelin expression was observed on the surface of untreated OVCAR-3 cells (red arrow in Figure 2C). During a 24 h MF-T incubation, mesothelin levels were reduced (Figure 2D) and the cell membrane appeared unstructured (blue arrow in Figure 2C). After 48 h, mesothelin expression on the cell surface increased again, supporting the hypothesis of internalization-induced degradation and subsequent re-synthesis of surface-localized mesothelin by MF-T.
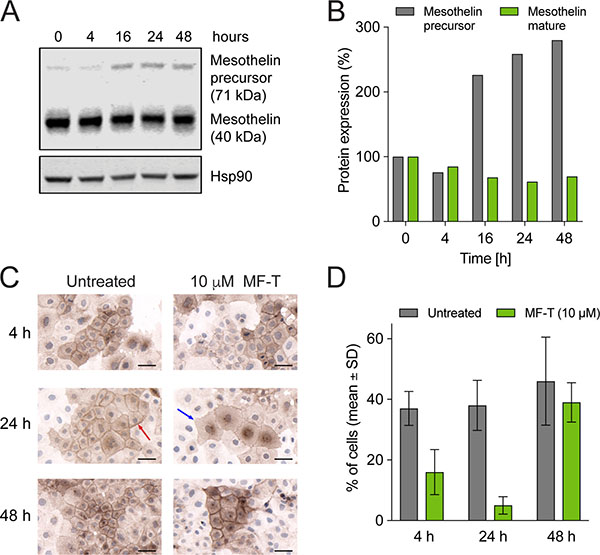
Figure 2: Mesothelin degradation and re-synthesis of surface mesothelin in OVCAR-3 human ovarian cancer cells. (A) Expression of mature mesothelin and mesothelin precursor was analyzed in OVCAR-3 cells treated with 100 nM anetumab ravtansine using Western blot. HSP90 served as a loading control. (B) The percentage of mesothelin expression compared to untreated cells as determined by Western blot described in panel (A). (C) Mesothelin expression in OVCAR-3 cells. OVCAR-3 cells were incubated with or without 10 μM anti-mesothelin antibody MF-T and fixed for staining with the anti-mesothelin K1 antibody. (D) Mesothelin expression on cell surface upon treatment with anetumab ravtansine. The number of OVCAR-3 cells with a clear membrane signal and the number of all cells per a microscope field were counted. The mean percentage of mesothelin-positive cells in 18 microscope fields per group is shown. All scale bars indicate 50 μm.
Anetumab ravtansine induces mitotic arrest, DNA damage and apoptosis in vitro
The cell damage-inducing capability of anetumab ravtansine was examined in OVCAR-3 cells by detecting markers for mitosis (phospho-histone H3), DNA damage (γH2AX) and apoptosis (caspase 7, PARP1). An increase in the phospho-histone H3 (pHH3) level was detected 16 h after the start of treatment, indicating mitotic arrest of the cells (Figure 3A). Furthermore, anetumab ravtansine treatment induced caspase 7 expression within 16 h after the start of treatment. As expected, the activation of caspase 7 was followed by cleavage of PARP1, one of the downstream targets of caspase 7 [35, 36], 48 h after the onset of anetumab ravtansine treatment. DNA damage was also induced, as indicated by an increased γH2AX signal at 16 h. A minor signal was seen already at 4 h, thus preceding the caspase 7 signal.
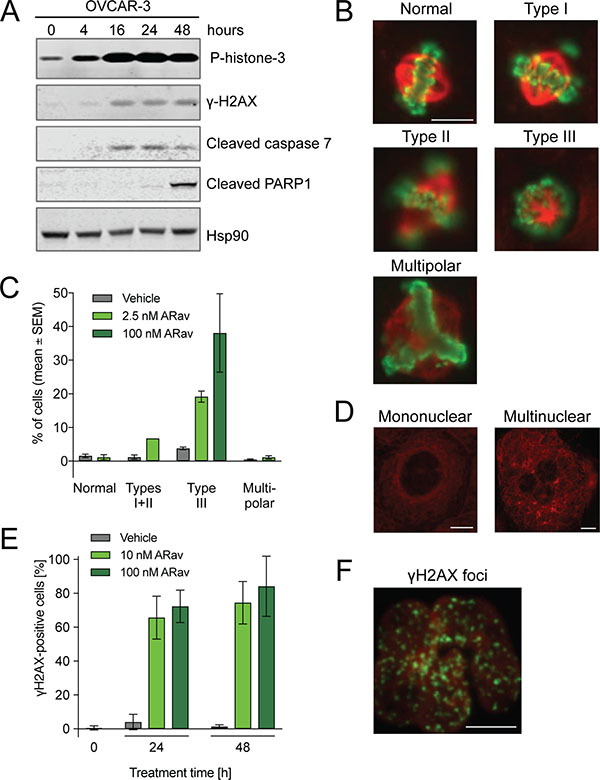
Figure 3: Analysis of anetumab ravtansine mode-of-action in OVCAR-3 human ovarian cancer cells. (A) OVCAR-3 cells treated with 100 nM anetumab ravtansine were assayed for phospho-histone H3, γH2AX, cleaved caspase 7, cleaved PARP1 and HSP90 by Western blot at indicated time points. (B) Representative images from the analysis of mitotic spindle assembly in OVCAR-3 cells treated with 2.5 or 100 nM anetumab ravtansine for 24 h. (C) The percentage of cells described in panel B exhibiting mitotic spindles categorized as normal, type I + II (abnormal bipolar mitotic spindles with uncongressed chromosomes), type III (monopolar spindles enclosed in a ball of chromosomes) or multipolar (multipolar spindles) per four microscopy fields. (D) Representation of mononuclear and multinuclear OVCAR-3 cells upon treatment with 100 nM anetumab ravtansine for 24 h. (E) OVCAR-3 cells were treated with 100 nM anetumab ravtansine for 24 or 48 h and γH2AX was detected by fluorescent microscopy. γH2AX-positive cells as a percentage of all cells (n = 6). (F) Representative fluorescent microscopy images of γH2AX (green) and DNA (red) staining in cells described in panel E. All scale bars indicate 10 μm. In panel B, the scale bar is representative for all images. ARav, anetumab ravtansine.
Next, the effects of anetumab ravtansine on microtubule (MT) organization and the cell cycle were investigated by fluorescence microscopy. In line with the Western blot results illustrated in Figure 3A, OVCAR-3 cells showed mitotic arrest, indicated by a notable increase in pHH3-positive cells after 24 h of 2.5 nM or 100 nM anetumab ravtansine treatment (Figure 3B). These pHH3-positive cells were frequently separated from each other and showed a round appearance. Of note, the maytansine payload of anetumab ravtansine did not result in the depolymerization of the MT network but rather in alterations of the mitotic spindle organization (Figure 3B–3D). Spindle structures were categorized (normal, type I-III, or multipolar) based on the degree of chromosome alignment [37]. At an anetumab ravtansine concentration of 2.5 nM, which is close to the anti-proliferative IC50, the cells showed an increase in chromosome aberration (type I-II) and type III monopolar spindles (Figure 3C). At a higher anetumab ravtansine concentration of 100 nM, almost all cells showed a pHH3-positive type III mitotic phenotype. Following drug exposure, cells in the interphase stage showed more MT bundles and were frequently multinucleated (Figure 3D). Fluorescent microscopy was used to characterize the γH2AX phenotype induced by anetumab ravtansine in OVCAR-3 cells (Figure 3E, 3F). In line with the published results for taxanes [38], we observed an increase of γH2AX foci in the OVCAR-3 cells treated with anetumab ravtansine, suggesting an induction of DNA damage.
Anetumab ravtansine shows potent in vitro efficacy in ovarian cancer cell lines
The antiproliferative activity of anetumab ravtansine was tested in a panel of ovarian cancer cell lines (Table 1). Anetumab ravtansine showed high potency in the tested cell lines, indicated by IC50 values in the nanomolar range. In line with in vitro data published for other ADCs [39], no linear correlation between the in vitro potency and surface mesothelin levels (determined by flow cytometry) could be established (Table 1).
Table 1: In vitro efficacy of anetumab ravtansine and cell surface mesothelin expression levels in a panel of ovarian cancer cell lines
Cell line |
IC50 [nM] |
Surface mesothelin level (antibodies bound per cell) |
|---|---|---|
A2780 |
20.7 |
952 |
AG6000 |
15.4 |
1105 |
BG1 |
5.9 |
900 |
EFO-21 |
20.8 |
9648 |
IGROV1 |
41.9 |
1942 |
NCI/ADR-RES |
42.4 |
53497 |
OVCAR-3 |
10.9 |
19998 |
OVCAR-5 |
11.9 |
1260 |
OVCAR-8 |
32.5 |
41887 |
SK-OV-3 |
4.1 |
3875 |
Potent in vivo efficacy of anetumab ravtansine in preclinical mesothelin-positive ovarian cancer models
Next, the in vivo antitumor efficacy of anetumab ravtansine was tested in two cell line- and eight patient-derived ovarian cancer models with varying histological backgrounds (Table 2). Anetumab ravtansine was clearly efficacious in five out of ten models tested; in the OVCAR-3 cell line-derived and ST103, ST081, ST207 and ST409 patient-derived xenograft models treatment/control (T/C) ratios between 0 and 0.36 were observed. Anetumab ravtansine resulted in total tumor eradication in ST081 and ST103 PDX models. Furthermore, a response of 100% was observed in ST081, ST103 and ST270 PDX models.
Table 2: In vivo efficacy of anetumab ravtansine as a monotherapy in a panel of ovarian cancer cell line- and patient-derived xenograft models
Xenograft |
CDX/PDX |
Ovarian cancer subtype |
T/C |
IHC score (0–3) |
H score (0–300) |
|---|---|---|---|---|---|
ST081 |
PDX |
High-grade serous ovarian cancer |
0a,* |
3 |
150 |
ST103 |
PDX |
High-grade serous ovarian cancer |
0a,* |
3 |
210 |
ST270 |
PDX |
High-grade serous ovarian cancer |
0.04a,* |
3 |
185 |
OVCAR-3 |
CDX |
High-grade serous ovarian cancer |
0.17* |
3 |
180 |
ST467 |
PDX |
Serous papillary carcinoma |
0.25 |
2 |
180 |
ST409 |
PDX |
High-grade serous ovarian cancer |
0.36* |
3 |
230 |
OVCAR-8 |
CDX |
High-grade serous ovarian cancer |
0.58* |
2 |
175 |
ST206B |
PDX |
Serous papillary carcinoma |
0.73 |
2 |
160 |
Ov6645 |
PDX |
Malignant mixed Müllerian carcinosarcoma |
1.83 |
0 |
0 |
ST2054 |
PDX |
High-grade serous ovarian cancer |
1.37 |
1 |
50 |
aAnetumab ravtansine administered at 15 mg/kg, Q2W
*Significantly different in comparison to vehicle control (p < 0.05, n = 3–10)
CDX, cell line-derived xenograft; IHC, immunohistochemistry; PDX, patient-derived xenograft; T/C, treatment/control
To investigate the correlation between the antitumor efficacy and mesothelin expression in the ovarian cancer tumor models, histological sections were analyzed by immunohistochemistry (Table 2 and Figure 4). Anetumab ravtansine showed potent anti-tumor activity in models with medium to high mesothelin expression (H-Score >50). In contrast, no efficacy was observed in the mesothelin-negative Ov6645 and ST2054 ovarian cancer PDX models (Table 2 and Figure 4).
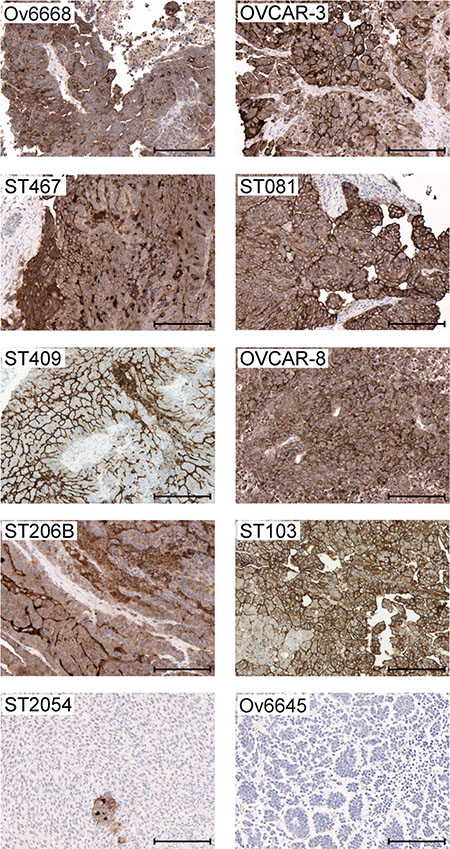
Figure 4: Mesothelin expression in ovarian cancer cell line- and patient-derived xenograft models. Representative images of IHC analyses in tumors derived from cell line- (OVCAR-3 and OVCAR-8) and patient-derived (Ov6668, ST467, ST081, ST409, ST206B, ST103, ST054 and Ov6645) xenografts using the anti-MSLN antibody SP74. Brown color indicates mesothelin expression. Scale bars indicate 200 μm.
Anetumab ravtansine exhibits improved potency in combination with doxorubicin
Pegylated liposomal doxorubicin (PLD) is the clinically most commonly used second-line treatment regimen for recurrent and platinum-resistant ovarian cancer patients. Combination therapies of DNA-damaging agents, such as PLD or carboplatin with microtubule-targeting agents (MTAs) are approved in the treatment of various malignancies including ovarian cancer [40]. Therefore we tested the potential of combining anetumab ravtansine with PLD in ovarian cancer models in vitro and in vivo. Since doxorubicin is the main active chemical ingredient of PLD and more directly metabolized by tumor cells in vitro, it was used in the in vitro combination studies. Anetumab ravtansine showed additive interaction with doxorubicin in the ovarian cancer cell line OVCAR-8, characterized by BRCA1 methylation [41], in vitro, as indicated by combination indices (CI) between 0.8 and 1.2 [42] in five repeated experiments (Figure 5A). The combination treatment with anetumab ravtansine and doxorubicin resulted in comparable levels of cleaved PARP1 and γH2AX compared to anetumab ravtansine alone (Figure 5B).
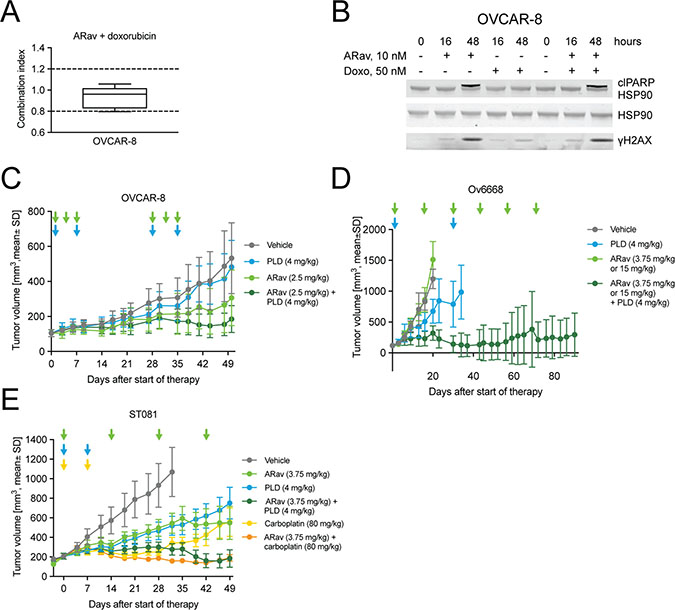
Figure 5: Antitumor efficacy of anetumab ravtansine in combination with doxorubicin/PLD or carboplatin in preclinical ovarian cancer models in vitro and in vivo. (A) Combination of anetumab ravtansine with doxorubicin in OVCAR-8 cells in vitro. The activity was determined as additive based on the determined combination indices (CI) between 0.8 and 1.2 (n = 5). (B) OVCAR-8 cell lysates were analyzed for γH2AX, cleaved PARP1 and HSP90 by Western blot at the indicated time points. (C) Tumor growth in the OVCAR-8 ovarian cancer model (n = 8). Treatments were initiated 21 days after tumor cell inoculation. (D) Tumor growth in the Ov6668 ovarian cancer PDX model (n = 9). Treatments were initiated 21 days after tumor inoculation. (E) Tumor growth in the ST081 ovarian cancer PDX model (n = 8). Treatments were initiated when tumors reached a size of 125–250 mm3. Anetumab ravtansine (i.v.), PLD (i.v.) and/or carboplatin (i.v.) were administered as indicated by arrows. ARav, anetumab ravtansine; Doxo, doxorubicin.
The antitumor efficacy of anetumab ravtansine in combination with PLD was further investigated in various ovarian cancer mouse xenograft models, including the OVCAR-8 cell line-derived model, as well as in the Ov6668 and ST081 PDX models. In OVCAR-8 mice, monotherapy with 2.5 mg/kg anetumab ravtansine or 4 mg/kg PLD showed no antitumor activity as indicated by T/C ratios of 0.58 and 0.91, respectively. Combination treatment with anetumab ravtansine and PLD showed improved antitumor activity, resulting in a T/C ratio of 0.35 (p < 0.001 vs PLD monotherapy; Figure 5C, Table 3) In the Ov6668 PDX model, the combination of anetumab ravtansine (first dose of 3.75 mg/kg followed by 15 mg/kg, Q2W) with 4 mg/kg PLD was synergistic with improved antitumor efficacy compared to vehicle (T/C = 0.27, p < 0.001, day 20) or either one as a monotherapy (anetumab ravtansine, T/C = 1.26; PLD, T/C = 0.56; both p < 0.001, day 20; Figure 5D). Five out of nine mice treated with combination therapy (55%) showed complete tumor eradication or tumor shrinkage of more than 30%, whereas no tumor shrinkage was observed in either monotherapy group. The benefit for the combination of anetumab ravtansine with PLD was confirmed in a second PDX model. In the ST081 PDX model, the combination of anetumab ravtansine (3.75 mg/kg) with PLD (4 mg/kg) showed improved antitumor efficacy compared to vehicle (T/C = 0.26, p = 0.003) or either of the agents alone (anetumab ravtansine, T/C = 0.51, p = 0.030; PLD, T/C = 0.49, p = 0.035; Figure 5E).
Table 3: In vivo efficacy of anetumab ravtansine in combination with other anticancer agents in ovarian cancer cell line- and patient-derived xenograft models
Xenograft model |
Treatmenta |
T/Cb |
Statisticsc |
Responsed |
Max. body weight loss (%)e |
|---|---|---|---|---|---|
Combination with PLD |
|||||
OVCAR-8 |
Vehicle |
1.00 |
10% SD (d50) |
0.0 |
|
ARav |
0.58 |
* |
0% (d50) |
0.0 |
|
PLD |
0.91 |
NS |
0% (d50) |
−0.7 |
|
ARav + PLD |
0.35 |
*** |
20% SD (d50) |
0.0 |
|
Ov6668 |
Vehicle |
1.00 |
0% (d40) |
0.0 |
|
ARav |
1.26 |
* |
0% (d40) |
0.0 |
|
PLD |
0.56 |
*** |
0% (d57) |
0.4 |
|
ARav + PLD |
0.27 |
*** |
11% CR, 44% PR, 11% SD (d110) |
0.0 |
|
ST081 |
Vehicle |
1.00 |
0% (d40) |
−2.3 |
|
ARav |
0.51 |
NS |
12.5% PR (d60) |
−1.5 |
|
PLD |
0.49 |
* |
0% (d60) |
−2.0 |
|
ARav + PLD |
0.26 |
** |
12.5% CR, 50% PR, 12.5% SD (d69) |
−4.4 |
|
Combination with copanlisib |
|||||
OVCAR-3 |
Vehicle |
1.00 |
0% (d71) |
−0.1 |
|
ARav |
0.17 |
*** |
11% PR, 33% (d71) |
−1.5 |
|
Copanlisib |
0.54 |
** |
0% (d71) |
−5.6 |
|
ARav + copanlisib |
0.10 |
*** |
56% PR, 22% SD (d71) |
−5.2 |
|
OVCAR-8 |
Vehicle |
1.00 |
10% SD (d50) |
0.0 |
|
ARav |
0.59 |
NS |
0% (d50) |
0.1 |
|
Copanlisib |
0.59 |
* |
0% (d50) |
−1.3 |
|
ARav + copanlisib |
0.46 |
*** |
0% (d50) |
−0.5 |
|
Combination with bevacizumab |
|||||
OVCAR-3 |
Vehicle |
1.00 |
0% (d71) |
−0.1 |
|
ARav |
0.17 |
*** |
11% PR, 33% SD (d71) |
−1.5 |
|
Bevacizumab |
0.23 |
*** |
22% SD (d71) |
−1.4 |
|
ARav + bevacizumab |
0.04 |
*** |
67% PR (d71) |
−1.3 |
|
Ov6668 |
Vehicle |
1.00 |
0% (d40) |
0.0 |
|
ARav |
1.26 |
NS |
0% (d40) |
0.0 |
|
Bevacizumab |
0.47 |
*** |
0% (d63) |
0.0 |
|
ARav + bevacizumab |
0.36 |
*** |
89% CR, 11% PR (d110) |
0.0 |
|
Combination with carboplatin |
|||||
ST081 |
Vehicle |
1.00 |
0% (d32) |
−2.3 |
|
ARav |
0.51 |
NS |
12.5% PR (d60) |
−1.5 |
|
Carboplatin |
0.32 |
** |
12.5% PR (d60) |
−2.6 |
|
ARav + carboplatin |
0.15 |
*** |
12.5% PR, 25% SD (d60) |
−4.4 |
|
aPlease see Materials and Methods for dosing and schedule.
bTreatment/control ratio, calculated from mean tumor volumes on the last day when the vehicle group remained in the study.
cStatistical significance in comparison to vehicle control on the last study day of the vehicle group. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant
dResponses calculated based on tumor volumes on the last day of each treatment group.
eThe maximum mean body weight loss expressed as a percentage of the animal weight at the start of the study. Weight loss greater than 20% is considered toxic.
ARav, anetumab ravtansine; PLD, pegylated liposomal doxorubicin; PR, partial response; SD, stable disease
Anetumab ravtansine exhibits improved potency in combination with carboplatin
Paclitaxel and carboplatin combination chemotherapy are used as standard of care in first-line therapy of ovarian cancer [43]. Anetumab ravtansine and paclitaxel are both MTAs. We therefore tested if anetumab ravtansine can replace paclitaxel in the treatment of ovarian cancer. The ST081 PDX model was treated with a combination of anetumab ravtansine (3.75 mg/kg) and carboplatin (80 mg/kg). The combination treatment demonstrated improved efficacy compared to vehicle (T/C = 0.15, p < 0.0001) or either one as a monotherapy (anetumab ravtansine, T/C = 0.51, p = 0.004; carboplatin, T/C = 0.32, p < 0.001; Figure 5E).
Anetumab ravtansine shows enhanced antitumor efficacy when combined with copanlisib
Copanlisib is a pan-class I phosphoinositide 3-kinase (PI3K) inhibitor with predominant activity on the α and δ isoforms [44, 45] and it was recently approved for the treatment of follicular lymphoma. The PI3K pathway has been identified as one of the most deregulated signaling pathways in many cancers, including ovarian cancer [46]. Anetumab ravtansine showed additive interaction with copanlisib in OVCAR-3 and OVCAR-8 cells in vitro (Figure 6A). Furthermore, the combination treatment with anetumab ravtansine and copanlisib resulted in higher effects on DNA damage and apoptosis compared to anetumab ravtansine alone, indicated by increased levels of cleaved PARP1 and γH2AX, respectively (Figure 6B).
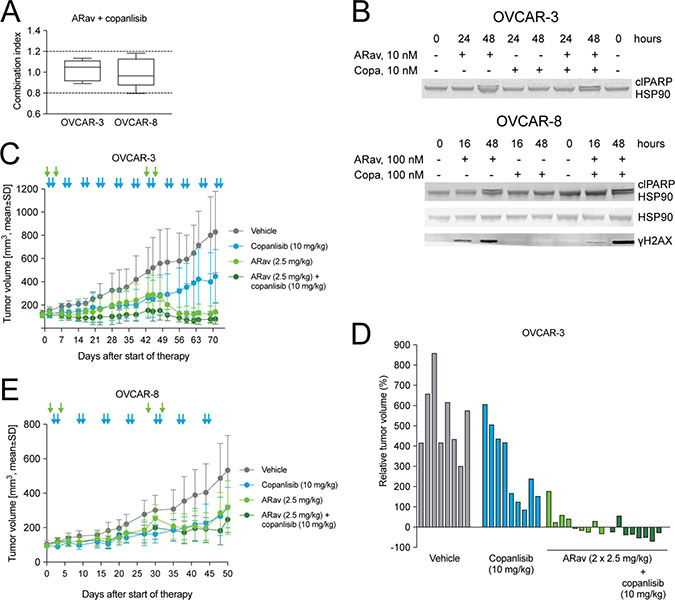
Figure 6: Antitumor efficacy of anetumab ravtansine in combination with copanlisib in preclinical ovarian cancer models in vitro and in vivo. (A) Combination of anetumab ravtansine with copanlisib in OVCAR-3 and OVCAR-8 cells in vitro. The activity was determined as additive based on the determined combination indices (CI) between 0.8 and 1.2 (n = 5). (B) Cell lysates were analyzed for γH2AX, cleaved PARP1 and HSP90 by Western blot at the indicated time points. (C) Tumor growth in the OVCAR-3 ovarian cancer model (n = 9). Treatments were initiated 43 days after tumor cell inoculation. (D) Changes in the relative volume of OVCAR-3 tumors in panel A on day 71 after start of therapy, represented as a percentage of the initial tumor volume in each individual mouse. (E) Tumor growth in the OVCAR-8 ovarian cancer model (n = 7–9). Treatments were initiated 21 days after tumor inoculation. Anetumab ravtansine (i.v.) and/or copanlisib (i.v.) were administered as indicated by arrows. ARav, anetumab ravtansine; Copa, copanlisib.
Next, the combination of anetumab ravtansine with copanlisib was tested in vivo using the OVCAR-3 xenograft model, characterized by an amplification of the AKT2 locus [47]. In this model, monotherapy with 2.5 mg/kg anetumab ravtansine showed efficacy with a T/C ratio of 0.17 (p < 0.001) and a stable disease in five out of nine mice (Figure 6C, 6D). The combination of anetumab ravtansine with copanlisib (10 mg/kg) resulted in improved efficacy, as indicated by a partial response in five out of nine mice and improved T/C ratios (Table 3). The combination potential of anetumab ravtansine with copanlisib was tested also in the OVCAR-8 xenograft model. In this study, the combination of 2.5 mg/kg anetumab ravtansine with 10 mg/kg copanlisib was more efficacious than the vehicle (T/C = 0.46, p < 0.001); however, no difference in comparison to respective monotherapies was observed (Figure 6E, Table 3).
Anetumab ravtansine shows improved potency in combination with bevacizumab
Bevacizumab is the only antiangiogenic therapy approved for the treatment of ovarian cancer patients. The combination of anetumab ravtansine with bevacizumab was tested in the OVCAR-3 and Ov6668 models. In OVCAR-3 mice, monotherapy with anetumab ravtansine or bevacizumab showed significant antitumor efficacy with T/C ratios of 0.17 and 0.23, respectively (both p < 0.001; Figure 7A, Table 3). The combination of anetumab ravtansine with bevacizumab was clearly synergistic compared to the respective monotherapies (both p = 0.003), resulting in total tumor eradication in eight out of nine mice (Figure 7B). The combination of anetumab ravtansine with bevacizumab was further explored in the Ov6668 PDX model. Bevacizumab (0.3 mg/kg) but not anetumab ravtansine (first dose of 3.75 mg/kg followed by 15 mg/kg) showed significant antitumor activity as monotherapy (T/C = 0.47, p < 0.001, day 20; Figure 7C). The combination of anetumab ravtansine with bevacizumab resulted in additive efficacy compared to the respective monotherapies (anetumab ravtansine, T/C = 1.26, p < 0.001; bevacizumab, T/C = 0.47, p = 0.003). To get a better understanding of the mechanism of action, tumors were harvested at the end of the experiment and stained with the endothelial cell marker CD31. In tumors from vehicle-treated animals numerous large CD31-positive blood vessels were detected. Contrarily, in tumors from bevacizumab-treated animals, fewer CD31-positive blood vessels were detected. Moreover, these vessels were smaller in size, translating into reduced vessel area per tumor area (Figure 7D).
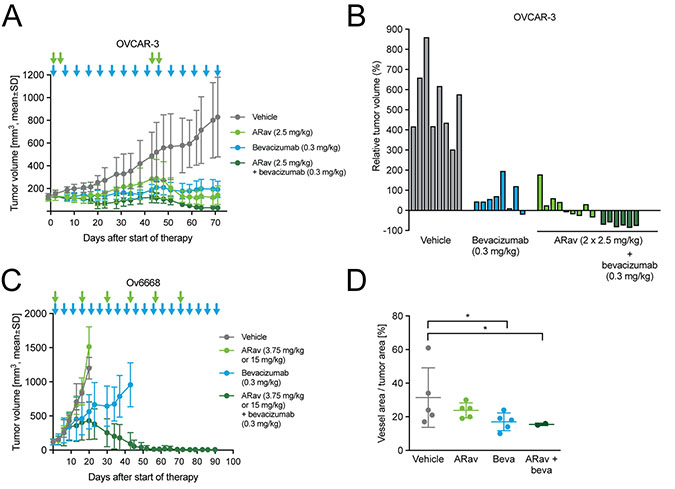
Figure 7: Antitumor efficacy of anetumab ravtansine in combination with bevacizumab in human ovarian cancer xenograft models in mice. Anetumab ravtansine (i.v.) and/or bevacizumab (i.p.) were administered as indicated by arrows. (A) Tumor growth in the OVCAR-3 ovarian cancer model (n = 9). Treatments were initiated 43 days after tumor cell inoculation. (B) Changes in the relative volume of OVCAR-3 tumors in panel A on day 71 after start of therapy, represented as a percentage of the initial tumor volume in each individual mouse. (C) Tumor growth in the Ov6668 ovarian cancer PDX model (n = 9). Treatments were initiated 21 days after tumor inoculation. (D) CD31-positive vessel area as a percentage of tumor area in OVCAR-3 mice described in panel A. ARav, anetumab ravtansine; Beva, bevacizumab.
All monotherapy and combination treatments were well-tolerated as indicated by less than 5% body weight losses in the mouse models tested (Table 3).
DISCUSSION
Cell surface glycoprotein mesothelin is highly expressed in ovarian cancer and its expression has been associated with poor prognosis [48]. In addition, the binding of mesothelin to CA125, an ovarian cancer biomarker, and upregulation of metalloproteinases has been reported to facilitate metastatic spread, making mesothelin an interesting target in ovarian cancer [18, 19, 49, 50]. The ADC anetumab ravtansine takes advantage of the tumor-specific expression of its target antigen mesothelin, thereby delivering the highly cytotoxic microtubule-targeting toxophore DM4 payload to tumor cells [28]. We have previously demonstrated that anetumab ravtansine exhibits superior antitumor efficacy compared to the standard of care cisplatin, resulting in complete tumor eradication in a preclinical ovarian cancer model [31]. Herein, the activity of anetumab ravtansine was investigated in a broader and more diverse set of ovarian cancer models, including patient-derived xenograft models with more relevant disease backgrounds.
Anetumab ravtansine was demonstrated to be rapidly internalized by cancer cells and exhibited the preferred intracellular trafficking route into lysosomes upon binding to mesothelin. Furthermore, anetumab ravtansine showed potent in vivo efficacy with T/C ratios less than 0.37 in several cell line- and patient-derived PDX ovarian cancer models. Efficacy was generally higher in tumors with strong mesothelin expression, while no activity was seen in mesothelin-negative ovarian cancer models. These data support the rationale of using mesothelin as a selection marker for patient stratification in clinical studies with anetumab ravtansine.
Despite the availability of platinum-based first-line therapies such as cisplatin and carboplatin for ovarian cancer patients, a high medical need remains for platinum-resistant recurrent ovarian cancer [9]. PLD is the most commonly utilized agent in recurrent ovarian cancer, and it has a response of approximately 26% [51, 52]. Many clinically established cancer therapy regimens combine a DNA-damaging agent with an MTA. MTAs cause prolonged mitotic arrest and partial activation of apoptotic pathways, and thereby DNA damage [38]. Furthermore, MTAs disrupt the intracellular trafficking of DNA repair proteins and thereby can enhance the toxicity of DNA-damaging agents such as PLD/doxorubicin or carboplatin [40]. In line with these reported findings, anetumab ravtansine induced DNA damage in OVCAR-3 and OVCAR-8 cells in vitro. The effects were of the same order of magnitude in cells treated with anetumab ravtansine alone or in combination with doxorubicin. Combined treatment with anetumab ravtansine and doxorubicin resulted in additive anti-proliferative activity in vitro, presumably through increased DNA damage or complementary effects on microtubule destabilization and DNA damage. The observed in vitro efficacy translated into improved in vivo efficacy in the anetumab ravtansine and PLD combination therapy in comparison to the respective monotherapies in the OVCAR-8, Ov6668 and ST081 ovarian cancer xenograft models.
A combination of paclitaxel with carboplatin has been the standard of care first-line therapy for ovarian cancer [43]. Based on similar mode-of-actions, there is a rationale to replace paclitaxel with anetumab ravtansine for front-line ovarian cancer therapy. Therefore, we also tested anetumab ravtansine in combination with carboplatin in the ST081 platinum-sensitive ovarian cancer PDX model. Anetumab ravtansine combined with carboplatin resulted in improved efficacy in comparison to the respective monotherapies in the ST081 model. Together, these results provide a rationale for the evaluation of anetumab ravtansine in combination with PLD or carboplatin in ovarian cancer patients.
Activation of the PI3K/AKT signaling pathway can mediate resistance to cytotoxic agents in ovarian cancer [53]. Preclinical data demonstrate the improved activity of paclitaxel, an MTA used as the first-line therapy in ovarian cancer [2], in combination with inhibitors of the PI3K/AKT pathway [53, 54]. In clinical studies, however, the combination of PI3K inhibitors and paclitaxel has been limited by the toxicity of these agents [55, 56]. ADCs are associated with less side effects and an improved risk-benefit ratio compared to paclitaxel, and therefore, present an alternative, more promising combination partner for PI3K inhibitors. Herein, the combination of anetumab ravtansine with copanlisib, a pan-class I PI3K inhibitor, showed improved antitumor activity compared to the respective monotherapies and was well-tolerated in the OVCAR-3 model, characterized by an amplification of the AKT2 locus [47]. In OVCAR-3 cells, combination treatment with anetumab ravtansine and copanlisib resulted in the induction of the apoptosis markers PARP and γH2AX in vitro, supporting PI3K-mediated inhibition of apoptosis as a potential mechanism of action.
Bevacizumab is a targeted anticancer therapy and currently the only antibody-based therapy approved for use in ovarian cancer [57]. Bevacizumab was approved based on improved progression-free survival; however, no advantage in overall survival has been demonstrated [58]. Combining bevacizumab with other targeted agents has been presented as an attractive approach to improve therapeutic efficacy [58], e.g. by improving the tumor penetration of antibody-based therapies by normalizing the aberrant structure and function of tumor vasculature [59]. In the present study, the combination of anetumab ravtansine with bevacizumab resulted in improved antitumor efficacy in the OVCAR-3 model. It is noteworthy that tumor shrinkage was seen in the combination group only. The superior activity of the combination treatment was confirmed in the Ov6668 ovarian cancer PDX model, supporting further exploration of the combination of these two targeted agents.
Overall, this work supports the development of anetumab ravtansine as monotherapy or in combination with various targeted agents and chemotherapy in the treatment of ovarian cancer. A phase 1b study (NCT02751918) exploring the pharmacokinetics and maximum tolerated dose of anetumab ravtansine in combination with PLD in ovarian cancer patients is currently ongoing.
MATERIALS AND METHODS
Cells and compounds
The ovarian cancer cell lines were obtained from ATCC, NCI, ECACC or Public Health England and authenticated by the DSMZ using short tandem repeat (STR) DNA fingerprinting. NCI-H322 human lung cancer cells were obtained from ATCC. Cells were passaged for up to 6 months after receipt or resuscitation and maintained at 37° C in a humidified atmosphere containing 5 % CO2. OVCAR-3 and OVCAR-8 cells were cultured in RPMI-1640 medium (Biochrom) supplemented with 10% (w/v) fetal calf serum (FCS). All other cell lines were cultured in DMEM/F12, GlutaMAX (Invitrogen) supplemented with 10% (v/v) heat-inactivated FCS (Sigma) and antibiotics (Invitrogen). Anetumab ravtansine (BAY 94–9343), the anti-mesothelin antibody MF-T (BAY 86–1903) and the transfected HT29-MSLN cell line stably expressing mesothelin were produced as described previously [31].
Internalization
To investigate the internalization mechanism of anetumab ravtansine, the targeting antibody moiety (MF-T, BAY 86–1903) of the ADC was coupled to a pH-sensitive fluorescent dye. Mesothelin-expressing HT29-MSLN cells were incubated for 6 h with the anti-mesothelin antibody MF-T or with an isotype control. Cells were fixed and permeabilized with methanol, followed by staining with antibodies against either the endocytosis marker clathrin or the lysosomal marker LAMP-1 (lysosome-associated membrane glycoprotein 1) and analyzed by fluorescence microscopy using the InCellAnalyzer 1000 (GE Healthcare).
Cell surface mesothelin expression was evaluated in NCI-H322 human lung cancer and OVCAR-3 human ovarian cancer cells endogenously expressing mesothelin. Cells were incubated with the anti-mesothelin antibody MF-T (10 μg/ml) for 0.5, 3 or 24 h and analyzed for mesothelin expression by flow cytometry (FACS).
To study the mechanism of action of anetumab ravtansine, OVCAR-3 human ovarian cancer cells endogenously expressing mesothelin were incubated with 100 nM anetumab ravtansine for 4, 16, 24 or 48 h and the expression level of mesothelin was detected by Western blot (please see details below).
Mitotic progression
To evaluate cell cycle stage and microtubule organization, 24 × 103 OVCAR-3 cells/well were seeded in chamber slides and incubated for 24 h. Cells were treated with anetumab ravtansine for 24 h, fixed in 4% paraformaldehyde for 20 min at 4° C and washed twice with cold PBS. Cells were stained with the PathScan Apoptosis and Proliferation Multiplex IF kit (Cell Signaling Technology) following the manufacturer’s instructions. The primary antibody was incubated overnight at 4° C. Stained cells were mounted using Fluorescent Mounting Medium (Dako) and imaged with an Axio Scan-Z1 (Zeiss) fluorescence microscope.
Proliferation assay
The analysis of anetumab ravtansine efficacy in a panel of ovarian cancer cell lines was performed at Charles River Discovery Research Services Germany GmbH. Ovarian cancer cells were seeded on 96-well plates, and after 24 h, the cells were treated with anetumab ravtansine (5 mg/ml, corresponding to 33.3 μM) or DMSO in 10 different concentrations with half-log increments up to 1000 pM. The cells were incubated for 72 h, and cell viability was determined using the CellTiter-Blue® Cell Viability Assay (Promega). The data was analyzed by a 4-parameter nonlinear curve fit (Oncotest Data Warehouse software).
In vitro combination assays
OVCAR-3 and OVCAR-8 cells (300 to 500 cells/well) were seeded in 384-well plates. Anetumab ravtansine (300 nM–0.41 nM) and copanlisib or doxorubicin (both 3 μM–0.41 nM) were in mixed compound ratios of 0.2, 0.4, 0.5, 0.6, 0.8 and 1. Cells were incubated for 72 h and viability was measured using Cell Titer-Glo® (Promega) according to the manufacturer’s instructions. IC50 values were calculated for each individual combination data point, and the respective isobolograms were generated. Combination indices (CI) were calculated according to the median-effect model [60]. Activity was determined as synergistic with CI < 0.8, additive with 0.8 < CI > 1.2 and antagonistic with CI > 1.2.
Detection of mesothelin expression
Mesothelin expression was determined immunohistochemically (IHC) in tumors fixed in neutral buffered formalin and embedded in paraffin. Following epitope retrieval at pH 9, freshly cut 3 μm slides were stained with 0.5 μg/mL anti-MSLN antibody SP74 (Spring Bioscience). Membrane mesothelin staining was scored by a trained pathologist. The percentage of tumor cells staining positive for mesothelin at each intensity level (0, 1, 2, or 3) and an H-score were calculated as described previously [61]. The K1 anti-mesothelin antibody (Thermo Fisher) was used for immunocytochemistry in OVCAR-3 cells.
Cell surface mesothelin expression levels were determined in a panel of ovarian cancer cell lines by FACS. Cells were seeded in 96-well round-bottom microtiter plates at a density of 3.6 × 105 cells/well and incubated with MF-T (0.1, 0.25, 0.5, 1, 3, 5 μg) or isotype control (5 μg) antibodies for 2 h at 4° C in the dark. After the incubation, the cells were washed with FACS staining buffer and resuspended for analysis. Instrument settings were set up with QuantiBRITE beads (BD Biosciences) according to the manufacturer´s instructions. Flow cytometric analysis was carried out on the Attune NxT Acoustic Focusing Cytometer and analyzed using the FlowJo software.
In the in vitro studies, protein expression was determined in OVCAR-3 and OVCAR-8 cells by Western blot. Cells were incubated with 10 or 100 nM anetumab ravtansine or copanlisib, or with 50 or 500 nM doxorubicin. In parallel, cells were treated with combinations of anetumab ravtansine (10 and 100 nM) and copanlisib (10 and 100 nM), or anetumab ravtansine (10 and 100 nM) and doxorubicin (50 and 500 nM). Culture medium was used as vehicle control. At 0,16, 24 and 48 h, cells were harvested for protein isolation. The cell extracts (10 μg of total protein) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were blocked in 5% skim milk (in TBS-Tween, 0.1 % (v/v)) and incubated with the indicated primary antibodies (mesothelin (D9R5G), phospho-histone H3 (Ser10), phospho-Akt (Ser473), cleaved PARP and phospho-p44/42MAPK (Erk1/2); all rabbit origin (Cell Signaling); HSP90 (mouse origin, BD Bioscience). Proteins were detected using IRDye-labeled secondary antibodies with reactivity against rabbit and mouse (IRDye 800CW anti-rabbit and IRDye 680LT; Licor) and visualized using the Odyssey infrared imaging system (Licor).
In vivo tumor models
All animal experiments were conducted in accordance with the German animal welfare law and approved by local authorities. For the OVCAR-3 and OVCAR-8 xenograft models, tumor cells (8 × 106 or 2 × 106, respectively) in Matrigel® (Basement Membrane Matrix, BD Biosciences) were inoculated subcutaneously to the right lower flank region of female nude/nude mice (Janvier Labs). The in vivo studies with the ovarian cancer patient-derived xenograft (PDX) models were performed at EPO Berlin-Buch GmbH, Germany (Ov6645 and Ov6668) or START, San Antonio, TX, USA (ST270, ST467, ST081, ST409, ST206B and ST2054). Ovarian cancer tissue pieces (2 × 2 mm), obtained from in vivo passage, were implanted subcutaneously in the inguinal region of female nude/nude or Scid (for the ST103 model) mice. Tumor volume [(length × width2)/2] and body weight were measured by caliper at least twice weekly, and the treatment response was defined using the RECIST criteria [62]. Progressive disease (PD) was defined as greater than 20% increase in tumor size. Partial response (PR) was defined as greater than 30% reduction in tumor size. Complete response (CR) was defined as an absence of any palpable tumor mass. No tumor growth or a slight reduction (<30%) or small increase (<20%) in tumor size was defined as stable disease (SD). Treatment-to-control (T/C) ratios were calculated on the basis of mean tumor volume. In the monotherapy experiments shown in Table 2, anetumab ravtansine was administered intravenously (i.v.) at 2.5 mg/kg three times every third day (Q3Dx3). For the in vivo combination studies with pegylated liposomal doxorubicin (PLD, Doxil®) or copanlisib (Bayer AG) in OVCAR-8 xenografts, anetumab ravtansine was administered i.v. at 2.5 mg/kg on days 1, 4, 7, 28, 32 and 35, or on days 1, 4, 28 and 35, respectively. For the in vivo combination studies in OVCAR-3 xenografts, anetumab ravtansine was administered i.v. at 2.5 mg/kg on days 1, 4, 43 and 46. For the Ov6668 xenografts, anetumab ravtansine was administered i.v. at 3.75 mg/kg on day 1 and at 15 mg/kg on days 16, 30, 43, 57 and 71. For the ST081 xenografts, anetumab ravtansine was administered i.v. at 3.75 mg/kg every second week (Q2W). PLD was administered i.v. at 4 mg/kg on days 1, 7, 28 and 35 (OVCAR-8 xenografts), on days 1 and 30 (Ov6668 xenografts) or on days 0 and 7 (ST081 xenografts). Carboplatin (Paraplatin®) was administered i.v. at 80 mg/kg QWx2. Copanlisib was administered at 10 mg/kg, 2 days on/5 days off, i.v., starting on day 2. Bevacizumab (Avastin®) was administered intraperitoneally (i.p.) at 0.3 mg/kg, every fifth day (Q5D).
Statistical analyses
All analyses were performed using the statistical programming language R (version 3.5.0) and results were considered significant at p-value < 0.05. Validity of the model assumptions was checked for each fitted statistical model. Analyses were performed using linear models estimated with generalized least squares that included separate variance parameters for each study group. For in vivo monotherapy studies, mean comparisons between the treatment and the control group were performed using the estimated linear model. For in vivo combination studies, all datasets were analyzed using second order linear mixed effects models with random intercepts and slopes for each subject. Pairwise contrasts were corrected for familywise false positive rate using Tukey’s, Sidak’s or Dunnet’s method where appropriate. All longitudinal models included the first and second order fixed time effects for each group and random intercepts and slopes for each subject.
Abbreviations
ADC: antibody drug-conjugate; ARav: anetumab ravtansine; ATCC: American Type Culture Collection; CA125: cancer antigen 125; CI: combination index; DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH; ECACC: European Collection of Authenticated Cell Cultures; EMA: European Medicines Agency; FCS: fetal calf serum; FACS: fluorescence-activated cell sorting; FDA: US Food & Drug Administration; IHC: immunohistochemistry; LAMP-1: lysosome-associated membrane glycoprotein 1; MF-T: mesothelin antibody; MSLN: mesothelin; MT: microtubule; MTA: microtubule-targeting agent; NCI: National Cancer Institute; PARP1: poly(ADP-ribose) polymerase 1; pHH3: phospho-histone H3; PDX: patient-derived xenograft; PI3K: phosphoinositide 3-kinase; PLD: pegylated liposomal doxorubicin; RPMI: Roswell Park Memorial Institute; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; SPECT-CT: single-photon emission computed tomography-computed tomography; STR: short tandem repeat; T/C: treatment/control; VEGF: vascular endothelial growth factor; Q3D: every third day; Q5D: every fifth day; Q2W: every second week; QW: once weekly.
Author contributions
MQ, UH, SZK, BSL, SG, CE, DM, KZ and CS and GTB participated in the design of the study. MQ, UH, SZK, BSL and CS carried out the experimental data acquisition. MQ, SZK and CS performed data analyses. MQ, UH, and CS wrote the manuscript. All authors read, critically revised, and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Jochen Hilbig, Manuela Steinbach, Oliver Gernetzki, Claudia Kamfenkel, Nicole Kahmann, Manuela Brand, Melanie Appel, Bettina Muchow and Henryk Bubik for excellent technical assistance. We thank EPO Berlin-Buch GmbH and START for the PDX experiments and Clara Schatz for help in counting CD31-positive cells. Aurexel Life Sciences Ltd. (https://aurexellifesciences.com/) is acknowledged for editorial support funded by Bayer AG.
CONFLICTS OF INTEREST
Conflicts of interest: Quanz, Hagemann, Zitzmann-Kolbe, Stelte-Ludwig, Golfier, Elbi, Mumberg, Ziegelbauer and Schatz are employees of Bayer AG, Cem Elbi is an employee of Bayer US LLS. Mumberg and Ziegelbauer have ownership interest as shares in Bayer AG.
FUNDING
Not applicable.
REFERENCES
1. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017; 14:9–32.
2. Herzog TJ, Monk BJ. Bringing new medicines to women with epithelial ovarian cancer: what is the unmet medical need? Gynecol Oncol Res Pract. 2017; 4:13.
3. Vaughan S, Coward JI, Bast RC Jr, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011; 11:719–25.
4. Kipps E, Tan DS, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer. 2013; 13:273–82.
5. Della Pepa C, Tonini G, Pisano C, Di Napoli M, Cecere SC, Tambaro R, Facchini G, Pignata S. Ovarian cancer standard of care: are there real alternatives? Chin J Cancer. 2015; 34:17–27.
6. Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat Rev Clin Oncol. 2013; 10:211–24.
7. Nick AM, Coleman RL, Ramirez PT, Sood AK. A framework for a personalized surgical approach to ovarian cancer. Nat Rev Clin Oncol. 2015; 12:239–45.
8. Markman M. Chemotherapy: limited use of the intraperitoneal route for ovarian cancer-why. Nat Rev Clin Oncol. 2015; 12:628–30.
9. Morgan RJ Jr, Armstrong DK, Alvarez RD, Bakkum-Gamez JN, Behbakht K, Chen LM, Copeland L, Crispens MA, DeRosa M, Dorigo O, Gershenson DM, Gray HJ, Hakam A, et al. Ovarian Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2016; 14:1134–63.
10. Rossi L, Verrico M, Zaccarelli E, Papa A, Colonna M, Strudel M, Vici P, Bianco V, Tomao F. Bevacizumab in ovarian cancer: A critical review of phase III studies. Oncotarget. 2017; 8:12389–405. https://doi.org/10.18632/oncotarget.13310.
11. Symeonides S, Gourley C. Ovarian Cancer Molecular Stratification and Tumor Heterogeneity: A Necessity and a Challenge. Front Oncol. 2015; 5:229.
12. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012; 366:1382–92.
13. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008; 44:46–53.
14. Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004; 10:3937–42.
15. Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A. 1996; 93:136–40.
16. Chang K, Pai LH, Batra JK, Pastan I, Willingham MC. Characterization of the antigen (CAK1) recognized by monoclonal antibody K1 present on ovarian cancers and normal mesothelium. Cancer Res. 1992; 52:181–86.
17. Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005; 13:243–47.
18. Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, Miyajima A. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004; 279:9190–98.
19. Gubbels JA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, Bera TK, Connor J, Sathyanarayana BK, Lee B, Pastan I, Patankar MS. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006; 5:50.
20. Cheng WF, Huang CY, Chang MC, Hu YH, Chiang YC, Chen YL, Hsieh CY, Chen CA. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br J Cancer. 2009; 100:1144–53.
21. Ma J, Tang WK, Esser L, Pastan I, Xia D. Recognition of mesothelin by the therapeutic antibody MORAb-009: structural and mechanistic insights. J Biol Chem. 2012; 287:33123–31.
22. Hassan R, Ebel W, Routhier EL, Patel R, Kline JB, Zhang J, Chao Q, Jacob S, Turchin H, Gibbs L, Phillips MD, Mudali S, Iacobuzio-Donahue C, et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007; 7:20.
23. Lindenberg L, Thomas A, Adler S, Mena E, Kurdziel K, Maltzman J, Wallin B, Hoffman K, Pastan I, Paik CH, Choyke P, Hassan R. Safety and biodistribution of 111In-amatuximab in patients with mesothelin expressing cancers using single photon emission computed tomography-computed tomography (SPECT-CT) imaging. Oncotarget. 2015; 6:4496–504. https://doi.org/10.18632/oncotarget.2883.
24. Hassan R, Cohen SJ, Phillips M, Pastan I, Sharon E, Kelly RJ, Schweizer C, Weil S, Laheru D. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010; 16:6132–38.
25. Fujisaka Y, Kurata T, Tanaka K, Kudo T, Okamoto K, Tsurutani J, Kaneda H, Okamoto I, Namiki M, Kitamura C, Nakagawa K. Phase I study of amatuximab, a novel monoclonal antibody to mesothelin, in Japanese patients with advanced solid tumors. Invest New Drugs. 2015; 33:380–88.
26. Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009; 15:5274–79.
27. Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, Le N, Ho M, Pastan I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006; 12:4695–701.
28. de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol. 2016; 40:14–23.
29. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012; 367:1783–91.
30. Moore KN, Matulonis UA, O’Malley DM, Konner JA, Martin LP, Perez RP, Bauer TM, Gilbert L, Seward SM, Oza AM. Mirvetuximab soravtansine (IMGN853), a folate receptor alpha (FRα)-targeting antibody drug-conjugate (ADC), in platinum-resistant epithelial ovarian cancer (EOC) patients (pts): Activity and safety analyses in phase I pooled expansion cohorts. American Society of Clinical Oncology. 2017.
31. Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, Mayer-Bartschmid A, Unterschemmann K, Bruder S, Linden L, Harrenga A, Hauff P, Scholle FD, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014; 13:1537–48.
32. Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M, Colombo N, Fowler JM, Argenta PA, De Geest K, Mutch DG, Burger RA, Swart AM, et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J Clin Oncol. 2009; 27:1419–25.
33. du Bois A, Weber B, Rochon J, Meier W, Goupil A, Olbricht S, Barats JC, Kuhn W, Orfeuvre H, Wagner U, Richter B, Lueck HJ, Pfisterer J, et al, and Arbeitsgemeinschaft Gynaekologische Onkologie, and Ovarian Cancer Study Group, and Groupe d’Investigateurs Nationaux pour l’Etude des Cancers Ovariens. Addition of epirubicin as a third drug to carboplatin-paclitaxel in first-line treatment of advanced ovarian cancer: a prospectively randomized gynecologic cancer intergroup trial by the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group and the Groupe d’Investigateurs Nationaux pour l’Etude des Cancers Ovariens. J Clin Oncol. 2006; 24:1127–35.
34. Ponte JF, Ab O, Lanieri L, Lee J, Coccia J, Bartle LM, Themeles M, Zhou Y, Pinkas J, Ruiz-Soto R. Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, Potentiates the Activity of Standard of Care Therapeutics in Ovarian Cancer Models. Neoplasia. 2016; 18:775–84.
35. Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salveson G, Eamshaw WC, Litwack G, Alnemri ES. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995; 55:6045–52.
36. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997; 326:1–16.
37. Ngan VK, Bellman K, Hill BT, Wilson L, Jordan MA. Mechanism of mitotic block and inhibition of cell proliferation by the semisynthetic Vinca alkaloids vinorelbine and its newer derivative vinflunine. Mol Pharmacol. 2001; 60:225–32.
38. Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012; 23:567–76.
39. Willuda J, Linden L, Lerchen HG, Kopitz C, Stelte-Ludwig B, Pena C, Lange C, Golfier S, Kneip C, Carrigan PE, Mclean K, Schuhmacher J, von Ahsen O, et al. Preclinical Antitumor Efficacy of BAY 1129980-a Novel Auristatin-Based Anti-C4.4A (LYPD3) Antibody-Drug Conjugate for the Treatment of Non-Small Cell Lung Cancer. Mol Cancer Ther. 2017; 16:893–904.
40. Poruchynsky MS, Komlodi-Pasztor E, Trostel S, Wilkerson J, Regairaz M, Pommier Y, Zhang X, Kumar Maity T, Robey R, Burotto M, Sackett D, Guha U, Fojo AT. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A. 2015; 112:1571–76.
41. Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, Thery J, Williams D, Potter J, Tran T, Korpanty G, Cremona M, Carey M, et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol. 2013; 7:567–79.
42. Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011; 731:421–34.
43. Marth C, Reimer D, Zeimet AG. Front-line therapy of advanced epithelial ovarian cancer: standard treatment. Ann Oncol. 2017; 28:viii36–viii39.
44. Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, Verhoef G, Linton K, Thieblemont C, Vitolo U, Hiemeyer F, Giurescu M, Garcia-Vargas J, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017; 28:2169–78.
45. Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K, Keng Magdalene KH, Tan SH, Lange M, Tan SY, Mumberg D, Lim ST, Ziegelbauer K, Liu N. Simultaneous Inhibition of PI3Kδ and PI3Kα Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-κB and AKT. Cancer Cell. 2017; 31:64–78.
46. Mazzoletti M, Broggini M. PI3K/AKT/mTOR inhibitors in ovarian cancer. Curr Med Chem. 2010; 17:4433–47.
47. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–83.
48. Hanaoka T, Hasegawa K, Kato T, Sato S, Kurosaki A, Miyara A, Nagao S, Seki H, Yasuda M, Fujiwara K. Correlation Between Tumor Mesothelin Expression and Serum Mesothelin in Patients with Epithelial Ovarian Carcinoma: A Potential Noninvasive Biomarker for Mesothelin-targeted Therapy. Mol Diagn Ther. 2017; 21:187–98.
49. Chang MC, Chen CA, Chen PJ, Chiang YC, Chen YL, Mao TL, Lin HW, Lin Chiang WH, Cheng WF. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem J. 2012; 442:293–302.
50. Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori J, Sima C, Rusch VW, Sadelain M, Adusumilli PS. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res. 2012; 18:2478–89.
51. Colombo N, Kutarska E, Dimopoulos M, Bae DS, Rzepka-Gorska I, Bidzinski M, Scambia G, Engelholm SA, Joly F, Weber D, El-Hashimy M, Li J, Souami F, et al. Randomized, open-label, phase III study comparing patupilone (EPO906) with pegylated liposomal doxorubicin in platinum-refractory or -resistant patients with recurrent epithelial ovarian, primary fallopian tube, or primary peritoneal cancer. J Clin Oncol. 2012; 30:3841–47.
52. Pisano C, Cecere SC, Di Napoli M, Cavaliere C, Tambaro R, Facchini G, Scaffa C, Losito S, Pizzolorusso A, Pignata S. Clinical trials with pegylated liposomal Doxorubicin in the treatment of ovarian cancer. J Drug Deliv. 2013; 2013:898146.
53. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002; 62:1087–92.
54. Jeong JY, Kim KS, Moon JS, Song JA, Choi SH, Kim KI, Kim TH, An HJ. Targeted inhibition of phosphatidyl inositol-3-kinase p110β, but not p110α, enhances apoptosis and sensitivity to paclitaxel in chemoresistant ovarian cancers. Apoptosis. 2013; 18:509–20.
55. Smyth LM, Monson KR, Jhaveri K, Drilon A, Li BT, Abida W, Iyer G, Gerecitano JF, Gounder M, Harding JJ, Voss MH, Makker V, Ho AL, et al. A phase 1b dose expansion study of the pan-class I PI3K inhibitor buparlisib (BKM120) plus carboplatin and paclitaxel in PTEN deficient tumors and with dose intensified carboplatin and paclitaxel. Invest New Drugs. 2017; 35:742–50.
56. Wheler J, Mutch D, Lager J, Castell C, Liu L, Jiang J, Traynor AM. Phase I Dose-Escalation Study of Pilaralisib (SAR245408, XL147) in Combination with Paclitaxel and Carboplatin in Patients with Solid Tumors. Oncologist. 2017; 22:377–e37.
57. Vetter MH, Hays JL. Use of Targeted Therapeutics in Epithelial Ovarian Cancer: A Review of Current Literature and Future Directions. Clin Ther. 2018; 40:361–71.
58. Mahner S, Woelber L, Mueller V, Witzel I, Prieske K, Grimm D, Keller-V Amsberg G, Trillsch F. Beyond Bevacizumab: An Outlook to New Anti-Angiogenics for the Treatment of Ovarian Cancer. Front Oncol. 2015; 5:211.
59. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307:58–62.
60. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006; 58:621–81.
61. Yamashita H, Otsuki Y, Matsumoto K, Ueki K, Ueki M. Fas ligand, Fas antigen and Bcl-2 expression in human endometrium during the menstrual cycle. Mol Hum Reprod. 1999; 5:358–64.
62. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–47.