
INTRODUCTION
Malignant pleural mesothelioma (MPM) is an aggressive cancer originating in the pleura. MPM is associated with poor prognosis: less than 5% of patients survive more than five years [1]. Surgery combined with radio-chemotherapy has shown benefit in patients presenting with early-stage disease [2], but most MPM are in advanced stage and show multi-focal growth at the time of diagnosis. The current treatment standard for advanced MPM - chemotherapy with cisplatin and pemetrexed or raltitrexed - is effective only in 25-30% of patients, and median survival is 12 months [3]. There is no approved salvage regimen after failure of first-line chemotherapy. Therefore, new treatment approaches are urgently needed. New drugs targeting aberrantly activated signaling molecules may be candidate therapeutics in MPM.
Receptor tyrosine kinases (RTKs) play a crucial role in tumor growth, transducing extracellular signals that regulate cellular proliferation and survival, in particular activating PI3K/AKT and RAS/MEK/MAPK signaling and STAT regulated transcription (Figure 1). The stringent control of RTKs is often abrogated in tumor cells: gene mutations, amplifications, overexpression and gene fusions result in constitutive activation. Specific tyrosine kinase inhibitors have shown remarkable clinical response in patients with tumors characterized by RTK activation, such as gefitinib and erlotinib in EGFR-mutated [4] and crizotinib in ALK- or ROS1-rearranged lung cancer [5, 6]. Crizotinib (PF02341066) is a potent, orally bioavailable, well tolerated, small molecule inhibitor of ALK, ROS1 and MET. It is approved for treatment of ALK- or ROS1-rearranged lung cancer and undergoing clinical trials in patients with other advanced solid tumors.
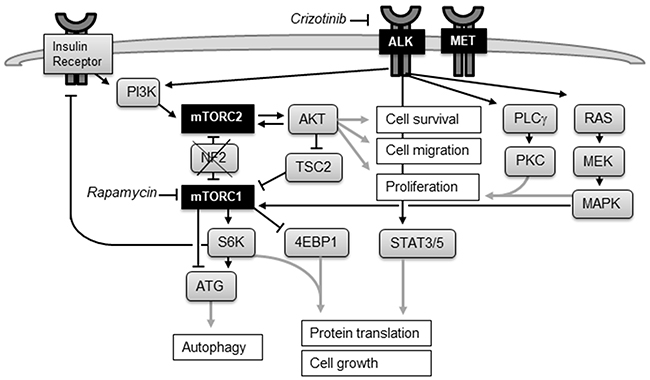
Figure 1: The mTOR and ALK signaling network. Activation of tyrosine kinase ALK through ligand binding stimulates downstream signaling through the PI3K/AKT, STAT3, and RAS/MAPK pathways. Deletion of NF2 leads in 40-50% of MPM to aberrant activation of mTOR. mTORC1 promotes protein synthesis through phosphorylation of S6K and 4EBP1 and inhibits autophagy. mTORC2 functions as an effector of PI3K signaling and controls proliferation and survival primarily by activating AKT. Positive feedback loop: partial activation of AKT promotes activation of mTORC2, which in turn phosphorylates and fully activates AKT. Negative feedback loop: mTORC1 suppresses mTORC2 activation through the inhibition of insulin/PI3K signaling. Inhibiting mTORC1 releases the negative feedback and increases AKT activation.
Various studies have reported on the expression and activation of RTKs in mesothelioma, including EGFR, MET, IGFR, FGFR1, and VEGFR [7–9]. Specific inhibition of VEGFR, FGFR1, MET and/or EGFR suppressed mesothelioma cell growth in vitro [7–12]. However, clinical studies with RTK inhibitors as single agents were disappointing. VEGFR inhibition, e.g. had minimal activity in MPM and was poorly tolerated [13, 14], and single-agent EGFR or MET inhibition was clinically ineffective in MPM patients, despite high expression of EGFR [15, 16] and MET (NCT01861301).
Deletion of the tumor suppressor NF2 seen in 40-50% of MPM leads to aberrant activation of the serine/threonine protein kinase mTOR. mTOR coordinates cell growth by regulating protein, lipid and nucleotide synthesis, cell proliferation, survival, and autophagy [17, 18]. mTOR forms the catalytic subunit of two distinct protein complexes, mTOR Complex 1 (mTORC1) and 2 (mTORC2). mTORC1 functions as a downstream effector for the oncogenic PI3K/AKT and RAS/MEK/MAPK pathway and regulates cell size and protein synthesis through its substrates S6K and 4EBP1. The mTORC2 complex regulates cell proliferation and survival through phosphorylation of AKT (Figure 1).
Inhibition of mTOR by rapamycin or its derivatives (sirolimus, temsirolimus, everolimus) suppressed mesothelioma cell growth in pre-clinical models [17, 19, 20], but was not effective in clinical trials: everolimus showed no therapeutic benefit in unselected MPM patients [21] and a small group of patients selected for NF2 deletion (NCT01024946). These disappointing results are most probably due to adverse AKT activation: Inhibiting mTORC1 releases the negative feedback on PI3K/AKT signaling and increases AKT activation (Figure 1), which may promote cell survival and prevent apoptosis [22, 23]. Moreover, mTORC1 inhibition induces autophagy, helping to maintain cancer cell survival [18].
We postulated, therefore, that co-targeting of mTOR and RTK signaling pathways may result in greater therapeutic benefit via simultaneous inhibition of mTORC1, RAS/MEK/MAPK and STAT signaling and simultaneous suppression of rapamycin-induced AKT activation. Consequently, we have elucidated the cellular basis of the combinatorial therapeutic potential of rapamycin and crizotinib in MPM. We performed a screen for aberrantly expressed crizotinib targets in a large panel of MPM tumors and have found ALK and mTOR as well as MET and mTOR co-expression in a subgroup of MPM. We also found that the combined use of rapamycin and crizotinib was more effective than rapamycin as single-agent in suppressing tumor growth in a patient-derived mesothelioma graft with co-activated ALK and mTOR pathways.
RESULTS
ALK/MET and mTOR kinases are co-expressed in a subset of primary mesotheliomas
To assess the frequency of co-activation in primary mesotheliomas, we examined the co-expression of mTOR and ALK, ROS1, and MET at both mRNA and protein levels in tumor samples by qRT-PCR and IHC, respectively.
We used recently developed qRT-PCR assays that reliably detect (1) ALK and ROS1 translocations by recognizing unbalanced expression of the ALK and ROS1 3’ parts encoding the kinase domain, while the 5’ parts remain unexpressed, and (2) upregulated, balanced ALK and ROS1 gene expression (Figure 2A) [24, 25]. ALK qRT-PCR was applied to 128 mesotheliomas and five normal pleura specimens. Unbalanced ALK transcript expression indicative of a gene rearrangement was not observed. Instead, 25 (19.5%) tumors showed upregulated balanced expression of ALK transcripts, while ALK was not expressed in normal pleura. ALK protein was detected by IHC in ten of the 25 samples with upregulated ALK transcript expression (Figure 2A, 2B). All ALK protein expressing tumors were confirmed to be negative for genomic ALK rearrangements and gene amplifications by FISH (ALK copy number: 1.30-1.68; Figure 2B). Because up-regulated ALK expression is frequently associated with mutations in the kinase domain in neuroblastoma [26], we examined ALK exons 20-29 for mutations in all cases with ALK protein expression. However, activating mutations in the ALK kinase domain were not found. As for ROS1, MET or mTOR genes, qRT-PCR analysis showed that ROS1 was not expressed in 126 MPM analyzed. Expression of MET or MTOR was significantly higher in tumor tissues than in normal pleura in 66/92 (71.7%) and 46/95 (48.4%) MPM, respectively. Immunostaining of ALK, MET and mTOR on tissue microarrays was interpretable in 101 mesotheliomas (Table 1). In this cohort, 10 cases (9.9%) were ALK positive. MET staining was detected in 83 of 101 tumors, of which 46 (45.5%) were moderately to strongly positive. Eighty-five mesotheliomas were immunoreactive for mTOR, of which 29 (28.7%) showed moderate to strong reactivity. Altogether, high-level ALK and mTOR or MET and mTOR co-expression - the basis for kinase co-activation – was observed in 5% and 15% MPM, respectively (Table 1, Figure 2B).
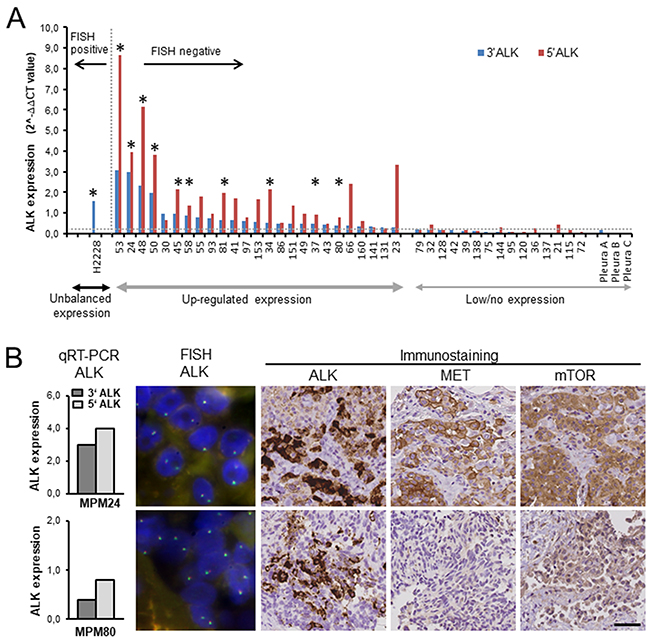
Figure 2: Expression of ALK, MET and mTOR in mesothelioma tumor tissue samples. (A) qRT-PCR analysis of ALK was performed relative to the PGK1 housekeeping gene. The horizontal dashed line indicates the cut-off value for altered ALK expression in ALK positive lung cancer (0.3); [24] blue bars, ALK 3‘ portion; red bars, ALK 5‘ portion. The asterisks point to tumors expressing ALK protein. Lung cancer cell line H2228 served as positive control showing an unbalanced expression of the ALK 3‘ portion due to EML4-ALK translocation. (B) Analysis of ALK, MET and mTOR expression in MPM24 and MPM80. Upregulated, balanced expression of ALK transcripts was independent of translocation, as confirmed by ALK break-apart FISH. Scale bar, 200μm.
Table 1: Expression of mTOR, ALK and MET in mesothelioma tumor tissue samples
n |
ALK positive1 |
ALK positive1 |
ALK negative2 |
ALK negative2 |
|
mTOR positive1 |
29 |
2 |
3 |
13 |
11 |
mTOR negative2 |
72 |
4 |
1 |
27 |
40 |
Total |
101 |
6 |
4 |
40 |
51 |
1Positive refers to moderate or strong IHC staining in at least 10% tumor cells.
2Negative refers to weak or no IHC staining in the tumor cells.
Correlation with clinicopathologial parameters revealed that age, sex, asbestos exposition, initial presentation with metastasis, and histological growth pattern (epithelioid, sarcomatoid, biphasic) were not significantly associated with ALK, MET or mTOR protein expression (Supplementary Table 1). In addition, overall survival did not differ between kinase-positive and kinase-negative MPM.
Combined therapy with rapamycin and crizotinib abrogates MPM growth in a xenograft model
To explore the anti-tumor efficacy of simultaneous inhibition of mTOR and ALK, we (1) determined the ALK and mTOR status in eight patient-derived mesothelioma grafts and (2) assessed the effect of rapamycin and crizotinib therapy on the growth of one graft (PDX680) with demonstrated co-activation of both mTOR and ALK. PDX680 overexpressed ALK transcripts independent of a gene rearrangement and without kinase-domain mutation and thus resembled ALK-expressing patient tumors (Figure 3A). ALK was activated in PDX680, evidenced by phosphorylation of ALK-Tyr1604 (Figure 3B). None of six further PDX tumors with protein lysates available was phospho-ALK positive. The other targets of crizotinib were inactive in PDX680: MET was expressed at low levels but not phosphorylated, and ROS1 was not expressed (Figure 3A, 3B). PDX680 is thus representative for MPM that express ALK without MET activation. This applies to (1) the four patient samples of our cohort that showed ALK expression, but were negative for MET (Table 1), and (2) MPM that express both kinases but do not activate MET. It is possible that the six patient tumors with co-expression of ALK and MET fall into the second group. However, appropriate (frozen) tissue to investigate phospho-MET in those tumors was not available.
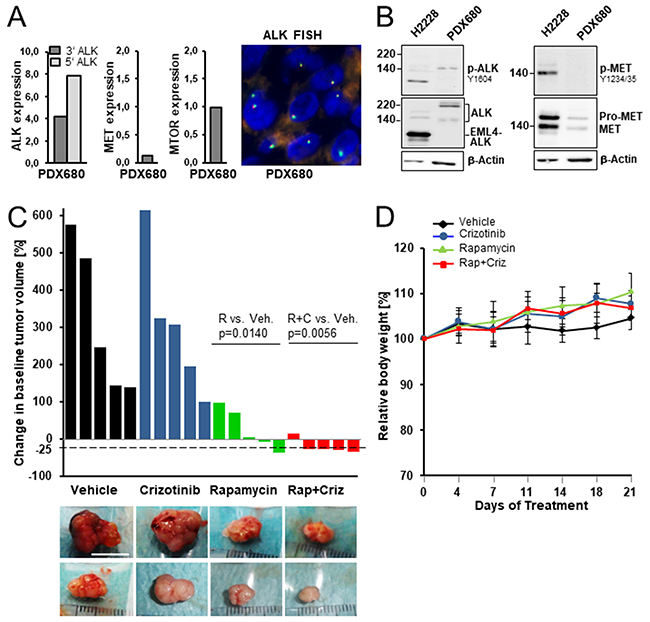
Figure 3: Preclinical efficacy of rapamycin, crizotinib and combined treatment in a mesothelioma xenograft model. (A) ALK, MET and MTOR transcript expression in xenograft PDX680. ALK break-apart FISH confirmed translocation-independent gene expression. (B) ALK was activated in PDX680, MET was expressed at low levels, but not phosphorylated demonstrated by immunoblot analysis using cell line H2228 as positive control for active EML4-ALK and MET. (C) Waterfall plots of tumoral response in mice treated with vehicle, crizotinib, rapamycin, or rapamycin and crizotinib. Each bar indicated percent change in the volume of an individual tumor on day 21 compared with day 0 of treatment. Statistically significant differences: vehicle vs. rapamycin (p=0.0140), vehicle vs. combined treatment (p=0.0056). Representative pictures of excised tumors from each treatment group; each row contains tumors that had almost identical volumes at day 0 of treatment: upper row, 160-200 mm3; lower row, 100-120 mm3. Scale bar, 10 mm. (D) Changes in the mean of body weight over the time course of the experiment.
As shown in Figure 3C, crizotinib alone did not exert anti-tumor growth activity on PDX680 grafts, but enhanced the anti-tumor effect of rapamycin. Rapamycin was effective in three of five tumors: Tumor shrinkage (35%-decrease in tumor volume) in one, growth stagnation in two cases. Rapamycin/crizotinib co-treatment was active in all five tumors: Tumor shrinkage (26-34%-tumor decrease) in four, growth stagnation in one case. Crizotinib, rapamycin and combined administrations were all well tolerated, as determined by stable body weights throughout the treatment period (Figure 3D).
Combined therapy suppresses tumor cell proliferation in a mesothelioma xenograft
We evaluated the anti-proliferative effects of crizotinib and rapamycin in xenografts by Ki-67 immunostaining of histological sections. We did not observe any significant change in the number of Ki-67 positive cells in the crizotinib or rapamycin treated groups, compared to the vehicle control group. Rapamycin/crizotinib co-treatment, however, distinctly decreased the expression of Ki-67 in mesothelioma grafts by 21.8%, compared to the control group (p=0.0065) (Figure 4A, 4B).
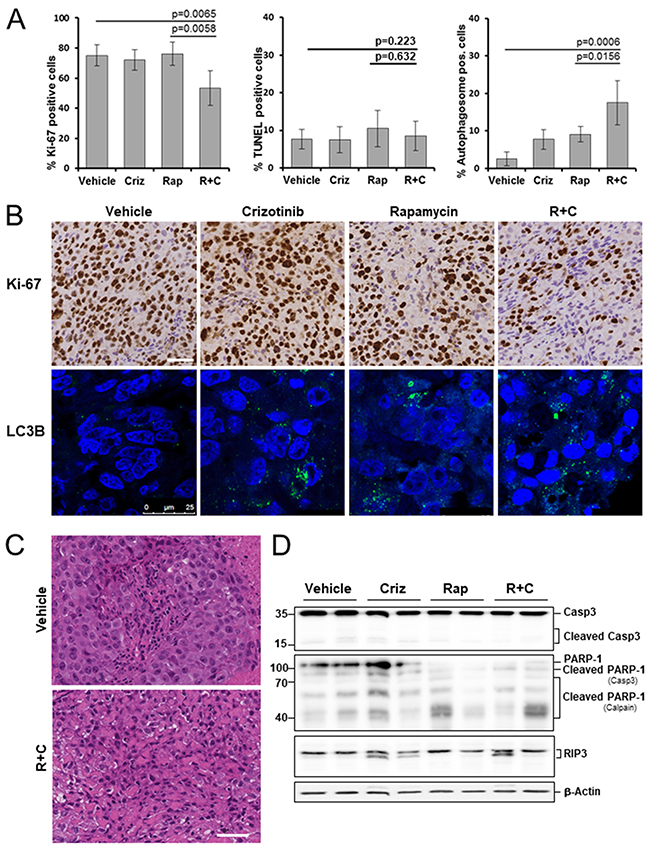
Figure 4: Effects of rapamycin, crizotinib and combined treatment on cell proliferation, apoptosis and autophagy in a mesothelioma xenograft model. Mice bearing PDX680 xenografts were treated with vehicle, crizotinib (Criz), rapamycin (Rap), or rapamycin and crizotinib (Rap+Criz) for 21 days. (A) Proliferative cells were visualized by immunohistochemical staining with anti-Ki-67, apoptotic cells by TUNEL assay, and autophagy by immunofluorescence monitoring of LC3B incorporation into autophagosomal membranes. Quantification of overall percentage of area positive for Ki-67 (left), TUNEL (middle), LC3B (right). Error bars represent s.d. between individual animals (n=5 per group). (B) Representative immunohistochemical images for Ki-67 (top) and immunofluorescence images for LC3B (bottom) of vehicle and treated samples. (C) Representative H&E images of tumors treated with vehicle or rapamycin and crizotinib. Note vacuolization, chromatin condensation and loss of cell membrane integrity as effects of combined treatment. (D) Immunoblots of PDX680 tumor cells isolated on day 21 after treatment of indicated vehicle or drug (two individual tumors per treatment group). Criz, crizotinib; Rap, rapamycin; R+C, rapamycin and crizotinib; scale bar, 200μm.
Combined therapy with crizotinib and rapamycin results in caspase-independent cell death
Next, we investigated the effect of different treatments on cell death. Graft tumor samples were examined for apoptosis by assessing DNA fragmentation (TUNEL assay), caspase 3 processing and cleavage of the caspase-3 substrate PARP-1. Surprisingly, treatment with crizotinib, rapamycin, and rapamycin/crizotinib for 21 days produced little apoptosis: 7.5%, 10.5%, and 8.5% of cells, respectively, were positive for the assay, not significantly different from the vehicle control (7.4%) (Figure 4A). Upon all treatments, only slight amounts of active caspase-3 and PARP-1 cleaved by caspase 3 were detected (Figure 4D). These observations indicate that apoptosis was not promoted by treatment. Interestingly, however, rapamycin and rapamycin/crizotinib co-treatment forced proteolytic cleavage of PARP-1 into 40-70-kD fragments, which appear specifically when PARP-1 is digested with calpain in the context of caspase-independent, necrotic cell death (Figure 4D) [27]. Indeed, histomorphological analysis revealed severe features characteristic of necrosis: Co-treatment induced loss of cell membrane integrity, chromatin condensation and vacuolization of the cytoplasm (Figure 4C). Pronounced RIP3 accumulation as a key feature of necroptosis was not observed (Figure 4D). Taken together, rapamycin/crizotinib combination therapy did not promote apoptosis, but induced caspase-independent, necrotic cell death.
Co-treatment with rapamycin and crizotinib synergistically induces autophagy in MPM cells
It has been reported that mTOR is the main negative regulator of autophagy [18, 28] and that crizotinib induces autophagy in ALK-positive tumors [29–31]. Indeed, we observed autophagy induction by rapamycin and crizotinib in the mTOR/ALK-positive mesothelioma graft model, evidenced by the accumulation of LC3B in autophagosomes: 9.1% and 7.7% autophagosome-positive cells in the rapamycin and crizotinib groups, respectively, versus 2.5% in the control group. There was a significant additive effect when both inhibitors were applied simultaneously: 17.5% (p=0.0156) (Figure 4A, 4B).
Effect of the co-treatment with rapamycin and crizotinib on downstream signaling pathways
To gain insight into the molecular mechanisms underlying the effects of rapamycin/crizotinib co-treatment, we evaluated ALK and MET activation in treated graft tumors. Phosphorylation of ALK (140-kD isoform) was not suppressed by crizotinib alone, but when applied in combination with rapamycin, MET was consistently expressed, but only in its non-phosphorylated, inactive form (Figure 5A). In order to clarify the activity of mTOR, we determined the phosphorylation status of the mTOR key substrates ribosomal S6 kinase (S6K), 4EBP1, and AKT. Rapamycin and combination treatment led to a robust dephosphorylation of S6K and its effector RPS6 (Figure 5B). Phospho-4EBP1 was insensitive to treatment in all groups. Rapamycin induced AKT activation, which was suppressed by simultaneous treatment with crizotinib, while crizotinib as a mono-drug did not affect AKT activity (Figure 5B). In addition, co-treatment significantly decreased STAT3 activity, whereas crizotinib alone had no effect (Figure 5B). Phosphorylation levels of MAPK showed no discernible reduction. Taken together, rapamycin/crizotinib co-treatment affected aberrant cell signaling in an mTOR/ALK-positive mesothelioma graft by simultaneous blocking of mTORC1, AKT and STAT3.
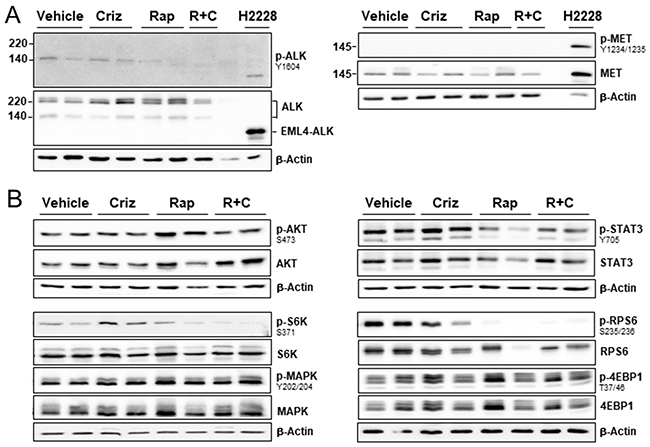
Figure 5: Effect of ALK/MET inhibitor crizotinib and mTOR inhibitor rapamycin on downstream signaling pathways in a mesotheliom xenograft model. Immunoblots of PDX680 tumor cells isolated from mice after treatment with vehicle, crizotinib (Criz), rapamycin (Rap), or rapamycin and crizotinib (R+C) for 21 days. (A) ALK and MET activity were assessed by using lung cancer cell line H2228 as positive control for the expression of EML4-ALK and active MET. Two tumors each treated with vehicle, crizotinib, rapamycin; one tumor treated with rapamycin/crizotinib. (B) Two tumors per treatment group were analyzed for AKT/p-AKT, STAT3/p-STAT3, S6K/p-S6K, and RPS6/p-RPS6 expression.
DISCUSSION
In this study, we have investigated the potential of simultaneous therapy of MPM with rapamycin and the ALK/ROS1/MET inhibitor crizotinib. Application of qRT-PCR and IHC analysis on a large cohort of MPM patient samples led to the detection of co-overexpression of mTOR and ALK or mTOR and MET in MPM subgroups of 5% and 15%, respectively. While MET expression had been previously documented [32], balanced full-length ALK expression in MPM is described here for the first time. ROS1 was not expressed in our MPM series.
In lung adenocarcinoma and anaplastic large-cell lymphoma, ALK is activated by the expression of chimeric proteins containing the ALK kinase domain, whereas ALK activation in neuroblastoma (NB) and rhabdomyosarcoma (RMS) results in the overexpression of wildtype or mutated transcripts [6]. Upregulated expression of non-rearranged transcripts was observed in 25 MPM (19.5%), of which ten expressed ALK protein. Resembling primary MPM, the patient-derived xenograft used in this study expressed non-rearranged ALK transcripts and ALK protein in its activated (Tyr1604-phosphorylated) form. Upregulated expression in our MPM cohort was independent of a copy number increase and no activating mutations were seen, while ALK activation in NB and RMS is often associated with copy number gains (NB [33], RMS [34, 35]) and kinase-domain mutations (NB [6]). Of interest, recent studies of NB tumor cells have demonstrated that (1) microRNAs (miR-96, miR-424-5p, miR-503-5p) were involved in regulating ALK expression [36, 37], and (2) intragenic non-CpG methylation was inversely associated with the amount of ALK transcripts [38]. Similarly, ALK overexpression in MPM might be mediated by microRNAs and/or demethylation of intragenic non-CpG sites. ALK-wildtype overexpression in NB [39, 40] and RMS [34, 35] primary tumors was associated with poor clinical outcome. In our MPM cohort, ALK expression did not correlate with clinicopathological parameters such as histological growth pattern, asbestos exposition, metastasis, and overall survival. In contrast to our observations, Varesano et al. [41] did not detect ALK protein expression in 63 malignant mesotheliomas by IHC using the antibody ALK1, which however is not as sensitive as the D5F3 antibody employed in our study. Recently, Salvi et al. [42] performed a FISH analysis of crizotinib target genes and found that none of 106 malignant mesotheliomas showed ALK gene rearrangement, which is in line with our finding that ALK overexpression was not caused by ALK gene rearrangement (proven by FISH).
Specific ALK inhibitors were shown to inhibit the growth of NB and RMS cells overexpressing wildtype or mutated ALK in vitro and in xenograft models [30, 33, 39, 43, 44], and crizotinib is currently being tested for the treatment of NB and RMS (NCT00939770, NCT01524926, NCT01121588). Therefore, it is tempting to speculate that upregulated ALK expression in mesothelioma might be of therapeutic relevance. In this study, crizotinib as mono-drug did not exert anti-tumor efficacy in an ALK-positive xenograft tumor, but was effective when applied simultaneously with rapamycin. In general, failure to achieve tumor growth inhibition by crizotinib could be due to mutations in the ALK kinase domain that effect inhibitor binding or increase ALK catalytic activity or ALK gene amplification both observed in therapy-resistant lung cancer [45]. However, neither ALK gene amplification nor mutation was detected in the graft tumor. Resistance could also be mediated by activation of ALK-independent pathways that hamper the effectiveness of crizotinib, including IGFR, RAS/SRC, EGFR and AKT/mTOR signaling [6, 46]. Indeed, we observed activated AKT/mTOR signaling in the graft tumor (indicated by phosphorylated AKT, S6K, RPS6). mTORC1 inhibition by rapamycin sensitized the tumor to crizotinib: Simultaneous treatment inhibited the activity of ALK and consequently its targets STAT3 and AKT, while crizotinib as single agent did not. In exactly the same way, rapamycin sensitized crizotinib-resistant EML4-ALK+ lung cancer cells to ALK inhibition: Crizotinib alone did not affect ALK and AKT activity, but blocked the feedback activation from mTOR to AKT, when administered together with rapamycin [47]. In those cells, the acquired crizotinib resistance was due to ligand-dependent EGFR activation, which was abrogated by targeting of mTOR by rapamycin (evidenced by decreased EGFR dephosphorylation). It has been reported that aberrant mTOR signaling increases the expression of various growth factors including RTK ligands [48]. Based on those observations, we hypothesize that in our ALK/mTOR positive MPM graft activated mTOR induced ligand-dependent bypass signaling leading to crizotinib resistance. Hence, it seems highly probable that mTOR inactivation sensitized the tumor to crizotinib by suppressing mTOR triggered bypass signaling. The resulting ALK inactivation was able to block the feedback activation from mTOR to AKT (see below).
The PI3K/AKT/mTOR pathway plays an important role in the genesis of various tumors, thus constituting a possible therapy target. Inhibition of mTOR by rapamycin and its derivatives, however, is complicated by adverse AKT activation triggered by release of the negative feedback from mTOR to PI3K. Indeed, increased AKT signaling has been observed in biopsies of cancer patients following everolimus treatment and may explain why rapamycin derivatives have largely cytostatic, but no cytotoxic effects on tumors [49]. Recently, it was shown that the combination of rapamycin or PI3K/mTOR inhibitors and one specific RTK inhibitor (EGFR, MET or IGF1R inhibitor) suppressed adverse AKT activation and hampered mesothelioma cell growth in vitro and in MPM xenografts [7, 10, 50].
Here, we have explored the anti-tumor effects of a simultaneous inhibition of mTOR and ALK on the growth of a patient-derived mesothelioma graft with co-activated mTOR and ALK. Our results show that rapamycin alone suppressed tumor growth, resulting in growth stagnation or tumor shrinkage in three of five tumors altogether. In line with other studies [51, 52], rapamycin decreased phosphorylation of the mTOR key substrate S6K and of RPS6, a critical effector of S6K regulated protein synthesis and proliferation. The phosphorylation of the mTORC1 effector 4EBP was insensitive to rapamycin - as described earlier for a variety of cell types [53–55]. While others report on G1 cell cycle arrest and proliferation inhibition induced by rapamycin [52], we did not observe decreased Ki-67 indices. However, we cannot exclude cell cycle arrest, which is not detectable by means of Ki-67 staining. As expected, AKT phosphorylation was enhanced by rapamycin.
Simultaneous treatment of the mTOR/ALK-positive graft with rapamycin and crizotinib was more effective than rapamycin as mono-drug: tumor shrinkage in four and growth stagnation in one of five MPM tumors was achieved. This result is the first of its kind for MPM: similar approaches used in recent studies successfully suppressed tumor growth, but could not induce significant MPM tumor shrinkage [10, 50]. Rapamycin/crizotinib co-treatment was well tolerated, which is in line with recent pre-clinical studies in MPM [50] and other cancers [56] when crizotinib and PI3K/mTOR inhibitors were applied.
Taken together, the combination of rapamycin with crizotinib had three major molecular effects: (1) rapamycin suppressed mTORC1 mediated signaling (indicated by dephosphorylation of S6K and RPS6); (2) rapamycin sensitized the tumor cells to crizotinib, leading to inactivation of ALK signaling (dephosphorylation of ALK, AKT and STAT3); (3) crizotinib blocked AKT feedback activation induced by rapamycin. Simultaneous blocking of mTORC1, STAT3 and AKT might be the underlying molecular mechanism for proliferation inhibition, induction of autophagy and caspase-independent cell death all observed in co-treated tumors, which in concert resulted in tumor shrinkage.
In our study, neither crizotinib nor rapamycin, nor their combination promoted apoptosis in MPM graft tumors. Instead, rapamycin and rapamycin/crizotinib co-treatment induced caspase-independent, necrotic cell death, indicated by (1) PARP-1 cleavage into 40-70-kD fragments, which appears specifically when PARP-1 is digested with calpain in the context of necrosis [27], and (2) histomorphological features characteristic of necrosis: loss of cell membrane integrity, chromatin condensation and vacuolization of the cytoplasm. In keeping with our data, Alvero et al. [57] observed induction of caspase-independent cell death by the mTOR inhibitor NV-128 through inhibition of mTOR and down-regulation of phosphorylated AKT and S6K.
Autophagy represents a pro-survival or pro-death response to several stresses, especially chemotherapy, radiotherapy and starvation, in tumor cells. Under moderate stress conditions, autophagy supplies cells with metabolic substrates, contributing to cell survival, tumor progression and therapeutic resistance. Excessive autophagy, however, is a cell death mechanism that can occur either in the absence of apoptosis or concomitantly with apoptosis [58]. Autophagy has a close relationship with the PI3K/AKT/mTOR pathway [18, 28]. Consistent with the observations made by others that specific PI3K/mTOR inhibitors induce autophagy in MPM cells [59], we also observed autophagy induction when mTOR/ALK-positive MPM tumors were treated with rapamycin. Simultaneous application of crizotinib could effectively block the activity of AKT and significantly enhanced autophagy. It is likely that extensive autophagy contributed to the necrotic cell death observed in tumors co-treated with rapamycin/crizotinib.
In conclusion, we have identified a subgroup of MPM characterized by overexpression of the tyrosine kinase ALK. Because ALK has been successfully targeted with specific inhibitors in other malignancies it is tempting to speculate that activated ALK in MPM might represent a therapeutic target. Indeed, our preclinical data presented here show that combination of the ALK inhibitor crizotinib and the mTOR inhibitor rapamycin results in rapid and robust abrogation of MPM tumor growth in vivo. Together, these results suggest that co-treatment with crizotinib and rapamycin should be further explored as a targeted therapeutic alternative in mesothelioma.
MATERIALS AND METHODS
Specimen collection
Formalin-fixed paraffin-embedded (FFPE) resection specimens from 144 patients diagnosed with MPM and five normal pleura specimens were included in this study. Triplicate 0.6 mm tumor cores had been inserted into tissue microarrays (TMA) used for all IHC analyses. Lung cancer cell line H2228 (CRL-5935, ATCC, Rockville, MD) served as positive control for EML4-ALK and MET expression; rhabdomyosarcoma cell line RH30 (DSMZ, Braunschweig, Germany) for the expression of non-rearranged ALK transcripts.
Antibodies and reagents
Antibodies to detect MET (D1C2), mTOR (7C10), LC3B, PARP, Caspase 3, Cyclin D1, phospho-ALK (Tyr1604), phospho-MET (Tyr1234/1235), AKT, phospho-AKT (Ser473), phospho-MAPK (Tyr202/204), 4EBP1, phospho-4EBP1 (Thr37/46), S6K, phospho-S6K (Ser371), RPS6, phospho-RPS6 (Ser235/236), STAT3, phospho-STAT3 (Tyr705) were from Cell Signaling (Danvers, MA, USA). Anti-RIP3 was from Santa Cruz Biotechnology (Dallas, TX, USA), Anti-Ki-67 from Cell Marque (Rocklin, CA, USA), and Anti-β-Actin from Sigma-Aldrich (München, Germany). ALK-1A4 (immunoblotting) was from Origene (Rockville, MD, USA), ALK-D5F3 (immunohistochemistry) was from Ventana Medical Systems (Tucson, AZ, USA). Crizotinib and rapamycin were purchased from Pfizer (Berlin, Germany) and ChemieTek (Indianapolis, IN, USA), respectively.
Quantitative reverse transcription PCR
Unbalanced ALK and ROS1 transcript expression indicative of ALK/ROS1 gene rearrangement and upregulated gene expression were measured by qRT-PCR amplification of the 5′ and 3′ portions of both genes as described [24, 25]. MET and MTOR expression was examined using one primer set each (Supplementary Table 2), and qRT-PCR was carried out as described [24]. Tumor expression data were normalized to PGK1 and compared to the mean value of all pleura samples; the cut-off values for significantly elevated MET and mTOR expression were 0.55 and 0.14, respectively. ALK expression values normalized to PGK1 were calculated relative to the average value in cell line RH30, which was arbitrarily defined as 1; the cut-off value for relevant ALK expression was 0.3.
PCR and DNA sequence analysis
The ALK kinase domain (exons 20-29) was searched for mutations by direct sequencing of PCR products amplified from genomic DNA as described using standard PCR conditions and cycle sequencing [24].
Fluorescence in situ hybridization
FISH was performed as described applying a break-apart probe specific for the ALK locus (ZytoLight SPEC ALK Dual Color Break Apart Probe; ZytoVision, Bremerhaven, Germany) [24].
Immunohistochemistry
IHC was accomplished on 3-μM tissue TMA or full tissue sections. For ALK expression analysis, the ALK D5F3 assay from Ventana was applied. MET and mTOR expressions were assessed with conventional DAB staining on a semi-automated autostainer. After heat-induced epitope retrieval at pH6, anti-mTOR (1:100) or anti-MET (1:100) were incubated for 30 minutes. ALK staining was classified according to the manufacturer’s instructions. MET and mTOR expressions were assessed by estimating the intensity of cytoplasmic and membranous staining: 0/negative, no staining; 1+, faint; 2+, moderate; 3+, strong staining intensity in at least 10% tumor cells.
Antitumor efficacy study
Eight MPM patient-derived xenograft models were established by Charles River Discovery Research Services (Freiburg, Germany). Tumor cells were implanted subcutaneously in NMRI nu/nu mice and allowed to grow to reach a tumor size of 50-200 mm3. Tumor-bearing mice were randomized into different treatment groups (n=5/group): Vehicle control (orally), crizotinib (100 mg/kg/d, orally), rapamycin (7.5 mg/kg/2d, i.p.), or combination treatment with crizotinib (100 mg/kg/d, orally) and rapamycin (7.5 mg/kg/2d, i.p.). The length (L) and width (W) of each tumor was measured with calipers, and the body weight was determined every 3 to 4 days. The tumor volumes were TV=L*W2*0.5; the relative tumor volumes of individual tumors were RTV=TV21/TV0*100. After 21 days of treatment, mice were sacrificed, solid tumors were excised and parts either processed for paraffin embedding or fresh-frozen for protein analysis.
Quantification of apoptosis
The TUNEL assay was accomplished on 3-μm FFPE sections employing the ApopTag-Plus Kit (Merck-Millipore, Darmstadt, Germany). After pretreatment (20 mg/ml proteinase) and quenching (3% H2O2) sections were treated with TdT enzyme and anti-digoxigenin-peroxidase, followed by DAB detection. Negative controls were incubated in medium lacking TdT enzyme. Immunoreactivity in nuclei and apoptotic bodies was considered positive. The apoptosis index corresponded to TUNEL-labelled cells among at least 900 cells per tumor and was expressed as the percentage of positive cells.
Analysis of necroptosis
Immunoblot analysis with anti-RIP3 (1:500) and anti-PARP (1:1000) was used to investigate cleavage products of RIP3 and PARP that are indicative of necroptotic cell death. Immunoblotting was performed as described below.
Quantification of proliferation
3-μm FFPE tissue sections were stained with Ki-67 antibody using conventional DAB staining on a semi-automated autostainer. After heat-induced epitope retrieval at pH9, anti-Ki-67 (1:100) was incubated for 30 minutes. Negative controls were obtained by omitting the primary antibody. Only nuclear Ki-67 immunoreactivity was considered positive. The proliferation index corresponded to the number of labelled Ki-67 cells among at least 670 cells per tumor and is expressed as percentage.
Quantification of autophagy
LC3B incorporation into autophagosomal membranes was monitored by immunofluorescence staining of 3-μM FFPE tissue sections. After heat-induced epitope retrieval at pH6, cells were incubated with anti-LC3B (1:200) overnight at 4°C and Alexa Fluor 488-conjugated anti-rabbit IgG (1:250; Thermo Fisher Scientific) for 1 h at room temperature. DAPI was used for nuclear staining. Images were taken on a confocal microscope Leica TCS SP8. The number of LC3B positive dots was quantified using ImageJ software (NIH, Bethesda, MD, USA), cells with more than five dots were considered positive. The autophagy index corresponded to the number of autophagosome positive cells among at least 580 cells per tumor and was expressed as percentage.
Gel electrophoresis and immunoblotting
Total protein extracts were prepared by lysing frozen PDX tumor cells in FastPrep lysing matrix tubes (MP Biomedicals, Illkirch, France) with lysis buffer containing cOmplete protease inhibitor cocktail, PhosSTOP phosphatase inhibitor (Roche, Mannheim, Germany). 30 μg protein were separated by SDS-PAGE and transferred onto nitrocellulose membranes. For immunoblotting, membranes were probed with primary antibodies overnight at 4°C, then incubated with anti-rabbit IgG-HRP (Cell Signaling). Signals were detected by enhanced chemiluminescence (SuperSignal WestDuraExtendedDurationSubstrate, Thermo Fisher Scientific) using CCD camera STELLA3200 (Raytest, Straubenhardt, Germany). Antibodies were used at a dilution of 1:1000, except anti-β-actin (loading control; 1:10000).
Statistical analysis
Differences between groups were assessed by two-tailed student’s t-test (continuous data) or Fisher’s exact test (categorial data). A P value less than 0.05 was considered significant. For all calculations, Analyse-it software (Leeds, United Kingdom) for Microsoft Excel was used.
Ethics statement
The investigations were conducted according to the ethical standards of the Declaration of Helsinki and according to national and international guidelines. This study was approved by the Ethics Committee of the University Hospital, Tübingen, Germany (344/2012BO2). Animal work was approved by the Regional Administrative Authority of Freiburg, Germany (G-13/13).
Author contributions
Conception or design of the work, DM, GO, CK; Tumor specimen characterization and data collection, DM, SB-E, JK, JS, CS, RF-G, SK, GF; Data analysis and interpretation, DM, GO, CK; Drafting the article, DM, GO, CK; Critical revision of the article, All.
ACKNOWLEDGMENTS
We thank Katja Bräutigam, IKP Stuttgart, and Daniela Pumm, RBK Stuttgart, for expert technical assistance.
CONFLICTS OF INTEREST
All authors have no conflicts of interest to declare.
GRANT SUPPORT
The study was supported by the Robert Bosch Stiftung, Stuttgart, Germany (Project 704).
REFERENCES
1. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet. 2005; 366:397–408.
2. Cao C, Tian D, Manganas C, Matthews P, Yan TD. Systematic review of trimodality therapy for patients with malignant pleural mesothelioma. Ann Cardiothorac Surg. 2012; 1:428–437.
3. Scherpereel A, Astoul P, Baas P, Berghmans T, Clayson H, de Vuyst P, Dienemann H, Galateau-Salle F, Hennequin C, Hillerdal G, Le Péchoux C, Mutti L, Pairon JC, et al. Guidelines of the european respiratory society and the european society of thoracic surgeons for the management of malignant pleural mesothelioma. Eur Respir J. 2010; 35:479–495.
4. Roengvoraphoj M, Tsongalis GJ, Dragnev KH, Rigas JR. Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non-small cell lung cancer: focus on epidermal growth factor receptor mutation testing and mutation-positive patients. Cancer Treat Rev. 2013; 39:839–850.
5. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, et al. Crizotinib in ROS1-rearranged non–small-cell lung cancer. N Engl J Med. 2014; 371:1963–1971.
6. Holla VR, Elamin YY, Bailey AM, Johnson AM, Litzenburger BC, Khotskaya YB, Sanchez NS, Zeng J, Shufean MA, Shaw KR, Mendelsohn J, Mills GB, Meric-Bernstam F, et al. ALK: a tyrosine kinase target for cancer therapy. Cold Spring Harb Mol Case Stud. 2017; 3:a001115.
7. Brevet M, Shimizu S, Bott MJ, Shukla N, Zhou Q, Olshen AB, Rusch V, Ladanyi M. Coactivation of receptor tyrosine kinases in malignant mesothelioma as a rationale for combination targeted therapy. J Thorac Oncol. 2011; 6:864–874.
8. Menges CW, Chen Y, Mossman BT, Chernoff J, Yeung AT, Testa JR. A phosphotyrosine proteomic screen identifies multiple tyrosine kinase signaling pathways aberrantly activated in malignant mesothelioma. Genes Cancer. 2010; 1:493–505. https://doi.org/10.1177/1947601910375273.
9. Marek LA, Hinz TK, von Mässenhausen A, Olszewski KA, Kleczko EK, Boehm D, Weiser-Evans MC, Nemenoff RA, Hoffmann H, Warth A, Gozgit JM, Perner S, Heasley LE. Nonamplified FGFR1 is a growth driver in malignant pleural mesothelioma. Mol Cancer Res. 2014; 12:1460–1469.
10. Kanteti R, Dhanasingh I, Kawada I, Lennon FE, Arif Q, Bueno R, Hasina R, Husain AN, Vigneswaran W, Seiwert T, Kindler HL, Salgia R. MET and PI3K/mTOR as a potential combinatorial therapeutic target in malignant pleural mesothelioma. PLoS One. 2014; 9:e0105919.
11. Kawaguchi K, Murakami H, Taniguchi T, Fujii M, Kawata S, Fukui T, Kondo Y, Osada H, Usami N, Yokoi K, Ueda Y, Yatabe Y, Ito M, et al. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis. 2009; 30:1097–1105.
12. Masood R, Kundra A, Zhu S, Xia G, Scalia P, Smith DL, Gill PS. Malignant mesothelioma growth inhibition by agents that target the VEGF and VEGF-C autocrine loops. Int J Cancer. 2003; 104:603–610.
13. Garland LL, Chansky K, Wozniak AJ, Tsao AS, Gadgeel SM, Verschraegen CF, DaSilva MA, Redman M, Gandara DR. Phase II study of AZD2171 in patients with malignant pleural mesothelioma: SWOG S0509. J Thorac Oncol. 2011; 6:1938–1945.
14. Laurie SA, Hao D, Leighl NB, Goffin J, Khomani A, Gupta A, Addison CL, Bane A, Seely J, Filion ML, Pond GR, Levine MN. A phase II trial of dovitinib in previously-treated advanced pleural mesothelioma: The ontario clinical oncology group. Lung Cancer. 2017; 104:65–69.
15. Govindan R, Kratzke RA, Herndon JE 2nd, Niehans GA, Vollmer R, Watson D, Green MR, Kindler HL; Cancer and Leukemia Group B (CALGB 30101). gefitinib in patients with malignant mesothelioma: a phase II study by the cancer and leukemia group B. Clin Cancer Res. 2005; 11:2300–2304.
16. Garland LL, Rankin C, Gandara DR, Rivkin SE, Scott KM, Nagle RB, Klein-Szanto AJP, Testa JR, Altomare DA, Borden EC. Phase II study of erlotinib in patients with malignant pleural mesothelioma: a southwest oncology group study. J Clin Oncol. 2007; 25:2406–2413.
17. López-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol. 2009; 29:4235–4249.
18. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017; 168:960–976.
19. Hartman ML, Esposito JM, Yeap BY, Sugarbaker DJ. Combined treatment with cisplatin and sirolimus to enhance cell death in human mesothelioma. J Thorac Cardiovasc Surg. 2010; 139:1233–1240.
20. Hoda MA, Mohamed A, Ghanim B, Filipits M, Hegedus B, Tamura M, Berta J, Kubista B, Dome B, Grusch M, Setinek U, Micksche M, Klepetko W, et al. Temsirolimus inhibits malignant pleural mesothelioma growth in vitro and in vivo: synergism with chemotherapy. J Thorac Oncol. 2011; 6:852–863.
21. Ou SH, Moon J, Garland LL, Mack PC, Testa JR, Tsao AS, Wozniak AJ, Gandara DR. SWOG S0722: phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J Thorac Oncol. 2015; 10:387–391.
22. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006; 66:1500–1508.
23. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008; 118:3065–3074.
24. Gruber K, Horn H, Kalla J, Fritz P, Rosenwald A, Kohlhäufl M, Friedel G, Schwab M, Ott G, Kalla C. Detection of rearrangements and transcriptional up-regulation of ALK in FFPE lung cancer specimens using a novel, sensitive, quantitative reverse transcription polymerase chain reaction assay. J Thorac Oncol. 2014; 9:307–315.
25. Kalla C, Gruber K, Rosenwald A, Kimmich M, Kohlhäufl M, Friedel G, Ott G. ROS1 gene rearrangement and expression of splice isoforms in lung cancer, diagnosed by a novel quantitative RT-PCR assay. J Mod Hum Pathol. 2016; 1:25–34.
26. Azarova AM, Gautam G, George RE. Emerging importance of ALK in neuroblastoma. Semin Cancer Biol. 2011; 21:267–275.
27. McGinnis KM, Gnegy ME, Park YH, Mukerjee N, Wang KK. Procaspase-3 and poly(ADP)ribose polymerase (PARP) are calpain substrates. Biochem Biophys Res Commun. 1999; 263:94–99.
28. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005; 12:1509–1518.
29. You L, Shou J, Deng D, Jiang L, Jing Z, Yao J, Li H, Xie J, Wang Z, Pan Q, Pan H, Huang W, Han W. Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. Oncotarget. 2015; 6:40268–40282. https://doi.org/10.18632/oncotarget.5592.
30. Megiorni F, McDowell HP, Camero S, Mannarino O, Ceccarelli S, Paiano M, Losty PD, Pizer B, Shukla R, Pizzuti A, Clerico A, Dominici C. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J Exp Clin Cancer Res. 2015; 34:112.
31. Mitou G, Frentzel J, Desquesnes A, Le Gonidec S, AlSaati T, Beau I, Lamant L, Meggetto F, Espinos E, Codogno P, Brousset P, Giuriato S. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget. 2015; 6:30149–30164. https://doi.org/10.18632/oncotarget.4999.
32. Jagadeeswaran R, Ma PC, Seiwert TY, Jagadeeswaran S, Zumba O, Nallasura V, Ahmed S, Filiberti R, Paganuzzi M, Puntoni R, Kratzke RA, Gordon GJ, Sugarbaker DJ, et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006; 66:352–361.
33. Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008; 455:967–970.
34. Lee JS, Lim SM, Rha SY, Roh JK, Cho YJ, Shin KH, Yang WI, Kim SH, Kim HS. Prognostic implications of anaplastic lymphoma kinase gene aberrations in rhabdomyosarcoma; an immunohistochemical and fluorescence in situ hybridisation study. J Clin Pathol. 2014; 67:33–39.
35. van Gaal JC, Flucke UE, Roeffen MH, de Bont ES, Sleijfer S, Mavinkurve-Groothuis AM, Suurmeijer AJ, van der Graaf WT, Versleijen-Jonkers YM. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol. 2012; 30:308–315.
36. Vishwamitra D, Li Y, Wilson D, Manshouri R, Curry CV, Shi B, Tang XM, Sheehan AM, Wistuba II, Shi P, Amin HM. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am J Pathol. 2012; 180:1772–1780.
37. De Mariano M, Stigliani S, Moretti S, Parodi F, Croce M, Bernardi C, Pagano A, Tonini GP, Ferrini S, Longo L. A genome-wide microRNA profiling indicates miR-424-5p and miR-503-5p as regulators of ALK expression in neuroblastoma. Oncotarget. 2017; 8:56518–56532. https://doi.org/10.18632/oncotarget.17033.
38. Gómez S, Castellano G, Mayol G, Suñol M, Queiros A, Bibikova M, Nazor KL, Loring JF, Lemos I, Rodríguez E, de Torres C, Mora J, Martín-Subero JI, et al. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics. 2015; 7:1137–1153.
39. Passoni L, Longo L, Collini P, Coluccia AML, Bozzi F, Podda M, Gregorio A, Gambini C, Garaventa A, Pistoia V, Del Grosso F, Tonini GP, Cheng M, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009; 69:7338–7346.
40. Schulte JH, Bachmann HS, Brockmeyer B, Depreter K, Oberthür A, Ackermann S, Kahlert Y, Pajtler K, Theissen J, Westermann F, Vandesompele J, Speleman F, Berthold F, et al. High ALK receptor tyrosine kinase expression supersedes ALK mutation as a determining factor of an unfavorable phenotype in primary neuroblastoma. Clin Cancer Res. 2011; 17:5082–5092.
41. Varesano S, Leo C, Boccardo S, Salvi S, Truini M, Ferro P, Fedeli F, Canessa PA, Dessanti P, Pistillo MP, Roncella S. Status of anaplastic lymphoma kinase (ALK) in malignant mesothelioma. Anticancer Res. 2014; 34:2589–2592.
42. Salvi S, Varesano S, Boccardo S, Ravetti JL, Canessa PA, Pistillo MP, Ferro P, Fedeli F, Roncella S. FISH analysis of crizotinib target genes ROS1/ALK/MET in malignant mesothelioma. J Thorac Oncol. 2017; 12:e116–118.
43. George RE, Sanda T, Hanna M, Fröhling S, Luther W 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008; 455:975–978.
44. van Gaal JC, Roeffen MH, Flucke UE, van der Laak JA, van der Heijden G, de Bont ES, Suurmeijer AJ, Versleijen-Jonkers YM, van der Graaf WT. Simultaneous targeting of insulin-like growth factor-1 receptor and anaplastic lymphoma kinase in embryonal and alveolar rhabdomyosarcoma: a rational choice. Eur J Cancer. 2013; 49:3462–7340.
45. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012; 18:1472–1482.
46. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012; 4:120ra17.
47. Oh SJ, Noh KH, Lee YH, Hong SO, Song KH, Lee HJ, Kim S, Kim TM, Jeon JH, Seo JH, Kim DW, Kim TW. Targeting stemness is an effective strategy to control EML4-ALK+ non-small cell lung cancer cells. Oncotarget. 2015; 6:40255–40267. https://doi.org/10.18632/oncotarget.5434.
48. Parker WE, Orlova KA, Heuer GG, Baybis M, Aronica E, Frost M, Wong M, Crino PB. Enhanced epidermal growth factor, hepatocyte growth factor, and vascular endothelial growth factor expression in tuberous sclerosis complex. Am J Pathol. 2011; 178:296–305.
49. Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, Ramon y Cajal S, Jones S, Vidal L, Shand N, Macarulla T, Ramos FJ, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008; 26:1603–1610.
50. Kanteti R, Riehm JJ, Dhanasingh I, Lennon FE, Mirzapoiazova T, Mambetsariev B, Kindler HL, Salgia R. PI3 kinase pathway and MET inhibition is efficacious in malignant pleural mesothelioma. Sci Rep. 2016; 6:32992.
51. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007; 26:1932–1940.
52. Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006; 66:7864–7869.
53. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008; 105:17414–17419.
54. Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009; 7:e38.
55. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009; 284:8023–8032.
56. Moore NF, Azarova AM, Bhatnagar N, Ross KN, Drake LE, Frumm S, Liu QS, Christie AL, Sanda T, Chesler L, Kung AL, Gray NS, Stegmaier K, et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget. 2014; 5:8737–8749. https://doi.org/10.18632/oncotarget.2372.
57. Alvero AB, Montagna MK, Chen R, Kim KH, Kyungjin K, Visintin I, Fu HH, Brown D, Mor G. NV-128, a novel isoflavone derivative, induces caspase-independent cell death through the Akt/mammalian target of rapamycin pathway. Cancer. 2009; 115:3204–3216.
58. Anding AL, Baehrecke EH. Autophagy in cell life and cell death. Curr Top Dev Biol. 2015; 114:67–91.
59. Echeverry N, Ziltener G, Barbone D, Weder W, Stahel RA, Broaddus VC, Felley-Bosco E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015; 6:e1757.