
INTRODUCTION
Interleukin-1 receptor-associated kinases (IRAK1, IRAK2, IRAK3 [IRAK-M], and IRAK4) are serine-threonine kinases that mediate toll-like receptor (TLR) and interleukin-1 (IL-1) signaling pathways [1–4]. Signaling through these pathways is key to the regulation of cellular processes that are critical to innate immunity and inflammation. Dysregulation of these pathways is known to be involved in numerous pathophysiologies, and studies in recent years have implicated IRAKs in neoplastic disorders, and metabolic, cardiovascular, and inflammatory diseases. Cellular and animal studies aimed at elucidating the roles of IRAKs have depended on tools such as genetic knockouts, endogenous down-regulators of IRAK expression (e.g., microRNAs) [5], increasingly optimized medicinal chemistry leads [6], and the unexpected discovery of potent IRAK1 activity in an advanced clinical stage investigational agent (pacritinib) [7]. These advances have paved the way for agents targeting IRAKs to enter the clinic for assessment in diverse indications.
Among members of the IRAK family, IRAK3, and arguably IRAK2, are pseudokinases that lack catalytic activity, although they may still play important roles in signaling cascades [2, 8]. The catalytically active IRAK1 and IRAK4 [9] bear many similarities in structure in their inhibitor-binding pockets [10, 11]. Both are key components of supramolecular organizing centers (SMOCs), involving the key adapter protein myeloid differentiation primary response protein (MyD88) that regulates cell survival and cytokine secretion, in which IRAK1 is phosphorylated by IRAK4. The development of selective inhibitors able to dissect the relative contributions of IRAK1 versus IRAK4 to downstream events is of great interest but to date has proven to be challenging.
This review focuses on the clinical potential for selective inhibition of IRAK1 [12]. We briefly describe its structure, known methods of knockdown and inhibition, the available evidence for its involvement in pathways relevant to diverse disease states, and the limited data available on selective interruption of IRAK1 signaling in a variety of disease states.
IRAK1 structure and inhibitors
Although serine-threonine kinases, IRAKs belong to the tyrosine-like kinase (TLK) family of the human kinome [13]. Structural similarities among IRAKs include a conserved N-terminal death domain and a central kinase domain. Until recently, crystal structures of only one IRAK family member, IRAK4, were available [11, 14]. Although IRAK1 and IRAK4 share only 31% sequence identity overall, sequence identity is >90% along the ATP binding pocket where inhibitors typically bind. In addition, both have the same “gatekeeper” tyrosine residue that blocks access to part of the binding pocket.
Most synthetic efforts to optimize inhibitors of IRAK kinase activity to date have focused on IRAK4 [15–22]. Unsurprisingly, many of these inhibitors had similar potencies against IRAK1 [23], for example, the widely used “IRAK1/4 Inhibitor I” (Figure 1) [24]. Efforts to optimize selectivity and in vivo properties of IRAK4 inhibitors have, however, proven successful, and two such selective inhibitors, Pf-06650833 (Pfizer) and CA-4948 (Curis/Aurigene) have reached clinical development, the former for rheumatoid arthritis (NCT02996500) and the latter for non-Hodgkin lymphoma (NHL; [NCT03328078]).
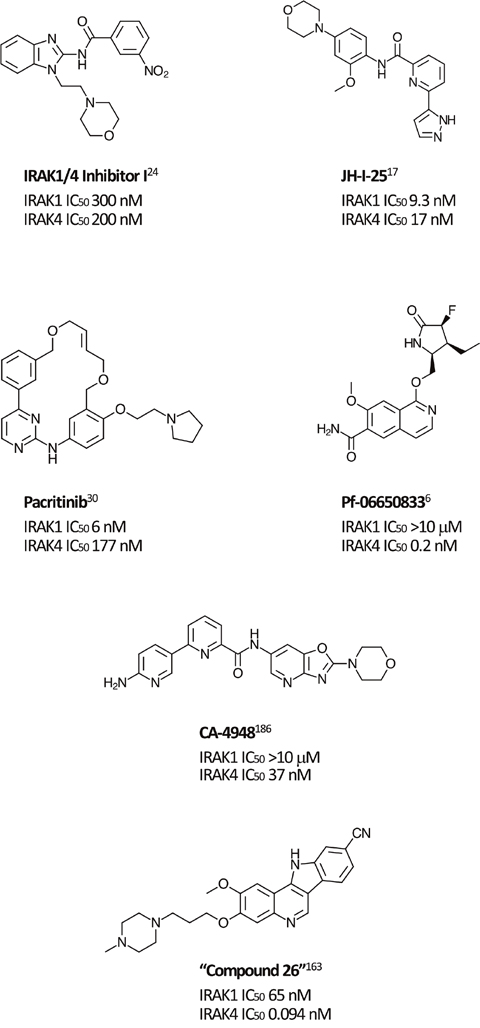
Figure 1: Selected IRAK1 and IRAK4 inhibitors. Small molecule inhibitors of IRAK1 and IRAK4 reported in the literature.
Although preclinical data suggest that there may be clinical advantages in selectively inhibiting IRAK1 [25], selective IRAK1 inhibitors have proven more elusive. Several naturally occurring compounds with anti-inflammatory properties have been reported to inhibit IRAK1 at micromolar concentrations [26, 27]. Medicinal chemistry efforts that began with nonselective lead compounds have only recently provided the first selective small molecule IRAK1 inhibitors, such as Jh-X-119-01 [28]. The structure of this compound has not yet been revealed, but it is reported to be an irreversible inhibitor, labeling the enzyme at C302, with IC50 values of 9.3 nM and >10 μM against IRAK1 and IRAK4, respectively. In addition, the crystal structure of the kinase domain of human IRAK1 binding with a small molecule inhibitor has recently being reported, providing a valuable tool for future efforts [10].
Unexpectedly, selective nanomolar IRAK1 inhibition was detected in a kinome-wide screening study for the JAK2/FLT3 inhibitor, pacritinib, which is in late stage development for patients with myelofibrosis (MF) and other myeloproliferative neoplasms (MPNs) [29–31]. In 2 randomized phase 3 studies in MF, pacritinib was associated with reductions in splenomegaly and symptoms and was well tolerated with limited myelosuppression [32, 33]. Anti-tumor activity was also detected in a Phase 1-2 study in relapsed/refractory non-Hodgkin lymphoma [34]. Although JAK2 inhibition provided the rationale for its identification and development, kinase profiling later uncovered nanomolar IRAK1 potency [7]. Follow-up screening found that pacritinib inhibits IRAK1 with moderate selectivity versus IRAK4 (IC50 6 nM and 177 nM, respectively) [35]. Computer modeling and site-directed mutagenesis studies subsequently validated its high-affinity binding to the IRAK1 kinase domain [36]. In keeping with the downstream effects of IRAK1 inhibition, in a human primary mononuclear cellular system at clinically relevant concentrations, pacritinib markedly reduced levels of the inflammatory cytokines sIL-17A, sIL-2, and sIL-6 and suppressed induced immunglobulin synthesis in normal human lymphocytes [37]. In normal human monocytes, it blocked LPS induction of inflammatory cytokines [35]. Pacritinib inhibited constitutively activated IRAK1 phosphorylation in AML cells [36] and in breast cancer cells with duplication of 1q23.1 [38]. Thus, pacritinib is currently the only clinical stage IRAK1 inhibitor with known clinical efficacy and acceptable safety even after prolonged administration. Further studies may elucidate the relative roles of its IRAK1 versus JAK2 suppression in its clinical effects in neoplastic and inflammatory diseases.
Preclinical experiments designed to elucidate IRAK1 roles have frequently made use of methods of perturbation other than small molecule inhibitors. Studies have reported the use of IRAK1 knockout mice [39, 40], knock-in mice having an inactive IRAK1 mutation [25, 41], and mutations or knockdowns of miR-146a, an endogenous IRAK1 synthesis inhibitor. Antisense oligonucleotides [42], short hairpin RNA (shRNA) [43, 44], microRNA (miRNA) [5, 45, 46], and small interfering RNA (siRNA) [47, 48] are among the many tools available to study IRAK1 function. Examples will be seen in many of the studies discussed herein.
Roles of IRAK1
IRAK1 functions as a key link in the chain of events initiated by the binding of ligands to IL-1R and TLRs [49, 50]. Activation of the IL-1 and TLR signaling pathways can be triggered by a variety of stimuli, including recognition of microbial pathogens/products (e.g., LPS), the presence of reactive oxygen species, recognition of DNA damage, abnormalities in the tissue matrix caused by chronic inflammation, and genetic factors, such as amplification of 1q21.3 and overproduction of S100A proteins [38, 51–54]. IL-1R/TLR signaling via the SMOC [55] known as the myddosome is critical to innate immunity [56], and controls a variety of cellular processes. Dysregulation of this signaling consequently plays a role in a number of diseases.
Binding of IL-1 to its cognate receptor, or of pathogen-associated molecular patterns (PAMPs) or lipid components like lipopolysaccharides (LPS) to TLRs initiates immune and inflammatory responses mediated by the myddosome complex (Figure 2) [53, 57]. The adaptor protein MyD88 is recruited to the cytosolic Toll/IL-1R (TIR) domain of the activated receptor, leaving the death domain (DD) of the oligomeric MyD88 available for further interactions. IRAK4 is then recruited to the DD, also as an oligomer, and it in turn recruits IRAK1 and/or IRAK2 [58, 59]. The structure of the resulting complex allows IRAK4 to phosphorylate IRAK1, leading to its activation and hyperphosphorylation, dissociation from the myddosome, and subsequent interaction with the E3 ubiquitin ligase, TNF receptor-associated factor 6 (TRAF6). The activated TRAF6 complex in turn drives downstream events, including the NF-κB [12, 60, 61] and MAPK pathways that ultimately result in upregulation of proinflammatory cytokines. In addition, IRAK1 plays a critical role in the induction of interferons via IRF7 (Figure 3) [62]. IRAK1 may also play a TRAF6-independent role in IL-1α-induced stabilization of downstream mRNAs that would otherwise be short-lived [63].
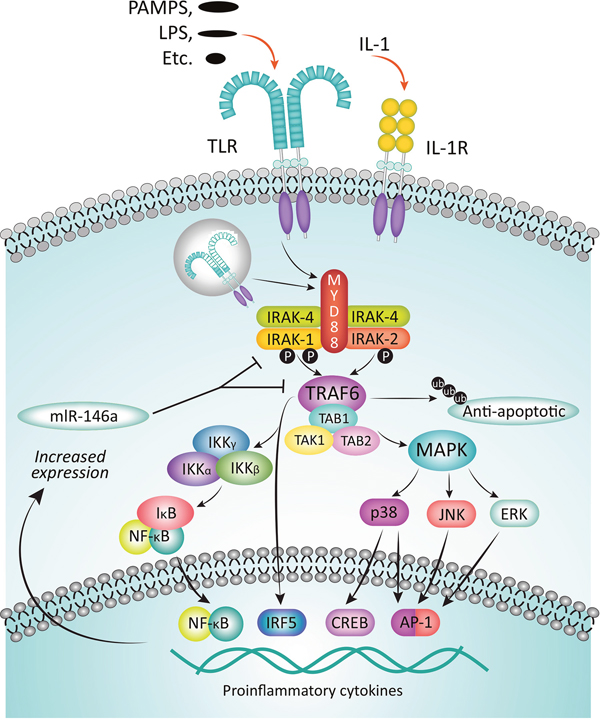
Figure 2: Function of IRAKs in the myddosome complex. Upon binding of cognate ligands, such as PAMPs and LPS, to TLRs or of IL-1 to the IL-1R, inflammatory response is mediated via the myddosome complex. Adapter protein MyD88 is recruited to the cytosolic receptor domain and IRAK4 is recruited, in turn attracting IRAK1 to the complex. IRAK4 phosphorylates IRAK1, thereby activating it, leading to its subsequent hyperphosphorylation, dissociation from the complex, and binding to TRAF6. The activated TRAF6 complex drives downstream gene transcription via multiple pathways, including the NF-kB pathway. Among the sequelae are an increased expression of inflammatory cytokines, but also miR-146a, which inhibits subsequent expression of IRAK-1 and TRAF6 proteins, thereby providing a negative feedback loop. Adapted from Jain A, Kaczanowska S, Davila E. IL-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance. Front Immunol. 2014;5:553-561. Used with permission.
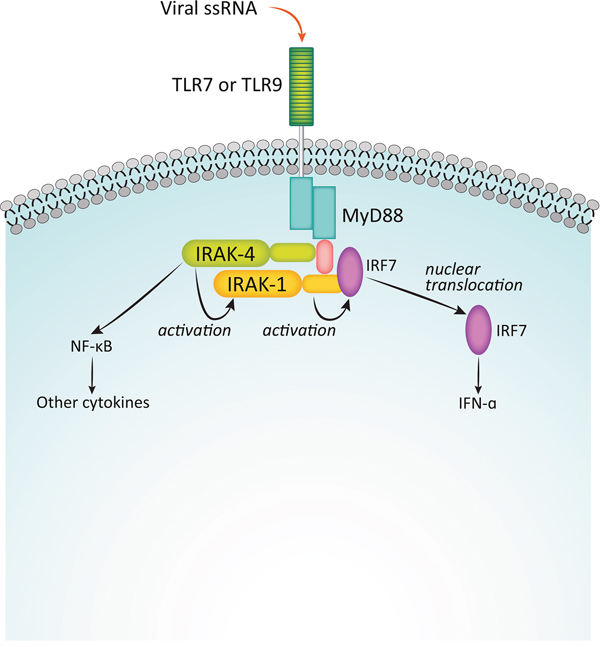
Figure 3: Function of IRAKs in interferon induction. Binding of viral single-stranded RNA to TLR7 or TLR9 initiates an innate immune response in an IRAK1-dependent process via the transcription factor IRF7. Adapted from Uematsu S, et al. J Exp Med. 2005;201:915-923. Used with permission.
Inhibition of either IRAK1 or IRAK4 kinase activity could therefore alter IL-1β and TLR-driven signaling. Observations to date suggest that the dominant kinase and the extent of its effects may depend on the particular receptor, species, and cell type. For example, it was recently demonstrated that in normal human macrophages, IRAK1 predominates, whereas IRAK2 and IRAK4 are more important in murine cells [64]. A recently published study of TLR signaling in cells from an infant with a fatal inherited IRAK1 deficiency found a poor fibroblast response to TLR agonists but an almost unimpaired response to IL-1β, in contrast to a normal response to both in PBMCs [47]. Another consideration for inhibitors of IRAK activity is the relative importance of catalytic activity versus structural roles [65]. This is exemplified by a study of kinase-inactive IRAK1 knock-in mice, which found that catalytic activity was not rate limiting for secretion of some inflammatory mediators (IL-6, TNFα, IL-10), but that IFN-β induction was greatly delayed by TLR agonists in dendritic cells from these animals [41].
Another potentially critical role that IRAK1 may play involves the generation of active IL-1, the “master cytokine in inflammation”[66]. Active IL-1β is produced in monocytes, macrophages, and dendritic cells in a cytosolic SMOC known as the inflammasome [67–70]. Among known types of inflammasomes, the Nod-like receptor protein 3 (NLRP3) inflammasome responds to the greatest variety of stress signals and is most frequently implicated in autoinflammatory and autoimmune disorders [71]. In the inflammasome, active IL-1β is produced by cleavage of pro-IL-1β by the cysteine protease caspase-1, which is itself produced through cleavage of its pro form [72]. Early phase, acute inflammasome activation by simultaneous TLR and NLRP3 activation, critical for pyroptosis and proinflammatory cytokine secretion, is regulated by IRAK1 [73]. Thus, IRAK1 also plays a role in innate immunity via IL-1β.
Its involvement in the NF-κB pathway and IL-1β secretion suggest that IRAK1 functions in regulating levels of other important proinflammatory cytokines. Interleukin-6 (IL-6) is a pleiotropic cytokine induced by IL-1β and is a downstream product of the NF-κB pathway [74]. Among the many effects of IL-6 is the promotion of differentiation of naïve T cells to Th17 effector cells, which in turn secrete other proinflammatory cytokines, such as interleukin-8 (IL-8) and transforming growth factor β (TGFβ). Inhibition of IL-6 via antibodies against IL-6 (siltuximab) or IL-6R (tocilizumab, sarilumab) is a proven therapeutic strategy in inflammatory diseases, such as rheumatoid arthritis, and is under investigation in cancer [75–77]. IL-8 is another downstream product of the NF-κB pathway. This proinflammatory cytokine is a critical mediator of neutrophil activation [78] and is of interest as a potential target in cancer [79].
Thus, multiple mechanisms by which perturbing IRAK1 function may have beneficial effects in inflammatory diseases and cancer may be posited, and several caveats (e.g., potential deleterious consequences of inhibiting innate immune response) may be foreseen. Additional studies are needed to clarify the potential utility of IRAK1 inhibition in specific disease states, and to differentiate effects of IRAK1 versus IRAK4 inhibition. Currently available data are summarized below.
IRAK1 in inflammatory diseases (Table 1)
Table 1: Experimental evidence for IRAK1 involvement in inflammatory and autoimmune diseases
Disease |
Experimental System |
Effector(s) |
Effects |
|---|---|---|---|
Sepsis [80] |
CLP IRAK1-/y mouse |
IRAK1 deficiency |
Reduced mortality (35% vs. 85%); reduced plasma IL-6, IL-10 @6 hrs. |
Sepsis [81] |
LPS IRAK1-/- mouse |
IRAK1 deficiency |
Significantly reduced neutrophil infiltration, and tubular changes in liver and kidney. |
Sepsis [85] |
CLP mouse |
MiRNA-146a |
Reduced inflammatory cytokine levels, neutrophil infiltration, and cardiac dysfunction in miRNA-146a myocardial-transfected animals. |
Sepsis [84] |
MiRNA-146a -/- mouse |
MiRNA-146a deficiency |
Mir-146a -/- mice are hypersensitive to LPS challenge. |
Sepsis [86] |
PB neutrophils from sepsis pts |
IRAK1 1595T haplotype |
Increased neutrophil NF-κB; increased shock (OR 2.9, P = 0.047); prolonged mechanical ventilation (OR 2.7, P = 0.04); higher 60 d mortality (OR 2.7, P = 0.05). |
Sepsis [87] |
IRAK1 1595T haplotype |
Increased need for prolonged mechanical ventilation (P = 0.02). |
|
Liver fibrosis [91] |
CCL4 rat |
miRNA-146a-5p |
Levels of miRNA-146a-5p correlate with fibrosis progression. |
Liver fibrosis [91] |
LX-2 cells |
miRNA-146a-5p |
MiRNA-146a-5p transfection reduced proliferation, HSC activation. |
Liver fibrosis [93] |
STAM mouse |
Pacritinib |
Significantly reduced liver fibrotic area (P < 0.01) and CK-18 fragment levels (P < 0.05). |
Phase 3 clinical trials |
Pacritinib |
Increased platelet count in pts with <50,000/mL at BL; significantly increased Hgb in pts with <10g/dL and no transfusion at BL; significantly increased proportion of RBC transfusion independent pts in those who were transfusion dependent at BL. |
|
GVHD [102] |
BALB/c mouse allograft |
Pacritinib |
Significantly reduced mortality, but role of IRAK1 vs. JAK2 inhibition unknown. |
GVHD [101] |
AlloSCT pt whole blood RNA |
MiRNA-146a |
Levels of miRNA-146a were significantly associated with acute GVHD incidence at day 28 post-transplantation (OR 0.15, P = 0.016). |
SLE [118] |
Human DNA |
IRAK1 rs1059702 SNP |
An IRAK1 rs1059702 SNP is associated with SLE susceptibility (OR 1.43). |
SLE [119] |
Human DNA; |
IRAK1 SNPs |
5 IRAK1 SNPs were associated with increased SLE risk (OR>1.5). IRAK1 deficiency abrogated lupus-associated phenotypes in congenic mouse models. |
SLE [25] |
ABIN1 x IRAK1[D359A]-knock-in mice |
Catalytically inactive IRAK1 |
Crossing ABIN1 mice, which have a phenotype similar to human lupus, with mice having catalytically inactive IRAK1 prevented splenomegaly, autoimmunity, and liver and kidney inflammation. |
SLE [48] |
SLE pt PBMCs; B6.lpr mouse |
IRAK1/4 inh 1; IRAK1 siRNA |
IRAK1/4 inh significantly reduced renal injury in the B6.lpr model; IRAK1 is overexpressed in PBMCs from pts with SLE, and levels of NF-κB phosphorylation were reduced by IRAK1/4 inh I or IRAK1 siRNA. |
Obesity [136] |
Human adipose tissue |
NA |
IRAK1 gene expression higher in adipose tissue from obese vs. nonobese participants (P = 0.01) and correlated with TNFα mRNA in both pts with and without diabetes |
Obesity [137] |
Human adipocytes, WAT |
MiRNA-146a |
MiRNA-146a is elevated in obesity, blunts inflammatory response as measured by IL-8 and MCP-1, and reduces JNK and p38 activation. |
T2D [39] |
IRAK1-/- mouse |
IRAK deficiency |
IRAK1 KO mice had improved glucose tolerance, and insulin-stimulated glucose disposal rates and uptake in muscle (but not liver). Effects varied between high- and low-fat diets. |
T2D [138] |
T2D pt, control blood |
MiRNA-146a |
MiRNA-146a expression levels decreased in PBMCs (P = 0.004) and plasma (P = 0.008) of T2D pts (n = 30) relative to controls (n = 30); IRAK1 mRNA expression was increased in T2D pts (P = 0.028). |
T2D [139] |
STZ rats, DPN rats |
T2D |
MiRNA-146a expression and NCV decreased in DPN vs. STZ and normal rats (P < 0.01); TNFα, IL-1β, NF-κB correspondingly increase (P < 0.01); mIRNA-146a level negatively correlated with cytokines. |
I/R [46] |
Liver I/R mouse |
MiRNA-146a |
MiRNA-146a was decreased, IRAK1 increased in mouse Kupfer cells after I/R. MiR-146a overexpression decreased IRAK1, attenuated proinflammatory cytokines in H/R-induced macrophages. |
I/R [140] |
LAD ligated mouse |
MiRNA-146a |
MiRNA-146a transfection suppressed IRAK1 and TRAF6 expression, decreased infarct size by 50%, attenuated apoptosis, and protected against myocardial injury and cardiac dysfunction after I/R. |
I/R [141] |
Intestinal I/R IRAK1-/- mouse |
MiRNA-146a |
Intestinal IRAK1 levels increased after I/R in normal mice; tissue damage was reduced in KO mice. Induction of miR-146a protects against intestinal I/R injury. |
I/R [142] |
MCAO rat |
IRAK1/4 inhibitor I |
IRAK1/4 inhibition reduced mortality, neurological deficits, and ischemic infarct volume in MCAO rat, and was anti-apoptotic in a cellular model of hypoxia. |
ABIN1, A20-binding inhibitor of NF-κB; BL, baseline; CLP, cecal ligation and puncture; DPN, diabetic peripheral neuropathy; GVHD, graft-versus-host disease; Hgb, hemoglobin; H/R, hypoxia/reperfusion; hrs, hours; inh, inhibitor; I/R, ischemia/reperfusion injury; LAD, left anterior descending artery; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion; NA, not applicable; NCV, nerve conduction velocity; OR, odds ratio; PBMC, peripheral blood mononuclear cells; pts, patients; RBC, red blood cell; STZ, streptozotocin-induced diabetic; WAT, white adipose tissue.
Sepsis
IRAK1 deficient mice have significantly increased survival relative to wild type (WT) mice in polymicrobial sepsis [80], and IRAK1 knockout mice treated with LPS had significantly less liver and kidney damage than did WT mice [81].
MicroRNA-146a (miR-146a) is an endogenous repressor of IRAK1 and TRAF6 that has been shown to suppress NF-κB activity and expression of its target genes, including IL-1β, IL-6, IL-8, and TNFα [82, 83]. MiR-146a knockout mice are hypersensitive to LPS challenge [84]. Lentivirus-expressing miR-146a delivered into the myocardium reduced IRAK1 expression and protected against cardiac dysfunction relative to untransfected mice in a murine model of polymicrobial sepsis [85]. Finally, genetic studies of patients with sepsis uncovered an IRAK1-1595C/T polymorphism for which the T haplotype was associated with greater NF-κB nuclear translocation upon ex vivo LPS stimulation, more severe organ dysfunction, the need for longer ventilation, and higher mortality [86, 87].
Thus, data, although limited, suggest that suppression IRAK1 pathway activation occurs during sepsis and over-activation can be associated with deleterious consequences. Modulation of signaling through IRAK1 may be beneficial in human sepsis, a condition considered a global health priority responsible for more than 5 million deaths per year worldwide [88].
Fibrotic diseases
Fibrosis is the end result of chronic inflammation and can affect single organs, such as the lung (pulmonary fibrosis), heart, kidney, liver (cirrhosis), bone marrow (myelofibrosis), or be systemic as in scleroderma. IL-1 through its role in induction of inflammatory cytokines including IL-8, TGF-β, and IL-17 has been implicated as a critical cytokine in several fibrotic diseases [89, 90], supporting the concept that IRAK1 inhibition may have a therapeutic effect. Liver fibrosis is characterized by the aberrant activation and proliferation of hepatic stellate cells (HSCs), believed to be driven in part by the NF-κB pathway. In a rat model of liver fibrosis, miR-146a-5p downregulation correlated with fibrosis progression. In a human HSC cell line, overexpression of miRNA-146a-5p downregulated IRAK1 and TRAF6, and suppressed proliferation and activation [91].
In a mouse model of liver fibrosis that recapitulates the stepwise, clinically observed progression from nonalcoholic fatty liver disease (NAFLD, an increasingly common, known risk factor for liver-related mortality [92]) to nonalcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC), pacritinib, presumably through IRAK1 signaling inhibition, significantly reduced liver fibrotic area compared with vehicle control without affecting steatosis [93]. Plasma CK-18 fragment levels, a clinical biomarker of liver cell necrosis, were also significantly reduced.
Clinical data for pacritinib are available in another fibrosing disease, myelofibrosis (MF). MF is a clonal multipoint stem cell disorder associated with progressive marrow fibrosis, resulting in increasing cytopenias over time. Although the cause of fibrosis remains to be fully elucidated, it is likely due to cytokine mediated inflammation within the marrow propagated by inflammatory cells, particularly abnormal megakaryocytes and macrophages derived from the neoplastic clone. The inflammatory process stimulates the proliferation of endothelial cells and fibroblasts and other structural cells leading to a fibrotic environment no longer capable of supporting hematopoiesis [94]. It has been posited that the inflammatory process due to other background mutational events may precede the mutations in the JAK2/STAT pathway associated with clinical MF, and thus suppressing the JAK/STAT pathway has not been demonstratred to control or reverse progressive fibrosis [95]. IL-1 produced by abnormal monocytes has a central role in MF in promoting high levels of TGFβ [96]. Recent clinical data indicate that mutational loss of miR-146a activity is associated with rapidly progressive myelofiobrosis, which suggests a central role for IRAK1 activation in this disease [97]. JAK1/2 inhibitors, such as the approved drug ruxolitinib inhibit signaling of some inflammatory cytokines such as IL-6 and other inflammatory or proliferation promoting cytokines that signal through JAK1 and/or JAK2, have been shown to suppress abnormal clonal expansion and reduce splenomegaly in MF, but do not substantially reduce the allelic burden of the mutated JAK clone or prevent or reverse bone marrow fibrosis in the majority of patients. To suppress fibrosis, it may be necessary to inhibit early phases of the inflammatory pathway mediated by IL-1. In data from phase 3 trials, pacritinib, which suppresses IL-1 production and signaling through its effects on IRAK1, was associated with improvements in platelet count and hemoglobin, and reductions in transfusion burden in some patients with baseline cytopenias, suggesting that marrow function may be improved possibly from reducing marrow fibrosis [32, 33]. Longer-term studies are needed to confirm potential antifibrotic effects of pacritinib in this disease.
Graft-versus-host disease (GVHD)
Chronic GVHD occurs in 30-70% of patients who undergo unmanipulated stem cell transplantation, and when severe, is a highly debilitating, multiorgan immuologic syndrome associated with tissue fibrosis, immune-incompetence and the production of auto-antibodies [98]. GVHD occurs due to donor cell recognition of antigenic differences in major or minor histocompatability loci of the transplant recipient. Multiple studies have confirmed the roles of IL-1β and IL-6 produced by multiple cell types during alloimmune reactions in mucosal barrier injury, an important first step in the pathogenesis of acute GVHD. IL-1β produced through activation in the inflammasome increases mucosal barrier permeability, facilitating the entry of microbes and microbe-associated molecular patterns (MAMPs) that further exacerbate inflammation and lead to a proinflammatory cytokine-positive feedback loop that further promotes the immune allograft reaction [67]. IL-17 is a critical effector molecule in GVHD [99, 100], and suppression of IL-17 and Th-17 cells by IRAK1 inhibitors, while also reducing IL-1β and IL-6 levels, suggests IRAK1 inhibition as a rational approach to treating this condition if corticosteroids fail to control the disease.
In patients who had undergone alloSCT, expression of miR-146a at day 28 post-transplantation significantly correlated with acute GVHD incidence [101]. Recently, pacritinib was reported to reduce GVHD and xenogeneic skin graft rejection in rodent models, while maintaining donor anti-tumor immunity [102]. Although one of the kinases inhibited by pacritinib, JAK2, has been implicated in the onset of GVHD and clinical trials of ruxolitinib to treat and prevent GVHD are underway, suppression of IRAK1 may also be an operative mechanism. Whether inhibition of IRAK1, which has been shown to increase T-regs and suppress IL-17 [103], will provide additional benefits in clinical GVHD is under study in a clinical trial (NCT02891603).
Inflammatory bowel disease/colitis
Increasing evidence implicates changes in innate immune response in the pathogenesis of inflammatory bowel disease. [104, 105] Because modulation of downstream effects of anti-TNF-α antibodies are associated with disease remission, blockading inflammatory pathways upstream via IRAK inhibition may be of benefit, but this remains to be determined by future research.
Autoimmune diseases
Rheumatoid arthritis
Genetic association studies have examined the potential role of SNPs in IRAK1 and miRNA-146 as risk factors for the development of arthritis [106–115], with rheumatoid arthritis most often examined. Some of these have reported a significant association between polymorphisms (most commonly IRAK1 rs3027898 SNPs) and increased risk, whereas others found no such associations.
Evidence of both upstream (IRAK4) inhibition effects and the clinical utility of downstream blockade via the anti-IL-6R antibody sarilumab suggest that IRAK1 inhibition may be worthy of investigation. An agent structurally similar to pacritinib with selective anti-IRAK1 activity, SB1578 [116], has been shown to have remarkable activity in a murine collagen- induced arthritis model [117]. A phase 1 PK/safety study of this agent in healthy volunteers (NCT01235871) has been completed, but results have not been reported and no other clinical trials are currently listed. An IRAK4 inhibitor, Pf-06650833, is in clinical development in refractory rheumatoid arthritis (NCT02996500).
Systemic lupus erythematosus (SLE)
A large (N=15783) genetic association study has linked an IRAK1 rs1059702 SNP, which results in S196F substitution and an increase in NF-κB activity, with increased risk of SLE in multiple ancestral groups [118]. Another genetic association study identified 5 IRAK1 SNPs (which do not include rs1059702) associated with increased susceptibility to SLE [119]. This study also investigated the functional relevance of IRAK1 in 2 congenic mouse models, finding that IRAK1 deficiency abrogated lupus-associated phenotypes. Another study used the A20-binding inhibitor of NF-κB (ABIN1[D485N]) knock-in mice, which display a phenotype resembling human lupus [25]. Crossing these mice with mice expressing catalytically inactive IRAK1 or IRAK4 mutants prevented the development of splenomegaly, autoimmunity, and liver and kidney inflammation, supporting the role of these enzymes as targets in human autoinflammatory diseases. The authors postulate that patients with SLE treated with IRAK inhibitors could have less risk of severe viral infections than those treated with the current standard, anti-interferon (IFN) therapy, and that selective IRAK1 inhibition may provide a reduced risk of microbial infections relative to treatment with IRAK4 or dual IRAK1/4 inhibitors. The involvement of IRAK1 in SLE is also supported by the increased incidence of SLE in individuals with downregulating miRNA-146a polymorphisms [120]. A recent study reported that IRAK1 was overexpressed and hyperactivated in peripheral blood mononuclear cells (PBMCs) from patients with SLE and found that IRAK1/4 inhibitor I significantly mitigated inflammatory response and renal injury in the B6.lpr mouse model of SLE, which overexpresses IRAK1 in splenic monocytes [48]. Additionally, levels of NF-κB phosphorylation in PBMCs from patients with SLE could be decreased through IRAK1/4 inhibition or IRAK1 small interfering RNA (siRNA). Collectively, these results suggest that further study of IRAK1 inhibition in SLE is warranted.
Schnitzler’s syndrome (SchS)
Schnitzler’s syndrome is a rare but underdiagnosed autoinflammatory disease characterized by monoclonal IgM (or IgG) gammopathy [121]. Patients experience urticarial rash, fever, pain, and other signs of inflammation, and are at risk of developing Waldenström macroglobulinemia (WM). Both IL-1β and IL-6 have been implicated in its pathogenesis [122], and antibodies targeting each of these cytokines have proven clinically effective [123]. As described below, mutations in the key adapter protein MyD88 resulting in constitutive activation are present and correlate with severity in WM [124], and these may play a similar role in SchS. Interference with IRAK1 may therefore represent another possible treatment approach for patients with SchS.
Cardiovascular disease (CVD)
A role for IRAK1 as a driver for the inflammatory component in atherosclerosis has been proposed. In an analysis of haplotypes of 996 Caucasian patients in the Diabetes Heart Study, the IRAK1 TCCG haplotype, present in 13%, was significantly (P = 0.0004) associated with greater CRP concentrations in women, but not in men [125].
Circulating monocytes are attracted to areas of vascular injury where they become resident macrophages [126] and undergo activation through TLR signaling, in turn activating the IRAK pathway [127]. IRAK1 activity leads to signal transducer and activator of transcription 3 (STAT3) activation [128]. In hepatocytes, in the presence of IL-6 (produced downstream of IRAK1 in the IL-1β pathway) activated STAT3 in turn activates C-reactive protein (CRP), a widely used biomarker for cardiovascular disease risk [129, 130]. STAT3 activation also leads to increased gene expression of interleukin-10 (IL-10) [131], the product of which is typically elevated in atherosclerosis. In addition, a recent study established a causal link between NLRP3-mediated overproduction of IL-1β and the development of atherosclerosis in a ten-eleven translocation 2 (TET2) mutant mouse model [132], which could be abrogated by an NLRP3 inflammasome inhibitor [133]. Thus, an ample mechanistic rationale exists for the study of IRAK1 inhibition in atherosclerosis.
Although IRAK1 inhibition has not been tested directly in clinical CVD, the positive results recently reported for IL-1β blockade in atherosclerosis lend support to the testing of this hypothesis. In the CANTOS trial, a multiyear study of >10,000 patients with previous myocardial infarction (MI) and high CRP levels, the anti-IL-1β antibody canakinumab provided dose-dependent reductions in CRP level and reduced the incidence of cardiovascular death [134]. A significant (P = 0.021) 15% reduction occurred in the risk of first occurrence of nonfatal MI, any nonfatal stroke, or CVD death relative to placebo. A subsequent analysis associated reductions in high-sensitivity CRP (hsCRP) levels to <2mg/L with the largest risk reductions, suggesting hsCRP level after a single canakinumab dose as a simple method of identifying patients most likely to derive the greatest benefit [135]. This provides a rationale and design for potential trials of IRAK1 inhibitors to prevent the progression of CVD in very high-risk patients.
Metabolic diseases
Suppression of IRAK1 increases glucose uptake into skeletal muscle and decreases insulin resistance. IRAK1 knockout mice had improved insulin sensitivity in muscle tissue relative to controls in a recent study [39]. In humans, IRAK1 expression is significantly higher in adipose tissue from obese versus nonobese individuals and correlates with degree of inflammation as measured by plasma markers [136].
MiR-146a, which affects IRAK1 and TRAF6, has been extensively employed to investigate the role of IRAK1 in metabolic diseases. It has been shown to reduce inflammatory response in human adipocytes [137]. A recent study examined miR-146a expression levels in 30 patients with type 2 diabetes (T2D) and 30 controls, finding that these levels were significantly decreased in PBMCs and plasma in patients with T2D relative to healthy participants [138]. Notably IRAK1 mRNA expression was significantly increased in the T2D group. A recent study also suggests that miRNA-146a is involved in the pathogenesis of diabetic peripheral neuropathy (DPN) [139]. Levels of this microRNA were significantly decreased, and levels of TNFα, IL-1β, and NF-κB were significantly increased in DPN rats relative to T2D or control rats. Collectively, evidence of IRAK1 involvement in insulin resistance, T2D, and NAFLD suggest that further studies of IRAK1 inhibitors in metabolic diseases are warranted.
Ischemia/reperfusion injury
MiR-146a has been reported to be protective in models of liver [46], myocardial [140], and intestinal [141] ischemia/reperfusion injury. In a model of hypoxia/ischemia-induced brain injury in the rat, IRAK1/4 inhibitor I was shown to be neuroprotective [142].
Role of IRAK1 in neoplasia
Inflammation and thus signaling through the TLR-IRAK pathways are important in promotion of mutagenesis and subsequent neoplastic transformation, tumor aggressiveness, propensity to become metastatic, and also may be involved in tumor resistance to therapy [143–145].
IRAK1 in oncology – solid tumors (Table 2)
Table 2: Experimental evidence for IRAK1 involvement in solid tumor malignancies
Tumor Type |
Experimental System |
Effector(s) |
Effects |
|---|---|---|---|
Breast [82] |
MDA-MB-231 cells |
MiRNA-146a |
MiRNA-146a transfection into BC cells downregulated IRAK1/TRAF6, reduced NF-κB target gene expression, impaired invasion and migration. |
Breast [147] |
TNBC cell lines; MB436 xenograft mouse |
IRAK1 shRNA; IRAK1/4 inhibitor I |
IRAK1 knockdown or inhibition reduced TNBC cell invasion, mammosphere formation; IRAK1 shRNA inhibits tumor growth in a TNBC mouse model; paclitaxel-treated TNBC cells acquire resistance that can be overcome by IRAK1/4 inhibitor + paclitaxel. |
Breast [38] |
Pt-derived tumorspheres; 1q21.3-amplified HCC70 xenograft mouse; other xenograft models |
Pacritinib |
Chromosome 1q21.3, encoding IRAK1, is amplified in recurrent BC; S100A7/8/9 and IRAK1 drive tumorsphere growth, disrupted by pacritinib via IRAK1, not JAK2; pacritinib TGI in xenograft models via IRAK1; pacritinib + paclitaxel caused durable tumor regressions in a neoadjuvant TNBC model. |
Endometrial [148] |
Pt tumor tissue; HEC-1B, JEC cells; BALB/c nude mouse |
IRAK1 siRNA |
IRAK1 tumor levels correlate with survival, stage, metastasis, invasion; knockdown in cells antiproliferative via cell cycle arrest, apoptosis; IRAK1 siRNA transducted cells less tumorigenic in mice. |
HCC [151] |
Pt FFPE liver tissue |
NA |
IRAK1 expression frequency increased in HCC progression, correlates with tumor size and metastasis; IRAK1 upregulation significantly predicts poor survival. |
HCC [152] |
Pt HCC tissue, HCC cell lines; SMMU-7721 xenograft mouse |
IRAK1/4 inhibitor I; IRAK1 siRNA |
IRAK1 overexpressed in HCC tissue; IRAK1 siRNA inhibits growth, augments cisplatin cytotoxicity in cells; IRAK1/4 inhibitor impedes proliferation, migration in cells, and is effective in a xenograft model of HCC. |
HCC [153] |
HCC pt macrophages; STK-/- DEN mouse |
IRAK1/4 inhibitor I |
Tumor suppressor STK4 levels inversely correlate with IRAK1 in HCC macrophages; IRAK1/4 inhibitor I had anti-tumor effects in STK KO HCC mice. |
HNSCC [43] |
UMSCC1, UMSCC6, UMSCC47 cell lines |
IRAK1/4 inhibitor I; IRAK1 shRNA |
IRAK1 overexpressed in 14% of HNSCC; IRAK1 inhibition via shRNA or IRAK1/4 inhibitor I induced apoptosis in HNSCC cell lines. |
NSCLC [157] |
Pt FFPE tumor samples |
NA |
Significantly higher cytoplasmic, lower nuclear IRAK1 expression in tumor cells than normal epithelium; overexpressed early in sequential preneoplastic evolution. |
Melanoma [158] |
Malme-3M, SK-MEL-2, WM115, C32, RPMI-7951, A375, G361 cell lines; A375 xenograft mouse |
IRAK1/4 inhibitor I |
p-IRAK1 constitutively expressed in 42% of cell lines; IRAK1/4 inhibition enhances vinblastine, 5-FU cell killing; combining IRAK1/4 inhibitor I with vinblastine improved TGI and survival in a xenograft mouse model. |
Melanoma [159] |
M4Beu, 7GP122, T1P26R, FM516-SV, WM239, C8161, MeWo cell lines; newborn immunosuppressed rat |
NA |
IRAK1 gene expression elevated in highly metastatic versus normal variants of melanoma cells grafted into immunosuppressed newborn rats. |
BC, breast cancer; DEN, diethylnitrosamine; FFPE, formalin-fixed and paraffin-embedded; HCC, hepatocellular carcinoma; NA, not applicable; NSCLC, non-small cell lung cancer; OS, overall survival; TGI, tumor growth inhibition.
Breast cancer (BC)
Several lines of evidence implicate IRAK1 in BC tumor growth and metastasis. In the metastatic human BC cell line MDA-MB-231, miRNA-146a downregulated NF-κB activity and markedly impaired invasion and cell migration [82]. The inflammasome/IL-1β pathway in the tumor microenvironment may be involved in IRAK1 effects. In the EO771 orthotopic BC mouse model, murine anti-IL-1R antibody significantly reduced tumor growth and metastasis [146].
A study of triple-negative BC (TNBC) correlated IRAK1 expression with poor survival (P = 0.0047) and found that IRAK1, but not IRAK4, overexpression conferred a TNBC growth advantage through an NF-κB-dependent mechanism, and that such cells were susceptible to genetic (shRNA) and pharmacologic (IRAKA1/4 inhibitor I) IRAK1 inhibition [147]. IRAK1 shRNA inhibition also impaired tumor growth and metastasis in TNBC xenograft models. In TNBC cell lines, paclitaxel treatment was found to increase IRAK1 phosphorylation, leading to acquired resistance and cancer stem cell enrichment; sensitivity to low doses of paclitaxel was restored by co-administration of IRAK1/4 inhibitor I, with the combination inducing massive apoptosis involving both NF-κB and p38-MCL-1 signaling pathways. A later study by the same group found that amplification of chromosome 1q21.3, which encodes S100 calcium-binding protein family members that activate TLRs and increase IRAK1 phosphorylation (in turn establishing a feedback loop that acts to increase production of both), was a trackable biomarker in BC. It was present in 10–30% of primary tumors but >70% of recurrent tumors regardless of subtype, and strongly correlated with early relapse and poor survival [38]. The study also demonstrated that IRAK1 drives the growth of patient-derived BC tumorspheres, and that BC growth could be disrupted by pacritinib both in in vitro and in vivo models. These results identified IRAK1 as an actionable target spanning different BC subtypes, and pacritinib as a potential agent for clinically testing this hypothesis in cancer types harboring 1q21.3 amplification. Amplification of 1q23 has been observed in other malignancies including multiple myeloma, hepatoma, acute myeloid leukemia (AML), and myelofibrosis.
Endometrial cancer (EC)
A recent study suggested a role for IRAK1 expression in EC tumorigenesis. The study found that IRAK1 levels are elevated in EC, with higher expression significantly correlating with poorer survival, and also correlating with higher tumor stage, lymph node metastasis, and myometrial invasion [148]. In EC cell lines, IRAK1 knockdown by siRNA induced cell cycle arrest and apoptosis, inhibiting proliferation, migration, and invasion. EC cells stably transducted with IRAK1 siRNA produced significantly lower tumor growth rates in nude mice compared with control cells.
Hepatocellular carcinoma (HCC)
Hepatic steatosis and nonalcoholic fatty liver disease (NAFLD) associated with obesity and type II diabetes have been linked with the increasing prevalence of HCC [149, 150]. IRAK1 expression may be diagnostic and prognostic in HCC [151]. An immunohistochemistry study (N = 171) found increasing IRAK1 expression in normal, cirrhotic, para-carcinoma, and HCC liver tissues. Rates of positive IRAK1 expression significantly correlated with large tumor size (P = 0.047) and the presence of metastasis (P = 0.041). IRAK1 upregulation predicted poorer survival in HCC (45.7 vs. 71.0 months; P = 0.008 for high- versus low-expression) in an analysis of The Cancer Genome Atlas (TCGA) database. Similarly, another study reported IRAK1 overexpression in clinical HCC specimens [152]. The authors reported that IRAK1 siRNA in HCC cell lines inhibited growth and augmented sensitivity to cisplatin-induced apoptosis, whereas IRAK1/4 inhibitor I impeded proliferation and migration. In an HCC xenograft model, it significantly (P < 0.05) reduced tumor volume and weight. Amplifcation of 1q23.1 (discussed in the context of breast cancer above) has also been observed in hepatocellular carcinoma [147].
Tumor suppressor serine/threonine kinase 4 (STK4) regulates TLR-mediated inflammatory responses in macrophages and protects against chronic inflammation-associated HCC [153]. STK4 binds to and phosphorylates IRAK1; its expression was markedly reduced in macrophages from HCC patients, inversely correlating with IRAK1 and IL-6 levels. In a diethylnitrosamine model, STK-/- mice developed more tumors than control mice did, but treatment with IRAK1/4 inhibitor I reduced liver tumor numbers to levels similar to levels in controls. Thus, macrophage STK4 may be protective in inflammation-induced HCC via IRAK1, and its deficiency mitigated by IRAK1 inhibition. In a murine hepatic steatosis and fibrosis model that leads to a high proportion of HCC, pacritinib was found to prevent fibrosis and to decrease serum levels of a marker for hepatocyte necrosis, cytokeratin 18, suggesting that suppression of IRAK1 may be beneficial in preventing premalignant fibrosis [93].
Head and neck squamous carcinoma (HNSCC)
IRAK1 is a transcriptional target of DEK, a protein known to promote the growth of both human papillomavirus (HPV) negative and HPV-positive HNSCC [43]. IRAK1 was overexpressed in 14% of HNSCC according to an analysis of TCGA data. Inhibiting IRAK1 via shRNA or IRAK1/4 inhibitor I induced apoptosis in HNSCC cell lines, which was enhanced by simultaneously targeting DEK.
Lung cancer (LC)
Elevated CRP level is a known risk factor for subsequent development of LC, implicating pulmonary inflammation in lung carcinogenesis [154]. Long-term (3.7 year) follow-up of the randomized phase 3 CANTOS trial of the anti-IL-1β antibody canakinumab in patients with atherosclerosis, prior MI, and high CRP level found a 77% reduction in risk of LC death relative to placebo in the canakinumab arm (HR 0.23; P = 0.0002) [155]. Additional support for a chemopreventative role for IL-1β inhibition in oncology comes from a phase 2 study of patients with smoldering or indolent multiple myeloma at high risk of progression to active myeloma, which found that the IL-1 receptor antagonist anakinra led to a chronic disease state and improved progression-free survival in responders [156].
Implicating IL-1β-mediated inflammation in cancer development raises the possibility that IRAK1, upstream of IL-1β, and with a role in the NF-κB pathway via TLR/IL-1R signaling, may represent a target for cancer prevention in patients known to be at high risk. Non-small cell lung cancer (NSCLC) is the most commonly occurring LC. IRAK1 has significantly higher cytoplasmic and lower nuclear expression in NSCLC tumors than in normal epithelium and is an early precursor of NSCLC development [157], making clinical studies of IRAK1 inhibitors in patients with elevated CRP who are at high risk for NSCLC of potential interest.
Melanoma
Phospho-IRAK1 is constitutively expressed in 42% of melanoma cell lines, and IRAK1/4 inhibition in combination with certain chemotherapeutic agents enhanced cell killing [158]. Similarly, in a xenograft mouse model of melanoma, IRAK1/4 inhibitor I in combination with vinblastine improved TGI and survival relative to either agent alone. In another study, IRAK1 gene transcript levels were found to be elevated in highly metastatic versus normal variants of human melanoma cells grafted into immunosuppressed newborn rats, suggesting a role for IRAK1 in melanoma metastasis [159].
IRAK1 in oncology – hematologic malignancies (Table 3)
Table 3: Experimental evidence for IRAK1 involvement in hematologic malignancies
Tumor Type |
Experimental System |
Effector(s) |
Effects |
|---|---|---|---|
AML [36] |
Primary AML cells |
Pacritinib |
Antiproliferative, cytotoxic effects in patient AML cells via IRAK1, not JAK2 or FLT3; IRAK1 kinase domain mutations conferred pacritinib resistance. |
AML [161] |
Patients with AML |
Pacritinib |
Clinical benefit (stable disease or better) in 3/7 (43%) patients with AML. |
T-ALL [44] |
Primary T-ALL cells; NSG T-ALL mouse model |
IRAK1/4 inhibitor I; IRAK1 shRNA, IRAK4 shRNA |
Primary AML cells have high IRAK1 mRNA levels; IRAK 1 and/or 4 inhibition is antiproliferative; IRAK1/4 inhibitor reduces tumor burden and prolongs survival when combined with Bcl-2 inhibitor or vinblastine. |
MLL [163] |
SEM(MLL-AFF1) cell line; MLL-AF9 leukemia mice |
IRAK1/4 inhibitor I; “Compound 26” [165] |
Both IRAK inhibitors impede proliferation of MLL leukemia cells; both delayed disease progression, improved survival in MLL mouse model. |
ABC-DLBCL [167] |
ABC- and GCB-DLBCL cell lines |
IRAK1/4 inhibitor I; IRAK1 shRNA, IRAK4 shRNA |
IRAK1/4 inhibitor I, IRAK1 shRNA, cytotoxic to ABC-DLBCL but not GCB-DLBCL cell lines; IRAK1 kinase activity not required for cytotoxicity. |
ABC-DLBCL [28] |
MyD88 L265P ABC-DLBCL cell lines |
Jh-X-119-01 |
Selective IRAK1 inhibition suppressed NF-κB activation, showed synergy with ibrutinib in killing MyD88 mutated ABC-DLBCL cells. |
CLL [169] |
Patient PBMCs |
Pacritinib |
Pacritinib significantly impaired monocyte and monocyte-derived macrophage viability, increased apoptosis, and significantly inhibited CLL cell viability, but contribution of IRAK1 inhibition unknown. |
WM [124] |
MyD88 L265P cell lines |
IRAK1/4 inhibitor I |
IRAK1/4 inhibitor I decreased nuclear NF-κB staining, phosphorylation of NF-κB and IκBα in mutated but not wild-type Myd88 cells. |
WM [173] |
MyD88 L265P cell lines |
IRAK1/4 inhibitor I |
IRAK1/4 inhibitor I was synergistic with ibrutinib in MyD88 L265P cells and enhanced cell killing relative to either single agent. |
WM [28] |
MyD88 L265P WM cell lines |
Jh-X-119-01 |
Selective IRAK1 inhibition suppressed NF-κB activation, and showed synergy with ibrutinib in killing MyD88 mutated WM cells. |
FL [174] |
Patient biopsy tissue |
N/A |
IRAK1 is 1 of the 2 most significant predictors of FL transformation to more aggressive disease. |
MDS [176] |
MiR-146a -/- mice |
MiR-146a |
MiR-146 knockout mice display an MDS-like phenotype. |
MDS [177] |
MiR-146a -/- mice |
MiR-146a |
MiR-146a regulates HSC homeostasis independently of Traf6 expression; Irak1 is a candidate. |
MDS [178] |
Patient tissue samples; MDS cell lines; NSG MDS xenograft mice |
NA |
IRAK1 overexpressed in 20–30% of patients, hyperactivated in BM samples in the majority of patients; IRAK1 expression correlates with poor survival (P = 0.035); IRAK1/4 inhibitor I is antiproliferative in MDS progenitor cells, ameliorates disease in MDS xenograft mouse model. |
PEL [186] |
KSHV-transfected SLK cells |
mIR-K5, mIR-K9 |
Both mRNAs downregulate IRAK1 and MyD88, and reduce IL-1α-induced IL-6, IL-8 levels. |
PEL [187] |
PEL cell lines; PEL patient exudates |
IRAK1 shRNA |
NGS identified common Phe196Ser IRAK1 mutation; IRAK1 shRNA abolished PEL cell growth in culture. |
ABC, activated B-cell like; GCB, germinal center B-cell like; HSC, hematopoietic stem cell; NA, not applicable; NGS, next-generation sequencing.
AML
The importance of IL-1 signaling in AML suggests IRAK1 as a potential intervention point. In patient samples, IL-1β promotes expansion of AML progenitor cells as well as growth and survival of AML cells, and IL1R1 null mice have improved survival relative to WT in an AML model [160].
Direct evidence for IRAK1 involvement in AML comes from a recent report confirming its overexpression and importance to survival signaling in primary AML cells [36]. The multikinase inhibitor pacritinib proved antiproliferative and cytotoxic in primary AML cells with a variety of genetic mutations. Primary AML samples were sensitive to pacritinib but not other JAK2 or FLT3 inhibitors, implicating IRAK1 inhibition as its operative mechanism of action in AML. IRAK1 kinase domain mutations engineered to resemble CDK2 conferred significant pacritinib resistance in AML cells, supporting this conclusion and confirming its mode of binding. Clinical evidence of pacritinib activity in AML has been shown. The dose-ranging portion of a phase 1/2 study reported a 43% clinical benefit rate (≥stable disease) for the 7 patients with AML who were treated with pacritinib [161]. Given the high unmet need in AML and results reported to date, IRAK1 inhibition appears worthy of further clinical investigation in this malignancy, particularly in combination with other active therapies.
T-acute lymphoblastic leukemia (T-ALL)
Patient-derived T-ALL cells have elevated levels of IRAK1 and IRAK4 mRNA [44]. IRAK1/4 inhibitor I, IRAK1 shRNA, and IRAK4 shRNA are antiproliferative in these cells through a mechanism involving destabilization of the anti-apoptotic protein induced myeloid leukemia cell differentiation protein 1 (MCL-1). In an NSG mouse model of T-ALL, IRAK1/4 inhibitor I was antiproliferative, but not cytotoxic. Combining it with the Bcl-2 inhibitor ABT-737 or vinblastine, however, markedly decreased tumor burden and prolonged survival relative to control.
Mixed lineage leukemias (MLLs)
MLLs, which include MLL-ALL and MLL-AML, represent the majority of leukemias in infants and, in contrast to most other pediatric leukemias, are associated with a dismal prognosis [162–164]. MLLs are characterized by the presence of MLL fusion proteins due to chromosomal transloactions at 11q23 that results in the destruction of normal MLL histone methyltransferase function. A recent study revealed that the chimeric MLL protein is more stable and therefore present at higher levels than the wild-type protein, and that degradation of the normal (but not the chimeric) protein is regulated by ubiquitin conjugating enzyme E2 O (UBE2O) in response to IL-1 signaling [163]. Using IRAK1/4 inhibitor 1 and a more IRAK4-specific inhibitor (“Compound 26”, Figure 1 [165]), investigators found that IRAK inhibition stabilized the MLL protein, allowing it to displace the MLL chimera from some of its target genes. IRAK1/4 inhibition thereby impeded proliferation in MLL cell lines, delayed progression, and improved survival in a murine MLL model. In addition to implicating IRAK inhibition in an area of high unmet need, the study raises the possibility that a similar strategy to improve protein stabilization may also apply to other cancers caused by translocations.
Activated B cell-like diffuse large B cell lymphoma (ABC-DLBCL)
The prognosis for patients diagnosed with DLBCL varies, with an estimated 5-year survival of 60% for those having the germinal center B-cell like (GCB) subtype, but only 35% for those with the ABC subtype [166]. The ABC subtype (~30% of DLBCL patients) is driven by MyD88 gain-of-function mutation that results in constitutive activation and depends upon IRAK1- and IRAK4-mediated signaling for survival [167]. IRAK1/4 inhibitor 1 and IRAK1 shRNA have been shown to be cytotoxic to ABC- but not GCB-DLBCL cell lines although IRAK1 kinase activity was not required for ABC-DLBCL cell survival. Recently, the selective, irreversible IRAK1 inhibitor JH-X-119-01 demonstrated cytotoxicity in MyD88 mutated cell lines that was synergistic with ibrutinib [28]. A recent report that ibrutinib resistance due to BTKCys481 mutations is driven by ERK1/2 activation with subsequent release of inflammatory cytokines including IL-6 and IL-10 provides further support for exploring upstream IRAK1 inhibition ABC-DLBCL and WM [168].
Chronic lymphocytic leukemia (CLL)
Monocyte-derived nurse-like cells (NLCs) play important roles in the CLL tumor microenvironment. NLC generation and CLL viability are dependent on colony-stimulating factor-1 receptor (CSF-1R). In patient-derived samples, including some with the difficult-to-treat 17p deletion, pacritinib significantly impaired macrophage viability and NLC survival. Pacritinib reduced CLL cell viability and increased apoptosis to a greater extent than ibrutinib did [169]. These effects were attributed to its CSF-1R and possibly JAK2 inhibition, but it remains to be determined whether its IRAK1 inhibition may have also contributed. Another study identified the IL-17/IL-6 axis as playing a crucial role in CLL [170]; as previously described, pacritinib has been shown to reduce levels of these cytokines in a cellular system.
Waldenström macroglobulinemia (WM)
An MyD88 L265P driver mutation that results in constitutive NF-κB activation is found in approximately 93% of WM patients [171]. In cell lines expressing this mutation, but not in WT cells, IRAK1/4 inhibitor I decreased nuclear NF-κB p65 staining, and phosphorylation of NF-κB and IκBα [124]. MyD88 L265P mutation signaling is via the TLR4 pathway mediated by Bruton tyrosine kinase; the small minority of WM patients with WT MyD88 do not achieve major responses to ibrutinib [172]. Combining ibrutinib with IRAK1/4 inhibitor I provided synergistic cell killing in MyD88 L265P cell lines and enhanced cell killing relative to either agent alone in primary MyD88 L265P cells [173]. Survival in MyD88 L265P mutant WM cell lines appeared to be more dependent on IRAK1 than IRAK4; selective IRAK1 inhibitor Jh-X-119-01 was synergistic with ibrutinib in cell lines with the mutation [28].
The MyD88 L265P mutation is also found in the majority of patients with immunoglobulin M monoclonal gammopathy of undetermined significance, which can progress to WM or other B-lymphoproliferative disorders [171]. Thus, clinical investigation of IRAK1 inhibition in combination with or after failure of a BTK inhibitor appears warranted.
Follicular lymphoma (FL)
IRAK1 is among the NF-κB-linked genes for which overexpression and high copy number in FL significantly (P < 0.05) correlate with transformation to more aggressive disease, most commonly DLBCL [174]. Along with TRIM7, IRAK1 is the most significant predictor of such transformations. In a phase 1 study of single-agent pacritinib in relapsed/refractory NHL, among 9 evaluable patients with FL, best response was 1 partial response (11%) and 7 stable disease (78%) [34]. Whether IRAK1 inhibition by pacritinib specifically contributed to this activity is unknown.
Myelodysplastic syndromes (MDSs)
A potential rationale for inhibiting IRAK1 in MDS emerged from its role as an activator of the NLRP3 inflammasome and evidence continues to accumulate. Activation of the NLRP3 inflammasome in HSCs and the resulting IL-18 production and pyroptosis have been shown to drive the MDS phenotype [175]. MiR-146a, an endogenous IRAK1 and TRAF6 regulator, is located within a chromosome 5 region frequently deleted in MDS (5q- syndrome), and miR-146a-deficient mice display an MDS-like phenotype [176]. MiR-146a has been shown to play a role in HSC homeostasis, potentially through IRAK1 [177].
IRAK1 is overexpressed in 20–30% of patients with MDS and correlates with poor survival; IRAK1 is hyperactivated in the majority of patient bone marrow samples [178, 179]. Inhibiting IRAK1 using IRAK1/4 inhibitor I blocks NF-κB activation, interferes with MDS progenitor cell function and growth, and ameliorates disease in an MDS xenograft model. Collectively, these data provide a strong rationale for targeting IRAK1 in MDS.
Myeloproliferative neoplasms (MPNs)
IRAK1 may play a role in MPNs (polycythemia vera [PV], essential thrombocythemia [ET], and myelofibrosis) via inflammatory cytokines, such as IL-6 and IL-8, that are downstream products of the dysregulated NF-κB pathway. Circulating IL-8 levels are increased in patients with MF and significantly correlate with poor survival (P < 0.001) in newly diagnosed disease [180]. Chronic inflammation has also been proposed as a driver for the development of atherosclerosis and second cancers in patients with MPNs via aberrant JAK-STAT signaling [95]. JAK inhibitors, such as ruxolitinib [181, 182] and pacritinib [32, 33, 183], have demonstrated clinical efficacy in MPNs. As previously described, pacritinib, but not ruxolitinib, also inhibits IRAK1 at clinically attainable levels, which may impart additional benefits [93]. Further studies, however, are needed to confirm such effects and determine whether its IRAK1 inhibition plays a role in its clinical profile.
KSHV-associated malignancies
Kaposi sarcoma-associated herpesvirus (KSHV, also called HHV8) is the causative agent of Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and KSHV-associated multicentric Castleman’s disease (MCD), all of which occur predominantly in HIV-positive patients [184, 185]. Among the micro-RNAs that KSHV expresses are mIR-K5 and mIR-K9. These have been shown to suppress IRAK1 and MyD88 expression and the subsequent downstream production of inflammatory cytokines that would normally be part of the immune response to the virus [186]. On the surface, this argues against further interference with IRAK1 as a therapeutic strategy. A later study, however, examined common missense mutations in PEL, finding that a single nucleotide variation (SNV) leading to an IRAK1 Phe196Ser driver mutation resulted in constitutive activation and survival in PEL cell lines [187]. A published correction to this report later noted that this SNV is relatively common as reported in various databases, and that the odds ratio for its association with PEL may have been as low as 8.4 using the limited number of available samples for this rare disease. Neverless, high levels of IL-1 and downstream cytokines are expressed in KSHV-associated neoplasia, suggesting that despite the induction of suppressing miRNAs, IRAK is likely activated by KSHV. Additional studies are necessary to clarify the effect of this virus on activation of the IRAK1 pathway.
CONCLUSIONS
As a key component of the myddosome and activator of the NLRP3 inflammasome, IRAK1 participates in multiple IL-1 and TLR-driven signaling processes that serve to regulate immunity and inflammation. Dysregulation of these processes have been implicated in multiple inflammatory diseases and appears to be involved in initiation and promotion of a number of cancer types. Since the discovery of IRAK1 in 1996, preclinical studies have established its involvement in a spectrum of inflammatory diseases, including fibrotic diseases, metabolic disorders, arthritis, lupus, and sepsis. Similarly, evidence suggests that IRAK1 plays a critical role in multiple solid tumor and hematologic malignancies, most notably in AML and breast cancer, indications with high unmet need. Tools used to probe the roles of IRAK1 have included genetic and RNA-based methods as well as nonselective inhibitors like IRAK1/4 inhibitor I, but selective IRAK1 inhibitors, unlike selective IRAK4 inhibitors, have yet to enter the clinic. The recent discovery that the JAK2/FLT3 inhibitor pacritinib inhibits IRAK1 at clinically relevant levels, along with clinical data indicating improvements in cytopenias in some patients with MF, and preclinical fibrosis data hint at a clinical potential for pacritinib as an IRAK1 inhibitor in other indications. With the availability of selective IRAK1 inhibitors suitable for clinical development, ongoing and future translational and clinical studies will determine the utility of IRAK1 inhibitors in the treatment of human diseases.
ACKNOWLEDGMENTS
This review was supported by CTI BioPharma. We thank Robert Rydzewski, MS, CMPP, and Stacey Rose, PhD, of Nexus Global Group Science for providing writing assistance.
CONFLICTS OF INTEREST
AF – Speakers bureau (Incyte), travel/accomodations (Incyte, Gilead)
AA – Research funding (CTI)
SAF- Employment, equity ownership, travel/accomodations (CTI)
JM – Consulting/advisory role (CTI), research funding (CTI, Incyte, Janssen, Novartis, Merck, Promedior)
QY – Nothing to disclose
JS – Employment, leadership role, equity ownership (CTI).
FUNDING
No grant funding to report.
REFERENCES
1. Cohen P. The TLR and IL-1 signalling network at a glance. J Cell Sci. 2014; 127:2383-2390.
2. Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010; 80:1981-1991.
3. Jain A, Kaczanowska S, Davila E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front Immunol. 2014; 5:553.
4. Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. Br J Cancer. 2015; 112:232-237.
5. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006; 103:12481-12486.
6. Lee KL, Ambler CM, Anderson DR, Boscoe BP, Bree AG, Brodfuehrer JI, Chang JS, Choi C, Chung S, Curran KJ, Day JE, Dehnhardt CM, Dower K, et al. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J Med Chem. 2017; 60:5521-5542.
7. Singer JW, Al-Fayoumi S, Ma H, Komrokji RS, Mesa R, Verstovsek S. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J Exp Pharmacol. 2016; 8:11-19.
8. Hammaren HM, Virtanen AT, Silvennoinen O. Nucleotide-binding mechanisms in pseudokinases. Biosci Rep. 2015; 36:e00282.
9. Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A. 2002; 99:5567-5572.
10. Wang L, Qiao Q, Ferrao R, Shen C, Hatcher JM, Buhrlage SJ, Gray NS, Wu H. Crystal structure of human IRAK1. Proc Natl Acad Sci U S A. 2017; 114:13507-13512.
11. Wang Z, Liu J, Sudom A, Ayres M, Li S, Wesche H, Powers JP, Walker NP. Crystal structures of IRAK-4 kinase in complex with inhibitors: a serine/threonine kinase with tyrosine as a gatekeeper. Structure. 2006; 14:1835-1844.
12. Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996; 271:1128-1131.
13. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002; 298:1912-1934.
14. Kuglstatter A, Villasenor AG, Shaw D, Lee SW, Tsing S, Niu L, Song KW, Barnett JW, Browner MF. Cutting Edge: IL-1 receptor-associated kinase 4 structures reveal novel features and multiple conformations. J Immunol. 2007; 178:2641-2645.
15. Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, et al. IRAK-4 inhibitors. Part II: a structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg Med Chem Lett. 2008; 18:3291-3295.
16. Buckley GM, Fosbeary R, Fraser JL, Gowers L, Higueruelo AP, James LA, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, et al. IRAK-4 inhibitors. Part III: a series of imidazo[1,2-a]pyridines. Bioorg Med Chem Lett. 2008; 18:3656-3660.
17. Buckley GM, Gowers L, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V, Fraser JL. IRAK-4 inhibitors. Part 1: a series of amides. Bioorg Med Chem Lett. 2008; 18:3211-3214.
18. Chaudhary D, Robinson S, Romero DL. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J Med Chem. 2015; 58:96-110.
19. Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, Srivastava R, Venkatesh K, Selvam S, Krishnamurthy P, Mariappan TT, Saxena A, Fan L, et al. Selective IRAK4 Inhibition Attenuates Disease in Murine Lupus Models and Demonstrates Steroid Sparing Activity. J Immunol. 2017; 198:1308-1319.
20. McElroy WT, Tan Z, Ho G, Paliwal S, Li G, Seganish WM, Tulshian D, Tata J, Fischmann TO, Sondey C, Bian H, Bober L, Jackson J, et al. Potent and Selective Amidopyrazole Inhibitors of IRAK4 That Are Efficacious in a Rodent Model of Inflammation. ACS Med Chem Lett. 2015; 6:677-682.
21. Scott JS, Degorce SL, Anjum R, Culshaw J, Davies RDM, Davies NL, Dillman KS, Dowling JE, Drew L, Ferguson AD, Groombridge SD, Halsall CT, Hudson JA, et al. Discovery and Optimization of Pyrrolopyrimidine Inhibitors of Interleukin-1 Receptor Associated Kinase 4 (IRAK4) for the Treatment of Mutant MYD88(L265P) Diffuse Large B-Cell Lymphoma. J Med Chem. 2017; 60:10071-10091.
22. Seganish WM, Fischmann TO, Sherborne B, Matasi J, Lavey B, McElroy WT, Tulshian D, Tata J, Sondey C, Garlisi CG, Devito K, Fossetta J, Lundell D, et al. Discovery and Structure Enabled Synthesis of 2,6-Diaminopyrimidin-4-one IRAK4 Inhibitors. ACS Med Chem Lett. 2015; 6:942-947.
23. Genung NE, Guckian KM. Small Molecule Inhibition of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4). Prog Med Chem. 2017; 56:117-163.
24. Powers JP, Li S, Jaen JC, Liu J, Walker NP, Wang Z, Wesche H. Discovery and initial SAR of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg Med Chem Lett. 2006; 16:2842-2845.
25. Nanda SK, Lopez-Pelaez M, Arthur JS, Marchesi F, Cohen P. Suppression of IRAK1 or IRAK4 Catalytic Activity, but Not Type 1 IFN Signaling, Prevents Lupus Nephritis in Mice Expressing a Ubiquitin Binding-Defective Mutant of ABIN1. J Immunol. 2016; 197:4266-4273.
26. Hossen MJ, Yang WS, Kim D, Aravinthan A, Kim JH, Cho JY. Thymoquinone: An IRAK1 inhibitor with in vivo and in vitro anti-inflammatory activities. Sci Rep. 2017; 7:42995.
27. Singh AK, Umar S, Riegsecker S, Chourasia M, Ahmed S. Regulation of Transforming Growth Factor beta-Activated Kinase Activation by Epigallocatechin-3-Gallate in Rheumatoid Arthritis Synovial Fibroblasts: Suppression of K(63) -Linked Autoubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6. Arthritis Rheumatol. 2016; 68:347-358.
28. Yang G, Hatcher J, Wang J, Liu X, Munshi M, Chen J, Xu L, Tsakmaklis N, Demos M, Kofides A, Chan G, Hunter Z, Patterson C, et al. A novel, highly selective IRAK1 inhibitor Jh-X-119-01 shows synergistic tumor cell killing with ibrutinib in MYD88 mutated B-cell lymphoma cells. Abstract 719. Oral presentation at: 59th Anual Meeting & Exposition for the American Society of Hematology; December 9-12, 2017; Atlanta, GA.
29. Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, William AD, Lee A, Poulsen A, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011; 25:1751-1759.
30. William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011; 54:4638-4658.
31. Poulsen A, William A, Blanchard S, Lee A, Nagaraj H, Wang H, Teo E, Tan E, Goh KC, Dymock B. Structure-based design of oxygen-linked macrocyclic kinase inhibitors: discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3). J Comput Aided Mol Des. 2012; 26:437-450.
32. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, Szoke A, Drummond M, Pristupa A, Granston T, Daly R, Dean JP, Al-Fayoumi S, et al. Results of the Persist-2 Phase 3 Study of Pacritinib (PAC) Versus Best Available Therapy (BAT), Including Ruxolitinib (RUX), in Patients (pts) with Myelofibrosis (MF) and Platelet Counts ≤100,000/μl. Abstract LBA-5. Oral presentation at: 58th Anual Meeting & Exposition for the American Society of Hematology; December 3-6, 2016; San Diego, CA.
33. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, Jakucs J, Perkins A, Prasad R, Mayer J, Demeter J, Ganly P, Singer JW, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 2017; 4:e225-e236.
34. Younes A, Romaguera J, Fanale M, McLaughlin P, Hagemeister F, Copeland A, Neelapu S, Kwak L, Shah J, de Castro Faria S, Hart S, Wood J, Jayaraman R, et al. Phase I study of a novel oral Janus kinase 2 inhibitor, SB1518, in patients with relapsed lymphoma: evidence of clinical and biologic activity in multiple lymphoma subtypes. J Clin Oncol. 2012; 30:4161-4167.
35. Unpublished data on file, CTI Biopharma.
36. Hosseini MM, Kurtz SE, Abdelhamed S, Mahmood S, Davare MA, Kaempf A, Elferich J, McDermott JE, Liu T, Payne SH, Shinde U, Rodland KD, Mori M, et al. Pacritinib inhibition of interleukin-1 receptor-associated kinase as a therapeutic strategy for acute myeloid leukemia. Leukemia. 2018 Mar 29. [Epub ahead of print].
37. Al-Fayoumi S, Watson R, O'Mahony A, Singer JW. Comparative biomarker profiles of pacritinib, momelotinib, pexidartinib, and ruxolitinib using BioMAP Diversity PLUS panel. European Journal of Cancer. 2016; 69:S136.
38. Goh JY, Feng M, Wang W, Oguz G, Yatim S, Lee PL, Bao Y, Lim TH, Wang P, Tam WL, Kodahl AR, Lyng MB, Sarma S, et al. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med. 2017; 23:1319-1330.
39. Sun XJ, Kim SP, Zhang D, Sun H, Cao Q, Lu X, Ying Z, Li L, Henry RR, Ciaraldi TP, Taylor SI, Quon MJ. Deletion of interleukin 1 receptor-associated kinase 1 (Irak1) improves glucose tolerance primarily by increasing insulin sensitivity in skeletal muscle. J Biol Chem. 2017; 292:12339-12350.
40. Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, Cao Z, Wasserman SA. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol. 1999; 163:978-984.
41. Pauls E, Nanda SK, Smith H, Toth R, Arthur JSC, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J Immunol. 2013; 191:2717-2730.
42. Guo F, Wu S. Antisense IRAK-1 oligonucleotide blocks activation of NF-kappa B and AP-1 induced by IL-18. Immunopharmacology. 2000; 49:241-246.
43. Adams AK, Bolanos LC, Dexheimer PJ, Karns RA, Aronow BJ, Komurov K, Jegga AG, Casper KA, Patil YJ, Wilson KM, Starczynowski DT, Wells SI. IRAK1 is a novel DEK transcriptional target and is essential for head and neck cancer cell survival. Oncotarget. 2015; 6:43395-43407. https://doi.org/10.18632/oncotarget.6028.
44. Li Z, Younger K, Gartenhaus R, Joseph AM, Hu F, Baer MR, Brown P, Davila E. Inhibition of IRAK1/4 sensitizes T cell acute lymphoblastic leukemia to chemotherapies. J Clin Invest. 2015; 125:1081-1097.
45. Hung PS, Liu CJ, Chou CS, Kao SY, Yang CC, Chang KW, Chiu TH, Lin SC. miR-146a enhances the oncogenicity of oral carcinoma by concomitant targeting of the IRAK1, TRAF6 and NUMB genes. PLoS One. 2013; 8:e79926.
46. Jiang W, Kong L, Ni Q, Lu Y, Ding W, Liu G, Pu L, Tang W, Kong L. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS One. 2014; 9:e101530.
47. Della Mina E, Borghesi A, Zhou H, Bougarn S, Boughorbel S, Israel L, Meloni I, Chrabieh M, Ling Y, Itan Y, Renieri A, Mazzucchelli I, Basso S, et al. Inherited human IRAK-1 deficiency selectively impairs TLR signaling in fibroblasts. Proc Natl Acad Sci U S A. 2017; 114:E514-E523.
48. Li M, Yu D, Ni B, Hao F. Interleukin-1 receptor associated kinase 1 is a potential therapeutic target of anti-inflammatory therapy for systemic lupus erythematosus. Mol Immunol. 2017; 87:94-101.
49. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018; 37:20-36.
50. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010; 3:cm1.
51. Vollmer S, Strickson S, Zhang T, Gray N, Lee KL, Rao VR, Cohen P. The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists. Biochem J. 2017; 474:2027-2038.
52. Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, Ek T, Weiss S, Gekara NO. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015; 42:332-343.
53. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010; 11:373-384.
54. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010; 10:712-723.
55. Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014; 14:821-826.
56. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. 2009; 284:25404-25411.
57. Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll-like receptors. Mol Cells. 2006; 21:174-185.
58. Brikos C, Wait R, Begum S, O'Neill LA, Saklatvala J. Mass spectrometric analysis of the endogenous type I interleukin-1 (IL-1) receptor signaling complex formed after IL-1 binding identifies IL-1RAcP, MyD88, and IRAK-4 as the stable components. Mol Cell Proteomics. 2007; 6:1551-1559.
59. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010; 465:885-890.
60. Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol. 2008; 28:3538-3547.
61. Swantek JL, Tsen MF, Cobb MH, Thomas JA. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J Immunol. 2000; 164:4301-4306.
62. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005; 201:915-923.
63. Hartupee J, Li X, Hamilton T. Interleukin 1alpha-induced NFkappaB activation and chemokine mRNA stabilization diverge at IRAK1. J Biol Chem. 2008; 283:15689-15693.
64. Sun J, Li N, Oh KS, Dutta B, Vayttaden SJ, Lin B, Ebert TS, De Nardo D, Davis J, Bagirzadeh R, Lounsbury NW, Pasare C, Latz E, et al. Comprehensive RNAi-based screening of human and mouse TLR pathways identifies species-specific preferences in signaling protein use. Sci Signal. 2016; 9:ra3.
65. Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark GR. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol Cell Biol. 1999; 19:4643-4652.
66. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012; 11:633-652.
67. de Mooij CEM, Netea MG, van der Velden W, Blijlevens NMA. Targeting the interleukin-1 pathway in patients with hematological disorders. Blood. 2017; 129:3155-3164.
68. Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. 2013; 191:3995-3999.
69. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002; 10:417-426.
70. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014; 157:1013-1022.
71. Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015; 4:296-307.
72. He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016; 41:1012-1021.
73. Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, Pasare C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2014; 111:775-780.
74. Oh K, Lee OY, Park Y, Seo MW, Lee DS. IL-1beta induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer. 2016; 16:724.
75. Kang S, Tanaka T, Kishimoto T. Therapeutic uses of anti-interleukin-6 receptor antibody. Int Immunol. 2015; 27:21-29.
76. Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. 2014; 2:288-294.
77. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014; 6:a016295.
78. Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992; 307:97-101.
79. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008; 14:6735-6741.
80. Chandra R, Federici S, Bishwas T, Nemeth ZH, Deitch EA, Thomas JA, Spolarics Z. IRAK1-dependent signaling mediates mortality in polymicrobial sepsis. Inflammation. 2013; 36:1503-1512.
81. Singh N, Li L. Reduced oxidative tissue damage during endotoxemia in IRAK-1 deficient mice. Mol Immunol. 2012; 50:244-252.
82. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008; 27:5643-5647.
83. Li L, Chen XP, Li YJ. MicroRNA-146a and human disease. Scand J Immunol. 2010; 71:227-231.
84. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011; 208:1189-1201.
85. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL, Li C. Attenuation of Cardiac Dysfunction in Polymicrobial Sepsis by MicroRNA-146a Is Mediated via Targeting of IRAK1 and TRAF6 Expression. J Immunol. 2015; 195:672-682.
86. Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, Abraham E. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med. 2006; 173:1335-1341.
87. Toubiana J, Courtine E, Pene F, Viallon V, Asfar P, Daubin C, Rousseau C, Chenot C, Ouaaz F, Grimaldi D, Cariou A, Chiche JD, Mira JP. IRAK1 functional genetic variant affects severity of septic shock. Crit Care Med. 2010; 38:2287-2294.
88. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing Sepsis as a Global Health Priority - A WHO Resolution. N Engl J Med. 2017; 377:414-417.
89. Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis. Am J Physiol Gastrointest Liver Physiol. 2009; 296:G1324-G1331.
90. Vesey DA, Cheung C, Cuttle L, Endre Z, Gobe G, Johnson DW. Interleukin-1beta stimulates human renal fibroblast proliferation and matrix protein production by means of a transforming growth factor-beta-dependent mechanism. J Lab Clin Med. 2002; 140:342-350.
91. Zou Y, Cai Y, Lu D, Zhou Y, Yao Q, Zhang S. MicroRNA-146a-5p attenuates liver fibrosis by suppressing profibrogenic effects of TGFbeta1 and lipopolysaccharide. Cell Signal. 2017; 39:1-8.
92. Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005; 129:113-121.
93. Al-Fayoumi S, Hashiguchi T, Shirakata Y, Mascarenhas J, Singer JW. Pilot study of the antifibrotic effects of the multikinase inhibitor pacritinib in a mouse model of liver fibrosis. J Exp Pharmacol. 2018; 10:9-17.
94. Le Bousse-Kerdiles MC, Martyre MC. Dual implication of fibrogenic cytokines in the pathogenesis of fibrosis and myeloproliferation in myeloid metaplasia with myelofibrosis. Ann Hematol. 1999; 78:437-444.
95. Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012; 119:3219-3225.
96. Rameshwar P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J Immunol. 2000; 165:2271-2277.
97. Ferrer F, Bellosillo B. RS2431697, a polymorphism of Mir-146a, is a marker for an early risk of progression to secondary myelofibrosis: new epigenetic regulation of JAK2/STAT3 signaling. 8th International Conference on Myeloproliferative Neoplasms, Dublin, Ireland May 15-17, 2018. 2018: Poster 7.
98. Zeiser R, Blazar BR. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N Engl J Med. 2017; 377:2565-2579.
99. van der Waart AB, van der Velden WJ, Blijlevens NM, Dolstra H. Targeting the IL17 pathway for the prevention of graft-versus-host disease. Biol Blood Marrow Transplant. 2014; 20:752-759.
100. Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, Holland AM, Grubin J, Mark NM, Liu C, Iwakura Y, Heller G, van den Brink MR. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood. 2009; 113:945-952.
101. Atarod S, Ahmed MM, Lendrem C, Pearce KF, Cope W, Norden J, Wang XN, Collin M, Dickinson AM. miR-146a and miR-155 Expression Levels in Acute Graft-Versus-Host Disease Incidence. Front Immunol. 2016; 7:56.
102. Betts BC, Bastian D, Iamsawat S, Nguyen H, Heinrichs JL, Wu Y, Daenthanasanmak A, Veerapathran A, O'Mahony A, Walton K, Reff J, Horna P, Sagatys EM, et al. Targeting JAK2 reduces GVHD and xenograft rejection through regulation of T cell differentiation. Proc Natl Acad Sci U S A. 2018; 115:1582-1587.
103. Maitra U, Davis S, Reilly CM, Li L. Differential regulation of Foxp3 and IL-17 expression in CD4 T helper cells by IRAK-1. J Immunol. 2009; 182:5763-5769.
104. Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000; 68:7010-7017.
105. Lu Y, Li X, Liu S, Zhang Y, Zhang D. Toll-like Receptors and Inflammatory Bowel Disease. Front Immunol. 2018; 9:72.
106. Atabaki M, Hashemi M, Daneshvar H, Alijani E. Association between interleukin-1 receptor associated kinase 1 rs3027898 A/C gene polymorphism and rheumatoid arthritis. Biomed Rep. 2017; 6:335-338.
107. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. The role of microRNA-146a (miR-146a) and its target IL-1R-associated kinase (IRAK1) in psoriatic arthritis susceptibility. Scand J Immunol. 2010; 71:382-385.
108. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. A polymorphism in the 3'-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility. Joint Bone Spine. 2010; 77:411-413.
109. Han TU, Cho SK, Kim T, Joo YB, Bae SC, Kang C. Association of an activity-enhancing variant of IRAK1 and an MECP2-IRAK1 haplotype with increased susceptibility to rheumatoid arthritis. Arthritis Rheum. 2013; 65:590-598.
110. Hassine HB, Boumiza A, Sghiri R, Baccouche K, Boussaid I, Atig A, Shakoor Z, Bouajina E, Zemni R. Micro RNA-146a But Not IRAK1 is Associated with Rheumatoid Arthritis in the Tunisian Population. Genet Test Mol Biomarkers. 2017; 21:92-96.
111. Khalifa O, Balandraud N, Lambert N, Auger I, Roudier J, Senechal A, Genevieve D, Picard C, Lefranc G, Touitou I, Mrenda BM, Benedito C, Pardoux E, et al. TMEM187-IRAK1 Polymorphisms Associated with Rheumatoid Arthritis Susceptibility in Tunisian and French Female Populations: Influence of Geographic Origin. J Immunol Res. 2017; 2017:4915950.
112. Singh S, Rai G, Aggarwal A. Association of microRNA-146a and its target gene IRAK1 polymorphism with enthesitis related arthritis category of juvenile idiopathic arthritis. Rheumatol Int. 2014; 34:1395-1400.
113. Song GG, Bae SC, Seo YH, Kim JH, Choi SJ, Ji JD, Lee YH. The association between susceptibility to inflammatory arthritis and miR-146a, miR-499 and IRAK1 polymorphisms. A meta-analysis. Z Rheumatol. 2015; 74:637-645.
114. Yang XK, Li P, Zhang C, Leng RX, Li S, Liu J, Li BZ, Pan HF, Ye DQ. Association between IRAK1 rs3027898 and miRNA-499 rs3746444 polymorphisms and rheumatoid arthritis : A case control study and meta-analysis. Z Rheumatol. 2017; 76:622-629.
115. Zhang H, Pu J, Wang X, Shen L, Zhao G, Zhuang C, Liu R. IRAK1 rs3027898 C/A polymorphism is associated with risk of rheumatoid arthritis. Rheumatol Int. 2013; 33:369-375.
116. William AD, Lee AC, Poulsen A, Goh KC, Madan B, Hart S, Tan E, Wang H, Nagaraj H, Chen D, Lee CP, Sun ET, Jayaraman R, et al. Discovery of the macrocycle (9E)-15-(2-(pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza- tetracyclo[18. 3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9, 14(26),15,17,20,22-nonaene (SB1578), a potent inhibitor of janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) for the treatment of rheumatoid arthritis. J Med Chem. 2012; 55:2623-2640.
117. Madan B, Goh KC, Hart S, William AD, Jayaraman R, Ethirajulu K, Dymock BW, Wood JM. SB1578, a novel inhibitor of JAK2, FLT3, and c-Fms for the treatment of rheumatoid arthritis. J Immunol. 2012; 189:4123-4134.
118. Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, Ojwang JO, Langefeld CD, Ziegler JT, Williams AH, Comeau ME, Marion MC, Glenn SB, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis. 2013; 72:437-444.
119. Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009; 106:6256-6261.
120. Lofgren SE, Frostegard J, Truedsson L, Pons-Estel BA, D'Alfonso S, Witte T, Lauwerys BR, Endreffy E, Kovacs L, Vasconcelos C, Martins da Silva B, Kozyrev SV, Alarcon-Riquelme ME. Genetic association of miRNA-146a with systemic lupus erythematosus in Europeans through decreased expression of the gene. Genes Immun. 2012; 13:268-274.
121. Simon A, Asli B, Braun-Falco M, De Koning H, Fermand JP, Grattan C, Krause K, Lachmann H, Lenormand C, Martinez-Taboada V, Maurer M, Peters M, Rizzi R, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy. 2013; 68:562-568.
122. de Koning HD, Schalkwijk J, Stoffels M, Jongekrijg J, Jacobs JF, Verwiel E, Koenen HJ, Preijers F, Holzinger D, Joosten I, van der Meer JW, Simon A. The role of interleukin-1 beta in the pathophysiology of Schnitzler's syndrome. Arthritis Res Ther. 2015; 17:187.
123. de Koning HD. Schnitzler's syndrome: lessons from 281 cases. Clin Transl Allergy. 2014; 4:41.
124. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, Arcaini L, Pinkus GS, Rodig SJ, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012; 367:826-833.
125. Lakoski SG, Li L, Langefeld CD, Liu Y, Howard TD, Brosnihan KB, Xu J, Bowden DW, Herrington DM. The association between innate immunity gene (IRAK1) and C-reactive protein in the Diabetes Heart Study. Exp Mol Pathol. 2007; 82:280-283.
126. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013; 13:709-721.
127. Lundberg AM, Hansson GK. Innate immune signals in atherosclerosis. Clin Immunol. 2010; 134:5-24.
128. Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem. 2004; 279:51697-51703.
129. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. Emerging Risk Factors Collaboration, C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010; 375:132-140.
130. Paffen E, DeMaat MP. C-reactive protein in atherosclerosis: A causal factor? Cardiovasc Res. 2006; 71:30-39.
131. Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996; 271:9503-9509.
132. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017; 355:842-847.
133. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015; 21:248-255.
134. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017; 377:1119-1131.
135. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ; CANTOS Trial Group. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018; 391:319-328.
136. Ahmad R, Shihab PK, Thomas R, Alghanim M, Hasan A, Sindhu S, Behbehani K. Increased expression of the interleukin-1 receptor-associated kinase (IRAK)-1 is associated with adipose tissue inflammatory state in obesity. Diabetol Metab Syndr. 2015; 7:71.
137. Roos J, Enlund E, Funcke JB, Tews D, Holzmann K, Debatin KM, Wabitsch M, Fischer-Posovszky P. miR-146a-mediated suppression of the inflammatory response in human adipocytes. Sci Rep. 2016; 6:38339.
138. Alipoor B, Ghaedi H, Meshkani R, Omrani MD, Sharifi Z, Golmohammadi T. The rs2910164 variant is associated with reduced miR-146a expression but not cytokine levels in patients with type 2 diabetes. J Endocrinol Invest. 2018; 41:557-566.
139. Feng Y, Chen L, Luo Q, Wu M, Chen Y, Shi X. Involvement of microRNA-146a in diabetic peripheral neuropathy through the regulation of inflammation. Drug Des Devel Ther. 2018; 12:171-177.
140. Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013; 97:432-442.
141. Chassin C, Hempel C, Stockinger S, Dupont A, Kubler JF, Wedemeyer J, Vandewalle A, Hornef MW. MicroRNA-146a-mediated downregulation of IRAK1 protects mouse and human small intestine against ischemia/reperfusion injury. EMBO Mol Med. 2012; 4:1308-1319.
142. Yang YF, Chen Z, Hu SL, Hu J, Li B, Li JT, Wei LJ, Qian ZM, Lin JK, Feng H, Zhu G. Interleukin-1 receptor associated kinases-1/4 inhibition protects against acute hypoxia/ischemia-induced neuronal injury in vivo and in vitro. Neuroscience. 2011; 196:25-34.
143. Bhatelia K, Singh K, Singh R. TLRs: linking inflammation and breast cancer. Cell Signal. 2014; 26:2350-2357.
144. Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014; 15:e493-503.
145. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013; 13:759-771.
146. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016; 6:36107.
147. Wee ZN, Yatim SM, Kohlbauer VK, Feng M, Goh JY, Bao Y, Lee PL, Zhang S, Wang PP, Lim E, Tam WL, Cai Y, Ditzel HJ, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015; 6:8746.
148. Wang Y, Wang Y, Duan X, Wang Y, Zhang Z. Interleukin-1 receptor-associated kinase 1 correlates with metastasis and invasion in endometrial carcinoma. J Cell Biochem. 2017; 119:2545-2555.
149. Smedile A, Bugianesi E. Steatosis and hepatocellular carcinoma risk. Eur Rev Med Pharmacol Sci. 2005; 9:291-293.
150. Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012; 56:1384-1391.
151. Ye ZH, Gao L, Wen DY, He Y, Pang YY, Chen G. Diagnostic and prognostic roles of IRAK1 in hepatocellular carcinoma tissues: an analysis of immunohistochemistry and RNA-sequencing data from the cancer genome atlas. Onco Targets Ther. 2017; 10:1711-1723.
152. Li N, Jiang J, Fu J, Yu T, Wang B, Qin W, Xu A, Wu M, Chen Y, Wang H. Targeting interleukin-1 receptor-associated kinase 1 for human hepatocellular carcinoma. J Exp Clin Cancer Res. 2016; 35:140.
153. Li W, Xiao J, Zhou X, Xu M, Hu C, Xu X, Lu Y, Liu C, Xue S, Nie L, Zhang H, Li Z, Zhang Y, et al. STK4 regulates TLR pathways and protects against chronic inflammation-related hepatocellular carcinoma. J Clin Invest. 2015; 125:4239-4254.
154. Chaturvedi AK, Caporaso NE, Katki HA, Wong HL, Chatterjee N, Pine SR, Chanock SJ, Goedert JJ, Engels EA. C-reactive protein and risk of lung cancer. J Clin Oncol. 2010; 28:2719-2726.
155. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ; CANTOS Trial Group. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017; 390:1833-1842.
156. Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, Geyer SM, Campbell ME, Kyle RA, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009; 84:114-122.
157. Behrens C, Feng L, Kadara H, Kim HJ, Lee JJ, Mehran R, Hong WK, Lotan R, Wistuba II. Expression of interleukin-1 receptor-associated kinase-1 in non-small cell lung carcinoma and preneoplastic lesions. Clin Cancer Res. 2010; 16:34-44.
158. Srivastava R, Geng D, Liu Y, Zheng L, Li Z, Joseph MA, McKenna C, Bansal N, Ochoa A, Davila E. Augmentation of therapeutic responses in melanoma by inhibition of IRAK-1,-4. Cancer Res. 2012; 72:6209-6216.
159. Boukerche H, Su ZZ, Kang DC, Fisher PB. Identification and cloning of genes displaying elevated expression as a consequence of metastatic progression in human melanoma cells by rapid subtraction hybridization. Gene. 2004; 343:191-201.
160. Carey A, Edwards DK 5th, Eide CA, Newell L, Traer E, Medeiros BC, Pollyea DA, Deininger MW, Collins RH, Tyner JW, Druker BJ, Bagby GC, McWeeney SK, et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017; 18:3204-3218.
161. Verstovsek S, Odenike O, Singer JW, Granston T, Al-Fayoumi S, Deeg HJ. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J Hematol Oncol. 2016; 9:137.
162. Grembecka J, Cierpicki T. Stabilizing the Mixed Lineage Leukemia Protein. N Engl J Med. 2017; 376:1688-1689.
163. Liang K, Volk AG, Haug JS, Marshall SA, Woodfin AR, Bartom ET, Gilmore JM, Florens L, Washburn MP, Sullivan KD, Espinosa JM, Cannova J, Zhang J, et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell. 2017; 168:59-72 e13.
164. Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009; 94:984-993.
165. Tumey LN, Boschelli DH, Bhagirath N, Shim J, Murphy EA, Goodwin D, Bennett EM, Wang M, Lin LL, Press B, Shen M, Frisbie RK, Morgan P, et al. Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4. Bioorg Med Chem Lett. 2014; 24:2066-2072.
166. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002; 346:1937-1947.
167. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011; 470:115-119.
168. Chen JG, Liu X, Munshi M, Xu L, Tsakmaklis N, Demos MG, Kofides A, Guerrera ML, Chan GG, Patterson CJ, Meid KE, Gustine J, Dubeau T, et al. BTK(Cys481Ser) drives ibrutinib resistance via ERK1/2, and protects BTK(Wild-Type) MYD88 mutated cells by a paracrine mechanism. Blood. 2018; 131:2047-2059.
169. Polk A, Lu Y, Wang T, Seymour E, Bailey NG, Singer JW, Boonstra PS, Lim MS, Malek S, Wilcox RA. Colony-Stimulating Factor-1 Receptor Is Required for Nurse-like Cell Survival in Chronic Lymphocytic Leukemia. Clin Cancer Res. 2016; 22:6118-6128.
170. Zhu F, McCaw L, Spaner DE, Gorczynski RM. Targeting the IL-17/IL-6 axis can alter growth of Chronic Lymphocytic Leukemia in vivo/in vitro. Leuk Res. 2018; 66:28-38.
171. Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, Morra E, Trojani A, Greco A, Arcaini L, Varettoni M, Brown JR, Tai YT, et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013; 121:2051-2058.
172. Treon SP, Xu L, Hunter Z. MYD88 Mutations and Response to Ibrutinib in Waldenstrom's Macroglobulinemia. N Engl J Med. 2015; 373:584-586.
173. Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, Patterson CJ, Buhrlage SJ, Gray N, Tai YT, Anderson KC, Hunter ZR, Treon SP. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013; 122:1222-1232.
174. Brodtkorb M, Lingjaerde OC, Huse K, Troen G, Hystad M, Hilden VI, Myklebust JH, Leich E, Rosenwald A, Delabie J, Holte H, Smeland EB. Whole-genome integrative analysis reveals expression signatures predicting transformation in follicular lymphoma. Blood. 2014; 123:1051-1054.
175. Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front Oncol. 2016; 6:151.
176. Zhao JL, Starczynowski DT. Role of microRNA-146a in normal and malignant hematopoietic stem cell function. Front Genet. 2014; 5:219.
177. Magilnick N, Reyes EY, Wang WL, Vonderfecht SL, Gohda J, Inoue JI, Boldin MP. miR-146a-Traf6 regulatory axis controls autoimmunity and myelopoiesis, but is dispensable for hematopoietic stem cell homeostasis and tumor suppression. Proc Natl Acad Sci U S A. 2017; 114:E7140-E7149.
178. Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, Mathews L, Ferrer M, Southall N, Guha R, Keller J, Thomas C, Beverly LJ, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013; 24:90-104.
179. Beverly LJ, Starczynowski DT. IRAK1: oncotarget in MDS and AML. Oncotarget. 2014; 5:1699-1700. https://doi.org/10.18632/oncotarget.1880.
180. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011; 29:1356-1363.
181. Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012; 366:787-798.
182. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366:799-807.
183. Komrokji RS, Seymour JF, Roberts AW, Wadleigh M, To LB, Scherber R, Turba E, Dorr A, Zhu J, Wang L, Granston T, Campbell MS, Mesa RA. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood. 2015; 125:2649-2655.
184. Bhutani M, Polizzotto MN, Uldrick TS, Yarchoan R. Kaposi sarcoma-associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin Oncol. 2015; 42:223-246.
185. Goncalves PH, Uldrick TS, Yarchoan R. HIV-associated Kaposi sarcoma and related diseases. AIDS. 2017; 31:1903-1916.
186. Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. Kaposi's sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol. 2012; 86:11663-11674.
187. Yang D, Chen W, Xiong J, Sherrod CJ, Henry DH, Dittmer DP. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma. Proc Natl Acad Sci U S A. 2014; 111:E4762-E4768.