
INTRODUCTION
mTOR is a highly conserved and widely expressed serine/threonine kinase, that is a member of the phosphatidylinositol-3 kinase–like kinase (PIKK) family, which also includes other protein kinases that regulate DNA damage responses, such as ATM (ataxia telangiectasia-mutated kinase) and ATR (ATM [ataxia telangiectasia-mutated]- and Rad3-related kinase) [1, 2].
mTOR plays a pivotal role in the PI3K/Akt/mTOR signaling pathway, which senses growth factor and serves as a central regulator of fundamental cellular processes such as cell growth/apoptosis, autophagy, translation, and metabolism [3, 4]. Activation of PI3K recruits cellular protein kinases that in turn activate downstream kinases, including the serine/threonine kinase Akt. Phosphorylation of Akt activates the mTOR complex 1 (mTORC1) and induces subsequent phosphorylation of S6K, and of the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). The activation of mTORC1 results in increased translation and protein synthesis [5]. A second complex of mTOR, known as mTORC2, has been more recently described and appears to act as a feedback loop via Akt [6].
Gene deletions/mutations and functional impairment of many proteins involved in this signaling pathway lead to a deregulation that results in different human cancers, including hematological malignancies. Furthermore hyperactivation of this pathway through loss of negative regulators, such as PTEN, or mutational activation of receptor tyrosine kinases upstream of phosphoinositide 3-kinase (PI3K) is a frequent occurrence in leukemia patients, where it negatively influences response to therapeutic treatments [7]. Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy and B-precursor acute lymphoblastic leukemia (B-pre ALL) is the most frequent pediatric ALL subtype, characterized by an aggressive neoplastic disorder of early lymphoid precursor cells [8, 9]. The treatment protocol for B-pre ALL includes an intense chemotherapy regimen with cure rates of 15–80% [10, 11].
In B-pre ALL many research efforts are currently devoted to the development of targeted therapies to limit side effects of chemotherapy and to increase treatment efficacy for poor prognosis patients, i.e. poor outcome following relapse [12, 13]. PI3K/Akt/mTOR pathway activation is a frequent feature in B-pre ALL [12] and therefore this pathway is an attractive target to efficiently treat this disease. A new class of ATP-competitive mTOR inhibitors, such as Torin-2, have been shown to potently target mTORC1 and mTORC2 [14]. Torin-2 is also a potent inhibitor of ATR, ATM, and DNA-PK. This compound exhibits an anti-tumour activity more broad-based and profound compared to the rapalogs that do not fully inhibit mTORC1 and are unable to inhibit mTORC2 [15].
We therefore hypothesized that dual inhibition of mTORC1 and mTORC2 by Torin-2 would provide a superior outcome in B-pre ALL as compared to inhibition of mTORC1 obtained with RAD001 [16]. We tested the cytotoxic activity of Torin-2 and its capability to prevent Akt reactivation after mTORC1 and mTORC2 inhibition. Furthermore we explored if dual targeting of mTORC1 and Akt, with RAD001 and MK-2206 respectively, might achieve results similar to those obtained with Torin-2 alone.
Torin-2 displayed a powerful cytotoxic activity with an IC50 in the nanomolar range, induced G0/G1 phase cell cycle arrest, modulated the PI3K/Akt/mTOR pathway and caused apoptosis and autophagy in a dose-dependent manner. Interestingly, feedback activation of PI3K/Akt was suppressed by Torin-2 alone, whereas RAD001 required the addition of MK-2206 to achieve the same efficacy. These findings indicates that mTORC1 and mTORC2 inhibition could be an attractive strategy to develop innovative therapeutic protocols for the treatment of B-pre ALL leukemia patients and to prevent Akt reactivation after mTORC1 targeting.
RESULTS
PI3K/Akt/mTOR pathway activation status in B-pre ALL cell lines
We first analyzed by Western blot the baseline expression of key components of the PI3K/Akt/mTOR pathway and their phosphorylation status in a panel of human B-pre ALL cell lines (NALM-6, SEM, REH, RS4;11, BV-173, SUP-B15, TOM-1). Three of these cell lines, BV-173, SUP-B15 and TOM-1 are Ph+, since they harbour the Bcr-Abl fusion protein. Despite some heterogeneity, all the B-pre ALL cell lines displayed phosphorylation at the Ser 2448 and Ser 2481 (readout for mTORC1 and mTORC2, respectively) residues of mTOR and at the Ser 473 residue of Akt, which are indicative of the constitutive activation of this signaling pathway (Fig. 1).
We further explored the basal condition of downstream targets of both kinases. In agreement with the hyperactivated status of mTORC1, the ribosomal protein S6 and the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) were phosphorylated at Ser 235/236 and Thr 37/46 respectively (Fig. 1). A readout for mTORC2 activity is represented by the phosphorylation at the Ser 473 site of Akt. Hyperactivation of Akt resulted in the phosphorylation at the Thr 32 site of Forkhead box O3A (FoxO3A) (Fig. 1).
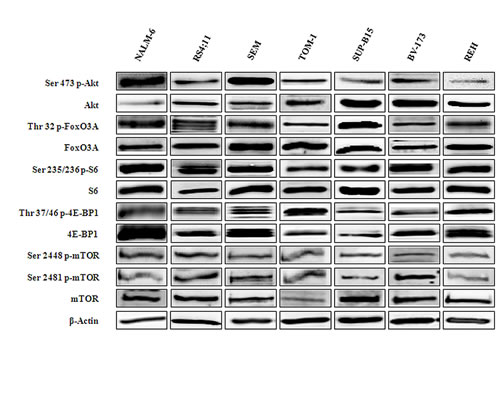
Figure 1: Expression and phosphorylation status of mTOR and Akt and their downstream targets in B-pre ALL cell lines. Western blot analysis of B-pre ALL cell lines to detect the expression and phosphorylation levels of Akt, mTOR and its downstream substrates. Twenty-five µg of protein were blotted to each lane. Antibody to β-actin served as a loading control.
Torin-2 induces cytotoxicity, blocks cell cycle progression at the G0/G1 phase and induces autophagy
To determine whether Torin-2 could affect viability of B-pre ALL cell lines, cells were incubated in the presence of increasing concentrations of Torin-2 for 48h and then analyzed by MTT assays. All cell lines resulted sensitive to the drug, in a concentration-dependent fashion, with the IC50 value that ranged between 0.07 µM and 0.19 µM (Fig. 2A). Given the importance of the PI3K/Akt/mTOR signaling pathway in the regulation of cell proliferation, the effects of Torin-2 on cell cycle progression were also investigated. SEM and BV-173 cell lines were treated with increasing concentrations of Torin-2 for 24h, then cells were harvested, fixed and stained with Propidium Iodide (PI) and analyzed with the MuseTM Cell Analyzer. The assay showed in both cell lines a concentration-dependent increase of G0/G1 phase of cell cycle and a simultaneous decrease in the S phase (Fig. 2B).
Autophagy can be a form of programmed cell death, but is also involved in protective mechanisms against apoptosis [17, 18]. To evaluate whether the treatment with Torin-2 could lead to autophagy, we detected the expression of LC3A/B I (non lipidated) and LC3A/B II (lipidated) by Western blot in BV-173, SEM and NALM-6 cells treated with increasing concentrations of Torin-2. The expression levels of LC3A/B II gradually increased in the three cell lines in a dose-dependent manner (Fig. 2C).
To verify whether autophagy was either a cell survival or a cell death mechanism, we used the autophagy inhibitor 3-MA (3-Methyladenine), which blocks an early stage of autophagy by inhibiting the class III phosphoinositide 3-kinase (PI3K) [19]. 3-MA alone did not affect cell growth, even at the concentration of 10 µM (cell viability was comparable to untreated cells), but cells treated with 3-MA become significantly more resistant to Torin-2 cytotoxic effect (Fig. 2D, upper panel). We also employed Bafilomycin A1, another well established autophagy inhibitor that inhibits vacuolar ATPase (V-ATPase) and promotes the accumulation of autophagic vacuoles [20]. As shown in the lower panel of Fig. 2D, the treatment with 4 µM Bafilomycin A1 confirmed that inhibition of autophagy significantly reduced the cytotoxic effect of Torin-2. These results indicated that autophagy is a critical determinant of the cytotoxic effects induced in B-pre ALL cells by Torin-2.
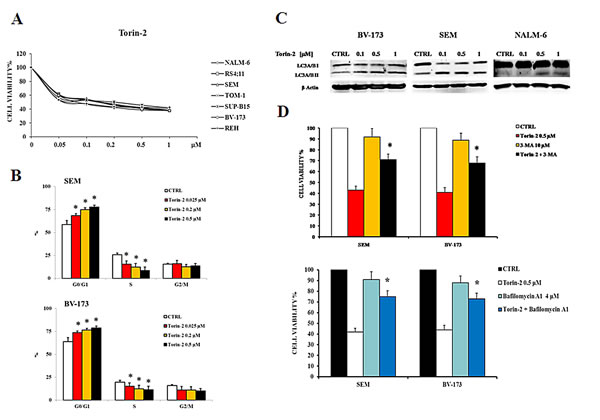
Figure 2: Torin-2 is cytotoxic, cytostatic and induces autophagy in B-pre ALL cell lines. A. MTT assay of B-pre ALL cell lines treated with increasing concentrations of Torin-2 for 48h. One representative experiment is shown. B. Flow cytometric analysis for SEM and BV-173 cells treated with increasing concentrations of Torin-2 for 24h. Asterisks indicate statistically significant differences with respect to untreated cells (*p<0.05). Cell lines displayed are representative of the cell panel used in this study. C. Effect of Torin-2 on autophagy in BV-173, SEM and NALM-6 cells, documented by the lipidation of the autophagy marker LC3A/B. Antibody to β-actin served as a loading control. D. The activity of 3-MA in combination with Torin-2 is reported in the upper histogram, after SEM and BV-173 treatment for 24h. Below, the effect on cell viability of SEM and BV-173 cells after treatment with Torin-2 and the autophagy inhibitor Bafilomycin A1 is reported. Results are the mean of three different experiments ± SD. Asterisks indicate statistically significant differences with respect to untreated cells (*p<0.05).
Torin-2 causes pro-apoptotic effects on B-pre ALL cell lines
To investigate whether the decreased viability was related to apoptosis, cells were treated for 24h with increasing concentrations of Torin-2 and analyzed by both Western blot and DNA staining. Cleavage of poly(ADP-ribose)polymerase (PARP) in BV-173, SEM and NALM-6 revealed the pro-apoptotic effect of Torin-2. REH and SUP-B15 cells were treated with Torin-2 at 0.5 µM for 24h (Fig. 3A). DAPI staining revealed the morphological changes associated with apoptosis, such as chromatin condensation and nuclear fragmentation (Fig.3B). Apoptosis was further investigated by flow cytometric analysis of Annexin V-stained samples, that showed in NALM-6 and TOM-1 a significant and concentration-dependent increase of apoptotic cells (Fig. 3C, left panel). To determine whether activated caspases are involved in the apoptotic action of Torin-2, we examined the effects of the broad spectrum caspase inhibitor Z-VAD-fmk on cell apoptosis as determined by annexin-V FITC binding. Torin-2-mediated apoptosis was blocked markedly by 25 μM Z-VAD-fmk (Figure 3C, right panel). Therefore these results showed that Torin-2 induced caspase-dependent cell death.
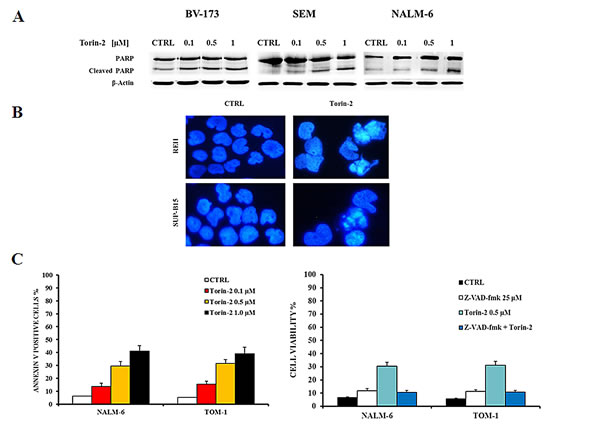
Figure 3: Torin-2 induces apoptosis. A. Western blot analysis documenting a Torin-2 concentration-dependent PARP cleavage in BV-173, SEM and NALM-6 cells. Antibody to β-actin served as a loading control. B. DNA staining of REH and SUP-B15 with the fluorescent dye DAPI is reported. In REH and SUP-B15 treated with 0.5 µM Torin-2 various aspects of nuclear shrinkage, fragmentation and chromatin margination, that are associated with the apoptotic mode of cell death, are observable. C. On the left, flow cytometric analysis of NALM-6 and TOM-1 cell lines treated with increasing concentrations of Torin-2. Samples were incubated with Annexin V-fluorescein isothiocyanate. On the right, MTT assay of NALM-6 and TOM-1 cells treated with Torin-2 and Z-VAD-fmk, a pan caspase inhibitor, is showed. Results are the mean of three different experiments ± SD.
Torin-2 affects the PI3K/Akt/mTOR pathway in B-pre ALL cells
To assess the effects of Torin-2 on the PI3K/Akt/mTOR signaling pathway, we studied the expression and activation status of critical components of the PI3K/Akt/mTOR cascade. NALM-6, RS4;11, SEM, TOM-1 and BV-173 cells were treated with increasing concentrations of Torin-2 for 2h and Western blot was then performed (Fig. 4).
Torin-2 decreased the phosphorylation levels of mTOR on both the Ser 2448 and Ser 2481 residues. It should be remembered that the phosphorylation of mTOR on Ser 2481 is a mTORC2-selective autophosphorylation site [21]. mTORC1 inhibition had functional effects on two well known mTORC1 substrates, S6 and 4E-BP1. S6 was completely dephosphorylated on the Ser 235/236 residue already at 50 nM concentration of Torin-2 in all cell lines, whereas 4E-BP1 was fully dephosphorylated on the Thr 37/46 site starting from the 100 nM concentration. Total levels of all these proteins were instead unaffected by Torin-2.
mTORC2 inhibition had a readout in Ser 473 Akt dephosphorylation and it was observable in all the cell lines starting from Torin-2 concentration of 50 nM. Despite some differences, also the Akt downstream substrate FoxO3A was dephosphorylated in a dose dependent fashion on its Thr 32 residue in all cell lines (Fig. 4).
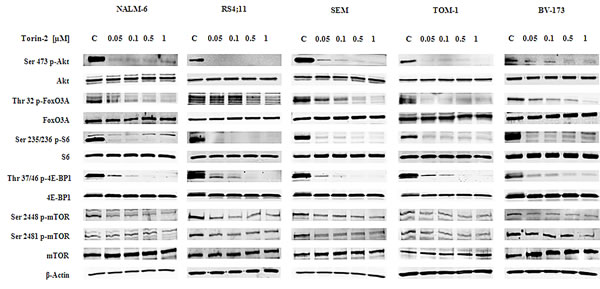
Figure 4: Torin-2 cytotoxicity is related to PI3K/Akt/mTOR signaling pathway inhibition. Western blot analysis for mTOR, Akt and their downstream targets S6, 4E-BP1 and FoxO3A in B-pre ALL cell lines. Twenty-five µg of protein were blotted to each lane. In all samples 2h of Torin-2 treatment with increasing concentrations was performed. β-actin served as a loading control.
Torin-2 prevents the reactivation of Akt upon mTOR inhibition in B-pre ALL cells
Since it has been previously described that in hematological malignancies [22, 23] and solid tumors [24, 25] with constitutive PI3K/Akt activation, the rapamycin derivative inhibitor everolimus (RAD001) increased Akt phosphorylation, we sought to explore if Torin-2 might prevent Akt re-activation after mTORC1 inhibition.
For this set of experiments we decided to use the concentration of 0.15 μM, that represents nearly the average IC50 of Torin-2 in the panel of cell lines employed. We prolonged the treatment with Torin-2 up to 48h and we compared it with the mTORC1 inhibitor, RAD001, employed at the concentration of 0.6 μM. This value has been chosen to mirror a concentration of a mTORC1 inhibitor (Temsirolimus) correspondent to the plasma concentration achievable in clinical trials [26]. As shown in Fig. 5A and B, in a range from 0.05 to 5 μM, RAD001 alone could not achieve the IC50. Thus, RAD001 had a putative IC50 higher than 5 µM, whereas Torin-2 displayed an IC50 value < 0.2 μM in all cell lines. Interestingly, we found that either mTORC1 and mTORC2 substrates, including Akt, after 48h of Torin-2 treatment remained dephosphorylated (Fig. 5C). On the contrary, samples treated with RAD001 already after 24h showed a re-phosphorylation of Akt. Direct (FoxO3A) or indirect (S6) downstream targets of Akt displayed the same behaviour (Fig. 5C).
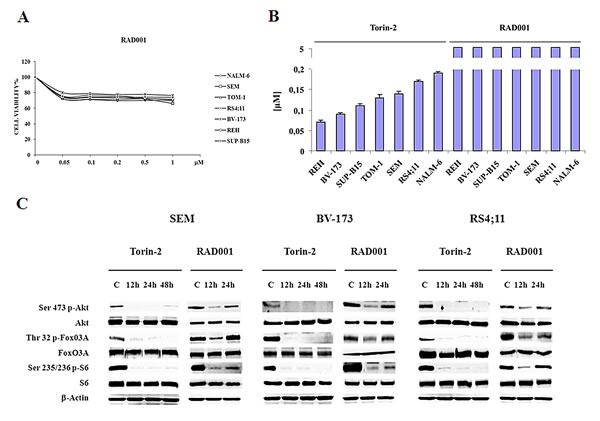
Figure 5: Torin-2 prevents Akt reactivation in B-pre ALL. A. MTT assay of B-pre ALL cell lines treated with increasing concentrations of RAD001 for 48h. One representative experiment of three is shown. B. IC50 values for Torin-2 and RAD001 on the viability of B-pre ALL cell lines after 48h. Results are the mean of three different experiments ± SD. C. Western blot analysis for PI3K/Akt/mTOR signaling pathway in SEM, BV-173 and RS4;11 cells. Cells were treated with 0.15 μM Torin-2 and 0.6 μM RAD001 for different times of incubation. In RAD001 treated samples after 24h is evident the re-phosphorylation of Akt, FoxO3A and S6. β-actin served as a loading control.
The allosteric Akt inhibitor MK-2206 synergizes with RAD001 but not with Torin-2
For therapeutic targeting of the PI3K/Akt/mTOR pathway, the combined inhibition at different points of the cascade often leads to more effective results than the use of a drug that acts on a single or dual targets [27]. To better assess the potential therapeutic value of Torin-2 in B-pre ALL, we analyzed its synergistic potential with MK-2206, an orally active, allosteric Akt inhibitor, which is currently tested in phase II clinical trials. We also compared this drug combination with a second one consisting of the association of MK-2206 and RAD001. The drugs were used at a fixed ratio (1:1 both for Torin-2/MK-2206 and RAD001/MK-2206).
After 48h of treatment, MTT assays were performed. The dual targeting of mTORC1/mTORC2 and Akt with Torin-2 and MK-2206 did not show a synergistic effect at any concentration, whereas the administration of RAD001 and MK-2206 together resulted in a relevant synergistic cytotoxic effect in RS4;11 and BV-173 cell lines (Fig. 6A). This phenomenon was more relevant in the range between 0.5 and 1 µM, as confirmed by the combination index (CI) values. Similar results were obtained with other B-pre ALL cell lines (supplementary Fig. 1). It should be noted that a comparison between the IC50 values obtained from the two different treatment at 48h (Torin-2/MK-2206 and RAD001/MK-2206), showed that the IC50 values were very similar and comparable in each cell line (Fig. 6B).
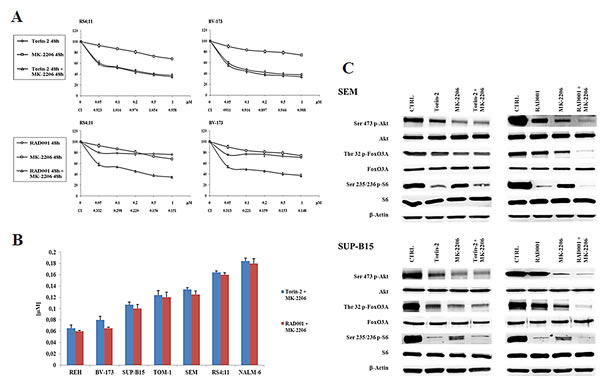
Figure 6: Dual administration of Torin-2 or RAD001 with MK-2206 in B-pre ALL cell lines. A: B-pre ALL cell lines were treated for 48h with Torin-2 or RAD001, either alone or in combination with MK-2206. Results are the mean of three different experiments ± SD. Combination index (CI) value for each data point was calculated with the appropriate software for dose effect analysis (Calcusyn). B: IC50 values for Torin-2 or RAD001 in combination with MK-2206 on the viability of B-pre ALL cell lines at 48h. Results are the mean of three different experiments ± SD. C: Western blot analysis for PI3K/Akt/mTOR in SEM and SUP-B15 cells. Cells were treated for 30 minutes with 0.15 μM Torin-2 or 0.6 μM RAD001 in combination with 0.5 μM MK-2206. β-actin served as a loading control.
We next studied the effects of the two drug combinations on the phosphorylation levels of Akt, FoxO3A and S6 protein. Torin-2 was used at 0.15 µM, RAD001 at 0.6 µM and MK-2206 at 0.5 µM for 30 minutes and then Western blot analysis was performed.
The concentration of MK-2206 was selected for either being in the range of maximal synergy and to be comparable to the plasma concentration that has been obtained in clinical trials in acute myelogenous leukemia [28]. Both SEM and SUP-B15 cell lines did not show a synergistic effect of Torin-2 and MK-2206 on the phosphorylation levels of Akt, FoxO3A and S6. Interestingly, there was an additional downregulation of protein phosphorylation only when the dual treatment RAD001/MK-2206 was administered, thus confirming the synergistic effect in modulating the PI3K/Akt/mTOR pathway (Fig. 6C). To further assess these findings, we explored the effects of dual treatments on cell cycle, by flow cytometric analysis of PI-stained samples in RS4;11 and BV-173 cells cultured for 24h. Both drug associations yielded comparable results. The treatment with Torin-2 and MK-2206 was similar to the single administration of Torin-2 alone. At variance, the dual administration of RAD001 and MK-2206 increased the percentage of cells in the G0/G1 phase of the cell cycle in comparison with single drug administration (Fig. 7A).
We also examined the pro-apoptotic effect of the two drug association on NALM-6 and TOM-1 cells. Cells were treated with 0.15 µM Torin-2 or 0.6 µM RAD001 and 0.5 µM MK-2206 for 24h and then analyzed by MTT assays. Using these drug concentrations, nearly 20% of apoptotic cells were observed in either cell line with both drug combinations. However, apoptosis induced by the treatment with Torin-2 and MK-2206 was comparable with that observable with the administration of Torin-2 alone, whereas the apoptosis obtained with the dual administration of RAD001 and MK-2206 was the consequence of a synergistic effect (Fig. 7B).
Finally, the effect of Bafilomycin A1 on dual targeting the PI3K/Akt/mTOR pathway with a combination of Torin-2/MK-2206 or RAD001/MK-2206 was analyzed. A consistent inhibition of autophagy increased the viability of B-pre ALL cells treated with Torin-2/MK-2206 (Fig. 7C, left panel) and to a lower extent with RAD001/MK-2206 (Fig. 7C, right panel). This difference may rely on the different mechanism through which autophagy is recruited by the drugs: those who act on mTOR, activated autophagy as a cell death mechanism [16],whereas the drug that inhibit Akt, induced autophagy as a cell protection mechanism [29].
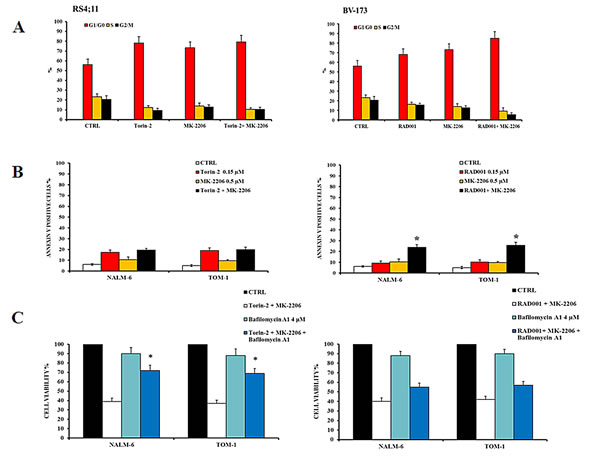
Figure 7: Effects of dual administration of Torin-2 or RAD001 with MK-2206 in B-pre ALL cell lines. A: Flow cytometric representation of the effects of Torin-2/MK-2206 and RAD001/MK-2206 combinations on the cell cycle of RS4;11 and BV-173 cell lines after 24h of treatment. Results are the mean of three different experiments ± SD. B: Flow cytometric analysis of NALM-6 and TOM-1 cell lines treated with increasing concentrations of Torin-2 or RAD001, alone and in combination with MK-2206, for 24h. Samples were incubated with Annexin V-fluorescein isothiocyanate and then analyzed for apoptosis. Results are the mean of three different experiments ± SD. Asterisks indicate statistically significant differences with respect to untreated cells (*p<0.05). C. MTT assay representation of the autophagy effects on NALM-6 and TOM-1 cells treated with Torin-2/MK-2206 or RAD001/MK-2206 combinations or plus the administration of the autophagy inhibitor Bafilomycin A1. Results are the mean of three different experiments ± SD. Asterisks indicate statistically significant differences with respect to untreated cells (*p<0.05).
Torin-2 suppresses doxorubicin-induced cell cycle checkpoint activation
When DNA is damaged by DNA intercalating agents, such as Doxorubicin, double stranded breaks (DSBs) trigger recruitment of ATM and ATR to the damage site which in turn phosphorylates histone H2AX leading to foci formation [30].
Torin-2 also exhibited potent biochemical and cellular activity against PIKK family kinases including ATM, ATR, and DNA-PK, whose inhibition sensitized cells to irradiation [15]. The ATR pathway is known to transmit DNA damage signals through the ATR-CHK1 kinase cascade and activation of cell cycle checkpoint regulators such as CHK1 and CHK2 has a critical role in promoting cell cycle arrest in response to cytotoxic agents, including doxorubicin [31].
We tested if the combination of Doxorubicin and Torin-2 may exert additional cytotoxic activity than the two drugs administered alone. In Figure 8A are shown the results of MTT assays of two representative cell lines (SEM and TOM-1) analyzed for cell viability after treatment with the drugs used either as single agents or combined together. In both cell lines, the drug combination induced a stronger decrease in cell viability (Fig. 8A). Doxorubicin alone induced phosphorylation of CHK1 (Ser 345, a marker for ATR activity) and CHK2 (Thr 68, a readout for ATM activity). In contrast, Torin-2 (0.25 µM) alone did not increase CHK1 and CHK2 phosphorylation, whereas it dramatically decreased the phosphorylation induced by Doxorubicin (Fig. 8B). One prominent chromatin modification in response to DNA damage is phosphorylation of histone H2AX on Ser 139, which is referred to as γ-H2AX [32].
Torin-2 inhibited the DNA damage response induced by Doxorubicin, as documented by the effects on the levels of phosphorylated γ-H2AX on the Ser 139 residue (Fig. 8B).
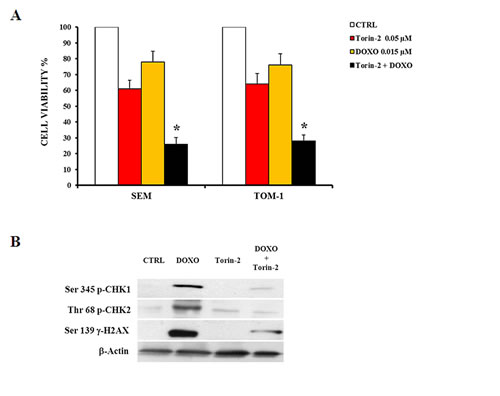
Figure 8: Torin2 suppresses Doxorubicin-induced cell cycle checkpoint activation. A: MTT assay showing the cytotoxic effect of Torin-2 and Doxorubicin (0.015 µM) alone or in combination in SEM and TOM-1 cells. DOXO, Doxorubicin-treated cells. Results are the mean of three different experiments ± SD. Asterisks indicate statistically significant differences with respect to untreated cells (*p<0.05). B: Western blot analysis of SEM cells showing the activation by Doxorubicin treatment of cell cycle checkpoint regulators Chk1 and Chk2, markers for ATR or ATM activity, respectively. Torin-2 alone did not modify CHK1 and CHK2 phosphorylation, whereas it dramatically decreased after Doxorubicin administration. Torin-2 also influenced the levels of phosphorylated γ-H2AX, inhibiting its phosphorylation after Doxorubicin treatment. β-actin served as a loading control.
DISCUSSION
Owing to the fundamental role of PI3K/Akt/mTOR pathway in tumor development and progression, there has been a significant interest in developing inhibitors against components of this pathway, up to having now many compounds currently under evaluation in clinical trials. ATP-competitive mTOR kinase inhibitors represent a promising new approach to target the PI3K/Akt/mTOR pathway with potentially greater tolerability than dual PI3K/mTOR inhibitors [5, 33, 34]. It has been previously reported that first generation mTOR kinase inhibitors had improved efficacy compared to rapamycin in models of Ph+ B-ALL [35].
We have recently explored the therapeutic potential of RAD001, an allosteric mTORC1 inhibitor in pre-clinical models of B-pre ALL [16]. We documented that RAD001 decreased cell viability, induced cell cycle arrest in G0/G1 phase and caused apoptosis in B-pre ALL cell lines. Autophagy was also induced, which was important for the RAD001 cytotoxic effect, as downregulation of Beclin-1 reduced drug cytotoxicity. RAD001, used in the micromolar range and administered 24h before MK-2206 showed the capacity to synergize with MK-2206 in both cell lines and patient samples [16]. In this study, we evaluated for the first time the efficacy of the novel mTORC1/mTORC2 second generation, ATP-competitive inhibitor, Torin-2 [14] in pre-clinical settings of Ph+ and Ph- B-pre ALL.
Torin-2 was both cytotoxic and cytostatic in a nanomolar range to B-pre ALL cell lines in a concentration dependent mechanism, as demonstrated by MTT assays, flow cytometric analysis of Annexin V-stained samples and of PI-stained samples. Apoptosis resulted to play a determinat role in the killing mechanism, since the treatment with a pan caspase inhibitor protected the cells from Torin-2 cytotoxic effect. These results are in agreement with those observed with other drugs in acute myeloid leukemia [36, 37].
Torin-2 also induced autophagy, as documented by increased expression of the lipidated form of LC3. In order to demonstrate whether autophagy was either a survival or a death mechanism, B-pre ALL were co-treated with the autophagic sequestration inhibitors, 3-MA and Bafilomycin A1. We found that either treatment with 3-MA and Bafilomycin A1, resulted in a lower sensitivity of both SEM and BV-173 cells to the cytotoxic effects of RAD001 and indicated that Torin-2-induced autophagy was very important for the cytotoxic effects of the drug. The phosphorylation status of the key elements of the PI3K/Akt/mTOR pathway, assessed by Western blot, was equally sensitive to Torin-2 inhibition in either cells harboring or not the Bcr-Abl fusion protein.
RAD001, when used in the nanomolar range against this panel of cell lines, was much less cytotoxic than Torin-2, as it displayed IC50s > 5 µM. This superior efficacy of the second generation inhibitors, has been also reported for MLN0128, another mTORC1/mTORC2 inhibitor, which displayed an improved anti-leukemic activity when compared to first generation inhibitors, in Ph- B-ALL derived from both adult and pediatric subjects [21]. It has been reported that in solid tumor models, activation of mTORC1 drives p70S6K-mediated degradation of the IR/IGF-1R adaptor protein IRS-1, and is therefore a negative regulator of PI3K [38]. Accordingly, drugs targeting mTORC1 block this feedback and trigger reactivation of the pathway and re-phosphorylation of Akt on Ser 473 residue in acute myelogenous leukemia cells [22, 39]. The issue of Akt reactivation in response to mTORC1 inhibition, has not been throughly investigated in B-pre ALL. It is worth highlighting that Torin-2 maintained a prolonged suppression of mTORC1/mTORC2 with a sustained anti-proliferative effect, overcoming the limitations of rapalogs such as RAD001, as demonstrated by our findings, which resulted in re/hyper-phosphorylation of Akt and could hamper their anti-tumor action and enhance resistance to antineoplastic therapy, thus resulting in a poor outcome [40].
Akt activity is directly downregulated on Thr 308 by the protein phosphatase PP2A [41], which is also critically involved in regulation of cell cycle progression [42] and DNA damage response [43]. Accumulating evidence indicates that PP2A acts as a tumor suppressor and impairment of PP2A activity may result in loss of this function [7]. The capacity of Torin-2, but not of RAD001, to inhibit both mTORC1 and 2 may represent a possible mechanism for why Akt is not re-phosphorylated with this drug on the Ser 473 residue and this phosphorylation may be independent form PP2A activity.
The Torin-2 prolonged suppression of Akt phosphorylation may overcome the occurrence of PP2A oncosuppressor altered function and may render Akt inhibition independent from the activity of PP2A, thus proposing this therapeutic options as a potential tool that could act functionally on either impaired Akt and PP2A functions.
Overall, Torin-2 alone was as potent as a combination consisting of RAD001 and MK-2206, in terms of reduction of cell viability, apoptosis induction, and cell cycle block. In contrast, when MK-2206 was combined with Torin-2, it did not display any synergistic effect. Interestingly, the presence of synergism or its lack was evident also when the phosphorylation status of key elements of the pathway was analyzed by Western blot.
Both ATM and ATR play a central role in coordinating the DNA damage response, including cell cycle checkpoint control and apoptosis [44]. Since the results by Liu et al. [15], using purified enzymes, suggested that Torin-2 was also an inhibitor of several PIKK family members including ATR, ATM, and DNA-PK, we investigated if this was also true in B-pre ALL cell lines. Western blot analysis documented that indeed Torin-2 inhibited ATM and ATR in intact cells and, by inhibiting DNA repair, potentiated the cytotoxic effect of Doxorubicin. Torin-2 displayed an obvious advantage over RAD001, which also induced a similar phenomenon, as Torin-2 could be used in the nanomolar range, whereas RAD001 required a concentration as high as 16 µM [45], which could not be attained in vivo. In addition, very recently it has been reported that Akt inhibition with CCT12893 increased the phosphorylation of CHK1/CHK2 and γ-H2AX [46].
These findings open a new very interesting field to be further explored in the future regarding the therapeutic effect of PI3K/Akt/mTOR inhibitors involving DNA damage sensors and cell cycle checkpoints such as CHK1 and CHK2.
In conclusion, our data indicate that the novel mTORC1/mTORC2 kinase inhibitor Torin-2 can suppress the growth of both Ph+ and Ph- B-pre ALL cells and extend the finding that the antiproliferative and proapoptotic effects of PI3K/Akt/mTOR pathway inhibitors are independent from ABL-translocation, as reported in long-term cultures of Ph+ and Ph- B-precursor ALL cells from patients [40]. Remarkably, Torin-2, even after a 48h incubation, blocked the reactivation of Akt, thus confirming the new therapeutic hopes that this second generation of inhibitors is developing. In addition, Torin-2 could be also effective in combination with chemotherapeutic DNA-damaging agents, in light of its capacity of blocking DNA repair.
Interestingly, with the aim of improving B-pre ALL treatment, we also came to the conclusion that low concentrations of RAD001 and MK-2206 (which can be attained in vivo) may achieve a therapeutic efficacy comparable to an mTORC1/mTORC2 inhibitor. These therapeutic strategies could be particularly effective when combined with targeted next generation sequencing of tumor samples, since genomic alterations can be detected and help to identify refractory patients with aberrations putatively activating the PI3K/Akt/mTOR pathway [47, 48]. These pharmacological options targeting PI3K/Akt/mTOR at different points of the signaling pathway cascade or in combination with conventional chemotherapy might represent a new therapeutic potential for treatment of B-pre ALL patients.
MATERIALS AND METHODS
Materials
Alpha-MEM, McCoy’s 5A and RPMI-1640 mediums, fetal bovin serum (FBS), penicillin and streptomycin were from Lonza (Lonza Milano SRL, Milan, Italy). Torin-2, RAD001, and MK-2206 were from Selleck Chemicals (Houston, TX, USA). For cell viability determination, Cell Proliferation Kit I (MTT) was purchased from Roche Applied Science (Basel, Switzerland). Annexin V/7-AAD detection kit was from Merck-Millipore (Darmstadt, Germany). Akt-1, Ser 473 p-Akt-1, and FoxO3A primary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) while all the other antibodies were from Cell Signaling Technology (Danvers, MA, USA), including the rabbit secondary antibody. The mouse secondary antibody, Bafilomycin A1, Z-VAD-fmk, 3-Methyladenine (3-MA), 1,4-Diazabicyclo[2.2.2]octane (DABCO) and 4′, 6 diamidino-2-pheny-lindole (DAPI) were from Sigma Aldrich (Milan, Italy). Signals were detected with the ECL Plus reagent purchased from Perkin Elmer (Boston, MA, USA).
Cell culture and Western blot analysis
All the B-pre acute lymphoblastic leukemia cell lines were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). SEM, REH, BV-173 and NALM6 were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS); RS4;11 cells were grown in Alpha-MEM medium with 10% FBS; TOM-1 cells were grown in RPMI 1640 medium with 20% FBS and SUP-B15 cells were grown in McCoy’s 5A medium with 20% FBS. All the media were supplemented with 100 units/ml penicillin and 100 mg/ml streptomycin. The cells were grown at a density of 0.5 to 2 x 106 cells/ml and were incubated at 37°C with 5% CO2. Western blot analysis was performed by standard methods as described elsewhere [49].
Cell viability analysis
MTT (3-(4,5-dimethylthythiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays were performed as previously described [50].
Cell cycle analysis
Cell cycle analysis was performed using the MuseTM Cell Analyzer (Merck Millipore, Milan, Italy) and/or propidium iodide (PI)/RNase A staining by flow cytometry according to standard techniques, as described elsewhere [51]. In brief, after 24h of drug treatment, cells were harvested, centrifuged at 300 x g for 5 min and washed once with 1X PBS. After fixing them with 70% ethanol at 20°C, cells were centrifuged at 300 x g for 5 min and washed once with 1X PBS. Then 200 μl of MuseTM Cell Cycle reagent or 100 μl of propidium iodide (PI)/RNase A staining was added to each tube with an incubation of 30 min at room temperature in the dark. Samples were then analyzed according to the manufacturer’s instructions.
PI/Annexin V assay
Apoptosis analysis was performed by staining with Annexin V/7-AAD, using the MuseTM Cell Analyzer in according to the manufacturer’s instructions. In brief, a 100 μl treated cell suspension was labeled for 20 min in the dark with the same volume of the MuseTM Annexin-V & Dead Cell reagent (Merck Millipore). Subsequently, quantitative detection of Annexin-V/7-AAD positive cells was performed with the MuseTM Cell Analyzer [51].
DAPI staining
Cell nuclear morphology was evaluated by fluorescence microscopy following DAPI staining. Cells were treated with Torin-2 for 24![]() h. The cells were washed with PBS (pH 7.4), cytocentrifuged, fixed with 4% paraformaldehyde/PBS and stained for 3 min with 1 μg/mL DAPI. The cells were then washed with PBS, specimens were embedded in glycerol with antifading agent (DABCO) and examined under Zeiss Axiophot fluorescence microscope (Zeiss, Germany).
h. The cells were washed with PBS (pH 7.4), cytocentrifuged, fixed with 4% paraformaldehyde/PBS and stained for 3 min with 1 μg/mL DAPI. The cells were then washed with PBS, specimens were embedded in glycerol with antifading agent (DABCO) and examined under Zeiss Axiophot fluorescence microscope (Zeiss, Germany).
Combined drug effect analysis
The combination effect and the potential synergy of Torin-2 and RAD001 with MK-2206 were evaluated from quantitative analysis of dose–effect relationship, as described previously [16]. For each Torin-2/MK-2206 or RAD001/MK-2206 combination experiment, a combination index (CI) number was calculated using the Biosoft CalcuSyn software (Biosoft, Cambridge, UK). This method of analysis generally defines CI values from 0.9 to 1.1 as additive, from 0.3 to 0.9 as synergistic and 0.3 as strongly synergistic, whereas values over 1.1 are considered as antagonistic.
Statistical evaluation
The data are presented as mean values from three separate experiments ± s.d. Data were statistically analyzed by a Dunnet test after one-way analysis of variance (ANOVA) at a level of significance of P<0.05 vs control samples.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Acknowledgments
This work was supported by a MIUR FIRB 2010 grant to SC (RBAP10Z7FS_002), MIUR FIRB 2010 grant to AMM (RBAP10447J_003), by a MIUR PRIN-2009 grant to SC and by current research funds IRCCS Burlo Garofolo to GZ.
References
1. Lovejoy CA and Cortez D. Common mechanisms of PIKK regulation. DNA repair. 2009; 8(9):1004-1008.
2. Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA repair. 2004; 3(8-9):883-887.
3. Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D and Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle. 2011; 10(13):2115-2120.
4. Francipane MG and Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014; 5(1):49-66.
5. Janes MR and Fruman DA. Targeting TOR dependence in cancer. Oncotarget. 2010; 1(1):69-76.
6. Eyre TA, Collins GP, Goldstone AH and Cwynarski K. Time now to TORC the TORC? New developments in mTOR pathway inhibition in lymphoid malignancies. British journal of haematology. 2014; 166(3):336-351.
7. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3(9):954-987.
8. Pui CH and Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Seminars in hematology. 2013; 50(3):185-196.
9. Inaba H, Greaves M and Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013; 381(9881):1943-1955.
10. Pui CH, Mullighan CG, Evans WE and Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012; 120(6):1165-1174.
11. Gaynon PS, Angiolillo AL, Carroll WL, Nachman JB, Trigg ME, Sather HN, Hunger SP and Devidas M. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children’s Oncology Group Report. Leukemia. 2010; 24(2):285-297.
12. Tasian SK, Teachey DT and Rheingold SR. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Frontiers in oncology. 2014; 4:108.
13. Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, Winick NJ, Hunger SP, Gaynon PS and Loh ML. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008; 22(12):2142-2150.
14. Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, Sabatini DM and Gray NS. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2( 1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. Journal of medicinal chemistry. 2011; 54(5):1473-1480.
15. Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P, Ercan D, Niepel M, Thoreen CC, Kang SA, Patricelli MP, Wang Y, et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer research. 2013; 73(8):2574-2586.
16. Neri LM, Cani A, Martelli AM, Simioni C, Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey JA and Capitani S. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia. 2014; 28(4):739-748.
17. Rubinsztein DC, Codogno P and Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews Drug discovery. 2012; 11(9):709-730.
18. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer research. 2014; 74(3):647-651.
19. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8(4):445-544.
20. Shacka JJ, Klocke BJ and Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006; 2(3):228-230.
21. Janes MR, Vu C, Mallya S, Shieh MP, Limon JJ, Li LS, Jessen KA, Martin MB, Ren P, Lilly MB, Sender LS, Liu Y, Rommel C and Fruman DA. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013; 27(3):586-594.
22. Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, Ifrah N, Dreyfus F, Mayeux P, Lacombe C and Bouscary D. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008; 111(1):379-382.
23. Advani AS, Mahfouz RZ, Maciejewski J, Rybicki L, Sekeres M, Tripp B, Kalaycio M, Bates J and Saunthararajah Y. Ribosomal S6 kinase and AKT phosphorylation as pharmacodynamic biomarkers in patients with myelodysplastic syndrome treated with RAD001. Clinical lymphoma, myeloma & leukemia. 2014; 14(2):172-177 e171.
24. Seront E, Pinto A, Bouzin C, Bertrand L, Machiels JP and Feron O. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. British journal of cancer. 2013; 109(6):1586-1592.
25. Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, Kwiatkowski D and Lane HA. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Molecular cancer therapeutics. 2009; 8(4):742-753.
26. Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G and Sherman ML. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. Journal of clinical oncology. 2004; 22(5):909-918.
27. Dienstmann R, Rodon J, Serra V and Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Molecular cancer therapeutics. 2014; 13(5):1021-1031.
28. Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G, Huang X, Kadia TM, Ruvolo PP, Feliu JB, Lu H, Debose L, Burger JA, Andreeff M, Liu W, Baggerly KA, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clinical cancer research. 2014; 20(8):2226-2235.
29. Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL, Melchionda F, Pagliaro P, Pession A, McCubrey JA, Capitani S and Martelli AM. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012; 26(11):2336-2342.
30. Reinhardt HC and Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Current opinion in cell biology. 2009; 21(2):245-255.
31. Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D, Poonepalli A, Hande MP, Liu H, He G, He L and Li B. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell death and differentiation. 2011; 18(1):5-15.
32. Neumann J, Boerries M, Kohler R, Giaisi M, Krammer PH, Busch H and Li-Weber M. The natural anticancer compound rocaglamide selectively inhibits the G1-S-phase transition in cancer cells through the ATM/ATR-mediated Chk1/2 cell cycle checkpoints. International journal of cancer. 2014; 134(8):1991-2002.
33. Vilar E, Perez-Garcia J and Tabernero J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Molecular cancer therapeutics. 2011; 10(3):395-403.
34. Wander SA, Hennessy BT and Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of clinical investigation. 2011; 121(4):1231-1241.
35. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C and Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nature medicine. 2010; 16(2):205-213.
36. Piedfer M, Dauzonne D, Tang R, N’Guyen J, Billard C and Bauvois B. Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. FASEB journal. 2011; 25(8):2831-2842.
37. Merhi F, Tang R, Piedfer M, Mathieu J, Bombarda I, Zaher M, Kolb JP, Billard C and Bauvois B. Hyperforin inhibits Akt1 kinase activity and promotes caspase-mediated apoptosis involving Bad and Noxa activation in human myeloid tumor cells. PLOS ONE. 2011; 6(10):e25963.
38. Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S and Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer discovery. 2011; 1(3):248-259.
39. Bertacchini J, Guida M, Accordi B, Mediani L, Martelli AM, Barozzi P, Petricoin E, 3rd, Liotta L, Milani G, Giordan M, Luppi M, Forghieri F, De Pol A, Cocco L, Basso G and Marmiroli S. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia. 2014: doi: 10.1038/leu.2014.123. [Epub ahead of print].
40. Badura S, Tesanovic T, Pfeifer H, Wystub S, Nijmeijer BA, Liebermann M, Falkenburg JH, Ruthardt M and Ottmann OG. Differential effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway in acute lymphoblastic leukemia. PLOS ONE. 2013; 8(11):e80070.
41. Eichhorn PJ, Creyghton MP and Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et biophysica acta. 2009; 1795(1):1-15.
42. Nolt JK, Rice LM, Gallo-Ebert C, Bisher ME and Nickels JT. PP2A (Cdc)(5)(5) is required for multiple events during meiosis I. Cell Cycle. 2011; 10(9):1420-1434.
43. Lee HJ, Hwang HI and Jang YJ. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle. 2010; 9(12):2389-2398.
44. Shiloh Y and Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013; 14(4):197-210.
45. Saunders PO, Weiss J, Welschinger R, Baraz R, Bradstock KF and Bendall LJ. RAD001 (everolimus) induces dose-dependent changes to cell cycle regulation and modifies the cell cycle response to vincristine. Oncogene. 2013; 32(40):4789-4797.
46. Wang FZ, Chang ZY, Fei HR, Yang MF, Yang XY and Sun BL. CCT128930 induces cell cycle arrest, DNA damage, and autophagy independent of Akt inhibition. Biochimie. 2014; 103:118-125.
47. Janku F, Kaseb AO, Tsimberidou AM, Wolff RA and Kurzrock R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 2014; 5(10):3012-3022.
48. Woo JS, Alberti MO and Tirado CA. Childhood B-acute lymphoblastic leukemia: a genetic update. Experimental hematology & oncology. 2014; 3:16.
49. Chiarini F, Lonetti A, Teti G, Orsini E, Bressanin D, Cappellini A, Ricci F, Tazzari PL, Ognibene A, Falconi M, Pagliaro P, Iacobucci I, Martinelli G, Amadori S, McCubrey JA and Martelli AM. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget. 2012; 3(12):1615-1628.
50. Buontempo F, Chiarini F, Bressanin D, Tabellini G, Melchionda F, Pession A, Fini M, Neri LM, McCubrey JA and Martelli AM. Activity of the selective IkappaB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia: involvement of FOXO3a. Cell Cycle. 2012; 11(13):2467-2475.
51. Simioni C, Martelli AM, Cani A, Cetin-Atalay R, McCubrey JA, Capitani S and Neri LM. The AKT inhibitor MK-2206 is cytotoxic in hepatocarcinoma cells displaying hyperphosphorylated AKT-1 and synergizes with conventional chemotherapy. Oncotarget. 2013; 4(9):1496-1506.