
INTRODUCTION
Gastric signet ring cell adenocarcinoma (SRCA) is characterized by the presence of isolated or small groups of malignant non-cohesive cells (>50%) containing intracytoplasmic mucin and exhibits diffuse growth and invasion without forming ducts [1]. Peritoneal invasion is the most frequent type of metastasis in patients with SRCA. It frequently occurs at the later stages of gastric carcinoma, especially after surgery and significantly contributes to gastric cancer-related mortality [2]. One of the characteristics of SRCA is its resistance to chemotherapy. Most tumor cells with multidrug resistance are characterized by the over-expression of multidrug resistance molecules such as P-glycoprotein (P-gp), lung resistance protein (LRP) and multidrug resistance-associated protein (MRP) [3, 4]. Another mechanism of chemoresistance is the epithelial-mesenchymal transition (EMT) [5]. EMT plays an essential role in tumor development namely cancer metastasis [6] and fibrosis [7]. During EMT, cells display on one hand, decreased expression of epithelial cell markers such as E-cadherin and on the other hand, increased expression of mesenchymal cell markers (Snail, Slug, Vimentin, α-SMA, fibronectin, collagen-I) and enhanced cell motility [8, 9]. SRCA was also characterized by the presence of tissues fibrosis [10]. EMT associated with hypersecretion of heparanase (HPSE) was reported in renal fibrosis. HPSE as a multitasking protein, characterized by enzymatic and non-enzymatic activities, modulates TGF-β-induced EMT and fibrosis [11, 12]. Enzymatically active HPSE binds to the cation-independent mannose 6-phosphate receptor (CD222) expressed on cell surfaces to degrade extracellular matrix [13] with heparan sulfate, a side chain of heparin sulfate proteoglycans (HSPGs) [14]. HSPGs store various cytokines and growth factors such as basic fibroblast growth factor-2 (FGF-2), vascular endothelial growth factor (VEGF), interferon-β (INF-β), and transforming growth factor-β (TGF-β) [15–17]. HPSE has also non-enzymatic functions [18]. The signaling activity is achieved by interacting with transmembrane proteins, leading to an activation of Akt and Src [19], or modulating the activity of factors such as FGF-2 and TGF-β [12, 20]. Recent analysis showed that degradation of heparan sulfate by HPSE increased permeability across the basal membrane and, thus, stimulated the release of cytokines and growth factors leading to tumor development via angiogenesis and metastasis [15, 17, 21]. Recently, protein or messenger RNA (mRNA) expression of HPSE has been identified in various cancer cells and the over-expression of HPSE protein or mRNA in tumor cells has been reported and correlated with the metastatic potential of tumor cells in vitro and in vivo as well as with poor prognosis [22, 23]. However, its role in SRCA is still not clearly clarified.
Suramin, a polysulfonated naphthylurea, was initially used to treat African parasitic infections, such as Rhodesian and Gambian trypanosomiasis [24]. Suramin, an inhibitor of HPSE and its analogues showed antiangiogenic and antiproliferative properties [25]. Increased levels of circulating glycosaminoglycans have been observed in suramin-treated cancer patients, suggesting that it might inhibit glycosaminoglycan catabolism [26]. Suramin has also been shown to inhibit HPSE in many human cancer cell lines by independent groups [27]. Despite the U.S. Food and Drug Administration disapproving the use of suramin at therapeutic concentrations, low or non-cytotoxic doses of suramin might be used as effective treatment in SRCA.
The aim of this study was to identify the position of HPSE in the SRCA malignancy and its inhibition by suramin.
RESULTS
Gastric signet ring cell adenocarcinoma nodules express HPSE
As presented in Figure 1A, high expression (p = 0.0327) of HPSE protein was found in the ascitic samples of primary gastric signet ring cell adenocarcinoma (SRCA) as compared to gastric non-SRCA and colic cancer by ELISA. Figure 1B shows that mRNA expression of HPSE is significantly higher in the tumoral zone of gastric SRCA than non-SRCA (p = 0.0002). HPSE mRNA was also found to be highly expressed in peritumoral zone of SRCA. As presented in Figure 1C, HPSE mRNA was found to be more (p = 0.015) in peritumoral-SRCA as compared to peritumoral non-SRCA. Relative expression of HPSE by several cancer cells such as primary SRCA cells isolated from peritoneal fluid of SRCA patients and various cell lines such as ovarian (OVCAR-3), gastric (AGS, KATO-III) presented in Figure 1D. Relative HPSE activity from the supernatants of ovarian (OVCAR-3) and gastric (KATO-III) cell lines presented in Figure 1E. The presence of HPSE was confirmed by immunohistochemistry in KATO-III cell line (Figure 1F). These results are in favor of high expression of HPSE mRNA and protein in gastric SRCA as found by qPCR and ELISA respectively in SRCA.
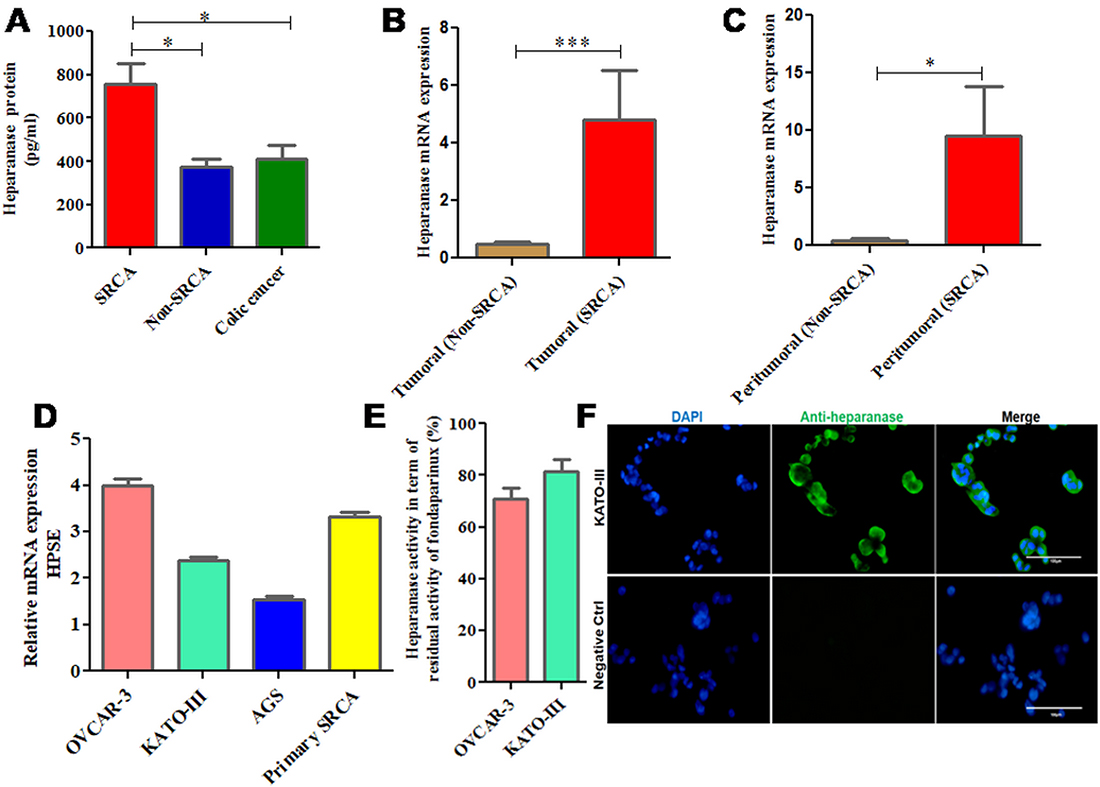
Figure 1: mRNA and protein expression of heparanase in clinical samples and cell lines including KATO-III. Heparanase protein was found in the ascitic samples of primary signet ring cell adenocarcinoma (SRCA) of stomach as compared to Non-SRCA of stomach and colic cancer by ELISA (SRCA n = 5, Non-SRCA n = 3 and colic cancer n = 6) (A) mRNA expression of heparanase was found higher in SRCA (n = 11) than non-SRCA (n = 10) (B) as well as in peritumoral-SRCA (n = 7) than peritumoral non-SRCA (n = 8) (C) Heparanase gene expressed by various cell lines ovarian (OVCAR-3), gastric (AGS, KATO-III), and primary SRCA (Primary GC) via RT-PCR, (D) Heparanase activity (evaluated by degradation of fondaparinux at pH 5) observed in supernatants of various cancer cell lines (OVCAR-3) and gastric (KATO-III), (E) Heparanase protein expression level in KATO-III by immunofluorescence is shown (F) The results are expressed as mean ± SEM of three independent experiments *P < 0.05, ***P < 0.001, statistically significant.
Gastric signet ring cell adenocarcinoma express EMT and multi drug resistance markers
Figure 2A presents the relative mRNA expression of several growth factors such as FGF-2, TGF-β1 and VEGF-A as well as EMT markers (E-cadherin, Snail, Slug, Vimentin, α-SMA and fibronectin) in tumoral tissues of SRCA as compared to the peripheral region of tumor. Except E-Cadherin and FGF-2, all other markers tested were expressed highly in tumor tissues as compared to periphery of tumors. These results suggest mesenchymal characteristics of tumor tissues and are also in favor of drug resistance in SRCA’s patients. When the drug transporter (MDR-1 (Pg-1), MDR-2, MDR-3, MDR-4, MDR-5, BCRP, MDR-1 and LRP) mRNA expression in tumor region, determined by RT-qPCR, as presented in Figure 2B, all ATP-binding cassette proteins as well as lung resistant protein were detected. However, among these drug transporters, MDR-1 and LRP were the most expressed.
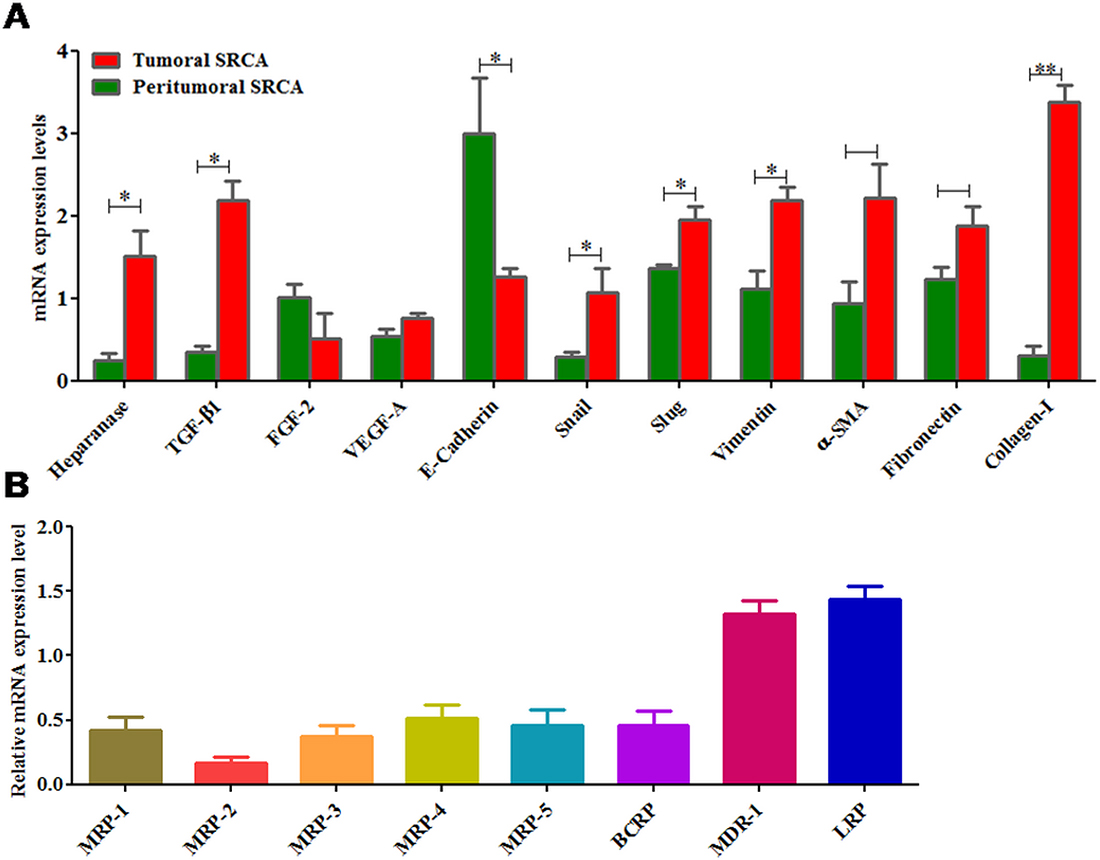
Figure 2: mRNA expression of heparanase, growth factors, EMT markers and drug transporters in clinical samples. Heparanase, growth factors (TGF-β1 and VEGF-A) except FGF-2 and epithelial marker like E-cadherin were found higher while mesenchymal markers (Snail, Slug, Vimentin, α-SMA and fibronectin) were lower in tumoral tissue of SRCA as compared to peritumoral tissue by qPCR (A). Similarly, of all the drug transporters (MDR-1, MDR-2, MDR-3, MDR-4, MDR-5, BCRP, MDR-1 and LRP) only two (MDR-1 and LRP) were found higher in SRCA tissue samples (B). The results are expressed as mean ± SEM of six independent experiments *P < 0.05, **P < 0.01, statistically significant.
KATO-III cell line formed spheroid clusters and expressed EMT markers in vitro
In culture medium, KATO-III cell line has two phenotypes; adherent and non-adherent cells (Figure 3A) and formed the spheroids. As presented in Figure 3B, micro-cinematographic studies of these cell clusters indicated that their diameters varied according to their origin (adherent and non-adherent cells). As presented in Figure 3C, the non-adherent cells are more spherogenic than adherent cells (p = 0.0001). Gene expression of EMT markers in both adherent and non-adherent KATO-III cells was studied. No alteration in gene expression (p = 0.124) of epithelial-mesenchymal transition (EMT)-related molecules was observed when adherent and non-adherent KATO-III cells were grown separately for one week (Supplementary Table 2).
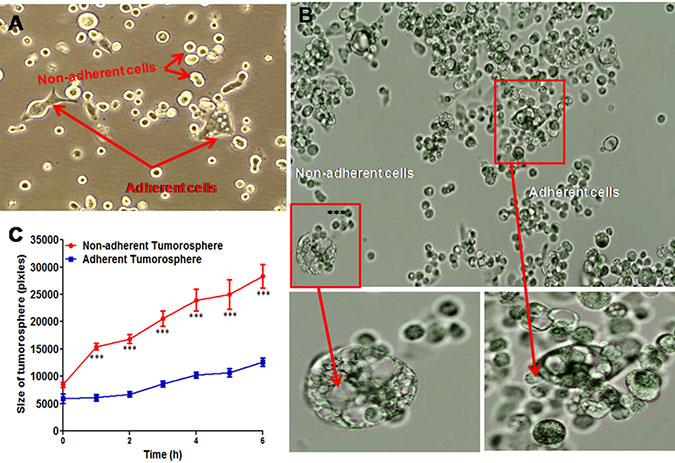
Figure 3: Spheroid cluster formation as well as cytokine array of both adherent and non-adherent KATO-III cell line. Adherent and non-adherent cells of KATOIII (A) Tumorosphere cells derived from adherent and non-adherent KATO-III (B) quantification of the maximal tumorosphere outgrowth diameter (pixels) during 6 hrs (C).
Suramin, an HPSE inhibitor down regulated TGF-β and collagen-1 mRNA KATO-III cells
As presented in Figure 4, when KATO-III cells were cultured in a medium containing suramin, there is a down regulation of TGFβ-1 and collagen-I expression in SRCA cell line in a time dependent manner. This result suggests that suramin simultaneously down regulates TGFβ-1 and collagen-1 expression in KATO-III cells.
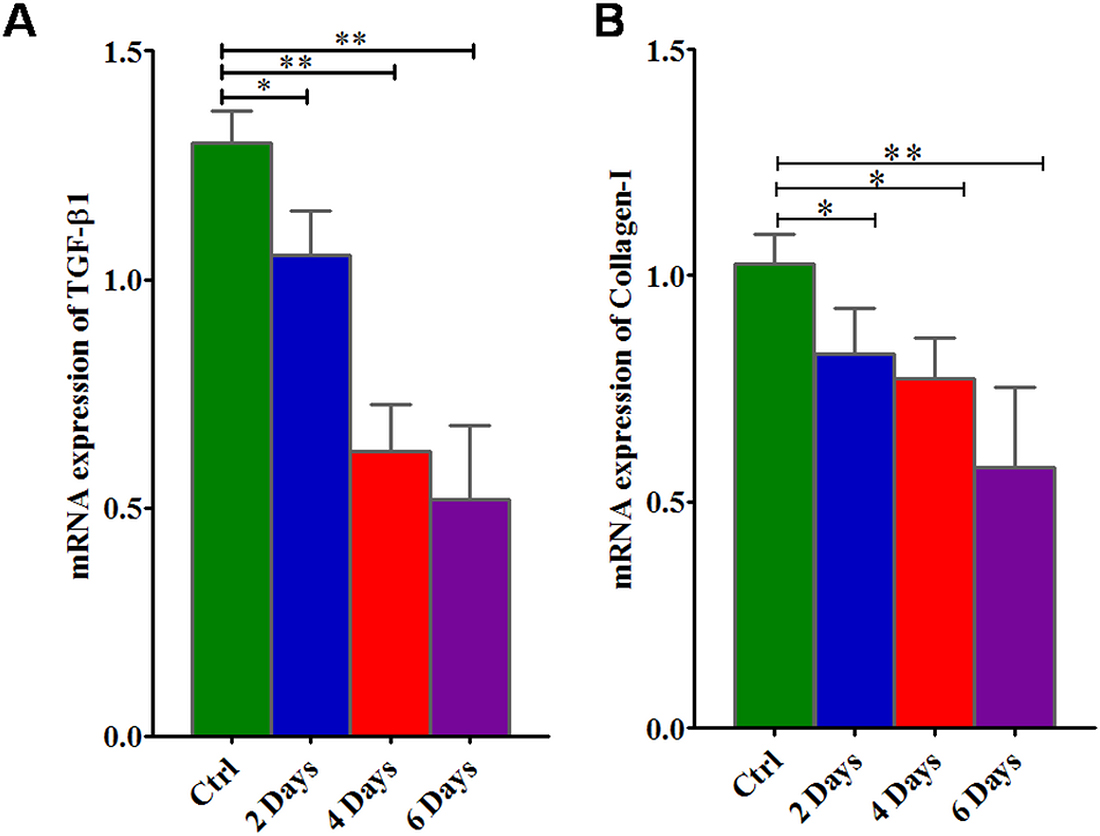
Figure 4: mRNA gene expression of TGFβ-1 and collagen-I in KATO-III cell line after treatment with suramin. Suramin lowers the expression of TGFβ-1 (A) and collagen-I (B) in KATO-III in a time dependent manner by qPCR. The results are expressed as mean ± SEM of six independent experiments *P < 0.05, **P < 0.01, statistically significant.
Suramin down regulates EMT and stem cell markers as well as inhibits cell cycle and proliferation of KATO-III cell line
As presented in Figure 5A, incubation of KATO-III cell line in a culture medium with suramin, down regulates HPSE expression in SRCA cell line in a time dependent manner. After 6 days incubation of these cells with suramin, mesenchymal markers such as Slug, Vimentin and α-SMA are significantly decreased (p = 0.005) while the E-cadherin is up regulated (p = 0.01) (Figure 5B). Suramin also decreased (Figure 5C) the expression of stem cell marker CXCR4, OCT3/4 and NANOG (p = 0.005). The inhibition of KATO-III cell proliferation was observed when these cells were incubated with suramin (p = 0.005) (Figure 5D). The influence of suramin on cell cycle (G1, M and G2) of KATO-III is presented in Figure 5E. The percentage of cells in each phase of the cell cycle is presented in Figure 5F). These results indicate that suramin via phase S pathway inhibits cancer cell proliferation.
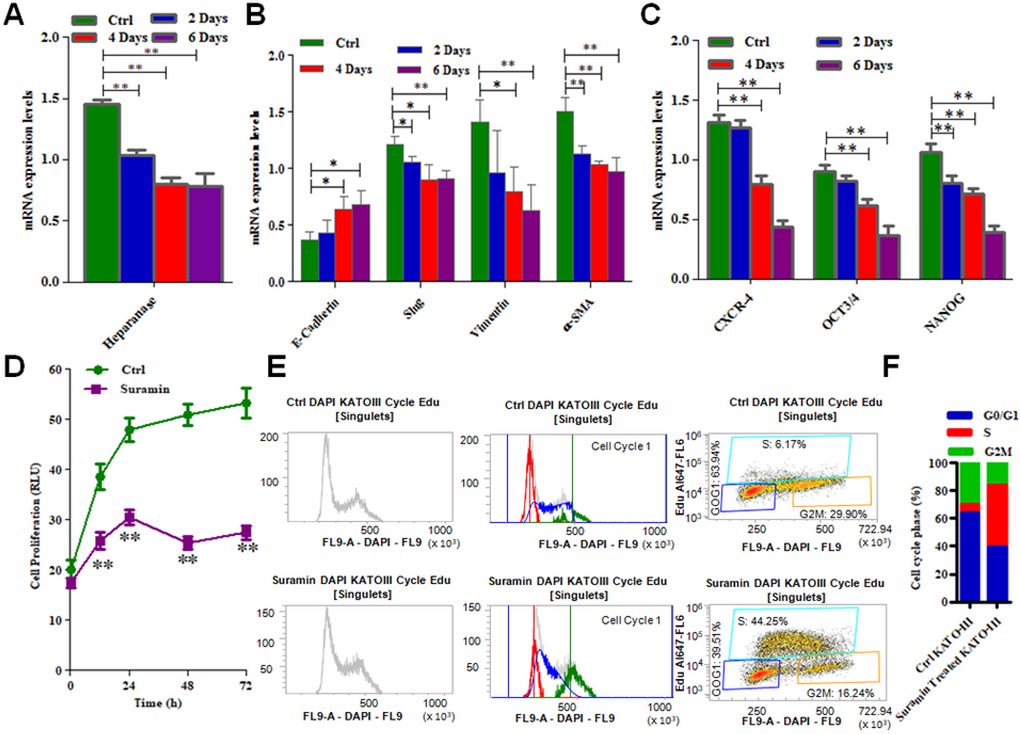
Figure 5: mRNA expression of EMT markers, stem cell markers as well as cell cycle arrest and cell proliferation in KATO-III after treatment with suramin. Expression of heparanase (A), mesenchymal markers (Slug, Vimentin and α-SMA) (B) and stem cell markers (CXCR-4, OCT3/4 and NANOG) (C) were found lower while epithelial marker (E-cadherin) was found higher (B) in KATO-III after treatment with suramin by qPCR. Suramin inhibits the proliferation (D) and cell cycle arrest (E) in KATO-III after treatment. The data indicates the percentage of cells in each phase of the cell cycle (F). The results are expressed as mean ± SEM of six independent experiments *P < 0.05, **P < 0.01, and are statistically significant.
Suramin modified phosphokinase activity pattern of KATO-III cells
As presented above, KATO-III cell line, in an autocrine manner or as a matter of fact by a paracrine pathway, secretes HPSE and influences endogenous kinase activity. The base line of phosphokinase activity of KATO-III cells (mean of three experiments) grown in the serum free culture medium presented is in Supplementary Figure 1A. These results indicate that some phosphokinase pathways such as GSK-3α/β, β-catenin, Chk-2, AMPKα-1, PRAS40 and C-Jun (more expressed proteins presented) are active in these cells. This pattern was modified when the cells were incubated in culture medium with the suramin. Results presented in Supplementary Figure 1B indicate that 6 different kinases were altered in KATO-III treated for 5 hrs as compared to the control. After treatment with suramin, two of the markers were upregulated (HSP60 and C-Jun) whereas 4 (GSK-3α/β, β-catenin, Chk-2 and AMPKα-1), were down regulated. These results correspond to the inhibition of secretion of non-activated HPSE and indicate the crucial role of HPSE in cell hemostasis.
These results so far presented (Figures 1–5 and Supplementary Figure 1) provide data that are important in elucidating the goal of the present work.
DISCUSSION
In our study, we have demonstrated high level of HPSE in SRCA (both tumor and ascitic fluid) and inhibition of HPSE induced EMT after addition of an inhibitor of HPSE (suramin).
HPSE is a multitasking protein characterized by enzymatic and non-enzymatic activities. Signaling activity of HPSE is achieved by interacting with transmembrane proteins, modulating the activity of factors such as FGF-2 and TGF-β [11].
HPSE expression was found high in the ascitic samples of the patient with SRCA of stomach as compared to that of patients with non-SRCA of stomach and colic cancer. Furthermore, high HPSE mRNA expression was found in primary gastric SRCA cells, KATO-III cell line as well as in an ovarian cancer cell line (OVCAR-3) as compared to gastric non-SRCA (AGS) cell lines. Active HPSE was also analyzed in the supernatants of cell lines including gastric (KATO-III). SRCA tissue of stomach showed more mRNA expression of HPSE, pro-fibrotic markers (TGF-β1, FGF-2), VEGF-A, mesenchymal markers (Slug, Vimentin) and fibrotic markers (fibronectin, α-SMA and Collagen-I) than those present in their corresponding peritumoral sites.
These findings allow concluding that SRCA are mesenchymal cells expressing high levels of HPSE and fibrotic markers. Our results are compatible with the report of David et al. who found HPSE association with intestinal fibrosis in vivo [28] and that of Mosala et al., who found FGF-2 dependent EMT related fibrosis in diabetic nephropathy [20]. Our results are also in accordance with the findings reported by the two authors cited previously.
FGF-2 was found absent in both forms of KATO-III cells (adherent and non-adherent) and in low amount from the tumoral tissues of patients with SRCA of stomach as compared to higher amount from peritumoral areas, suggesting its effect emanating from peritumoral sites. The interference of FGF-2 with HPSE [29] as well as TGF-β [30] and IGF-2 [31] for CD222 activity in the tumor microenvironments may modulate epithelial-mesenchymal interactions [20]. We also observed high mRNA expression of IGF2 in gastric SRCA tumoral tissue than peritumoral tissues (p = 0.004) as well as gastric non-SRCA (p = 0.007) (results not shown). It also affects TGF-β [32] and FGF-2 [33] related functions and inhibits multilineage differentiation of mesenchymal cells [34] as well as promotes tissue fibrosis, cancer expansion and malignancy.
Multi-drug resistance (MDR) remains a great obstacle to effective chemotherapy for SRCA gastric cancer [35]. The present work also indicates, for the first time that in SRCA tumoral cells, LRP and MDR-1 proteins expression is higher than other resistance proteins (MRP1–5, BCRP).
We showed also that KATO-III cells were capable of conversion between two distinct forms (adherent and non-adherent cells), a transition attributed to a reversible adaptive plasticity. We found no alteration in gene expression of epithelial-mesenchymal transition (EMT)-related molecules when adherent and non-adherent cells were grown separately for one week. This finding is consistent with the observation of Jun-Jun She et al. who found no difference in tumorigenicity in vivo when “side population” and “non-side population” of KATO-III cells were injected subcutaneously in nude mice [36].
In addition, we observed spheroid clusters of KATO-III that originated from both adherent and non-adherent cells. Size of these spheroid clusters that originated from non-adherent cells was found to be significantly larger than those that originated from adherent cells. Multiple evidences suggested the existence of spheroid clusters after isolation of stem cells from a variety of normal and tumor tissues [37–39] indicating spheroid formation, a common growth characteristic of SRCA cell line. LRP and MDR-1 expression by SRCA tumor, an EMT characteristic of this tumor, indicated the chemoresistance of SRCA patients to treatments.
Clinical trials at non-cytotoxic suramin levels in combination with chemotherapeutic agents have been conducted to treat metastatic breast cancer [40] and advanced non-small cell lung cancer [41], with discernible antitumor activity being noted in the latter. After finding HPSE mRNA and protein in KATO-III cells, we treated these cells with non-cytotoxic dose of suramin (200 μM) and observed increased phosphorylation of HSP60 and c-Jun while GSK-3α/β, β-catenin, Chk-2 and AMPkα-1 decreased. These results suggest the crucial role of HPSE in KATO-III cell homeostasis. GSK-3 has been associated with tumor progression by stabilizing components of the β-catenin complex [42]. Chk-2 as a kinase is involved in cell differentiation [43] and may influence SRCA differentiation and cell fibrosis while activation of AMPkα-1 acts to maintain cellular energy stores, switching on catabolic pathways that produce ATP for cell homeostasis [44]. C-jun phosphorylation is required for maintaining sufficient cyclin D1 kinase activity and for allowing cell cycle progression and apoptosis [45, 46].
Our interest was to focus on the regulation of EMT and fibrosis by altering the expression of EMT markers as well as TGF-β and collagen-I expression respectively using suramin. This was investigated in KATO-III cells in which we showed significant inhibitory effect of suramin on HPSE and collagen-I related fibrosis. Our findings demonstrate that suramin exerts a considerable degree of inhibition on EMT and proliferation in KATO-III cells. Similarly, suramin also induced a G0/G1 cell cycle block of up to 39% and inhibited S phase up to 98% of the cell population. These findings are similar to those reported by HuaPing Li et al. who found inhibition of cell proliferation by suramin in ovarian and cervical cancer by down regulating HPSE expression [47]. These results are in good correlation with our results showing that HPSE regulated EMT is inhibited by suramin, an HPSE inhibitor.
We have demonstrated increased HPSE level in SRCA microenvironment, which is responsible for pro-fibrotic factor dependent EMT leading to cell malignancy and fibrosis. The expression level of LRP and MDR-1 was found higher in SRCA that contributes to the chemoresistance observed in this malignancy. Suramin has been shown to be effective in the prevention and treatment of the EMT correlated with the metastatic propensity of tumor cells. HPSE could therefore be an interesting pharmacological target for the treatment of SRCA.
MATERIALS AND METHODS
Materials
Cell lines and reagents
Human cancer cell lines used, ovarian (OVCAR-3) and gastric (AGS, KATO-III) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA).
Ascitic fluids from 14 cancer patients of the Hospital Lariboisière (Paris, France) were collected. As ascitic fluid evacuation is a part of routine management of patients, only oral consents were obtained from them. Cells from ascitic fluids were pelleted by a short spin at 1000 rpm and supernatants were collected after a 10 min centrifugation.
Drug used in this study was suramin (Sigma Chemical Co, St. Louis, MO, USA).
Culture
Cells were cultured in RPMI 1640 medium or IMDM medium containing 10% heat-inactivated fetal bovine serum (FBS), 50 ug/ml of streptomycin, 50 IU/ml of penicillin and 2 nM of L-glutamine (Gibco, Saint Aubin, France). Cells were incubated at 37° C in a humidified atmosphere containing 5% CO2.
Tissues
Tumor and corresponding normal gastric tissue specimen (SRCA tumoral, SRCA peritumoral, Non-SRCA tumoral and Non-SRCA peritumoral) were obtained from 21 patients with signet ring cell adenocarcinoma from the General and Digestive Tract Surgery Department at Lariboisière Hospital in Paris (France). Informed consent was obtained from each patient prior to surgery. All of the tumor and macroscopically normal gastric tissue samples were obtained at the time of surgery. These tissue samples were rapidly frozen in liquid nitrogen and stored at −80° C until analysis. Tissue samples were histologically confirmed by hematoxylin and eosin staining.
Evaluation of HPSE by ELISA
After centrifugation of ascitic fluids from 14 cancer patients (SRCA n = 5, Non-SRCA n = 3, Colic carcinoma n = 6) at 1200 rpm for 5 minutes at room temperature, the supernatants were collected. The HPSE was quantified by using the commercially available HepAnalyze™ HPSE ELISA Kit (InSight Biopharmaceuticals Ltd. Rehovot, Israel) according to the manufacturer’s instructions. The results were expressed in pg/ml.
Immunocytochemistry
KATO-III cells grown on multichamber slides were fixed at 25° C for 15 min with 4% paraformaldehyde in PBS (Phosphate Buffered Saline), and rinsed 3 times with PBS. Cells were permeabilized with 0.3% triton for 15 min and then rinsed 3 times with PBS at 25° C. The slides were incubated for 20 min with 1% (w/v) bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA) in PBS to block non- specific binding sites and then with anti HPSE polyclonal (1:50 in 1% BSA; Bioss antibodies, Woburn, USA) overnight at 4° C, protected from light. After washing with PBS, the slides were further incubated with a secondary anti- Ig rabbit antibody coupled to FITC (1/500 in 1% BSA) to maximize coloring, for 1 hour at room temperature
Fluorometric assays
A substrate based activity assay kit (Kaivogen, Turku, Finland) that determines HPSE activity in term of fondaparinux® degradation was utilized according to the manufacturer’s instructions. Briefly, various cancer cell lines (106 cancer cells) ovarian (OVCAR-3) and gastric (KATO-III) were incubated in serum-free culture medium at 37° C in a humidified atmosphere of 5% CO2 for 24 hrs. 15 μl of each supernatant diluted to one half of fondaparinux (50 μg/ml) with 135 μl of buffer pH 5.2 were incubated for 2 hrs at 37° C. HPSE activity in term of residual activity of fondaparinux was then evaluated by adding factor Xa and its substrate in each supernatant mixture using STA Compact Max® (Gennevilliers, France) with excitation at 490 nm and emission at 520 nm. Results are expressed in % age.
Cell viability assay
Cell viability was assayed by Real Time-Glo™ MT Cell Viability Assay. In brief, cells (3 × 103/well) were seeded on 96-well plates, followed 24 hrs later by treatment with drug (or vehicle control) for 96 hrs. Bioluminescence was measured with the spectrofluorometer SAFAS Xenius XC. Cell viability was expressed as the percentage of absorbance of the drug-treated cells relative to that of the vehicle-treated cells.
RNA isolation, RT and real-time PCR
Gastric tissue specimens were homogenized with a polytron tissue homogenizer. Total RNA in cells and tissues was extracted using the Qiagen RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s instruction. RNA samples were transcribed to cDNA in a 20 μl volume by using the QuantiTect reverse transcription kit (Qiagen).
The thermal cycling comprised of the real time PCR as per following conditions: 95° C for 5 min, followed by 40 cycles (denaturation for 15 sec at 95° C, annealing for 20 sec at 60° C and extension for 20 sec at 72° C). The primer sequences and PCR product size for the target and reference genes are listed in Supplementary Table 1.
mRNA expression levels of different markers were detected by real-time PCR with ß-actin as internal reference, using Mesa Blue qPCR Master Mix Plus for SYBR assay (Eurogentec) on the Mastercycler Realplex2 (Eppendorf).
Relative quantitation was calculated by using the comparative threshold cycle (CT) method with realplex software. Mean CT of triplicate measurements was used to calculate ΔCT as the difference in CT for target and internal reference (ß-actin) genes. The difference between the ΔCT of the control experiment (KATO-III) and the ΔCT of each sample were calculated to give ΔΔCT. Fold increase in mRNA was calculated by 2−ΔΔCT.
The PCR products of cell lines and tissue samples after real-time PCR were electrophoresed by E-Gel Precast Agarose Electrophoresis System.
Human phosphokinase array
A membrane-based antibody array (R&D Systems, Raffles, China) that determines the relative levels of 45 different human phosphorylated protein kinases was used according to the manufacturer’s instructions. Briefly, equal amounts of cell lysates of KATO-III cell line treated with or without 200 μM Suramin (Sigma Chemical Co, St. Louis, MO, USA) into FBS free IMDM medium along with control for 5 hrs were incubated overnight with the phosphokinase array membrane. The array was washed to remove unbound proteins followed by incubation with a mixture of biotinylated detection antibodies. Streptavidin-HRP and chemiluminescent detection reagents were applied to visualize the signal produced at each capture spot corresponding to the amount of phosphorylated protein bound with densitometry by using a photosensitive film (Kodak, X-OMAT, AR, USA).
Analysis of cell cycle in KATO-III after treatment with suramin
Apoptosis assay was performed by EdU staining using a Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Life Technologies), according to the manufacturer’s protocol. KATO-III cell line was synchronized with IMDM medium having 5% FBS for 24 hrs. The following day, cells were incubated for 4 days with or without 200 μM suramin and then trypsinized. These harvested cells were incubated in culture medium with 15 μM EdU for 2 hrs. After incubation, cells were washed with 1% BSA in PBS and 100 μl of Click-iT fixative was added for 15 minutes at room temperature. After washing, cells were incubated in Click-iT Plus reaction cocktail including fluorescent dye (Alexa Fluor 647 picolyl azide) for 30 minutes. The flow cytometric cell analysis was performed, using a BD LSR II analytical flow cytometer (Becton Dickinson, San Jose, CA). MultiCycle AV (Phoenix Flow Systems) DNA analysis software enabled determination of the phase of cell cycle arrest by comparing percentages of each cell stage between the control and treatment groups (G1, S, G2/M).
ACKNOWLEDGMENTS
We would like to thank Professor Amu Therwath for his valuable assistance.
CONFLICTS OF INTEREST
The authors have no financial conflicts of interest.
REFERENCES
1. Otsuji E, Kuriu Y, Okamoto K, Ochiai T, Ichikawa D, Hagiwara A, Yamagishi H. Outcome of surgical treatment for patients with scirrhous carcinoma of the stomach. Am J Surg. 2004; 188:327–32.
2. Yonemura Y, Endou Y, Sasaki T, Hirano M, Mizumoto A, Matsuda T, Takao N, Ichinose M, Miura M, Li Y. Surgical treatment for peritoneal carcinomatosis from gastric cancer. Eur J Surg Oncol. 2010; 36:1131–38.
3. Huh HJ, Park CJ, Jang S, Seo EJ, Chi HS, Lee JH, Lee KH, Seo JJ, Moon HN, Ghim T. Prognostic significance of multidrug resistance gene 1 (MDR1), multidrug resistance-related protein (MRP) and lung resistance protein (LRP) mRNA expression in acute leukemia. J Korean Med Sci. 2006; 21:253–58.
4. Vtorushin SV, Khristenko KY, Zavyalova MV, Perelmuter VM, Litviakov NV, Denisov EV, Dulesova AY, Cherdyntseva NV. The phenomenon of multi-drug resistance in the treatment of malignant tumors. Exp Oncol. 2014; 36:144–56.
5. Sui H, Zhu L, Deng W, Li Q. Epithelial-mesenchymal transition and drug resistance: role, molecular mechanisms, and therapeutic strategies. Oncol Res Treat. 2014; 37:584–89.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
7. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–28.
8. Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011; 27:347–76.
9. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–90.
10. Yashiro M, Matsuoka T. Scirrhous gastric cancer: a critical review. OA Cancer. 2014; 2:4.
11. Secchi MF, Masola V, Zaza G, Lupo A, Gambaro G, Onisto M. Recent data concerning heparanase: focus on fibrosis, inflammation and cancer. Biomol Concepts. 2015; 6:415–21.
12. Masola V, Zaza G, Secchi MF, Gambaro G, Lupo A, Onisto M. Heparanase is a key player in renal fibrosis by regulating TGF-β expression and activity. Biochim Biophys Acta. 2014; 1843:2122–28.
13. Wood RJ, Hulett MD. Cell surface-expressed cation-independent mannose 6-phosphate receptor (CD222) binds enzymatically active heparanase independently of mannose 6-phosphate to promote extracellular matrix degradation. J Biol Chem. 2008; 283:4165–76.
14. Nakajima M, Irimura T, Nicolson GL. Heparanases and tumor metastasis. J Cell Biochem. 1988; 36:157–67.
15. Vlodavsky I, Miao HQ, Medalion B, Danagher P, Ron D. Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev. 1996; 15:177–86.
16. Cohen T, Gitay-Goren H, Sharon R, Shibuya M, Halaban R, Levi BZ, Neufeld G. VEGF121, a vascular endothelial growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J Biol Chem. 1995; 270:11322–26.
17. Vlodavsky I, Bar-Shavit R, Ishai-Michaeli R, Bashkin P, Fuks Z. Extracellular sequestration and release of fibroblast growth factor: a regulatory mechanism? Trends Biochem Sci. 1991; 16:268–71.
18. Goldshmidt O, Zcharia E, Cohen M, Aingorn H, Cohen I, Nadav L, Katz BZ, Geiger B, Vlodavsky I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003; 17:1015–25.
19. Fux L, Ilan N, Sanderson RD, Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009; 34:511–19.
20. Masola V, Gambaro G, Tibaldi E, Brunati AM, Gastaldello A, D’Angelo A, Onisto M, Lupo A. Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J Biol Chem. 2012; 287:1478–88.
21. Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Büchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001; 61:4655–59.
22. Wei RR, Sun DN, Yang H, Yan J, Zhang X, Zheng XL, Fu XH, Geng MY, Huang X, Ding J. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol Sin. 2018; 39:1326–37.
23. Nadir Y, Brenner B. Heparanase multiple effects in cancer. Thromb Res. 2014; 133:S90–94.
24. Steverding D. The development of drugs for treatment of sleeping sickness: a historical review. Parasites & Vectors. 2010; 3:15.
25. Jia L, Ma S. Corrigendum to: recent advances in the discovery of heparanase inhibitors as anti-cancer agents [Eur. J. Mech. 121 (2016) 209-220]. Eur J Med Chem. 2017; 125:793–94.
26. Nakajima M, DeChavigny A, Johnson CE, Hamada J, Stein CA, Nicolson GL. Suramin. A potent inhibitor of melanoma heparanase and invasion. J Biol Chem. 1991; 266:9661–66.
27. Marchetti D, Reiland J, Erwin B, Roy M. Inhibition of heparanase activity and heparanase-induced angiogenesis by suramin analogues. Int J Cancer. 2003; 104:167–74.
28. Davids JS, Carothers AM, Damas BC, Bertagnolli MM. Chronic cyclooxygenase-2 inhibition promotes myofibroblast-associated intestinal fibrosis. Cancer Prev Res (Phila). 2010; 3:348–58.
29. Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006; 8:596–606.
30. Zhang J, Wong MG, Wong M, Gross S, Chen J, Pollock C, Saad S. A cationic-independent mannose 6-phosphate receptor inhibitor (PXS64) ameliorates kidney fibrosis by inhibiting activation of transforming growth factor-β1. PLoS One. 2015; 10:e0116888.
31. Leksa V, Pfisterer K, Ondrovičová G, Binder B, Lakatošová S, Donner C, Schiller HB, Zwirzitz A, Mrvová K, Pevala V, Kutejová E, Stockinger H. Dissecting mannose 6-phosphate-insulin-like growth factor 2 receptor complexes that control activation and uptake of plasminogen in cells. J Biol Chem. 2012; 287:22450–62.
32. Bailey KL. Understanding the Relationship between TGF-Beta and IGF-1R Signaling in Colorectal Cancer. Theses & Dissertations. 190. 2017. Available at: https://digitalcommons.unmc.edu/etd/190.
33. Brahmkhatri VP, Prasanna C, Atreya HS. Insulin-like growth factor system in cancer: novel targeted therapies. BioMed Research International. 2015; 2015:538019.
34. Lai WT, Krishnappa V, Phinney DG. Fibroblast growth factor 2 (Fgf2) inhibits differentiation of mesenchymal stem cells by inducing Twist2 and Spry4, blocking extracellular regulated kinase activation, and altering Fgf receptor expression levels. Stem Cells. 2011; 29:1102–11.
35. Wu J, Qin H, Li T, Cheng K, Dong J, Tian M, Chai N, Guo H, Li J, You X, Dong M, Ye M, Nie Y, et al. Characterization of site-specific glycosylation of secreted proteins associated with multi-drug resistance of gastric cancer. Oncotarget. 2016; 7:25315–27. https://doi.org/10.18632/oncotarget.8287.
36. She JJ, Zhang PG, Wang X, Che XM, Wang ZM. Side population cells isolated from KATO III human gastric cancer cell line have cancer stem cell-like characteristics. World J Gastroenterol. 2012; 18:4610–17.
37. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003; 17:1253–70.
38. Toma JG, Akhavan M, Fernandes KJ, Barnabé-Heider F, Sadikot A, Kaplan DR, Miller FD. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001; 3:778–84.
39. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003; 63:5821–28.
40. Lustberg MB, Pant S, Ruppert AS, Shen T, Wei Y, Chen L, Brenner L, Shiels D, Jensen RR, Berger M, Mrozek E, Ramaswamy B, Grever M, et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother Pharmacol. 2012; 70:49–56.
41. Lam ET, Au JL, Otterson GA, Guillaume Wientjes M, Chen L, Shen T, Wei Y, Li X, Bekaii-Saab T, Murgo AJ, Jensen RR, Grever M, Villalona-Calero MA. Phase I trial of non-cytotoxic suramin as a modulator of docetaxel and gemcitabine therapy in previously treated patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2010; 66:1019–29.
42. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014; 5:2881–911. https://doi.org/10.18632/oncotarget.2037.
43. Takagaki K, Katsuma S, Kaminishi Y, Horio T, Tanaka T, Ohgi T, Yano J. Role of Chk1 and Chk2 in Ara-C-induced differentiation of human leukemia K562 cells. Genes Cells. 2005; 10:97–106.
44. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009; 458:1056–60.
45. Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999; 18:188–97.
46. Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997; 16:1695–709.
47. Li H, Li H, Qu H, Zhao M, Yuan B, Cao M, Cui J. Suramin inhibits cell proliferation in ovarian and cervical cancer by downregulating heparanase expression. Cancer Cell Int. 2015; 15:52.