
INTRODUCTION
Primary glioblastoma multiforme (GBM) is the most common and malignant subtype of glial tumors [1]. From the clinical and biological point of view, GBM includes a rather heterogeneous group of tumors that vary by site of origin, histophatological features, tumor microenvironment [2] and genetics [3]. They are usually resistant to radio/chemotherapy and show overall survival (OS) rates of a few months to years, making them unvariably lethal [4, 5]. Criteria used for the histological classification and grading of GBM have been recently revised in the 4th World Health Organization (WHO) classification of GBM [1]; however, this classification still fails in distinguishing subgroups (or variants) of primary GBM that display clearly distinct clinical and biological behaviours, despite sharing similar histopathological features [6–9].
Besides histopathology, molecular genetics data has also confirmed the high heterogeneity of GBM, both at the intertumoral and intratumoral levels [3, 10–12]; in addition, molecular genetics data also proved useful for improving the diagnosis, classification, and prognostic stratification of GBM [13, 14], with therapeutic implications [14–16]. However, despite all advances achieved via the study of the methylation status, gene mutations (e.g. IDH1 or PT53 genes) [17] and affected oncogenic pathways [11, 18–20], the precise mechanisms involved in the pathogenesis of GBM still remain far from being fully understood. In turn, routine implementation of molecular genetics into the diagnostic classification of GBM still remains limited, due to the complexity of the genetic findings involved.
Among other genetic abnormalities, the copy number aberration (CNA) profile of tumor cells, over the copy number variation (CNV) of individual patients, has been recognized [21–24] as a useful prognostic tool in GBM [14]. Thus, some copy number aberrations involving one or multiple genes that affect a significant fraction of the tumors, (e.g. CNA associated with either gains of chromosome 7 and/or amplification of the epidermal growth factor receptor (EGFR) gene [18], and losses of DNA sequences in chromosome 10 [19], together with other less frequent alterations involving DNA sequences in chromosomes 12q13-15 [25], 4q and 1q [26], have all been associated with the outcome of GBM patients. Despite this, risk stratification of GBM into classical and non-classical GBM, based on gene expression profiling (GEP) data, was first proposed [27]. Later on, Phillips et al [13] defined three molecular subtypes of GBM according to a combination of GEP data and numerical alterations of chromosomes 7 and 10: proneural, proliferative and mesenchymal GBM. This classification was subsequently redefined by the Cancer Genome Atlas Consortium (TCGA) [14] into four subtypes – proneural, proliferative, mesenchymal and neural GBM, using a combination of GEP data and CNA, together with the pattern of somatic mutations. Despite overlapping data is used in the later two classifications, both approaches are not equivalent, at the same time they are rather complex to be reproducibly applied in routine laboratory diagnostics. In addition, the prognostic impact of both approaches remains controversial because e.g. the proneural subgroup of Philips [13] has a longer survival, while proneural tumors defined according to the TCGA classification [14], have a poorer outcome. In turn, despite EGFR amplification is a defining event for the classical subtype of GBM, in the TCGA classification it appears in >95% of the neural and mesenchymal tumor subtypes, but also in 54% of the proneural subtype; similarly, the same EGFR mutations (e.g. the EGFRvIII variant) can also be detected across all above subtypes of GBM; in addition, neither the criteria used to define the proneural class based on focal amplification of the 4q12 locus harboring the PDGFRA gene (with or without EGFR amplification), nor the evaluation of the expression of the NEFL, GABRA1, SYT1 and SLC12A5 genes, are currently applied for routine diagnostic classification of GBM. Altogether this hampers fast and reproducible risk-stratification of GBM patients at diagnosis, based on this classification.
Here we investigated the gene amplification profiles (GAP) across the whole tumor genome of a series of 80 GBM tumors, as detected by high-density single-nucleotide polymorphism (SNP)-arrays, and evaluated their impact on overall survival (OS) of GBM patients. Based on the results obtained, a GAP-based risk-stratification model was built and validated in a series of 267 GBM tumors from a total of 7 GBM series publicly available at the GEO and ArrayExpress databases and/or whose data was kindly provided by the authors [15, 24, 28–32], in addition to our own cases.
RESULTS
Gene amplification profiles and chromosomal regions involved in GBM
SNP-arrays showed CNA containing > 7 DNA copies of the same DNA sequence for ≥1 chromosome/chromosomal region in all (80/80) cases analyzed from series 1. In the majority of patients (45/80; 56%) such CNA involved genetic amplification of DNA sequences from ≥1 chromosomal region (Table 1 and Figure 1). As expected, DNA sequences at chromosome 7p11.2 containing the EGFR gene, were the most frequently amplified sequences (30/80 cases; 38%), followed by DNA sequences at the 12q (14/80; (18%), 4q (8/80; 10%) and 1q (5/80; 6%) chromosomal regions.
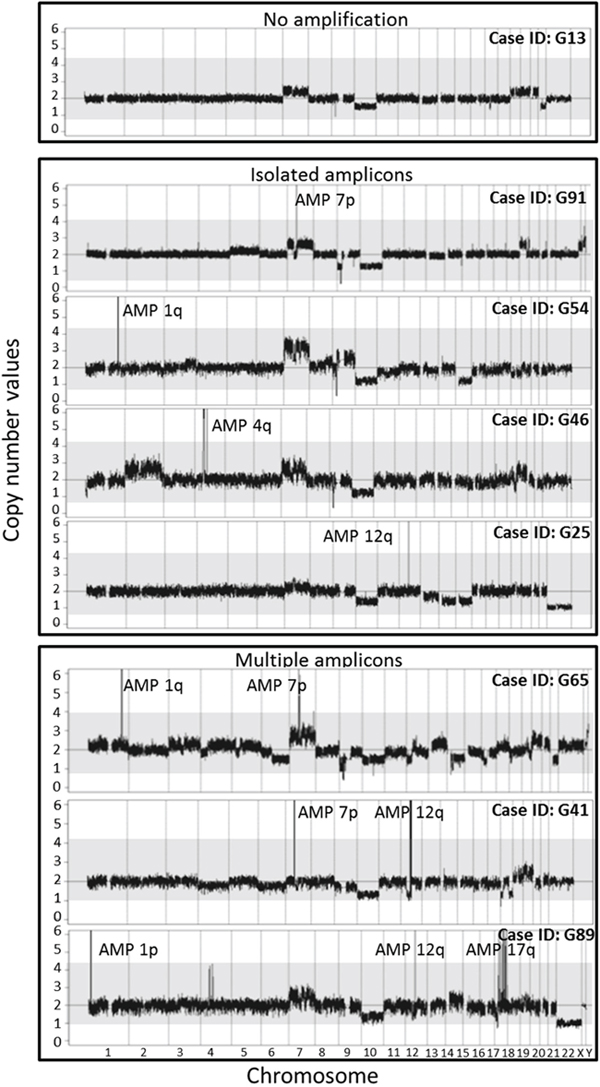
Figure 1: Illustrating examples of the cytogenetic profiles representative of the distinct patterns of gene amplification identified in GBM tumors from series 1, used for the definition of the 5 different subgroups of GBM based on their gene amplification profiles.
Table 1: Major subsets of GBM that carried different gene amplification profiles, as identified in our series (series 1) of GBM patients (n=80) grouped according to the location, type and number of chromosomal regions involved (n=45/80 tumors)
Tumor group |
|
Tumor ID |
Amplified chromosome(s) |
N. of amplified chromosomal band(s)/genes |
Amplified Chromosomal bands |
|---|---|---|---|---|---|
Gene amplification at a single chromosomal region |
EGFR gene involved (n=17;21%) |
G94 |
7p |
2/4 |
7p11.2/7p12.1# |
EGFR gene not involved (n=10;13%) |
G73 |
4q |
3/35 |
4q11/4q12/4q13.3 |
|
Gene amplification at multiple |
EGFR gene & other amplicons involved (n=13;16%) |
G39ϑ |
7p, 12q |
1/2, 2/9 |
7p11.2/12q13.3/12q14.1 |
ID: Case identification code. Most frequently involved genes per chromosomal band included: 1q32.1, MDM4; 4q12, PDGFRA; 12q14.1, CDK4; 12q15, MDM2; 7q31.2,MET. #: Amplified chromosomal region without any annotated gene in it; ϑ: EGFR gene mutation studied in this sample.
Amplification of DNA sequences at chromosome 7p11.2 was found either alone (17/80; 21%) or in combination with amplification of DNA sequences at other regions in chromosomes 1q, 4q, 5q, 6q, 11p, 11q, 12q, 17p and 17q (13/80; 16% of cases) (Table 1). The most frequent combination of simultaneously amplified DNA sequences at chromosomal regions involving 7p11.2 were those of 7p and 12q (7/80 cases; 9%) and of 7p and 11p (2/80 cases; 3%) (Table 1).
Isolated amplification of DNA sequences at chromosomal regions other than 7p11.2 was found in 10/80 cases (13%); this included isolated amplification of 4q12 (where the PDGFRA gene is coded) in 4/80 cases (5%), 12q14.1 (where the CDK4 gene is coded) in 3/80 cases (4%), 1q32.1 (this chromosomal region contains the MDM4 gene) in another 2/80 tumors (3%) and 16q in one tumor (1%) without any annotated gene being coded in the amplified DNA sequences from this later chromosomal region (Table 1).
In turn, genetic amplification at 12q was detected in 14/80 cases (18%) including isolated amplification in 3 GBM and combined 12q gene amplification with gene amplification at other chromosome regions (1p, 1q, 4q, 11p, 11q, 7q, or 17q) in 11 tumors (Table 1), including amplification at chromosome 7p in 7 cases (9%). Gene amplification at 4q was found in 8/80 cases (10%) either alone (4/80; 5% or in association with amplification of DNA sequences at the 12q and/or 7p (4/80; 5%) chromosomal regions (Table 1).
Overall, coexistence of genetically amplified DNA sequences at ≥ 2 chromosomal regions in the same tumor (median of 2 altered chromosomal regions; range: 2-4 regions) was found in 18 cases (23%). In 13 of these 18 tumors, the EGFR gene was involved (Table 1). Of note, gene amplification involving regions at chromosomes 7q (4%), 11p (3%), 17q (3%), 5q (1%), 6q (1%), 11q (1%) and 17p (1%) included variable chromosomal patterns and numbers of genes involved (Table 1 and Table 2). Although gene amplification was frequently associated with chromosomal gains (polyploidy), there were also cases with polyploidies in the absence of gene amplification and vice versa.
Table 2: Frequency of genetic amplification involving different chromosomal regions in the test (series 1) vs the validation series (series 2-8) of GBM patients analyzed
Number of amplified regions |
Chromosomal regions involved |
Amplified GBM cases |
|
|---|---|---|---|
Series 1 |
Series 2-8 |
||
One |
7p |
17 (21%) |
46(17%) |
Subtotal |
27 (34%) |
73 (27%) |
|
Two |
7p,12q |
4 (5%) |
4 (1%) |
Subtotal |
11 (14%) |
28 (10%) |
|
≥Three |
1p,1q,10q |
(0) |
1 (0.4%) |
Subtotal |
7 (9%) |
10 (4%) |
|
Results expressed as number (percentage) of cases showing gene amplification/chromosome arm.
Tumors with isolated amplification of DNA sequences at a single chromosomal region had a median of 9 genes involved (range: 1 to 40 genes). Gene amplification at 7p11.2 included the smallest number of affected genes (median: 5 vs 16 genes for gene amplification involving DNA sequences at chromosomes 12q, 1q and 4q). In turn, cases showing amplified DNA sequences at several chromosomal regions had a median of 19 genes involved (range: 1 to 45 genes). The most commonly (>5% of cases) amplified genes per chromosomal region included: i) 9 genes at chromosome 1q32.1 (SOX13, ETNK2, REN, KISS1, GOLT1A, PLEKHA6, PIK3C2B, MDM4 and LRRN2); ii) 4 genes at chromosome 4q12 (SCFD2, FIP1L1, PDGFRA and KIT); iii) 5 genes in chromosome 7p11.2 (EGFR, LANCL2, VSTM2A, VOPP1 and SEC61G), and; iv) multiple genes at chromosome 12 which had 3 cytobands involved, i.e. 12q14.1 (CDK4, METTL1, CYP27B1, AVIL, CTDSP2, METT21B, AGAP2, OS9, TSFM), 12q13.3 (B4GALNT1, KIF5A, PIP4K2C, DTX3, SLC26A10, MARS, DCTN2, ARHGEF25) and 12q15 (ATP23, MDM2, CPM) (Table 3). When individually considered, the EGFR gene (38%) together with the LANCL2 gene (23%), coded also at chromosome 7p11.2, were the two most frequently amplified genes (Table 3). Of note, amplification of none of the genes detected here had been previously described in healthy individuals [33] and/or publicly available GBM databases.
Table 3: Frequency and chromosomal localization of recurrently amplified genes (> 5% of cases) in GBM from series 1 (n=45/80 tumors), and other previously reported series of GBM (n=111/267 tumors) as detected by SNP-arrays
Amplified genes |
|||||
|---|---|---|---|---|---|
Amplified chromosomal regions |
Symbol |
Frequency of gene amplification |
|||
Chr |
Amplified/Total cases |
Cytoband |
Series 1 |
Series 2-8 |
|
Chr 7 |
119/347 |
7p11.2 |
EGFR |
67% |
57% |
|
|
12q14.1 |
CDK4 |
29% |
14%* |
Chr 12 |
34/347 |
12q13.3 |
B4GALNT1 |
16% |
14%* |
|
|
12q15 |
ATP23 |
16% |
14%* |
Chr 4 |
26/347 |
4q12 |
SCFD2 |
18% |
7% |
Chr 1 |
14/347 |
1q32.1 |
SOX13 |
11% |
5% |
#genes included in the SNP6 array; *The incidence of amplified genes localized in the 12q14.1, 12q13.3 and 12q15 cytobands was calculated using only the 250k-SNP_Nsp and/or STY arrays (n=50) due to the absence of probes for these specific genes in the 50k array.
Classification of GBM tumors based on their gene amplification profiles (GAP)
Based on the presence of gene amplification, their specific subtype and the number of chromosomal regions affected by gene amplification (Figure 1), GBM tumors from group 1 (series 1) were divided into five different subgroups: i) tumors which had no gene amplification (n=35; 44%); ii) tumors with isolated amplification of DNA sequences at chromosome 7p including the EGFR gene (n=17; 21%); iii) GBM with isolated amplification of DNA sequences at a chromosomal region different from chromosome 7p (n=10; 13%); iv) tumors with amplifications of DNA sequences from multiple (≥2) chromosomal regions including that of the EGFR gene (n=13; 16%), and; v) tumors with amplification of DNA sequences at ≥2 chromosomal regions which did not include amplification of the EGFR gene (n=5; 6%) (Table 4 and Figure 2).
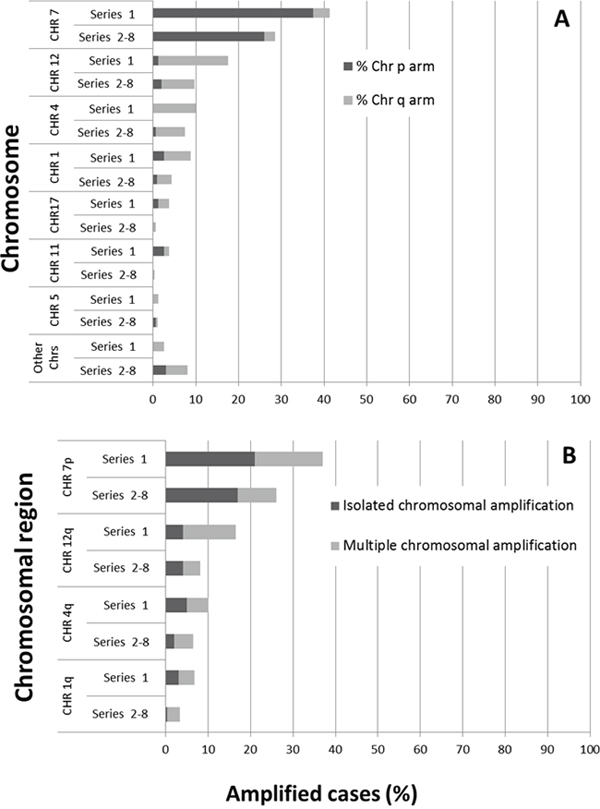
Figure 2: Distribution of the most frequently altered chromosomes (A) and chromosomal regions (B) showing isolated or multiple amplifications both in cases from series 1 (n=45/80) and in patients (111/267) from the seven distinct series previously reported in the literature by others. Results are shown as percentage values from all cases analyzed.
Table 4: Overall survival and genetic features of GBM patients from our series (series 1; n=80) and the seven series of GBM patients previously reported in the literature (series 2-8; n=267) and included in this study for a total of 347 GBM investigated
Variables |
GBM patient series |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Series 1 |
Series 2 |
Series 3 |
Series 4 |
Series 5 |
Series 6 |
Series 7 |
Series 8 |
|||
Total N. of cases |
80 |
24 |
120 |
15 |
12 |
53 |
13 |
30 |
||
N. of cases with |
80 |
24 |
104 |
15 |
NA |
50 |
NA |
NA |
||
Median OS |
15 |
15 |
18 |
10 |
NA |
17 |
NA |
NA |
||
N. of SNP probes investigated |
5-18x105 |
5x105 |
1-2.5x105 |
2.5x105 |
0.5x105 |
0.5-2.5x105 |
0.5x105 |
2.5x105 |
||
New genetical subsets |
NO gene AMP |
35 |
11 |
75 |
9 |
7 |
31 |
7 |
16 |
|
Isolated EGFR AMP |
17 |
7 |
20 |
1 |
2 |
10 |
2 |
4 |
||
Isolated non-EGFR AMP |
10 |
1 |
11 |
3 |
2 |
7 |
1 |
2 |
||
Multiple AMP including EGFR |
13 |
3 |
8 |
1 |
0 |
3 |
2 |
6 |
||
Multiple AMP without EGFR |
5 |
2 |
6 |
1 |
1 |
2 |
1 |
2 |
||
* 23 additional GBM tumors not available in the GEO data repository were hybridized with the Cytoscan750K (n=11 samples) and Cytoscan HD (n=12 samples) SNP-arrays; AMP: genetic amplification; GAP: genetic amplification profile; GSE: genomic repository series code; OS: overall survival; N: number; NA: not available.
The above GAP were further investigated in the other 7 series of GBM used for validation purposes, for a total of 267 primary GBM patients (Table 4). Around half of these cases ˗156/267 (58%)˗ did not show genetic amplification at any of the chromosomal regions investigated (Table 4); in contrast, 69 (26%) tumors had gene amplification at chromosome 7p11.2 amplification –46/267 cases (17%) with isolated EGFR amplification and 23/267 cases (9%) showed amplification of DNA sequences at multiple chromosomal regions including amplification of the EGFR gene– (Table 2 and Table 4). Similarly to what was observed in the test series, gene amplification at 7p, in association with gene amplification at chromosome 12q, was found in 6 tumors (2%) and gene amplification at chromosome 7p together with amplification of DNA sequences at chromosomal regions other than 12q was found in another 17 cases (6%), –e.g. gene amplification at the 1q or 4q chromosomal regions in 3/267 (1.1%) and 4/267 (1.5%) cases, respectively, and at 7q or 3q in 3/267 (1.1%) and 2/267 (0.8%) tumors, respectively – (Table 2). DNA sequences from other chromosomal regions which were also found to be amplified at lower frequencies than those amplified at 7p, included DNA sequences at chromosomes 12q (10/267; 4%) and 4q (6/267; 2%) (Table 2). Genetic amplification at other chromosomal regions such as 7q, 12p and 13q was found in two tumors each, and gene amplification at the 6p, 8q, 15q and 17q chromosomal regions was found in a single tumor each (Table 2).
Upon merging the test and validation series (series 1 to 8), the following distribution into the 5 subgroups of GBM defined by their distinct GAP (Table 4), was found: 191 cases (55%) had no gene amplification (group 1), 63/347 cases (18%) showed isolated amplification of EGFR) (group 2); 37/347 (11%) displayed isolated amplification of genes other than EGFR (group 3); 36/347 (10%) had genetic amplification at multiple chromosomal regions, including amplification of the EGFR gene (group 4); and 20/347 (6%) showed multiple amplified DNA sequences at ≥ 2 distinct chromosomal regions, which did not include amplification of the EGFR gene (group 5) (Table 4).
Prognostic impact of gene amplification profiles in GBM
From the prognostic point of view, the above defined GAP showed a significant impact on OS of GBM patients, both in the test series (series 1) alone (p<0.001; Figure 3A), and in the whole cohort (series 1-8; p<0.001) (Figure 3C). In detail, cases that did not show gene amplification or that displayed amplification of the EGFR gene (alone or in combination with amplification of genes in other chromosomal regions) (groups 1, 2, and 4, respectively) showed a significantly better outcome than patients with isolated amplification of genes other than the EGFR gene (group 3) and cases with amplification of multiple chromosomal regions which did not involve the EGFR region (group 5): median OS of 14, 18 and 14 months vs 6 (p=0.001, p<0.001 and p=0.007, respectively) and 8 months (p=0.03, p<0.001 and p=0.003, respectively) in the test series alone, and of 14, 18 and 14 months vs 8 (p=0.03, p=0.003 and p=0.16, respectively) and 8 (p<0.001, p<0.001 and p=0.001, respectively) months in the whole cohort, respectively (Figure 3, panels A and C, respectively). When cases were re-grouped according to i) the absence of gene amplification or presence of EGFR gene amplification vs ii) occurrence of other GAP, the prognostic impact of the re-grouped GAP was enhanced, both when the test series alone and the whole cohort of patients analyzed, were considered: median OS rates of 15 vs 6 months (p< 0.001) and of 15 vs 8 months (p<0.001), respectively (Figure 3 panels B and D, respectively). Subsequent multivariate analysis of prognostic factors showed that the tumor GAP (p<0.001), together with the administration of chemotherapy (p<0.001) were the best combination of independent prognostic factors to predict patient OS (Table 5).
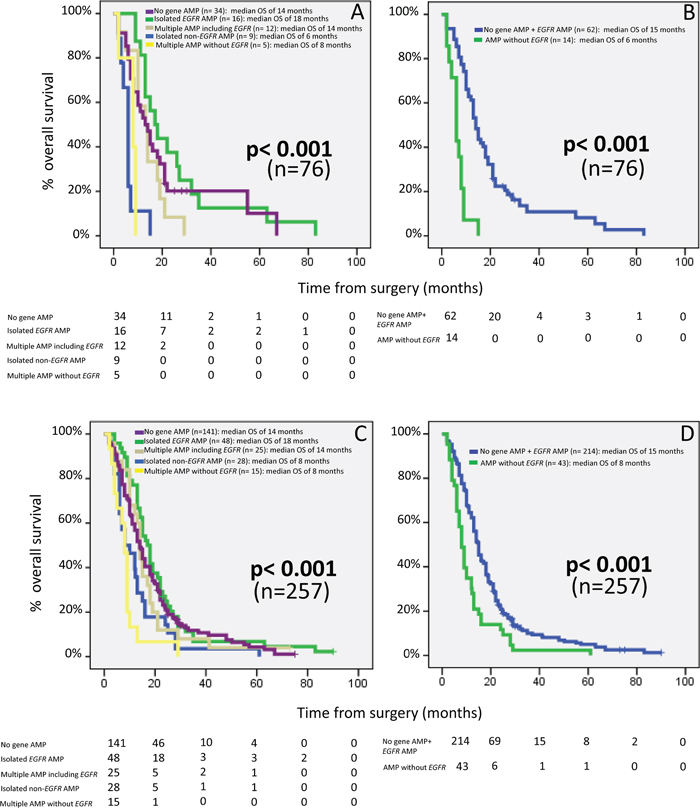
Figure 3: Prognostic impact on overall patient survival of distinct gene amplification profiles of GBM patients from the test cohort – series 1 alone (n=76); panels (A) and (B) – and after pooling our cases with 181 additional cases from seven series of GBM patients previously reported in the literature by other groups (panels (C) and (D); n= 257 GBM). In panels (A) and (C), GBM patients were grouped as having i) no gene amplification; ii) isolated EGFR amplification; iii) gene amplification at multiple chromosomal regions including EGFR gene amplification; iv) isolated amplification of DNA sequences from a single chromosomal region other than 7p11.2 (i.e not including amplification of the EGFR gene), and; v) gene amplification at multiple chromosomal regions in the absence of EGFR amplification. In panels (B) and (D), patients were grouped as: i) cases showing either no gene amplification or having EGFR gene amplification and ii) patients showing genetic amplification at one or more chromosomal regions which did not involve the EGFR gene. Median overall survival is expressed in months and was calculated for 257 patients from series 1-8. Cases who were alive but had a follow-up of less than 18 months and/or died within the 1st month after surgery (n= 16) were excluded from OS analyses.
Table 5: Clinical characteristics of the GBM patients included in the series 1 (n=76) and their association to disease outcome
Clinical/genetic characteristics |
Patient distribution |
Overall survival |
||||
|---|---|---|---|---|---|---|
Univariate analysis |
Multivariate analysis |
|||||
Median (range) |
p-value |
Hazard Ratio (95% CI) |
p-value |
|||
Age |
>30 |
2 (2%) |
67 (2.7-67) |
<.001 |
|
|
|
31-45 |
9 (12%) |
15 (8-21) |
|
|
|
|
46-60 |
21 (28%) |
14 (2-63) |
|
|
|
|
61-75 |
34 (45%) |
13 (2-83) |
|
|
|
|
>75 |
10 (13%) |
6 (2-11) |
|
|
|
Karnofsky Index # |
>70 |
48 (64%) |
15 (2-83) |
.004 |
|
|
|
≤70 |
27 (36%) |
10 (2-30) |
|
|
|
Type of Surgery |
Complete resection |
24 (32%) |
15 (2-83) |
.012 |
|
|
|
Partial resection |
41 (54%) |
13 (2-67) |
|
|
|
|
No resection |
11 (14%) |
6 (2-21) |
|
|
|
Adjuvant chemotherapy# |
Stupp |
43 (67%) |
18 (2-83) |
<.001 |
3 (2-4) |
<.001 |
|
BCNU |
10 (16%) |
13 (5-67) |
|
|
|
|
No chemotherapy |
11 (17%) |
6 (2-11) |
|
|
|
Gene amplification profile |
No gene AMP |
34 (45%) |
13 (2-67) |
<.001 |
|
|
Isolated EGFR AMP |
16 (21%) |
17 (9-83) |
|
|
|
|
Multiple AMP including EGFR |
12 (16%) |
13 (2-29) |
|
|
|
|
Isolated non-EGFR AMP |
9 (12%) |
6 (2-15) |
|
|
|
|
Multiple AMP without EGFR |
5 (6%) |
8 (2-9) |
|
|
|
|
No gene AMP+EGFR AMP |
62 (82%) |
14 (2-83) |
<.001 |
6 (2-12) |
<.001 |
|
AMP without EGFR |
14 (18%) |
6 (2-15) |
|
|
|
|
CI: confidence interval; #: Data of Karnofsky index and adjuvant chemotherapy were only available in only 75 and 65 GBM patients respectively; Stupp: radiotherapy plus temozolamide; BCNU: carmustine; AMP: genetic amplification.
DISCUSSION
Despite histopathology remains the gold standard for the diagnosis of GBM, it provides limited information about patient outcome. Consequently, new classifications have been proposed in recent years for GBM in which molecular genetics data derived from chromosomal alteration profiles, DNA mutational status and GEP data, is used for the definition of tumor subgroups of distinct embryonic tissue origins [13, 14]. However, these classification models are difficult to implement in routine diagnostics and/or remain of relatively limited prognostic value [34–36].
Among other cytogenetic/molecular alterations, gene amplification, particularly gene amplification involving the EGFR gene, represents one of the most common genetic changes in GBM [11, 20-22, 37]. Thus, EGFR amplification at chromosome 7p11.2 can be found in between one third and half of all GBM patients as the only chromosomal region amplified – including a variable number of amplified genes – or it can be find in the same tumor in combination with amplification of genes located at other chromosomal regions, such as the MDM2, MDM4, PDGFRA and CDK4 genes encoded in chromosomes 12q15, 1q32.1, 4q12 and 12q14.1 [11, 14, 38], respectively. Of note, gene amplification is a common genetic alteration across different malignancies and it usually involves (wild-type or mutated) genes that show oncogenic potential (i.e are capable of reproducing the tumor); thereby, it might confer a malignant phenotype associated with a variable outcome, depending on the specific genes amplified and/or overexpressed. Despite this, no study has been reported so far in which the impact of different GAP on OS has been investigated in a large series of GBM patients.
Here we investigated the GAP across the whole GBM tumor genome and analyzed their impact on patient OS, based on one of the largest series of GBM patients reported so far in the literature. SNP-arrays were used for both sensitive identification of CNA involving specific DNA sequences across the whole tumor genome and detailed delineation of the amplified genes; in order to avoid CNV due to germinal single nucleotide polymorphisms, insertions and deletions potentially associated with an increased predisposition to GBM (e.g. the rs1801320 SNP in the RAD51 DNA repair gene [39]), paired tumor and peripheral blood (PB) samples were analyzed per patient.
Overall, our results showed the presence of gene amplification in the majority (>50%) of tumors investigated. As expected, genetic amplification involving DNA sequences at the 7p11.2 chromosomal region was by far the most frequent alteration, followed by genetic amplification at the 12q, 4q12 and 1q32.1 chromosomal regions, and to a lower extent also, at 1q, 5q, 6q, 7q and at regions in both arms of chromosomes 11 or 17. These results confirm and extend on previous observations by our [20–22] and other groups [40, 41] which indicate that EGFR is the most frequently amplified oncogene in GBM, where it is detected in up to 40% of primary GBM tumors in association with a better outcome, compared to cases that show either no gene amplification or amplification of genes other than EGFR [20, 37]. At present, it is well-established that activation of the EGFR gene via gene amplification and/or mutations, up-regulates the RAS/RAF/MAPK and PI3K signaling pathways, translating into a tumor phenotype consisting of: i) abnormally high cell proliferation and ii) survival of tumor cells, and iii) an increased angiogenesis [11, 42]. Of note, here we confirm via mapping of the amplified region in chromosome 7p11.2, that this alteration frequently includes also the LANCL2 gene, in addition to other genes adjacent to the EGFR and LANCL2 genes [20, 43]. Although the LANCL2 gene codes for a protein involved in up-regulation of AKT and cell survival, and an increased cell sensitivity to adriamycin [44, 45], its precise mechanism of action remains elusive.
In line with previous observations [11, 38, 46], EGFR gene amplification at the 7p11.2 chromosomal region, was found either as the only amplified DNA sequence, or in association with amplification of DNA sequences at other chromosomal regions and genes such as the PDGFRA, MDM2, MDM4 and CDK4 genes. Such combined pattern of amplification of multiple genes at distinct chromosomal regions might lead to unique malignant transformation profiles for which the underlying mechanisms are still poorly understood; however, in our series, it did not prove to confer a more adverse outcome vs isolated amp 7p11.2. In contrast, isolated amplification of DNA sequences at chromosomal regions other than that containing the EGFR gene, were associated with a significantly shorter OS of GBM patients. Of note, these later alterations most frequently affected genes encoded in the 12q, 4q and 1q chromosomal regions, and they typically involved multiple genes [15]. Thus, amplification of DNA sequences at the 12q13-14 chromosomal region usually included the CDK4 gene together with the METTL1, CYP27B1, AVIL, CTDSP2, METT21B, AGAP2 and OS9 genes, while genetic amplification at 12q15 affected the MDM2 oncogene in a significant fraction of all (primary) GBM tumors analyzed. CDK4 is a member of the Ser/Thr protein kinase family, required for the cell cycle transition from the G1 to S-phase [47]; thus, CDK4 phosphorylates the Rb gene product leading to its inactivation and the release of proteins required for cell cycle progression, at the same time it also down-regulates TP53 [47]. In turn, AVIL binds actin and promotes the development of neuronal cells, while the MDM2 gene codes for a nuclear-localized E3-ubiquitin ligase whose transcription is recognized as the main p53 negative regulator [48]. In our series, genetic amplification at the 4q11-12 chromosomal region, systematically affected the PDGFRA gene together with the SCFD2 and FIP1L1 genes. PDFGRA [11] codes for a tirosine-protein kinase cell surface receptor of the PDGF growth factor, which promotes cell proliferation and migration. Finally, amplification of DNA sequences at chromosome 1q32.1 involved the MDM4 gene together with the SOX13, ETNK2, KISS1, GOLT1A, PLEKHA6, REN and PIK3C2B genes. MDM4 inhibits TP53 and TP73, mediating cell cycle arrest via binding to their transcriptional activation domain, at the same time it inhibits degradation of MDM2 [48], whereas PIK3C2B belongs to the PI3K gene family and activates signaling for cell proliferation, survival and migration [49]. In turn, the DTX3 ubiquitin ligase gene probably acts both as a positive and negative regulator of Notch, depending on the developmental stage and cell context [50].
Altogether, these findings indicate that the distinct GAP here reported for GBM tumors might confer a distinct biological and (also) clinical behavior to these tumors. Thus, based on the presence vs absence of gene amplification and its subtypes, five different patterns were defined among our GBM patients, which showed an association with OS. From the prognostic point of view, these five GAP could be further re-grouped into two major risk-groups including: i) patients with either no gene amplification or EGFR gene amplification associated with or without amplification of genes coded at chromosomal regions other than chromosome 7p11.2, with a significantly longer OS; and, ii) cases presenting with amplification of one or more chromosomal regions that did not include EGFR gene amplification, and that were associated with a significantly poorer outcome. The prognostic impact of this later classification was further confirmed in a larger cohort of GBM patients previously reported in the literature and, together with the type of treatment administered, emerged as the most powerful combination of independent prognostic factor for GBM patients. However, the precise mechanisms involved in determining the distinct survival rates of these two molecular groups of patients, still remain to be elucidated and deserve further investigations.
Despite several classification models have been previously proposed which address the genomic heterogeneity of GBM [13, 14, 27] and identify tumors with different cellular origins, so far they have proven to be of limited prognostic value [34-36, 51] and/or difficult to be used in routine diagnostics due to the complexity of the information they require to classify GBM patients at diagnosis. In contrast, here we propose a relatively simple prognostic stratification model for GBM tumors based on their gene amplification profiles that might be easily implemented in routine laboratory diagnostics, and that will potentially contribute to a better management of the patients. In line with this, we have recently patented an array containing this combination of probes [52] to assess the above referred GAP, and that we hope can be commercially available for routine diagnostics soon.
MATERIALS AND METHODS
Patients and samples
Overall, 347 GBM tumors were studied. These included two groups of adult patients: the first group consisted of 80 caucasian GBM patients (group 1, series 1) with histological diagnosis of primary GBM based on the WHO criteria (38 males and 42 females; mean age of 62±13 years, ranging from 24 to 84 years) (Table 6). Fifty-seven of these 80 patients (71%) were admitted to the University Hospital of Coimbra (Coimbra, Portugal) and their genomic data has been deposited in the genomic repository GEO (series code number: GSE42631) and 23 (29%) were from the University Hospital of Salamanca (Spain). Each patient from series 1 gave his/her informed consent prior to entering the study, and the study was approved by the local Ethics Committees of both institutions, according to the Declaration of Helsinki. For each patient within this first group, tumor samples containing representative areas of (fresh) tumor tissues were obtained by surgical resection, immediately (<30 min) snap-frozen in liquid nitrogen, and stored at -80ºC for further SNP-array studies; in parallel, a PB sample was also collected from each patient. Prior to the SNP-array studies, a section was cut from the stored tissue blocks and assessed by conventional histopathological procedures for its tumoral cell contents. Specimens with ≥75% tumoral cells, in the absence of significant contamination by normal brain parenchyma and tumoral necrosis, were selected for further DNA and RNA extraction.
Table 6: Clinical and biological characteristics of GBM patients from series 1 who were analyzed by single-nucleotide polymorphism arrays in this study (n=80)
Case ID |
Age |
Gender |
Karnofsky |
Location |
Brain hemisphere |
Overall survival or follow-up |
Exitus |
Treatment |
|
|---|---|---|---|---|---|---|---|---|---|
Type of surgery |
Chemotherapy |
||||||||
GBM1 |
80 |
M |
ND |
Temporal |
L |
6 |
Yes |
T |
ND |
GBM2 |
75 |
M |
80 |
Fronto-temporal |
R |
22 |
Yes |
T |
Stupp |
GBM3 |
61 |
F |
90 |
Parietal |
R |
83 |
Yes |
T |
Stupp |
GBM4 |
73 |
M |
100 |
Temporal |
L |
19 |
Yes |
P |
Stupp |
GBM5 |
38 |
M |
90 |
Frontal |
R |
15 |
Yes |
T |
Stupp |
GBM6 |
49 |
F |
60 |
Frontal |
R |
30* |
No |
P |
Stupp |
GBM7 |
41 |
M |
40 |
Temporal |
R |
11 |
Yes |
P |
Stupp |
GBM8 |
57 |
M |
90 |
Tempo-parietal |
R |
6 |
Yes |
T |
Stupp |
GBM9 |
72 |
M |
90 |
Temporal |
R |
28 |
No |
P |
Stupp |
GBM10 |
62 |
M |
100 |
Parietal |
R |
28 |
No |
T |
Stupp |
GBM11 |
71 |
M |
80 |
Temporal |
R |
27 |
Yes |
P |
Stupp |
GBM12 |
50 |
F |
100 |
Temporal |
R |
13 |
Yes |
T |
Stupp |
GBM13 |
72 |
F |
70 |
Temporal |
R |
9 |
Yes |
P |
Stupp |
GBM14 |
78 |
F |
100 |
Frontal |
R |
6 |
Yes |
T |
- |
GBM15 |
61 |
F |
100 |
Frontal |
R |
25 |
No |
T |
Stupp |
GBM16 |
54 |
M |
100 |
Fronto-parietal |
R |
2 |
Yes |
T |
Stupp |
GBM17 |
52 |
M |
80 |
Frontal |
R |
63 |
Yes |
P |
Stupp |
GBM18 |
57 |
F |
90 |
Temporal |
R |
10 |
Yes |
P |
Stupp |
GBM19 |
68 |
M |
90 |
Occipital |
L |
10 |
Yes |
T |
Stupp |
GBM20 |
82 |
F |
80 |
Frontal |
L |
7 |
Yes |
T |
- |
GBM21 |
77 |
M |
70 |
Temporal |
R |
6 |
Yes |
P |
- |
GBM22 |
69 |
F |
100 |
Frontal |
L |
8 |
Yes |
T |
Stupp |
GBM23 |
24 |
F |
80 |
Frontal |
L |
21 |
Yes |
P |
Stupp |
G97 |
53 |
M |
80 |
Temporal |
R |
21 |
Yes |
T |
Stupp |
G94 |
79 |
F |
80 |
Temporal |
R |
9 |
Yes |
P |
- |
G93 |
63 |
M |
80 |
Occipital |
R |
29 |
Yes |
T |
Stupp |
G92 |
54 |
F |
80 |
Parietal |
R |
15 |
Yes |
T |
Stupp |
G91 |
73 |
F |
60 |
Occipital |
R |
13 |
Yes |
P |
Stupp |
G90 |
57 |
F |
60 |
Parietal |
L |
5 |
Yes |
B |
- |
G89 |
51 |
M |
80 |
Temporal |
R |
2 |
Yes |
P |
- |
G88 |
71 |
M |
80 |
Parietal |
R |
8 |
Yes |
P |
Stupp |
G87 |
45 |
M |
80 |
Temporal |
L |
16 |
Yes |
P |
Stupp/ Sequential |
G83 |
75 |
M |
70 |
Temporal |
R |
10 |
Yes |
P |
- |
G82 |
78 |
M |
70 |
Frontal |
R |
2 |
Yes |
B |
- |
G81 |
62 |
F |
70 |
Frontal |
R |
13 |
Yes |
P |
Stupp |
G80 |
43 |
M |
80 |
Frontal |
R |
18 |
Yes |
T |
Stupp |
G79 |
71 |
F |
60 |
Occipital |
R |
6 |
Yes |
B |
- |
G73 |
78 |
F |
60 |
Parietal |
L |
4 |
Yes |
B |
- |
G72 |
77 |
F |
70 |
Temporal |
L |
1 |
Yes |
P |
- |
G71 |
66 |
F |
60 |
Parietal |
R |
10 |
Yes |
P |
Sequential |
G70 |
56 |
F |
80 |
Occipital |
L |
21 |
Yes |
P |
Stupp |
G68 |
72 |
M |
70 |
Insular |
L |
26 |
Yes |
T |
Stupp |
G67 |
68 |
F |
80 |
Parietal |
R |
35 |
Yes |
P |
Stupp |
G66 |
60 |
M |
80 |
Occipital |
R |
14 |
Yes |
T |
Stupp |
G65 |
69 |
F |
60 |
Parietal |
L |
1 |
Yes |
P |
- |
G64 |
57 |
M |
60 |
Occipital |
L |
8 |
Yes |
P |
Sequential |
G63 |
61 |
F |
60 |
Insular |
R |
13 |
Yes |
P |
Sequential |
G62 |
57 |
F |
90 |
Occipital |
R |
18 |
Yes |
T |
Stupp |
G57 |
34 |
M |
90 |
Frontal |
R |
8 |
Yes |
T |
Stupp |
G56 |
65 |
M |
80 |
Frontal |
L |
13 |
Yes |
P |
Stupp |
G55 |
54 |
F |
80 |
Frontal |
R |
17 |
Yes |
P |
Stupp |
G54 |
65 |
F |
60 |
Parietal |
L |
6 |
Yes |
P |
- |
G53 |
74 |
M |
60 |
Frontal |
L |
29 |
Yes |
T |
Stupp |
G52 |
56 |
M |
90 |
Frontal |
L |
21 |
Yes |
B |
Stupp |
G51 |
60 |
M |
60 |
Temporal |
R |
2 |
Yes |
B |
- |
G50 |
84 |
M |
70 |
Temporal |
R |
11 |
Yes |
P |
- |
G46 |
62 |
M |
60 |
Frontal |
L |
3 |
Yes |
P |
- |
G45 |
76 |
F |
60 |
Temporal |
R |
10 |
Yes |
P |
- |
G44 |
48 |
M |
80 |
Frontal |
L |
22 |
Yes |
P |
PCV |
G43 |
67 |
F |
70 |
Temporal |
R |
7 |
Yes |
P |
- |
G42 |
67 |
M |
80 |
Temporal |
R |
2 |
Yes |
P |
- |
G41 |
44 |
F |
60 |
Frontal |
R |
14 |
Yes |
B |
Sequential |
G40 |
45 |
F |
80 |
Frontal |
R |
15 |
Yes |
P |
BCNU+TMZ |
G39 |
70 |
F |
50 |
Frontal |
R |
18 |
Yes |
P |
Stupp |
G37 |
70 |
M |
80 |
Temporal |
R |
32 |
Yes |
T |
Stupp |
G35 |
50 |
F |
80 |
Frontal |
L |
2 |
Yes |
P |
- |
G34 |
69 |
M |
60 |
Temporal |
R |
5 |
Yes |
B |
- |
G31 |
71 |
F |
90 |
Frontal |
R |
7 |
Yes |
P |
- |
G30 |
71 |
F |
70 |
Temporal |
R |
9 |
Yes |
B |
- |
G29 |
49 |
F |
80 |
Parietal |
L |
12 |
Yes |
B |
Sequential |
G25 |
68 |
M |
80 |
Frontal |
L |
6 |
Yes |
P |
Stupp |
G23 |
50 |
F |
70 |
Frontal |
R |
14 |
Yes |
B |
Stupp |
G17 |
30 |
F |
90 |
Temporal |
R |
67 |
Yes |
P |
Sequential |
G15 |
79 |
M |
80 |
Parietal |
L |
5 |
Yes |
T |
Sequential |
G14 |
69 |
F |
70 |
Frontal |
R |
0 |
Yes |
B |
- |
G13 |
39 |
F |
90 |
Frontal |
R |
20 |
Yes |
P |
Sequential |
G12 |
74 |
M |
70 |
Temporal |
R |
1 |
Yes |
B |
- |
G10 |
35 |
F |
80 |
Temporal |
L |
15 |
Yes |
P |
Stupp |
G8 |
67 |
F |
90 |
Deep |
NA |
9 |
Yes |
P |
Stupp |
G6 |
70 |
F |
80 |
Temporal |
R |
19 |
Yes |
P |
Stupp |
The second group of GBM patients included 267 unselected cases from 7 different series of GBM (series 2 to 8; group 2) previously reported in the literature [15, 24, 28–32]. Data from cases included in one of these series (series 4; n=15 cases) were kindly provided by Bralten et al [32], while data about the patients and tumor samples from the other six series was accessed from publicly available data bases – GSE19612 (series 2; 24 cases); GSE19399 and GSE9635 (series 3; 104 and 16 cases, respectively); GSE14804 (series 5; 12 cases); E-MEXP1330 (series 6, 53 cases); GSE10922 (series 7; 13 cases) and GSE13021 (series 8; 30 cases)–.
DNA extraction and identification of copy number alterations by SNP-arrays
DNA from frozen tumor samples (n=80 tumors from series 1) was purified using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA), according to the instructions of the manufacturer. The yield and purity of the extracted DNA were determined using a NanoDrop-1000 spectrophotometer (Nano-Drop Technologies Inc, Wilmington, DE, USA), and they systematically showed absorbance (A) values >1.5 at 260/230nm and ≥1.8 at 260/280nm wavelengths, respectively. DNA integrity was evaluated by conventional electrophoretic procedures in a 1% agarose gel. RNA extraction and cDNA synthesis were performed following the standard operating procedures (SOPs) of the Spanish DNA Bank Carlos III (University of Salamanca, Salamanca, Spain) (http://www.bancoadn.org).
For the investigation of CNAs by SNP-arrays, DNA from frozen tumor tissues and their paired PB samples was used in order to exclude individual CNV due to germline SNPs, small insertions and deletions. Briefly, extracted DNA (250ng per array) was digested with restriction enzymes and ligated to the corresponding adaptors, following conventional Affymetrix procedures (Affymetrix Inc, Thermo-Fisher Scientific, Waltham, MA, USA). A generic primer that recognizes the adaptor sequence was used in triplicate, to amplify adaptor-ligated DNA fragments via polymerase chain reaction (PCR). The amplified DNA was then fragmented, labeled, and hybridized to the corresponding SNP-array (please see below). After hybridization, chips were washed in an Affymetrix Fluidics Station 450 (Affymetrix) and the hybridized sequences were labeled using streptavidin-phycoerythrin, and assayed by fluorescence detection using a GeneChip Scanner 3000 (Affymetrix). The allelotype at a locus was then determined based on probe-associated fluorescence intensity data for oligonucleotides complementary to the reference sequences that covered the corresponding SNP position.
In the test series (series 1), 4 different types of SNP-arrays were used. These included: i) the GeneChip Human Mapping 500K Array Set (n=35 tumors), which provides information about >500,000 SNPs according to the NCBI/hg19 assembly (262,264 SNPs in the Nsp array and 238,304 SNPs in the Sty array); ii) the Genome-Wide Human SNP Array 6.0 (n=22 cases), which contains probes for 906,600 SNPs and 945,826 non-polymorphic probes featuring a total of >1.8 million probes (Affymetrix); and, iii) the CytoScan 750K and Cytoscan HD arrays (n=23 tumors) which contain probes for 200,436 SNPs and 743,304 non-polymorphic probes (Affymetrix). Data about DNA probes was analyzed with the Console Genotyping software (version 3.0.2; Affymetrix). In addition, the dChip 2010 software (http://www.dchip.org; Dana Farber Cancer Institute, Boston, MA, USA) was used to calculate CNA values. To plot CNAs according to their chromosomal location, the Chromosome Analysis Suite (CHAS) was used. The Hg 19 human genome sequence was used as reference to name the amplified genes, as defined by CNAs values > 4.8 (arbitrary units) typically corresponding to > 7 DNA copies (in order to exclude polyploidies) [29].
For the validation series, data on 7 different SNP-array chips (Affymetrix 50K, 100K, 250K and/or 500K SNP-arrays) previously reported by others [15, 23, 24, 28–32] were used for the analysis of CNA. The number of common SNPs for the 100K and 500k arrays, and for the 500K and cytoscan HD arrays, was of 21,144 and 65,535 SNPs, respectively, for a total of around 11,000 SNPs in common to all SNPs-arrays used.
Mutational analyses
Analysis of IDH1 and IDH2 gene mutations was based on DNA extracted from formalin-fixed and paraffin-embebed tissues (n=54) using the QIAamp DNA Mini kit (Qiagen, Germany) according to the instructions of the manufacture. Exon 4 DNA of both the IDH1 and the IDH2 genes was amplified by PCR and sequenced on a capillary automated sequencer (CEQ 8000; Beckman-Coulter, Hialeah, FL, USA); mutational analysis of the sequence data was performed using the Sequencher, (version 4.7) software (Genes Codes, Ann Arbor, MI, USA). None of the 54 primary GBM cases analyzed showed IDH1 or IDH2 mutations.
Statistical analysis
The statistical significance of differences observed between groups was assessed by the Student T and the Mann-Whitney U tests, for parametric and non-parametric (continuous) variables, respectively; for categorical variables, the X2 test was used. Overall, 257 GBM who survived for >1 month after surgery and had a minimum follow-up of 18 months (for patients remaining alive) were included in OS analyses. Survival curves were plotted according to the method of Kaplan and Meier, and the (two-sided) log-rank test was used to assess the statistical significance of differences in OS among distinct groups of patients. Multivariate analysis of prognostic factors for OS was performed using the Cox stepwise regression model. In this part of the study, only those variables showing a significant association with RFS in the univariate analysis were included (Table 5). P-values <0.05 were considered to be associated with statistical significance. For all statistical analysis, the SPSS software (SPSS 17.0, IBM SPSS, Armonk, NY, USA), was used.
Author contributions
Specific contributions of each author are: Álvaro Otero, Pablo Sousa, Hermínio Tão, Olinda Rebelo, Marcos Barbosa and Pim J French have provided samples and clinical or histopathological data; María González-Tablas, Inês Crespo, Ana Luísa Vital and María Dolores Tabernero have processed the samples; Luis Antonio Corchete, Ana Belen Nieto and María Carmen Patino-Alonso have analyzed SNP-data; María Dolores Tabernero, Alberto Orfao, Maria Celeste Lopes and María González-Tablas have design the study, drafted and writing the manuscript. Maria Rosario Almeida and Ana Filipa Guedes analyzed the IDH1 and IDH2 mutational status. All authors have contributed significantly to this article and all of them have read and approved the manuscript.
CONFLICTS OF INTEREST
The authors declared that they have no conflicts of interest.
FUNDING
This work was supported by RETICC RD06/0020/0035, RD06/0020/0059 and RD12/0036/0048 grants from Red Temática de Investigación Cooperativa en Cáncer (RTICC), Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, (Madrid, Spain and FONDOS FEDER), AES PI16/000476 (Instituto de Salud Carlos III, Madrid, Spain and FONDOS FEDER), GRS909A14 (JCYL) and CB16/12/00400 grant (CIBER-ONC, Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain and FONDOS FEDER).
REFERENCES
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803-20.
2. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012; 60:502-14.
3. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013; 110:4009-14.
4. Lee CY. Strategies of temozolomide in future glioblastoma treatment. Onco Targets Ther. 2017; 10:265-70.
5. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010; 60:166-93.
6. Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010; 9:135.
7. Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, De Moor B, Eilers PH, van der Spek PJ, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009; 69:9065-72.
8. Cloughesy TF, Mischel PS. New strategies in the molecular targeting of glioblastoma: how do you hit a moving target? Clin Cancer Res. 2011; 17:6-11.
9. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015; 129:829-48. https://doi.org/10.1007/s00401-015-1432-1.
10. Virk SM, Gibson RM, Quinones-Mateu ME, Barnholtz-Sloan JS. Identification of variants in primary and recurrent glioblastoma using a cancer-specific gene panel and whole exome sequencing. PLoS One. 2015; 10:e0124178.
11. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, Leversha MA, Mikkelsen T, Brennan CW. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012; 109:3041-6
12. The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061-8.
13. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9:157-73.
14. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98-110.
15. Yin D, Ogawa S, Kawamata N, Tunici P, Finocchiaro G, Eoli M, Ruckert C, Huynh T, Liu G, Kato M, Sanada M, Jauch A, Dugas M, et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009; 7:665-77.
16. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155:462-77.
17. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009; 11:341-7.
18. Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010; 12:675-84.
19. Rong Y, Belozerov VE, Tucker-Burden C, Chen G, Durden DL, Olson JJ, Van Meir EG, Mackman N, Brat DJ. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res. 2009; 69:2540-9.
20. Crespo I, Tao H, Nieto AB, Rebelo O, Domingues P, Vital AL, Patino Mdel C, Barbosa M, Lopes MC, Oliveira CR, Orfao A, Tabernero MD. Amplified and homozygously deleted genes in glioblastoma: impact on gene expression levels. PLoS One. 2012; 7:e46088.
21. Crespo I, Vital AL, Nieto AB, Rebelo O, Tao H, Lopes MC, Oliveira CR, French PJ, Orfao A, Tabernero MD. Detailed characterization of alterations of chromosomes 7, 9, and 10 in glioblastomas as assessed by single-nucleotide polymorphism arrays. J Mol Diagn. 2011; 13:634-47.
22. Vital AL, Tabernero MD, Crespo I, Rebelo O, Tao H, Gomes F, Lopes MC, Orfao A. Intratumoral patterns of clonal evolution in gliomas. Neurogenetics. 2010; 11:227-39.
23. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899-905.
24. Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, Hariono S, Silber J, Feiler HS, Gray JW, Spellman PT, Vandenberg SR, Berger MS, et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro Oncol. 2009; 11:477-87.
25. Reifenberger G, Ichimura K, Reifenberger J, Elkahloun AG, Meltzer PS, Collins VP. Refined mapping of 12q13-q15 amplicons in human malignant gliomas suggests CDK4/SAS and MDM2 as independent amplification targets. Cancer Res. 1996; 56:5141-5.
26. Riemenschneider MJ, Knobbe CB, Reifenberger G. Refined mapping of 1q32 amplicons in malignant gliomas confirms MDM4 as the main amplification target. Int J Cancer. 2003; 104:752-7.
27. Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C, McLaughlin ME, Batchelor TT, Black PM, von Deimling A, Pomeroy SL, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003; 63:1602-7.
28. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS, Modrusan Z, Nacu S, Guerrero S, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010; 17:362-75.
29. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007; 104:20007-12.
30. Kuga D, Mizoguchi M, Guan Y, Hata N, Yoshimoto K, Shono T, Suzuki SO, Kukita Y, Tahira T, Nagata S, Sasaki T, Hayashi K. Prevalence of copy-number neutral LOH in glioblastomas revealed by genomewide analysis of laser-microdissected tissues. Neuro Oncol. 2008; 10:995-1003.
31. Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, Ressom H, Jean W, Bigner D, Yan H, Samuels Y, Waldman T. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008; 68:10300-6.
32. Bralten LB, Kloosterhof NK, Gravendeel LA, Sacchetti A, Duijm EJ, Kros JM, van den Bent MJ, Hoogenraad CC, Sillevis Smitt PA, French PJ. Integrated genomic profiling identifies candidate genes implicated in glioma-genesis and a novel LEO1-SLC12A1 fusion gene. Genes Chromosomes Cancer. 2010; 49:509-17.
33. Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015; 16:172-83.
34. Lin N, Yan W, Gao K, Wang Y, Zhang J, You Y. Prevalence and clinicopathologic characteristics of the molecular subtypes inmalignant glioma: a multi-institutional analysis of 941 cases. PLoS One. 2014; 9:e94871.
35. Yan W, Zhang W, You G, Zhang J, Han L, Bao Z, Wang Y, Liu Y, Jiang C, Kang C, You Y, Jiang T. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 2012; 14:1432-40.
36. Crisman TJ, Zelaya I, Laks DR, Zhao Y, Kawaguchi R, Gao F, Kornblum HI, Coppola G. Identification of an Efficient Gene Expression Panel for Glioblastoma Classification. PLoS One. 2016; 11:e0164649.
37. Hobbs J, Nikiforova MN, Fardo DW, Bortoluzzi S, Cieply K, Hamilton RL, Horbinski C. Paradoxical relationship between the degree of EGFR amplification and outcome in glioblastomas. Am J Surg Pathol. 2012; 36:1186-93.
38. Furgason JM, Koncar RF, Michelhaugh SK, Sarkar FH, Mittal S, Sloan AE, Barnholtz-Sloan JS, Bahassi el M. Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience. 2015; 2:618-28. https://doi.org/10.18632/oncoscience.178.
39. Franceschi S, Tomei S, Mazzanti CM, Lessi F, Aretini P, La Ferla M, De Gregorio V, Pasqualetti F, Zavaglia K, Bevilacqua G, Naccarato AG. Association between RAD 51 rs1801320 and susceptibility to glioblastoma. J Neurooncol. 2016; 126:265-70.
40. Jeuken J, Sijben A, Alenda C, Rijntjes J, Dekkers M, Boots-Sprenger S, McLendon R, Wesseling P. Robust detection of EGFR copy number changes and EGFR variant III: technical aspects and relevance for glioma diagnostics. Brain Pathol. 2009; 19:661-71.
41. Srividya MR, Thota B, Arivazhagan A, Thennarasu K, Balasubramaniam A, Chandramouli BA, Hegde AS, Santosh V. Age-dependent prognostic effects of EGFR/p53 alterations in glioblastoma: study on a prospective cohort of 140 uniformly treated adult patients. J Clin Pathol. 2010; 63:687-91.
42. Turner KM, Sun Y, Ji P, Granberg KJ, Bernard B, Hu L, Cogdell DE, Zhou X, Yli-Harja O, Nykter M, Shmulevich I, Yung WK, Fuller GN, et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc Natl Acad Sci U S A 2015; 112:3421-6.
43. Eley GD, Reiter JL, Pandita A, Park S, Jenkins RB, Maihle NJ, James CD. A chromosomal region 7p11.2 transcript map: its development and application to the study of EGFR amplicons in glioblastoma. Neuro Oncol. 2002; 4:86-94.
44. Zeng M, van der Donk WA, Chen JR. Lanthionine synthetase C-like protein 2 (LanCL2) is a novel regulator of Akt. Mol Biol Cell. 2014; 25:3954-61.
45. Landlinger C, Salzer U, Prohaska R. Myristoylation of human LanC-like protein 2 (LANCL2) is essential for the interaction with the plasma membrane and the increase in cellular sensitivity to adriamycin. Biochim Biophys Acta. 2006; 1758:1759-67.
46. Chakravarty D, Pedraza AM, Cotari J, Liu AH, Punko D, Kokroo A, Huse JT, Altan-Bonnet G, Brennan CW. EGFR and PDGFRA co-expression and heterodimerization in glioblastoma tumor sphere lines. Sci Rep. 2017; 7:9043.
47. Brookes S, Gagrica S, Sanij E, Rowe J, Gregory FJ, Hara E, Peters G. Evidence for a CDK4-dependent checkpoint in a conditional model of cellular senescence. Cell Cycle. 2015; 14:1164-73.
48. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012; 3:228-35. https://doi.org/10.18632/oncotarget.443.
49. Katso RM, Pardo OE, Palamidessi A, Franz CM, Marinov M, De Laurentiis A, Downward J, Scita G, Ridley AJ, Waterfield MD, Arcaro A. Phosphoinositide 3-Kinase C2beta regulates cytoskeletal organization and cell migration via Rac-dependent mechanisms. Mol Biol Cell. 2006; 17:3729-44.
50. Kanu OO, Hughes B, Di C, Lin N, Fu J, Bigner DD, Yan H, Adamson C. Glioblastoma Multiforme Oncogenomics and Signaling Pathways. Clin Med Oncol. 2009; 3:39-52.
51. Le Mercier M, Hastir D, Moles Lopez X, De Nève N, Maris C, Trepant AL, Rorive S, Decaestecker C, Salmon I. A simplified approach for the molecular classification of glioblastomas. PLoS One. 2012; 7:e45475.
52. Tabernero MD. A method for the global detection of chromosomal gains and losses, and an array for assessing genetic abnormalities. Spanish patent nº 2012318929 and European Trade Mark Register “FullChromArray” nº 11057627 and United States TM number 85826459. 2012 Nov 27.