
Introduction
Almost 50% of metastatic melanoma patients harbor a BRAFV600 mutation and the introduction of BRAF inhibitors (BRAFi) has improved their treatment options [1-3]. BRAFi vemurafenib and dabrafenib have been approved for the treatment of BRAFV600-mutated metastatic melanoma. In a recent update of the Phase III study of vemurafenib [2], median overall survival (OS) and progression-free survival (PFS) were significantly longer in the vemurafenib group than in the dacarbazine group (OS: 13.6 vs 9.7 months; PFS: 6.9 vs 1.6 months). In the Phase III study of dabrafenib vs dacarbazine [3], median PFS was 6.9 months compared with 2.7 months and also median OS was significantly longer (18.2 vs 15.6 months). However, most patients treated with BRAFi develop mechanisms of acquired resistance and about 15% of them do not achieve tumor regression at all [2,3].
The purpose of this paper is to review the mechanisms of resistance to therapy with BRAFi and to discuss the strategies to overcome them based on pre-clinical and clinical evidence.
Signaling Pathways in BRAF-mutated Melanoma
The RTK-RAF-MEK-ERK signaling cascade (Figure 1). The mitogen-activated protein kinase (MAPK) pathway plays an important role in the pathogenesis of melanoma. RAF kinases ARAF, BRAF and CRAF are key components of the pathway, which is physiologically activated when extracellular signals bind to their cognate membrane receptor, typically a receptor tyrosine kinases (RTK). In the absence of such signal, RAF kinases are in an inactive conformation, with the N terminus inhibiting the catalytic C terminus. In the presence of an upstream stimulus, RAS-GTP binds RAF at its N-terminus, relieving its auto-inhibition. The activity of wild-type RAF proteins requires the formation of homo- and heterodimers, which is promoted by RAS activation. In addition, RAF proteins require chaperone or scaffold proteins, such as heat shock protein 90 and the adaptor protein 14-3-3, which stabilize their structure [4].
Activated RAF kinases phosphorylate and activate MEK1/2, which in turn activate ERK1/2. ERK regulates cellular proliferation, survival and differentiation; the activation of ERK is regulated by a complex network of negative-feedback interactions through direct phosphorylation of the components of the RTK-RAS-MAPK cascade by ERK and through the induction of genes which inhibit activation of the pathway, such as Sprouty (SPRY) proteins and dual-specifity phosphatases (DUSPs) [4].
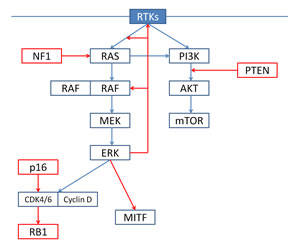
Figure 1: Under physiological conditions, ERK signaling is regulated by extracellular signals binding to receptor tyrosine kinases (RTKs). Activated RTKs promote RAS-mediated dimerization of RAF; wild-type RAF, as hetero- or homodimers, phosphorylate and activate MEK1/2, which in turn phosphorylate and activate ERK1/2. Activated ERK promote cell cycle progression and proliferation and negatively regulates upstream signaling components (negative feedback). RTKs also regulate the PI3K-AKT-mTOR pathway. The two pathways interact at multiple points: most importantly, RAS directly binds and activates PI3K.
BRAFV600 mutations (Figure 2). About 50% of melanomas harbor an activating mutation in BRAF, the most common being BRAFV600E [1], which renders the kinase constitutively active. In contrast to wild-type BRAF, the mutated forms of BRAF are active as monomers. In BRAF-mutated melanomas, RAS is negatively regulated by ERK-dependent feedback: BRAF-mutated cells have hyperactive ERK signaling and elevated ERK-dependent transcriptional output, including negative-feedback components. Feedback suppression takes place at multiple levels downstream of RTKs: as a result, RAS expression is low in BRAF-mutated cells and BRAFV600E exists mainly as a monomer, which is not dependent on RAS-GTP induced dimerization [5]. In addition, ERK negative feedback substantially attenuates PI3K/AKT signaling [5].
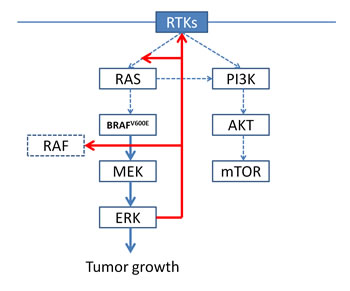
Figure 2: In BRAF-mutated cells, BRAFV600E is constitutively active as a monomer, leading to high ERK signaling and elevated ERK-dependent transcriptional output, including negative-feedback components. As a result, RAS expression is low and does not promote RAF dimerization, and PI3K/AKT signaling is substantially attenuated.
BRAF inhibition. BRAFi vemurafenib inhibits different RAF kinases with different half-maximum inhibitory concentrations: 10 nM for BRAF V600E, 15 nM for CRAF, 35 nM for ARAF and 40 nM for wild-type BRAF [6]. Vemurafenib inhibits ERK signaling only in BRAF-mutated tumors, while wild-type BRAF tumors and, especially, those with RAS mutations show a paradoxical activation of ERK due to the transactivation of RAF dimers [6]. In fact, in wild-type cells, BRAF and CRAF form homo- and heterodimers on RAS activation; vemurafenib binding to one member of the dimer causes an allosteric transactivation of the drug-free protomer and activation of MEK/ERK. The activation of MEK/ERK is enhanced when RAS is overexpressed [6]: this is consistent with the pre-clinical evidence that activated RAS promotes the dimerization of RAF and with the clinical evidence of RAS mutations in most cutaneous squamous cell carcinomas and keratoacanthomas which develop in patients treated with BRAFi [7]. In contrast, the activity of RAS in BRAF-mutated cells is low due to ERK negative-feedback signaling and insufficient to promote RAF dimerization: as a result, RAF exists predominantly as a monomer which responds to selective BRAFi. The relief of ERK negative-feedback during treatment with BRAFi may play a role in the mechanisms of resistance to BRAF inhibition and it will be discussed later.
The PI3K/AKT/mTOR pathway (Figure 1). The PI3K/AKT/mTOR kinase cascade is triggered by RTKs and G-protein-coupled receptors situated at the cell surface. When binding their extracellular ligands, these receptors sequester the regulatory subunit of PI3K, allowing the catalytic subunit to catalyze the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 activates downstream signaling components, including the protein kinase AKT, and it is antagonized by the tumor suppressor phosphatase and tensin homolog (PTEN). AKT phosphorylates many survival, proliferation and motility factors, including the tuberous sclerosis protein complex 1 (TSC1) and TSC2, which in turn lead to activation of mTOR complex 1 (mTORC1), a key regulator of cellular growth and protein synthesis [8,9].
PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathway cross-talk. The PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways interact at multiple points, resulting in cross-activation, cross-inhibition, and pathway convergence. Most importantly, RAS directly binds and activates PI3K; in addition, ERK phosphorylates TSC2, activating mTORC1. Cross-inhibition between the two pathways may occur under certain circumstances; for example, under high insulin-like growth factor 1 (IGF1) stimulation, AKT can inhibit RAF activity by phosphorylating its regulatory domain [8,9].
Mechanisms of Resistance
Numerous mechanisms of resistance have been predicted and detected based on in vitro and in vivo models and many of them have been confirmed on pre- and post-treatment tumor samples (Table 1). Resistant tumors may arise under selective pressure of therapy from pre-existing resistant subclones or as a result of an evolutionary process during treatment, or a combination of both. A detailed understanding of the causes of resistance to BRAFi is necessary to develop more effective treatment strategies. These mechanisms are largely classifiable as either primary/intrinsic, when no clinical benefit is achieved, or secondary/acquired, when progressive disease is observed after a clinical benefit. Moreover, mechanisms of adaptive resistance arise early during exposure to BRAFi and may explain why clinical responses to therapy are mostly partial responses, with complete response rate being in the range of only 3-6% in the Phase III studies [2,3].
Table 1: Mechanisms of Resistance to BRAF inhibition.
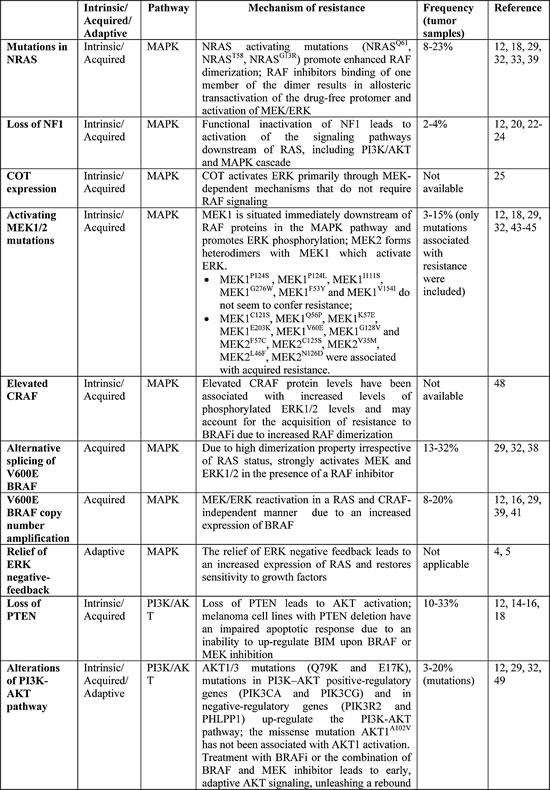
Mechanisms of Primary/Intrinsic Resistance
RAC1P29S mutations. RAC1 is a RAS-related GTPase that regulates cell proliferation and migration [10,11]; RAC1P29S is a recurrent UV-signature mutation in cutaneous non-acral melanomas and was the third most frequent activating mutation (9.2%) after those of BRAF and NRAS in a large cohort (n=147) of exome-sequenced melanomas [11]. The RAC1P29S mutation was less frequent, but not mutually exclusive, in NRAS or BRAF mutated melanomas (6.2% vs 12.5%).
Clinical evidence suggests that activating RAC1 mutations may confer resistance to BRAFi: in a cohort of 45 patients treated with BRAFi, among 14 patients exhibiting intrinsic resistance, three pretreatment sample harbored RAC1P29S mutations, and in one of them there was no other alteration known to confer resistance to therapy; no patient who achieved response to therapy harbored this mutation (P=0.026) [12].
RAC1 effectors include various protein kinases, offering a target for pharmacological inhibition, which may be of therapeutic value in the treatment of melanomas harboring the RAC1P29S mutations, although experimental evidence in support of this hypothesis is necessary.
Loss of PTEN. PTEN functions as a tumor suppressor by inhibiting PI3K signaling [13]. Loss of PTEN, which is observed in 10-33% of melanoma specimens [14-16], may contribute to intrinsic resistance to BRAFi via increased PI3K/AKT signaling when BRAF is inhibited and suppression of apoptosis mediated by proapoptotic protein BIM, member of the Bcl-2 protein family [14]. It is likely that loss of PTEN alone is not sufficient to confer resistance to BRAFi, but only when it is concurrent with other alterations. In fact, even if AKT activation is sufficient to provide resistance to vemurafenib-mediated apoptosis [17], PTEN loss is not always well correlated with increased AKT activation [14] and responses have been observed even in patients with complete loss of PTEN [12,18]. Nevertheless, cell lines with functional inactivation of PTEN seem to be less sensitive to BRAFi than wild-type PTEN melanoma cells and this was also observed in the clinical setting. In a study by Nathanson et al. [16], patients with wild-type PTEN treated with BRAF inhibitor dabrafenib had longer PFS compared with patients with at least one mutated allele of PTEN (32.1 vs 18.3 weeks; p=0.066), and a modest association between low expression of PTEN and lower response was observed in the phase 2 study of vemurafenib as well [18].
Dual BRAF/PI3K inhibition restores apoptosis in PTEN null cells [14], suggesting that such combination may overcome this mechanism of resistance.
Dysregulation of cyclin-dependent kinase 4 (CDK4) and/or cyclin D1. Cyclin D1 regulates proliferation binding both CDK4 and CDK6, which in turn phosphorylate the retinoblastoma protein and lead to progression through the cell cycle. Under physiologic conditions, p16INK4A (encoded by CDKN2A) negatively regulates CDK4 function [19]. Smalley and colleagues [19] found that CDK4 mutations alone did not alter responsiveness to BRAF inhibition, while cyclin D1 overexpression alone increased resistance. Cell lines harboring both a CDK4 mutation and a CCND1 (cyclin D1) amplification were the most resistant to therapy with BRAFi. This is clinically relevant given that CCND1 is amplified in 17% of BRAFV600E-mutated metastatic melanoma samples [19]. This pre-clinical model is supported by some clinical evidence: higher copy number of CCND1 (P=0.009) and lower copy number of CDKN2A (P=0.012) at baseline were significantly and independently associated with decreased PFS upon treatment with dabrafenib [16], evidencing the importance of the RB1 (retinoblastoma protein) pathway, which may become a target within a combination approach.
Loss of NF1. NF1 functions as a tumor suppressor by inhibiting RAS. Functional inactivation of NF1 leads to activation of the signaling pathways downstream of RAS, including PI3K/AKT and MAPK. Inactivating mutations in NF1 are present in 4% of BRAF-mutated melanomas [20]. NF1 may not only cooperate with BRAF mutations to drive melanomagenesis, but also have a role in the mechanism of primary and secondary resistance to BRAF inhibitors [21-24]. In vivo studies suggest that combined MEK and mTOR inhibition [23] and the use of ERK and irreversible RAF inhibitors (such as AZ628) [22] may be strategies to overcome or delay this mechanism of resistance.
COT expression. COT activates ERK primarily through MEK-dependent mechanisms that do not require RAF signaling. COT over-expression was identified as a driver of primary and secondary resistance to BRAF inhibition in cell lines and in progressing tumors of patients treated with BRAFi [25].
Alterations in RTK signaling (stromal secretion of HGF). The addition of hepatocyte growth factor (HGF) to BRAF-mutated melanoma cell lines confer resistance to BRAFi [26], hence stromal cells producing large amounts of HGF may be responsible for intrinsic resistance to therapy with BRAFi [27]. This mechanism of resistance is mediated by the activation of HGF receptor c-MET and subsequent activation of both the MAPK and PI3K-AKT signaling pathways and is sensitive, in vitro and in a xenograft model, to c-MET and HGF inhibition [26,27]. The combination of a BRAFi with a MEK inhibitor is unlikely to overcome this mechanism of resistance, since the PI3K-AKT pathway is involved as well, whereas the addition of an AKT inhibitor led to the suppression of the majority of HGF-induced resistance in vitro [27]. Patients with high baseline HGF serum levels have reduced response rate, PFS and OS [26,27].
HOXD8 mutations. HOXD8 is a homeobox transcription factor dysregulated in multiple cancers [12,28]. The detection in a non-responder patient treated with BRAF inhibitors of a nonsense mutation in the HOXD8 gene in the absence of other known resistance-associated alterations suggested that inactivation of this transcription factor may be a cause of intrinsic resistance.
Mechanisms of Secondary/Acquired Resistance
Most mechanisms of acquired resistance involve a reactivation of the MAPK pathway due to events that can occur upstream, downstream or at the level of BRAF; the PI3K-PTEN-AKT pathway constitutes a second core resistance pathway, which often overlaps with the MAPK pathway. Notably, no gatekeeper mutations have been identified as drivers of acquired resistance. Among 56 progressive tumors samples, deep sequencing of all 18 BRAF exons revealed no BRAFV600E/K secondary mutations and confirmed the persistence of the same BRAFV600E/K mutation in all progressive tumors, demonstrating that BRAFi did not select for minor, preexisting wild-type clones [29]; this was confirmed by another study [30] demonstrating intrapatient homogeneity of BRAFV600E assessed with immunohistochemistry in 171 tumors from 64 patients.
BRAF-mutant melanomas may develop multiple mechanisms of resistance simultaneously, even within a single cell line, and some of them may drive resistance to multiple MAPK inhibitors [31]. In a study on 100 resistant tumor samples from 44 patients [29], an alteration in the MAPK pathway was detected in 70% of the progressive tumors, while alterations of the PI3K –AKT pathway were detected in 22%; in 20% of patients, at least two mechanisms of resistance were detected in the same patient, and the alterations involved both pathways in all cases except for one; 13/16 patients, from whom multiple progressive biopsies were available, harbored multiple mechanisms of resistance. In another study [12], 3/45 patients harbored multiple independent mechanisms of resistance within the same tumor biopsy.
No association was observed between clinical outcome (best response and PFS) and specific mechanisms of resistance [32].
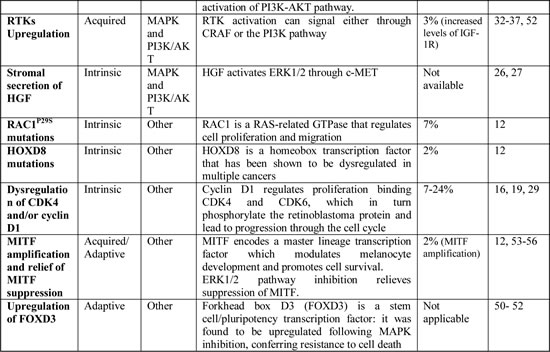
Upregulation and activation of the RTKs. Activation of RTKs may drive resistance through the activation of parallel pathways or increasing RAS activity. Upregulation and activation of the platelet-derived growth factor receptor b (PDGFRb) was one of the first alteration identified as an acquired mechanism of resistance to BRAFi treatment [33]. In vitro, the PDGFRb cells were insensitive to the PDGFR inhibitor imatinib [34]; however, resistant cells should be sensitive to the combined inhibition of BRAF and the RTK-PI3K-AKT-mTORC pathway [35].
Moreover, immunohistochemistry analysis on 16 pre- and post-treatment samples of metastatic melanoma patients receiving BRAFi (15/16) or MEKi (1/16) revealed that 6 progressing tumor samples acquired EGFR expression. EGFR is not expressed, in general, by melanoma cells, and in vitro studies concluded that EGFR expression is disadvantageous for BRAFV600E melanoma cells in the absence of BRAF or MEK inhibitor drugs, but it confers a selective advantage in the presence of these drugs. The addition of an EGFR inhibitor to vemurafenib did not lead to inhibition of cell proliferation, as other drivers of drug resistance, such as RTKs PDGFRB and ERBB3, were implicated. Many RTKs share the RAS-MEK-ERK and the PI3K-AKT signaling pathways: consistent with this, dual inhibition of the two pathways could restore sensitivity to BRAFi in cells with high EGFR expression [36,37]. Acquired EGFR expression is reversed in the absence of the BRAFi, providing evidence that a drug holiday could restore sensitivity when resistance is driven by this mechanism [36].
NRAS Mutations. MAPK reactivation due to high levels of activating mutations of NRAS was identified in 2010 by Nazarian and colleagues [33]. This model was validated by the detection of NRAS mutations in 4 of 19 tumor samples with acquired resistance to vemurafenib [38], underlying the importance that this mechanism has in the clinical setting. A combination strategy targeting downstream of NRAS, such as MEK and ERK, and the PI3K/AKT pathway should overcome this resistance mechanism.
Alternative splicing of V600E BRAF. Poulikakos et al. [38] detected from in vitro-resistant cell lines a 61-kDa form of V600E BRAF (p61BRAFV600E), which lacked exons 4–8, a region encoding the RAS-binding domain of BRAF, critical for RAF activation. In cells expressing p61BRAFV600E ERK signaling is resistant to BRAFi. The resistance of p61BRAFV600E to vemurafenib is not due to its inability to bind the inhibitor, but because this variant forms dimers in a RAS-independent manner, strongly activating MEK and ERK in the presence of BRAFi [38]. The use of BRAFi in combination with MEK or ERK inhibitors should delay or prevent this mechanism of resistance, as p61BRAFV600E cells should retain sensitivity to inhibitors of downstream components of the pathway. Nevertheless, an alternative splice variant of BRAF has also been detected in 1/5 patients after treatment with dabrafenib and trametinib in combination [39], raising the notion that this kind of combination may not be effective in this subset of patients. Pre-clinical evidence indicates that PLX7904, a next-generation BRAFi, blocks the growth of vemurafenib-resistant BRAFV600E cells harboring distinct BRAFV600E splice variants [40], so this drug may become a treatment option in this subset of resistant tumors.
BRAFV600E copy number amplification. BRAFV600E copy number gain, which results in BRAFV600E over-expression, is sufficient to lead to ERK reactivation in a RAS and CRAF-independent manner [41]. This alteration has been detected in 8-20% of tumor samples after PD with BRAFi [12,16,29,41]. ERK reactivation is saturable with higher doses of BRAFi and is sensitive in vitro to MEK inhibition and combined BRAF and MEK inhibition. Nonetheless, BRAF amplification has also been detected in patients after treatment with BRAF and MEK inhibitors in combination [39], pointing out that a combination approach with only these two drugs may not be sufficient to overcome this mechanism of resistance in the clinical setting. Dose-escalation may re-achieve disease control in patients with BRAF copy number gain; since the maximum tolerated dose of dabrafenib has not been determined, with doses up to 300 mg twice daily being tested in a Phase 1 study [42], the feasibility of dose escalation trials with dabrafenib in combination with a MEKi should be assessed in this subset of patients.
Activating MEK1/2 mutations. Activating MEK1 and MEK2 mutations may confer resistance in a small percentage of cases [12,18,32,43-45]: a number of different mutations have been identified and only some of them are capable to drive resistance to BRAFi, while the others did not alter sensitivity to BRAFi in vitro and did not prevent tumor regressions in the clinical setting (see Table 1 for details). MEK mutations driving resistance to BRAFi may as well confer cross-resistance to MEKi, raising the possibility that BRAF and MEK inhibitors in combination may have limited efficacy in this setting [12,39], while most of these mutations remain sensitive to ERK inhibitors (ERKi) in vitro [12,46-47].
Elevated CRAF levels. Elevated CRAF protein levels have been associated with increased levels of phosphorylated ERK1/2 levels and may account for the acquisition of resistance to BRAFi due to increased RAF dimerization [48]. Elevated CRAF levels seem to reflect a post-transcriptional regulatory mechanism rather than CRAF gene amplification [48]. In a small subset of BRAF mutated melanoma patients, this alteration may similarly contribute to intrinsic resistance [48].
Mutations in PI3K-AKT pathway genes. AKT1/3 mutations and mutations in PI3K-AKT positive-regulatory genes (PIK3CA and PIK3CG) and negative-regulatory genes (PIK3R2 and PHLPP1) may up-regulate the PI3K-AKT pathway [12,29,32], driving resistance to BRAFi.
Mechanisms of Adaptive Resistance to BRAF inhibition
Early adaptive responses to BRAFi, in addition to limiting the initial efficacy leading to incomplete responses, may favor the selection of a sub-population of resistant clones and the acquisition of alterations that lead to secondary resistance, ultimately causing tumor regrowth and progressive disease.
Relief of ERK negative-feedback. In BRAF-mutated melanoma, ERK-dependent feedback suppress RAS activation and BRAF exists predominantly as an active monomer. These cells have decreased sensitivity to growth factors and the transduction of signal from RTKs is suppressed. The exposure to a BRAFi produces a relief of ERK negative feedback, with a consequent enhanced ability of ligands, including growth factors, to activate signaling and an increased expression of RAS-GTP, which promotes the formation of RAF dimers. BRAFi bind to one component of the dimer and cause an allosteric activation of the other one. As a result, the relief of ERK-dependent negative feedback restores sensitivity to growth factors and may diminish the effect of BRAFi through the activation of RAS; however, negative-feedback pathways are partially restored over time, leading to the formation of a new steady state of reactivated ERK signaling, and the level of activation of RAS is variable in different cell lines and, in most melanomas, is not enough to cause resistance in the absence of other activating signals, but can cooperate with mechanisms requiring the presence of active RTK [4]. In addition, it provides a partial explanation to the variability of responses observed in patients treated with BRAFi and it suggests that a combination including an inhibitor of ERK rebound may be necessary to achieve more durable and complete responses.
Alterations in the PI3K-PTEN-AKT pathway. The inhibition of the MAPK pathway leads to early, adaptive AKT signaling, unleashing a rebound activation of PI3K-AKT pathway [49]. AKT activation is normally limited by the activity of PTEN [13], supporting the hypothesis that PTEN loss of function has an important role in the failure of BRAFi therapy. This PI3K-AKT-dependent adaptive response to BRAFi, along with the high frequency of melanomas with loss of function of PTEN [14-16] and other alterations linked to this pathway [12,29,32,49], indicate that the addition of a PI3K/AKT inhibitor to MAPK inhibitors may be necessary to obtain a long-term clinical benefit.
Upregulation of FOXD3. Forkhead box D3 (FOXD3) is a stem cell/pluripotency transcription factor: it was found to be upregulated following MAPK inhibition, conferring resistance to cell death [50]. The combination of BRAFi with integrin inhibitors should overcome this mechanism of resistance, as the blockade of signals from the extracellular matrix through treatment with integrin inhibitors attenuates the upregulation of FOXD3 in vitro [51]. Moreover, since ERBB3 was identified as a direct transcriptional target of FOXD3, a combination with ERBB signaling inhibitors may also have therapeutic value in this setting [52].
Upregulation of mitochondrial synthesis and oxidative metabolism. ERK1/2 pathway inhibition relieves suppression of MITF (microphthalmia-associated transcription factor), a promoter for cell survival expressed exclusively in the melanocyte lineage, leading to the upregulation of mitochondrial synthesis and oxidative metabolism through PGC1α (peroxisome proliferator-activated receptor γ coactivator 1α) and promoting survival in the presence of the inhibitor [53-56]. Moreover, amplification of MITF was reported as a mechanism of secondary resistance in a resistant tumor biopsy [12]. Overexpression of MITF desensitizes BRAF-mutated melanoma cells to BRAFi, but MITF expression can be impaired following treatment with histone deacetylase inhibitors (HDACi), suggesting the potential benefit of a combination strategy with MAPK inhibitors. Furthermore, since BRAFi-induced oxidative metabolism renders melanoma cells highly dependent on antioxidant enzymes to survive, the combination with pro-oxidative agents may be a rational strategy to overcome this mechanism of resistance [54].
Discussion
Combination versus Sequential strategies
The approval of BRAFi vemurafenib and dabrafenib and MEKi trametinib was a breakthrough in the treatment of metastatic melanoma, even if their efficacy is limited by the development of resistance; these inhibitors now represent key elements to build more effective strategies, such as sequential and combination regimens with other inhibitors and/or immunotherapy.
The understanding of the mechanisms of resistance to BRAFi suggests that stronger anti-tumoral activity may be achieved targeting multiple pathways. The combination of dabrafenib with trametinib significantly increased PFS over dabrafenib alone in a phase II study [57]. Nevertheless, in vitro experiments suggest that resistance to BRAFi may confer cross-resistance to MEKi [9,58] and that most mechanisms of resistance involve an activation of additional pathways, such as PI3K/AKT/mTOR, thus the combination of BRAFi only with MEKi, which targets a single pathway, is unlikely to provide long-term disease control. In fact, in the phase III study of dabrafenib+trametinib vs dabrafenib monotherapy, PFS was only slightly better in the combination arm (9.3 vs 8.8 months) (Long, ASCO 2014: Abstract No. 9011). This implies that multiple pathways may be needed to be targeted either in parallel, if not limited by toxicity, or in series, as part of an intermittent dosing schedule.
Resistant tumors, as mentioned earlier, often show an activation of the PI3K/AKT/mTOR pathway; in fact, in addition to the intrinsic and acquired activating alterations, the cross talk between the MAPK and PI3K/AKT pathways results in activation of one pathway when the other is suppressed [8,59], suggesting that a strategy to effectively counteract resistance must rely, at least, on the targeting of both the MAPK/ERK and PI3K/AKT/mTOR pathways. Pre-clinical studies pointed out the synergistic nature of targeting the PI3K/AKT pathway in combination with either BRAF or MEK inhibitors [35,60] and Phase I/II trials are evaluating such combination regimens in patients (Table 2): preliminary data show that these combinations are tolerated and active in patients with BRAF-mutated metastatic melanoma [9].
Table 2: Ongoing Phase I-II clinical trials including BRAF-mutated melanoma patients (www.clinicaltrials.gov accessed on 30th May 2014).
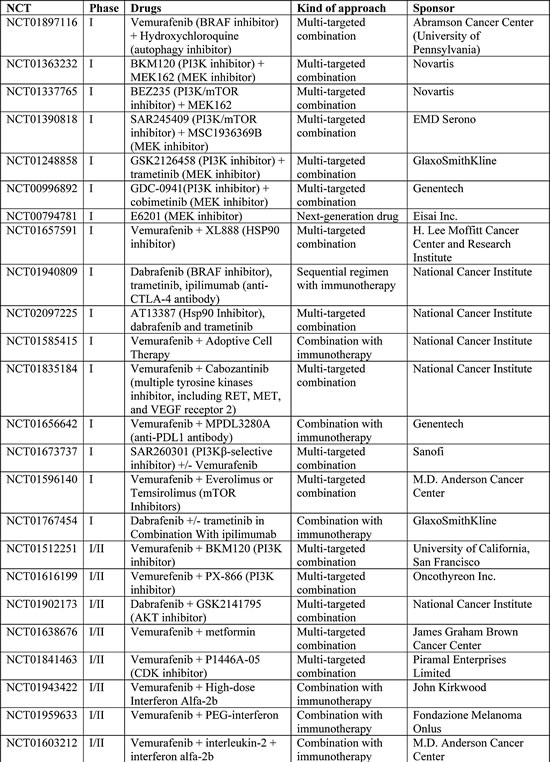
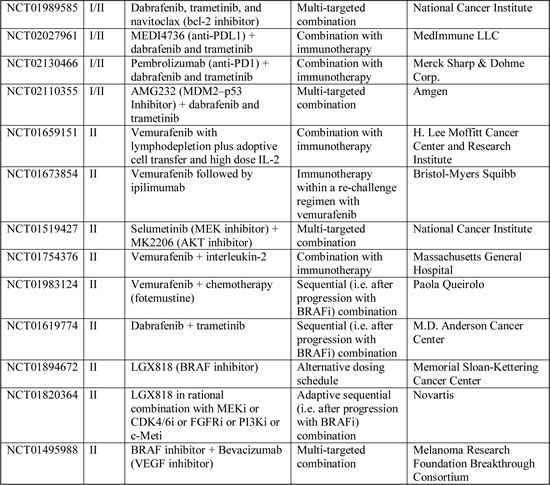
ERKi are in early phase clinical trials and may have a value for overcoming resistance mechanisms relying on ERK re-activation. ERKi were more effective than MEKi at overcoming BRAFi resistance conferred by a number of mechanisms (BRAF splice variants, NRAS mutations, MEK mutations, BRAF amplification, RTKs activation) in vitro [46,61]; however, ERKi did not overcome resistance mediated by RTK activation in all studies [61], even if a higher level of MAPK suppression was evidenced compared to MEKi. ERKi target wild-type kinases, so they are likely to have a narrow therapeutic index: clinical studies are underway to determine whether ERKi can be delivered at concentrations that are clinically effective (NCT01781429, NCT01358331). Nevertheless, ERKi are likely to have limited efficacy as monotherapy or as sequential therapy after progression with BRAFi; ERK inhibition, in fact, causes the relief of the ERK-dependent negative feedback, leading to activated RAS and PI3K signaling. Pre-clinical evidences support the notion that only a combinatorial strategy targeting both ERK and PI3K/mTOR, if clinically deliverable, may circumvent BRAFi resistance [61].
Anticipating the emergence of multiple mechanism of resistance by targeting multiple pathways in a combinatorial approach may have more success than an adaptive sequential approach. A mathematical model developed by Bozic et al. [62] has shown that concurrent combination therapy is more potent than sequential combination treatment, even when there are no possible mutations that can confer cross-resistance; when there are potential mutations conferring cross-resistance to two or more agents, combination therapy offers some chance while there is no chance with a sequential strategy [62,63]. Although preclinical data suggest that a subset of resistance mechanisms should respond to MEKi, this mathematical model is supported in the clinical setting by the lack of clinical activity of MEKi and BRAFi plus MEKi in patients previously treated with BRAFi. In fact, in the phase II study of MEKi trametinib in BRAF-mutated patients [64], only minimal clinical activity was observed as sequential therapy in patients previously treated with a BRAFi. The lack of clinical activity of MEKi after progression on single agent BRAFi was confirmed in another cohort of patients who were treated after progression with a MEKi or combined BRAF and MEK inhibitors. No objective responses were observed, including patients whose mechanism of resistance would predict a response to downstream inhibition, such as alternative splicing and amplification of BRAF [32]. The limited clinical activity of this sequential regimen is most likely due to intra-patient heterogeneity of resistance [12,29,65-66], cross-resistance [9,58] and alternative pathways activation. Heterogeneous tumor clones probably exist before treatment and cases of tumors developing multiple mechanisms of resistance have been reported [33,65,67]. Intratumor and intrapatient heterogeneity is a challenge for personalized targeted therapies: single tumor samples may lead to underestimation of the tumor genomics landscape and adaptive clinical trials involving the selection of sequential targeted drugs based on the molecular characteristics of a single progressing biopsy are unlikely to provide durable responses [68].
Tumor sampling will have an increasing influence on therapeutic strategies [69]. The analysis of circulating tumor cells or circulating tumor-derived DNA may provide a more accurate and complete genetic profile of patient tumors compared to single tumor biopsies, allowing a better choice of treatments for individual patients [13]. Nevertheless, a highly effective method for the detection and molecular analysis of melanoma circulating cell is yet to be standardized [70,71].
Until the development of more effective diagnostic strategies, the detection of the mechanisms of resistance in clinical practice relies on tumor biopsies. An adaptive sequential strategy using such an approach will be investigated in the clinical setting in a phase II study (NCT01820364) in patients with melanoma who progress on the BRAFi LGX818. Resistant tumors will be biopsied and compared with a pretreatment biopsy to identify the mechanism of resistance. On the basis of the alterations identified in the tumor samples, a second targeted agent from a list of MEK, CDK4/ 6, FGFR, PI3K, and c-MET inhibitors will be added to the regimen.
Treatment beyond progression versus drug holiday/re-challenge
Tumor heterogeneity may also explain the apparent contrast between the clinical evidences of continued anti-proliferative activity of BRAFi in BRAF-resistant tumors [72] and the pre-clinical models predicting that in some cases cessation of BRAFi may lead to regression or slower progression of resistant tumors [73-75].
Treatment beyond PD may achieve clinical benefits because sensitive and resistant tumor sub-clones probably coexist, and the discontinuation of treatment would lead to faster progression as a result of the proliferation of sensitive clones [76]. Treatment with BRAFi continued after PD was associated with prolonged OS compared with ceasing treatment in a series of 114 patients treated with BRAFi within Phase I-II-III-IV trials [72]; moreover, in the Phase I study of vemurafenib, 14 patients with isolated PD in a site suitable for local therapy continued treatment beyond progression until systemic progression and, some of them, achieved long-term survival (Kim, Society for Melanoma Research 2012 Congress). These data may be biased by patient selection and future prospective randomized trials are needed to assess the efficacy of treatment beyond progression.
In contrast, apparently, with these observations, Das Thakur et al. demonstrated that some vemurafenib-resistant melanoma cell lines expressing BRAF alternative splicing variants or amplified BRAF may become drug dependent for their proliferation, and that cessation of vemurafenib exposure may lead to regression of these tumors. A discontinuous dosing strategy using either a 4 weeks on/2 weeks off or an individualized regimen, exploiting the fitness disadvantage displayed by drug-resistant cells in the absence of the drug, delayed the onset of drug-resistant disease in two primary human xenograft models. Even if the pause from BRAF inhibition allowed sensitive tumours to re-grow, these cells remained responsive to vemurafenib re-administration. Repeated cycles of vemurafenib treatment using these regimens controlled tumor growth over the course of 7 months of treatment, whereas mice treated on a continuous cycle developed resistance as soon as 2 months after initiation of therapy [73-75]. Other studies seem to support these findings: cells expressing distinct mutant BRAF splice variants grew more efficiently in vitro and in vivo in the presence rather than in the absence of the vemurafenib analog PLX4720 and, after a drug free interval, became re-sensitized to BRAF inhibition; a drug holiday was also shown to be effective in a melanoma cell line in which resistance was mediated by BRAF copy number amplification [77] and acquired EGFR expression [36]. These findings further provide a rationale to investigate an intermittent regimen with BRAFi: a Phase II clinical study is underway evaluating intermittent dosing of a BRAFi (LGX818) in patients with BRAF-mutated metastatic melanoma (NCT01894672). LGX818 will be administered on a daily schedule dosing for the first 6 weeks; this will be followed by a 2 week break and, thereafter, patients will resume LGX818 on a 2 weeks on/2 weeks off schedule (Table 2).
To what degree these observations made in preclinical models translate into patients is an open issue. In fact, as already mentioned, continued treatment beyond progression with BRAFi seems to be associated with increased survival and no tumor regressions have been reported after treatment discontinuation, suggesting that many and more complicated factors are involved in determining the response of resistant cells to BRAFi in the clinical setting. Nevertheless, anecdotal clinical evidence already exist for the successful application of intermittent dosing or re-challenge of BRAF/MEK inhibitors in melanoma patients. Two patients were successfully rechallenged after progression with vemurafenib or dabrafeib+trametinib [78], and a melanoma patient in which vemurafenib induced proliferation of a previously undetected NRAS-mutant chronic myelomonocytic leukemia was successfully treated using an adjusted intermittent schedule of vemurafenib guided by changes in white cell counts [79].
In clinical practice, until further investigations and availability of new drugs, patients who progress rapidly and in multiple sites during treatment are unlikely to benefit from continuation of MAPK inhibitors and should switch to another line of therapy, while patients with isolated progression, suitable for local treatment, may benefit from continuation of BRAFi treatment, as resistance mechanisms are not always shared by all metastasis [65]. Local treatments may also have a role in case of partial responses with low and accessible tumor burden, when residual lesions, which are a potential evolutionary reservoir of resistant tumor cells, may be removed radically.
Combinations with immunotherapy
Even if a multi-targeted treatment approach and individualized regimens may improve response rate and their duration, tumor heterogeneity remains a barrier to obtain complete and long-term responses; a combination strategy including the use of an immunotherapeutic agent may provide more durable responses and long-term survival.
There is strong rationale for combined BRAF-targeted therapy and immunotherapy for melanoma. Most studies have supported the idea that BRAFi are unlikely to impair the immune system [80] [81]; on the contrary, these agents may have immunomodulatory properties and enhance immune activation, as treatment with MAPK inhibitors is associated with enhanced expression of melanocytic antigens, antigen recognition by T cells and an influx of cytotoxic T lymphocytes [82] [83-86]. BRAFi paradoxically activate ERK in wild-type cells and this effect was also observed in T cells: BRAFi enhance T cell activation in a concentration-dependent fashion [87]. Moreover, emerging evidence suggests that oncogenic BRAF is immunosuppressive [88], further supporting the rationale for the development of combined targeted therapies with immunotherapy. The first attempt combining BRAFi vemurafenib with anti-CTLA-4 antibody ipilimumab failed due to toxicity issues [89]. Nevetheless, new immunotherapeutic drugs are about to be approved, such as anti-PD-1 antibodies nivolumab and pembrolizumab, which seem to be more tolerated than ipilimumab and, at the same time, to have higher response rates. Increased PD-L1 expression by cancer cells is an escape mechanism from host immunity and reactivation of MAPK seems to be associated with increased expression of PD-L1 [90,91]. Anti-PD-1 agents block the ligation of PD-L1, expressed on cancer and antigen-presenting cells, to PD-1, expressed on T cells, preventing it to suppress T-cell activation and proliferation and to induce T-cell apoptosis. Moreover, tumor expression of PD-L1 is a biomarker of clinical activity of anti-PD-1 agents, further supporting a combination with such drugs. Clinical studies are underway to assess the feasibility and clinical activity of combination therapy with BRAFi, not only with anti-PD-1 and anti-PDL-1 agents, but also with interleukin, adoptive cell therapy and interferon (Table 2). As a matter of fact, the combination of targeted therapy with BRAF inhibitors and interferon has a strong rationale [92]. Interferon receptor subunit IFNAR1 is down-regulated in BRAF-mutated melanoma cells, while BRAF inhibition up-regulates its expression; moreover, vemurafenib and IFNα-2b combination enhances HLA class I antigen expression, which is associated with an increased recognition of melanoma cells by cognate T cells. The combination of vemurafenib and interferon significantly prolonged survival in mice grafted with BRAF-mutated melanoma cells, and two phase I-II studies are evaluating in humans the safety and activity of the combination of vemurafenib plus low-dose peg-interferon (NCT01959633) or high-dose interferon (NCT01943422).
Pre-clinical and clinical evidence suggest that immunotherapy should be incorporated early in the course of MAPK inhibitor treatment, in combination or within an intermittent regimen. In fact, laboratory data demonstrate immune cell infiltration into tumors soon after treatment commencement [82,85] and clinical data available on ipilimumab show that immunotherapy has limited efficacy in almost half patients failing BRAFi treatment [93,94].
Conclusion
The successful development of BRAFi is an example of translation of molecular biology for cancer personalized treatment. However, the benefit provided by BRAFi is limited by resistance and several challenges must be addressed to develop more effective therapeutic strategies. Multiple treatment modalities, including other targeted therapies and immunotherapy, are now available for testing and combination. The understanding of the molecular basis of BRAFi drug resistance may provide insights useful to design the best strategies to prevent or delay the emergence of resistant clones; paradoxically, drug resistance may even be exploited to selectively kill resistant cells [95].
Growing evidence of intra-patient heterogeneity of resistance, cross-resistance and alternative pathways activation highlight the need to perform dedicated molecular analysis of resistance and support the rationale that stronger and longer anti-tumor activity will be obtained when multiple pathways are targeted, either in combination or as part of an intermittent dosing regimen. Moreover, combinatorial and sequential approaches which merge the high response rate and rapidity of tumor regression of BRAFi–based treatments with the long-term responses of immunotherapy may be effective strategies to obtain long-term survival in patients with BRAF-mutated metastatic melanoma, although toxicity issues may arise and should be carefully evaluated in clinical trials.
References
1. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W. Mutations of the BRAF gene in human cancer. Nature 2002; 417(6892):949–954.
2. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, Garbe C, Testori A, Maio M et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014; 15(3):323–332.
3. Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller Jr WH, Kaempgen E. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. The Lancet 2012; 380(9839):358–365.
4. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013; 19(11):1401–1409.
5. Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, Chandarlapaty S et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012; 22(5):668–682.
6. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464(7287):427–430.
7. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012; 366(3):207–215.
8. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 2011; 36(6):320–328.
9. Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013; 71(6):1395–1409.
10. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, DiCara D, Ramos AH, Lawrence MS et al. A Landscape of Driver Mutations in Melanoma. Cell 2012; 150(2):251–263.
11. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012; 44(9):1006–1014.
12. Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014; 4(1):94–109.
13. Smalley KSM, Weber JS. Up Close and Personal: The Challenges of Precision Medicine in Melanoma. JNCI J. Natl. Cancer Inst. 2014; 106(2):djt443–djt443.
14. Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson ARA, Ribas A, Palma MD, Nathanson KL et al. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011; 71(7):2750–2760.
15. Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, Gallardo H, Liu C, Merghoub T, Hefter B. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring V600EBRAF. Oncogene 2012; 31(4):446–457.
16. Nathanson KL, Martin A-M, Wubbenhorst B, Greshock J, Letrero R, D’Andrea K, O’Day S, Infante JR, Falchook GS, Arkenau H-T, Millward M, Brown MP, Pavlick A et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013; 19(17):4868–4878.
17. Shao Y, Aplin AE. Akt3-Mediated Resistance to Apoptosis in B-RAF-Targeted Melanoma Cells. Cancer Res. 2010; 70(16):6670–6681.
18. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim KB, Weber JS, Hersey P, Long GV, Lawrence D et al. Pharmacodynamic Effects and Mechanisms of Resistance to Vemurafenib in Patients With Metastatic Melanoma. J. Clin. Oncol. 2013; 31(14):1767–1774.
19. Smalley KS, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S, Hansson J, Wu H, King AJ, Van Belle P. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E–mutated melanomas. Mol. Cancer Ther. 2008; 7(9):2876–2883.
20. Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, Behren A, Cebon JS, Wolfe R et al. BRAF/NRAS Wild-Type Melanomas Have a High Mutation Load Correlating with Histologic and Molecular Signatures of UV Damage. Clin. Cancer Res. 2013; 19(17):4589–4598.
21. Gibney GT, Smalley KSM. An Unholy Alliance: Cooperation between BRAF and NF1 in Melanoma Development and BRAF Inhibitor Resistance. Cancer Discov. 2013; 3(3):260–263.
22. Whittaker SR, Theurillat J-P, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA. A Genome-Scale RNA Interference Screen Implicates NF1 Loss in Resistance to RAF Inhibition. Cancer Discov. 2013; 3(3):350–362.
23. Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, Wargo JA, Marais R, Cichowski K. Elucidating Distinct Roles for NF1 in Melanomagenesis. Cancer Discov. 2013; 3(3):338–349.
24. Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, Merghoub T, Ribas A, Chapman PB et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014; 74(8):2340–2350.
25. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468(7326):968–972.
26. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012; 487(7408):505–509.
27. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487(7408):500–504.
28. Spitz F, Gonzalez F, Peichel C, Vogt TF, Duboule D, Zákány J. Large scale transgenic and cluster deletion analysis of the HoxD complex separate an ancestral regulatory module from evolutionary innovations. Genes Dev. 2001; 15(17):2209–2214.
29. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014; 4(1):80–93.
30. Menzies AM, Lum T, Wilmott JS, Hyman J, Kefford RF, Thompson JF, O’Toole S, Long GV, Scolyer RA. Intrapatient Homogeneity of BRAFV600E Expression in Melanoma. Am. J. Surg. Pathol. 2014.
31. Gowrishankar K, Snoyman S, Pupo GM, Becker TM, Kefford RF, Rizos H. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J. Invest. Dermatol. 2012; 132(7):1850–1859.
32. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, Howle J, Saw R, Thompson JF et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 2014; 20(7):1965–1977.
33. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee M-K, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468(7326):973–977.
34. Poulikakos PI, Rosen N. Mutant BRAF Melanomas—Dependence and Resistance. Cancer Cell 2011; 19(1):11–15.
35. Shi H, Kong X, Ribas A, Lo RS. Combinatorial Treatments That Overcome PDGFR -Driven Resistance of Melanoma Cells to V600EB-RAF Inhibition. Cancer Res. 2011; 71(15):5067–5074.
36. Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014; 508(7494):118–122.
37. Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, Lorigan P, Springer C, Larkin J et al. Inhibiting EGF Receptor or SRC Family Kinase Signaling Overcomes BRAF Inhibitor Resistance in Melanoma. Cancer Discov. 2013; 3(2):158–167.
38. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011; 480(7377):387–390.
39. Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discov. 2014; 4(1):61–68.
40. Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014; 27(3):479–484.
41. Shi H, Moriceau G, Kong X, Lee M-K, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012; 3:724.
42. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. The Lancet 19; 379(9829):1893–1901.
43. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, MacConaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011; 29(22):3085–3096.
44. Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, Bacchiocchi A, Dahlman KB, Chmielowski B, Sosman JA, Halaban R, Kefford RF, Long GV et al. Preexisting MEK1 Exon 3 Mutations in V600E/KBRAF Melanomas Do Not Confer Resistance to BRAF Inhibitors. Cancer Discov. 2012; 2(5):414–424.
45. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. 2009; 106(48):20411–20416.
46. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, DiNunzio E et al. Discovery of a Novel ERK Inhibitor with Activity in Models of Acquired Resistance to BRAF and MEK Inhibitors. Cancer Discov. 2013; 3(7):742–750.
47. Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, Soriano R, Forrest WF, Heldens S, Chen H, Toy K, Ha C, Zhou W et al. ERK Inhibition Overcomes Acquired Resistance to MEK Inhibitors. Mol. Cancer Ther. 2012; 11(5):1143–1154.
48. Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008; 68(12):4853–4861.
49. Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, Scolyer RA, Kefford RF, Ribas A et al. A Novel AKT1 Mutant Amplifies an Adaptive Melanoma Response to BRAF Inhibition. Cancer Discov. 2014; 4(1):69–79.
50. Abel EV, Aplin AE. FOXD3 Is a Mutant B-RAF-Regulated Inhibitor of G1-S Progression in Melanoma Cells. Cancer Res. 2010; 70(7):2891–2900.
51. Basile KJ, Abel EV, Aplin AE. Adaptive upregulation of FOXD3 and resistance to PLX4032/4720-induced cell death in mutant B-RAF melanoma cells. Oncogene 2011; 31(19):2471–2479.
52. Abel EV, Basile KJ, Kugel CH, Witkiewicz AK, Le K, Amaravadi RK, Karakousis GC, Xu X, Xu W, Schuchter LM, Lee JB, Ertel A, Fortina P et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Invest. 2013; 123(5):2155–2168.
53. Haq R, Yokoyama S, Hawryluk EB, Jonsson GB, Frederick DT, McHenry K, Porter D, Tran T-N, Love KT, Langer R, Anderson DG, Garraway LA, Duncan LM et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. 2013; 110(11):4321–4326.
54. Corazao-Rozas P, Guerreschi P, Jendoubi M, André F, Jonneaux A, Scalbert C, Garçon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget. 2013; 4(11):1986-98.
55. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Pätzold S, Villanueva J et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 2013; 23(6):811–825.
56. Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, Garraway LA. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013; 504(7478):138–142.
57. Menzies AM, Long GV. Dabrafenib and Trametinib, Alone and in Combination for BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2014; 20(8):2035–2043.
58. Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012; 11(4):909–920.
59. Sánchez-Hernández I, Baquero P, Calleros L, Chiloeches A. Dual inhibition of V600EBRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK-independent mechanism. Cancer Lett. 2012; 314(2):244–255.
60. Lassen A, Atefi M, Robert L, Wong DJ, Cerniglia M, Comin-Anduix B, Ribas A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 2014; 13(1):83.
61. Carlino MS, Todd JR, Gowrishankar K, Mijatov B, Pupo GM, Fung C, Snoyman S, Hersey P, Long GV, Kefford RF, Rizos H. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 2014; 8(3):544–554.
62. Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013.
63. Kirk R. Targeted therapies: The maths behind combination therapy. Nat. Rev. Clin. Oncol. 2013; 10(9):488–488.
64. Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, Gonzalez R, Ott PA, Long GV et al. Phase II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients With Metastatic BRAF-Mutant Cutaneous Melanoma Previously Treated With or Without a BRAF Inhibitor. J. Clin. Oncol. 2013; 31(4):482–489.
65. Romano E, Pradervand S, Paillusson A, Weber J, Harshman K, Muehlethaler K, Speiser D, Peters S, Rimoldi D, Michielin O. Identification of Multiple Mechanisms of Resistance to Vemurafenib in a Patient with BRAFV600E-Mutated Cutaneous Melanoma Successfully Rechallenged after Progression. Clin. Cancer Res. 2013; 19(20):5749–5757.
66. Wilmott JS, Menzies AM, Haydu LE, Capper D, Preusser M, Zhang YE, Thompson JF, Kefford RF, von Deimling A, Scolyer RA, Long GV. BRAFV600E protein expression and outcome from BRAF inhibitor treatment in BRAFV600E metastatic melanoma. Br. J. Cancer 2013; 108(4):924–931.
67. Wilmott JS, Tembe V, Howle JR, Sharma R, Thompson JF, Rizos H, Lo RS, Kefford RF, Scolyer RA, Long GV. Intratumoral Molecular Heterogeneity in a BRAF-Mutant, BRAF Inhibitor-Resistant Melanoma: A Case Illustrating the Challenges for Personalized Medicine. Mol. Cancer Ther. 2012; 11(12):2704–2708.
68. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012; 366(10):883–892.
69. Griewank KG, Scolyer RA, Thompson JF, Flaherty KT, Schadendorf D, Murali R. Genetic Alterations and Personalized Medicine in Melanoma: Progress and Future Prospects. JNCI J. Natl. Cancer Inst. 2014; 106(2):djt435–djt435.
70. Rodic S, Mihalcioiu C, Saleh RR. Detection methods of circulating tumor cells in cutaneous melanoma: A systematic review. Crit. Rev. Oncol. Hematol. 2014. doi:10.1016/j.critrevonc.2014.01.007.
71. Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, Hoang MP, Brannigan BW, Lawrence DP, Flaherty KT, Sequist LV, McMahon M, Bosenberg MW et al. Isolation and Molecular Characterization of Circulating Melanoma Cells. Cell Rep. 2014; 7(3):645–653.
72. Chan MMK, Haydu LE, Menzies AM, Azer MWF, Klein O, Lyle M, Clements A, Guminski A, Kefford RF, Long GV. The nature and management of metastatic melanoma after progression on BRAF inhibitors: Effects of extended BRAF inhibition: BRAF Inhibitor Progression in Melanoma. Cancer 2014:n/a–n/a.
73. Thakur MD, Stuart DD. The Evolution of Melanoma Resistance Reveals Therapeutic Opportunities. Cancer Res. 2013; 73(20):6106–6110.
74. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013; 494(7436):251–255.
75. Thakur MD, Stuart DD. Molecular Pathways: Response and Resistance to BRAF and MEK Inhibitors in BRAFV600E Tumors. Clin. Cancer Res. 2014; 20(5):1074–1080.
76. Carlino MS, Gowrishankar K, Saunders CAB, Pupo GM, Snoyman S, Zhang XD, Saw R, Becker TM, Kefford RF, Long GV, Rizos H. Antiproliferative Effects of Continued Mitogen-Activated Protein Kinase Pathway Inhibition following Acquired Resistance to BRAF and/or MEK Inhibition in Melanoma. Mol. Cancer Ther. 2013; 12(7):1332–1342.
77. Hartsough EJ, Basile KJ, Aplin AE. Beneficial Effects of RAF Inhibitor in Mutant BRAF Splice Variant-Expressing Melanoma. Mol. Cancer Res. 2014; 12(5):795–802.
78. Seghers AC, Wilgenhof S, Lebbé C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012.
79. Abdel-Wahab O, Klimek VM, Gaskell AA, Viale A, Cheng D, Kim E, Rampal R, Bluth M, Harding JJ, Callahan MK, Merghoub T, Berger MF, Solit DB et al. Efficacy of Intermittent Combined RAF and MEK Inhibition in a Patient with Concurrent BRAF- and NRAS-Mutant Malignancies. Cancer Discov. 2014; 4(5):538–545.
80. Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, Legos JJ, Blackman S, Scarmadio A, Kurzrock R, Lizee G, Hwu P. BRAF(V600) Inhibitor GSK2118436 Targeted Inhibition of Mutant BRAF in Cancer Patients Does Not Impair Overall Immune Competency. Clin. Cancer Res. 2012; 18(8):2326–2335.
81. Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The Oncogenic BRAF Kinase Inhibitor PLX4032/RG7204 Does Not Affect the Viability or Function of Human Lymphocytes across a Wide Range of Concentrations. Clin. Cancer Res. 2010; 16(24):6040–6048.
82. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013; 19(5):1225–1231.
83. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw C-NJ, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010; 70(13):5213–5219.
84. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y, Frederick DT, Cooper ZA, Mbofung RM et al. BRAF Inhibition Increases Tumor Infiltration by T cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clin. Cancer Res. 2013; 19(2):393–403.
85. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF Inhibitors Induce Marked T-cell Infiltration into Human Metastatic Melanoma. Clin. Cancer Res. 2012; 18(5):1386–1394.
86. Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW, McMahon M, Flaherty KT, Fisher DE et al. Response to BRAF Inhibition in Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade. Cancer Immunol. Res. 2014; 2(7):643–654.
87. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J, Gupta A, Wigginton JM, Rosen N et al. Paradoxical Activation of T Cells via Augmented ERK Signaling Mediated by a RAF Inhibitor. Cancer Immunol. Res. 2014; 2(1):70–79.
88. Sullivan RJ, LoRusso PM, Flaherty KT. The Intersection of Immune-Directed and Molecularly Targeted Therapy in Advanced Melanoma: Where We Have Been, Are, and Will Be. Clin. Cancer Res. 2013; 19(19):5283–5291.
89. Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with Combination of Vemurafenib and Ipilimumab. N. Engl. J. Med. 2013; 368(14):1365–1366.
90. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression That Is Reversible by MEK and PI3K Inhibition. Clin. Cancer Res. 2013; 19(3):598–609.
91. Atefi M, Avramis E, Lassen A, Wong DJL, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B, Ribas A. Effects of MAPK and PI3K Pathways on PD-L1 Expression in Melanoma. Clin. Cancer Res. 2014; 20(13):3446–3457.
92. Ascierto P, Grimaldi A, Acquavella N, Borgognoni L, Calabro L, Cascinelli N, Cesano A, Del Vecchio M, Eggermont A, Faries M, Ferrone S, Fox B, Gajewski T et al. Future perspectives in melanoma research. Meeting report from the “Melanoma Bridge. Napoli, December 2nd-4th 2012.”J. Transl. Med. 2013; 11(1):137.
93. Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be more beneficial than vice versa for the majority of patients with advanced melanoma. Cancer 2014; 120(11):1617–1619.
94. Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP, Gunturi A, Flaherty KT, Hodi FS, Kefford R, Menzies AM, Atkins MB, Long GV et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer 2014; 120(11):1695–1701.
95. Blagosklonny MV. Why therapeutic response may not prolong the life of a cancer patient: selection for oncogenic resistance. Cell Cycle. 2005; 4(12):1693-8.