
INTRODUCTION
Classical Hodgkin lymphoma (cHL) is the commonest lymphoid malignancy of young adults [1]. While cHL results from the neoplastic transformation of germinal centre B-cells, the characteristic Hodgkin and Reed-Sternberg (HRS) cells are typically seen at very low levels (1–10%), with most of the tumour consisting of a pleomorphic infiltrate but predominantly T-lymphocytes [2–4].
Epstein-Barr virus (EBV) infection is causally related to a number of cases [3, 5], but there is little evidence to support the involvement of other environmental or lifestyle risk factors. A role for inherited susceptibility to cHL is provided by familial risk [6, 7], including a high concordance in monozygotic twins [8]. Although a strong human leukocyte antigen (HLA) association for cHL risk is established [9–14], this only accounts for <30% of the familial risk [15]. Direct evidence for the role of non-HLA genetics in cHL has come from recent genome-wide association studies (GWAS), which have identified common variants at 13 loci associated with the risk of sporadic cHL [9, 14, 16, 17]. In addition to common genetic variation influencing cHL, reports of familial aggregation raise the possibility of the existence of Mendelian susceptibility to the disease, caused by the inheritance of high-impact mutations [18]. A number of these multiple-case families have been the subject of linkage searches and various putative linkage signals reported; notably at 3p21.31 implicating germline variation in the kelch protein gene KLHDC8B [19].
Nodular sclerosis Hodgkin lymphoma (NSHL), the most common histological subtype of cHL in developed countries, is more frequent in young adults and women [4]. With the aim of furthering our understanding of cHL susceptibility, we describe a family in which five members have been diagnosed with the cHL subtype, NSHL. Seeking to identify a major risk locus responsible for cHL in the family, we conducted a genome-wide linkage scan and whole exome sequencing (WES) of affected family members. To complement the analysis, we also searched for evidence of association at regions of linkage by analysing a large GWAS dataset.
RESULTS
Linkage and mutation analysis
No region of the genome showed evidence for linkage with cHL in the family at genome-wide significance (i.e. logarithm of odds (LOD) score >3.0). Three regions of the genome, however showed evidence of linkage with a LOD score greater than 2.0: 2q35-37 (220-237Mb), 3p14-22 (42-66Mb), 21q22 (42-47Mb) (Supplementary Figure 1). Linkage at 6p21 (HLA) was lacking and no shared HLA class I allele was shown between affected family members (Table 1). Previous studies have demonstrated a strong association between HLA class II alleles with EBV-negative NSHL. Only the HLA-DPB1*04:01 allele (Table 1), which has population frequency (>40%), was shared between affected family members. 712 coding variants were shared by affected family members (III-a, IV-b, IV-f and IV-g). Of these 712, only 37 mapped to the three regions of linkage. Restricting our analysis to those variants with a population frequency <1% and those predicted to impact on the protein function by both SIFT [20] and POLYPHEN-2 [21] algorithms, we identified p.(Arg76Gln) in FAM107A and p.(Thr220Ala) in SLC26A6 mapping to 3p21 as two plausible candidate variants causal for cHL in the family. We confirmed the fidelity of WES for both variants by Sanger sequencing. Finally, we did not identify any rare sequence variants in the previously identified candidate cHL susceptibility gene in this region, KLHDC8B, in any of the affected family members.
Table 1: Clinico-pathological details of affected family members
ID |
Stage |
Treatment |
Age at diagnosis |
HLA type |
Subtype, EBV status |
|---|---|---|---|---|---|
IV-f |
2A |
6 × ABVD |
22 |
A*02:01,A*02:01, B*07:02,B*13:02, C*06:02,C*07:02, DRB1*07:01,DRB1*15:01, DRB4*01, DRB5*01, DQB1*02:02,DQB1*06:02, DPB1*04:01:01,DPB1*04:02 |
NSHL, LMP1 negative. |
IV-b |
2A |
6 × ABVD, radiotherapy |
18 |
A*01:01,A*02:01, B*08:01,B*44:02, C*05:01,C*07:01, DRB1*01:01,DRB1*03:01, DRB3*01, DQB1*05:01,DQB1*02:01, DPB1*04:01:01 |
NSHL, unknown |
III-g |
2A |
3 × ABVD, radiotherapy |
31 |
A*03:01, B*07:02,B*49:01, C*07:01,C*07:02, DRB1*04:01,DRB1*15:01, DRB4*01, DRB5*01, DQB1*06:02,DQB1*03:01, DPB1*04:01 |
NSHL, unknown |
IV-g |
4B |
2 × ABVD, 6 × BEACOPPe, IFRT |
25 |
A*03:01,A*25:01, B*07:02,B*18:01, C*07:02,C*12:03, DRB1*15:01, DRB5*01:01, DQB1*06:02, DPB1*04:01,DPB1*145:01 |
NSHL, EBER negative. |
III-a |
NA |
NA |
33 |
NA |
NA |
ABVD, adriamycin, bleomycin, vinblastine, dacarbazine; BEACOPP, bleomycin, etoposide, cyclophosphamide, adriamycin, vincristine, procarbazine, prednisolone; IFRT, involved-field radiotherapy, LMP1, latent membrane protein 1; EBV, Epstein–Barr virus; EBER, - Epstein–Barr virus-encoded small RNAs; NSHL, nodular sclerosis Hodgkin lymphoma; NA, not available.
Analyses of candidate genes
SLC26A6 expression was lower in lymphoblastoid cell lines from family members carrying the T220A mutation compared to unrelated healthy individuals (Figure 1). FAM107A expression was undetectable in lymphoblastoid cell lines. Expression of FAM107A and SLC26A6 was low or absent in HRS cell lines (Figure 2) and in HRS cells in Hodgkin lymphoma (Figure 3). Finally, expression levels of KLHDC8B were similar between lymphoblastoid cell lines from mutation carriers and non-carriers as well as between HRS cell lines and other cell lines (Figures 1 and 2).
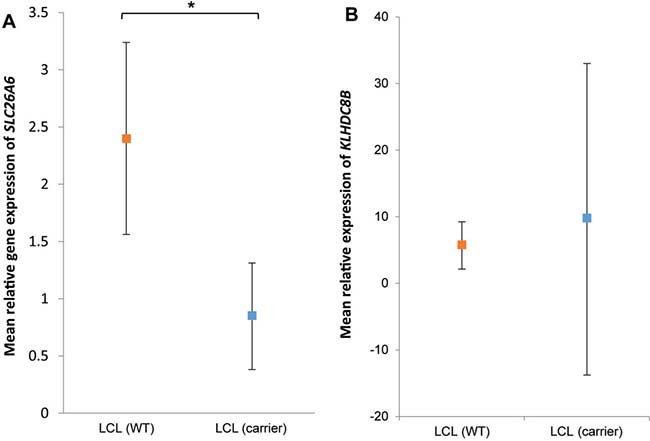
Figure 1: (A) SLC26A6 and (B) KLHDC8B expression in lymphoblastoid cells from patients and controls. Mean relative expression in lymphoblastoid cell lines derived from mutations carriers III-g, IV-f and IV-g (blue) and ten non-carriers (red). Cell lines assayed in triplicate and normalized against GAPDH expression. Values are mean of replicates and carrier status. Error bars denote 95% confidence intervals.*P < 0.05.
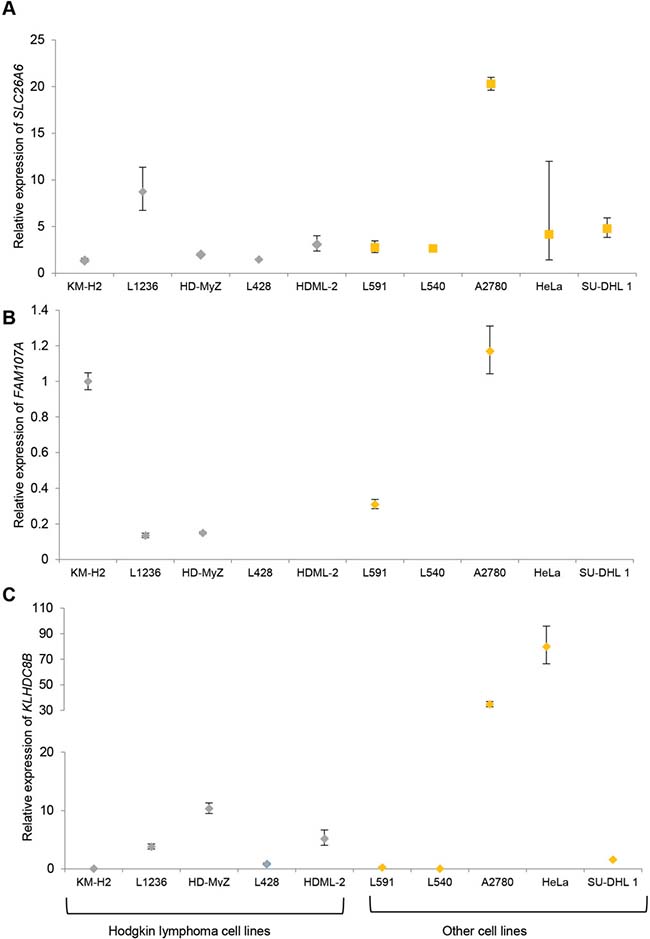
Figure 2: (A) SLC26A6 (B) FAM107A (C) KLHDC8B expression in established cell lines. Hodgkin lymphoma cell lines: KM-H2, L1236, HD-MyZ, L428, HDML-2. Each cell line was assayed in triplicate and normalized against GAPDH expression. Expression of FAM107A in L428, HDML-2, L540, HeLa and SU-DHL1 was below the limit of detection. Data are shown as mean (± standard error of the mean).
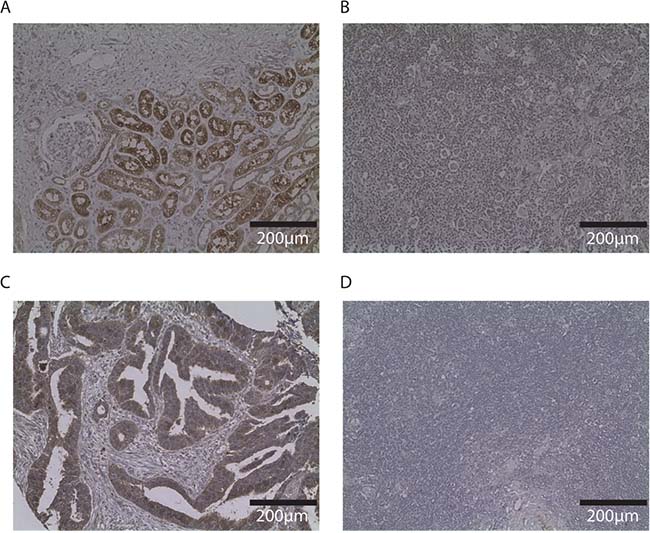
Figure 3: Immunohistochemistry of SLC26A6 and FAM107A in other tissues and Hodgkin lymphoma node biopsy. Photos (20×) of representative examples illustrating SLC26A6 staining of renal biopsy (A) and Hodgkin node (B); FAM107A staining of colorectal cancer tissue (C) and Hodgkin node (D). Immunostaining indicated by DAB, counterstained with haematoxylin.
Association analysis of 2q35-37, 3p14-22, 21q22
To explore the possibility that variants with frequency greater than 1% might contribute to a relationship between 2q35-37, 3p14-22, 21q22 and cHL risk, we analysed directly typed and imputed genotypes in our large case-control series. Across the three regions we examined the relationship for 107,472 SNPs and cHL risk (Supplementary Figure 2). The strongest associations at 2q35-37, 3p14-22, 21q22 were provided by rs6714255 (P = 5.79 × 10–6), rs115918946 (P = 1.70 × 10–4) and rs2839384 (P = 1.31 × 10–3), which, after adjustment for multiple testing, were not statistically significant (i.e. P < 4.65 × 10–7, 0.05/107,472). Furthermore, these common genetic variants were not in linkage disequilibrium with the candidate genetic mutations in FAM107A and SLC26A6.
DISCUSSION
It is notable that we identified the class II HLA allele (DPB1*0401) as being shared between affected individuals. Although a HLA class II association is well established for NSHL, there is limited evidence to implicate this specific HLA allele. Excluding the possibility of multiplicative interaction with a non-HLA linked allele, the pattern of inheritance of cHL in the family we describe is parsimonious with autosomal inheritance with incomplete penetrance. Predicated on this assumption, we sought to identify a disease locus for cHL in the family by performing a linkage scan. Although not statistically significant per se, we found suggestive linkage at 3p21, a region previously implicated by Salipante et al. [19], which in a combined analysis would provide a LOD score of >3.0.
By performing WES of affected individuals we identified two rare protein-disrupting mutations in SLC26A6 and FAM107A on chromosome 3p21, as possible candidates for cHL risk. In contrast, we did not identify any mutations in KLHDC8B, which has been previously suggested as a cause of familial cHL [19], and which lies within the linkage peak on 3p21 only ~500kb from SLC26A6.
SLC26A6 is expressed in many tissues, consistent with its role as an anion transporter [22, 23]. SLC26A6 has so far not been directly implicated in cancer. FAM107A, originally identified in a commonly deleted region on 3p21 in renal cell carcinoma, appears to function as a tumour suppressor [24–26]. Loss of heterozygosity at 3p21 is common in human malignancies [24, 27–30], although the region has not been shown to be recurrently lost or gained in primary HRS cells [31, 32]. However, both cHL and non-Hodgkin lymphoma have been reported to display chromosomal rearrangements involving this region by fluorescent in situ hybridization [33]. Furthermore, the International Cancer Genome Consortium report that mutations in SLC26A6 and FAM107A occur in 3.7% (9/241) and 12.9% (31/241) respectively of germinal centre B-cell derived lymphomas [34]. Although we were able to demonstrate lower SLC26A6 mRNA expression in LCLs of affected individuals, we have been unable to provide a mechanistic basis by which identified variants in either SLC26A6 or FAM107A contributes to lymphomagenesis.
Although WES is a well-recognised strategy to identify disease-causing mutations [35, 36], it does not identify pathogenic non-coding abnormalities, which are increasingly recognised to be important [37, 38]. Hence, we cannot exclude the possibility that susceptibility to cHL in the family might be mediated through non-coding changes within the linked region. Reference to GWAS data has however provided evidence that common genetic variation does not underscore the linkage signal at 3p21. In addition, this study would be further enhanced by analysis of data relating to unaffected individuals in this family.
In summary, our analysis of this family support the existence of a susceptibility locus on chromosome 3p21 for cHL, with exome sequencing suggesting SLC26A6 and FAM107A as possible candidate genes.
MATERIALS AND METHODS
Hodgkin lymphoma family
Collection of blood samples and clinical information from subjects was undertaken with informed consent and relevant ethical review board approval (North of Scotland regional ethics committee (12/NS/0105), in accordance with the tenets of the Declaration of Helsinki.
A family segregating cHL was identified through a female patient (IV-f) diagnosed with NSHL at age 22 under the clinical care of the Department of Haematology at Aberdeen Royal Infirmary, UK (Figure 4). A detailed family history taken from this index case revealed that three members of her extended family had also been diagnosed with NSHL (III-a; III-g, IV-b), all before age 35 (Table 1). During the time the family has been under investigation, IV-g was diagnosed with stage 4B cHL at age 25. For four of the five affected family members (III-g, IV-f, IV-b, IV-g) the diagnosis of NSHL was confirmed by histological and immunohistochemical studies through reference to patient notes and diagnostic pathology. Tumors from IV-f and IV-g were EBV-negative (Table 1) by immunohistochemistry (LMP1) or in situ hybridization (EBER). In all family members, complete remission was attained with first-line standard treatment (i.e. chemotherapy ± radiotherapy). None had a significant medical history or past history of cancers and IV-f was found to have a normal constitutional chromosome karyotype.
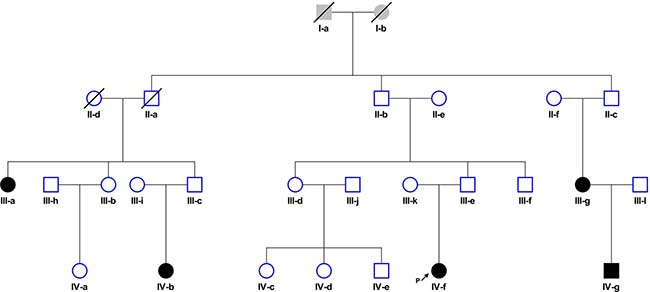
Figure 4: Family segregating cHL: Circles, female and squares, male. Filled-in symbols represent individuals with nodular sclerosis Hodgkin lymphoma. Slash indicates individual is deceased. Arrow indicates proband.
Genetic analysis of affected family members
Constitutional DNA was extracted from EDTA-venous blood or saliva samples using either QiaAMP DNA mini kits (Qiagen, UK) or prep-IT purification (DNAGenotek, Canada) kits and quantified by Quant-IT Picogreen (Invitrogen, UK). Family members were genotyped using either Affymetrix 6.0 chips (III-g, IV-b, IV-f, IV-g) or Affymetrix 500K (III-a) mapping arrays. CEL intensity files were processed using Affymetrix Power Tools (APT v1.16.1) and PLINK used to harmonise genotypes. Mendelian errors were removed and SNPs with high heterozygosity spaced ~0.3Mb apart selected using linkdatagen were used for linkage analysis [39]. Non-parametric linkage analysis was performed using MERLIN [40].
Typing of classical HLA alleles was performed using SeCore sequencing (Life Technologies, UK) in conjunction with Applied Biosystems 3130XL technology and analysed using uTYPE 6.0 software (Life Technologies) with IMGT as reference (version 3.14.0).
For WES of germline DNA from III-a, IV-b, IV-f and IV-g was fragmented using a Covaris E Series instrument (Covaris, United States). Indexed paired-end libraries were prepared using the SureSelect Human All Exon 50Mb (Agilent, United States) and 2 × 100 bp sequencing performed using Illumina HiSeq2000 technology (Illumina, United States). Paired end fastq files were extracted using CASAVA software (v.1.8.1, Illumina) and aligned to build 37 (hg19) of the human reference genome using Stampy and BWA software [41, 42]. Alignments were processed using the Genome Analysis Tool Kit (GATK) pipeline [43]. We imposed GATK internal calling thresholds and required a genotyping quality (GQ) of ≥ 30. Only non-silent variants were considered for analysis (i.e. missense, nonsense, frameshift, in-frame insertion/deletions, splice donor/acceptors, and initiator codon variants). Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP) [44], 1000 genomes project [45] and dbSNP [46] were used as population frequency references. Functional consequences of missense changes were predicted using SIFT [20] and POLYPHEN [21] algorithms. Sanger confirmation of sequence changes was carried out by using Big-Dye Ver 3.1 chemistry implemented on an ABI3730xl (Applied Biosystems, Foster City, USA).
Detailed studies of SLC26A6 and FAM107A
Immunohistochemistry of SLC26A6 and FAM107A were performed on formalin-fixed, paraffin-embedded material lymph node biopsies from IV-f and sporadic cHL (n = 5) patients using primary antibodies to SLC26A6 (Sigma) or FAM107A (Biorbyt) and DAB chromogen (Dako).
SLC26A6, KLHDC8B and FAM107A RNA expression was evaluated in lymphoblastoid cell lines (LCLs) derived from III-g, IV-f and IV-g and healthy controls (n = 10), HL cells (L428, L1236, HD-MyZ, KM-H2, HDML-2) and other cell lines (L540, L591, A2780, HeLa, SU-DHL1). RNA was extracted using the RNeasy mini kit (Qiagen, UK) and qRT-PCR performed using a Roche LightCycler 480 thermal cycler. Assays were performed in triplicate with GAPDH as internal control.
Association at 2q35-37, 3p14-22, 21q22
To evaluate the association between common genetic variation at chromosomes 2q35-37, 3p14-22, 21q22 and cHL we made use of data previously generated on two non-overlapping case-control series of Northern European ancestry, which had been the subject of a previous GWAS [14]. The UK GWAS was based on 589 cases ascertained through Royal Marsden Hospitals National Health Service Trust Family History study during 2004–2008. Individuals from the 1958 Birth Cohort and National Blood Service served as controls [47]. The UK-NSHLG GWAS was based on 1,612 cases ascertained through the National Study of Hodgkin lymphoma Genetics [14]. Individuals from the BCAC and PRACTICAL consortia served as controls [48]. Collection of blood samples and clinical information from subjects was undertaken with informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki. Full details of the genotyping and quality control are detailed in previously published work [14]. Briefly, we have previously confirmed an absence of systematic genetic differences between cases and controls and shown no significant evidence of population stratification in these sample sets. GWAS data were imputed to >10 million SNP with IMPUTE2 v2.392 software [49], using a merged reference panel consisting of data from 1000 Genomes Project (phase 1 integrated release 3, March 2012) [45] and UK10K (ALSPAC, EGAS00001000090/EGAD00001000195 and TwinsUK EGAS00001000108/EGAS00001000194 studies) [50]. Tests of association between SNP genotype and cHL were performed under an additive genetic model in SNPTESTv2.5 [51]. Meta-analyses were performed under a fixed-effects model using META v1.6101 [52].
CONFLICTS OF INTEREST
None.
REFERENCES
1. Curado M, Edwards B, Shin H, Storm H, Ferlay J, Heanue M, Boyle P. Cancer Incidence in Five Continents Vol. IX: WHO Press. 2008.
2. Mathas S, Hartmann S, Kuppers R. Hodgkin lymphoma: Pathology and biology. Semin Hematol. 2016; 53:139–47. https://doi.org/10.1053/j.seminhematol.2016.05.007.
3. Kuppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009; 9:15–27. https://doi.org/10.1038/nrc2542.
4. Cartwright R, McNally R, Roman E, Simpson J, Thomas J. Incidence and time trends in Hodgkin’s disease: from parts of the United Kingdom (1984–1993). Leuk Lymphoma. 1998; 31:367–77. https://doi.org/10.3109/10428199809059230.
5. Weiss LM, Strickler JG, Warnke RA, Purtilo DT, Sklar J. Epstein-Barr viral DNA in tissues of Hodgkin’s disease. Am J Pathol. 1987; 129:86–91.
6. Kharazmi E, Fallah M, Pukkala E, Olsen JH, Tryggvadottir L, Sundquist K, Tretli S, Hemminki K. Risk of familial classical Hodgkin lymphoma by relationship, histology, age, and sex: a joint study from five Nordic countries. Blood. 2015; 126:1990–5. https://doi.org/10.1182/blood-2015-04-639781.
7. Paltiel O, Schmit T, Adler B, Rachmilevitz EA, Polliack A, Cohen A, Haim N, Shachar MB, Epelbaum R, Barchana M, Cohen R, Ben Yehuda D. The incidence of lymphoma in first-degree relatives of patients with Hodgkin disease and non-Hodgkin lymphoma: results and limitations of a registry-linked study. Cancer. 2000; 88:2357–66.
8. Mack TM, Cozen W, Shibata DK, Weiss LM, Nathwani BN, Hernandez AM, Taylor CR, Hamilton AS, Deapen DM, Rappaport EB. Concordance for Hodgkin’s Disease in Identical Twins Suggesting Genetic Susceptibility to the Young-Adult Form of the Disease. N Engl J Med. 1995; 332:413–9. https://doi.org/10.1056/nejm199502163320701.
9. Urayama KY, Jarrett RF, Hjalgrim H, Diepstra A, Kamatani Y, Chabrier A, Gaborieau V, Boland A, Nieters A, Becker N, Foretova L, Benavente Y, Maynadie M, et al. Genome-wide association study of classical Hodgkin lymphoma and Epstein-Barr virus status-defined subgroups. J Natl Cancer Inst. 2012; 104:240–53. https://doi.org/10.1093/jnci/djr516.
10. Enciso-Mora V, Broderick P, Ma Y, Jarrett RF, Hjalgrim H, Hemminki K, van den Berg A, Olver B, Lloyd A, Dobbins SE, Lightfoot T, van Leeuwen FE, Forsti A, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet. 2010; 42:1126–30. http://www.nature.com/ng/journal/v42/n12/abs/ng.696.html.
11. Amiel J. (1967). Study of the leukocyte phenotypes in Hodgkin’s disease.
12. Diepstra A, Niens M, Vellenga E, van Imhoff GW, Nolte IM, Schaapveld M, van der Steege G, van den Berg A, Kibbelaar RE, te Meerman GJ, Poppema S. Association with HLA class I in Epstein-Barr-virus-positive and with HLA class III in Epstein-Barr-virus-negative Hodgkin’s lymphoma. Lancet. 2005; 365:2216–24. https://doi.org/10.1016/s0140-6736(05)66780-3.
13. Moutsianas L, Enciso-Mora V, Ma YP, Leslie S, Dilthey A, Broderick P, Sherborne A, Cooke R, Ashworth A, Swerdlow AJ, McVean G, Houlston RS. Multiple Hodgkin lymphoma-associated loci within the HLA region at chromosome 6p21.3. Blood. 2011; 118:670–4. https://doi.org/10.1182/blood-2011-03-339630.
14. Sud A, Thomsen H, Law PJ, Försti A, da Silva Filho MI, Holroyd A, Broderick P, Orlando G, Lenive O, Wright L, Cooke R, Easton D, Pharoah P, et al. Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat Commun. 2017; 8:1892. https://doi.org/10.1038/s41467-017-00320-1
15. Thomsen H, da Silva Filho MI, Forsti A, Fuchs M, Ponader S, von Strandmann EP, Eisele L, Herms S, Hofmann P, Sundquist J, Engert A, Hemminki K. Heritability estimates on Hodgkin’s lymphoma: a genomic- versus population-based approach. Eur J Hum Genet. 2015; 23:824–30. https://doi.org/10.1038/ejhg.2014.184.
16. Cozen W, Timofeeva MN, Li D, Diepstra A, Hazelett D, Delahaye-Sourdeix M, Edlund CK, Franke L, Rostgaard K, Van Den Berg DJ, Cortessis VK, Smedby KE, Glaser SL, et al. A meta-analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus. Nat Commun. 2014; 5: 3856. https://doi.org/10.1038/ncomms4856.
17. Frampton M, da Silva Filho MI, Broderick P, Thomsen H, Forsti A, Vijayakrishnan J, Cooke R, Enciso-Mora V, Hoffmann P, Nothen MM, Lloyd A, Holroyd A, Eisele L, et al. Variation at 3p24.1 and 6q23.3 influences the risk of Hodgkin’s lymphoma. Nat Commun. 2013; 4: 2549. https://doi.org/10.1038/ncomms3549.
18. Rotunno M, McMaster ML, Boland J, Bass S, Zhang X, Burdette L, Hicks B, Ravichandran S, Luke BT, Yeager M, Fontaine L, Hyland PL, Goldstein AM, et al. Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene. Haematologica. 2016; 101:853–60. https://doi.org/10.3324/haematol.2015.135475.
19. Salipante SJ, Mealiffe ME, Wechsler J, Krem MM, Liu Y, Namkoong S, Bhagat G, Kirchhoff T, Offit K, Lynch H, Wiernik PH, Roshal M, McMaster ML, et al. Mutations in a gene encoding a midbody kelch protein in familial and sporadic classical Hodgkin lymphoma lead to binucleated cells. Proc Natl Acad Sci U S A. 2009; 106:14920–5. https://doi.org/10.1073/pnas.0904231106.
20. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003; 31:3812–4. https://doi.org/10.1093/nar/gkg509.
21. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet. 2013; Chapter 7:Unit7.20. https://doi.org/10.1002/0471142905.hg0720s76.
22. Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J. Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics. 2000; 70:102–12. https://doi.org/10.1006/geno.2000.6355.
23. Lohi H, Lamprecht G, Markovich D, Heil A, Kujala M, Seidler U, Kere J. Isoforms of SLC26A6 mediate anion transport and have functional PDZ interaction domains. Am J Physiol Cell Physiol. 2003; 284: C769–C79. https://doi.org/10.1152/ajpcell.00270.2002.
24. Wang L, Darling J, Zhang JS, Liu W, Qian J, Bostwick D, Hartmann L, Jenkins R, Bardenhauer W, Schutte J, Opalka B, Smith DI. Loss of expression of the DRR 1 gene at chromosomal segment 3p21.1 in renal cell carcinoma. Genes Chromosomes Cancer. 2000; 27:1–10.
25. Yamato T, Orikasa K, Fukushige S, Orikasa S, Horii A. Isolation and characterization of the novel gene, TU3A, in a commonly deleted region on 3p14.3—>p14.2 in renal cell carcinoma. Cytogenet Cell Genet. 1999; 87:291–5. https://doi.org/15452.
26. van den Boom J, Wolter M, Blaschke B, Knobbe CB, Reifenberger G. Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int J Cancer. 2006; 119:2330–8. https://doi.org/10.1002/ijc.22108.
27. Chan AS, To KF, Lo KW, Mak KF, Pak W, Chiu B, Tse GM, Ding M, Li X, Lee JC, Huang DP. High frequency of chromosome 3p deletion in histologically normal nasopharyngeal epithelia from southern Chinese. Cancer Res. 2000; 60:5365–70.
28. Maitra A, Wistuba II, Washington C, Virmani AK, Ashfaq R, Milchgrub S, Gazdar AF, Minna JD. High-resolution chromosome 3p allelotyping of breast carcinomas and precursor lesions demonstrates frequent loss of heterozygosity and a discontinuous pattern of allele loss. Am J Pathol. 2001; 159:119–30. https://doi.org/10.1016/s0002-9440(10)61679-3.
29. Oh JJ, Razfar A, Delgado I, Reed RA, Malkina A, Boctor B, Slamon DJ. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res. 2006; 66:3419–27. https://doi.org/10.1158/0008-5472.can-05-1667.
30. de Oliveira MM, de Oliveira SF, Lima RS, de Andrade Urban C, Cavalli LR, de Souza Fonseca Ribeiro EM, Cavalli IJ. Differential loss of heterozygosity profile on chromosome 3p in ductal and lobular breast carcinomas. Hum Pathol. 2012; 43:1661–7. https://doi.org/10.1016/j.humpath.2011.12.008.
31. Hartmann S, Martin-Subero JI, Gesk S, Husken J, Giefing M, Nagel I, Riemke J, Chott A, Klapper W, Parrens M, Merlio JP, Kuppers R, Brauninger A, et al. Detection of genomic imbalances in microdissected Hodgkin and Reed-Sternberg cells of classical Hodgkin’s lymphoma by array-based comparative genomic hybridization. Haematologica. 2008; 93:1318–26. https://doi.org/10.3324/haematol.12875.
32. Steidl C, Telenius A, Shah SP, Farinha P, Barclay L, Boyle M, Connors JM, Horsman DE, Gascoyne RD. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood. 2010; 116:418–27. https://doi.org/10.1182/blood-2009-12-257345.
33. Szeles A. Fluorescence in situ hybridization (FISH) in the molecular cytogenetics of cancer. Acta Microbiol Immunol Hung. 2002; 49:69–80. https://doi.org/10.1556/AMicr.49.2002.1.7.
34. Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, Liang Y, Rivkin E, Wang J, Whitty B, Wong-Erasmus M, Yao L, Kasprzyk A. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database (Oxford). 2011; 2011: bar026. https://doi.org/10.1093/database/bar026.
35. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010; 42:790–3. http://www.nature.com/ng/journal/v42/n9/abs/ng.646.html.
36. O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, MacKenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011; 43:585–9. http://www.nature.com/ng/journal/v43/n6/abs/ng.835.html.
37. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339:959–61. https://doi.org/10.1126/science.1230062.
38. Lupiáñez Darío G, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz John M, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell. 2015; 161:1012–25. https://doi.org/10.1016/j.cell.2015.04.004.
39. Bahlo M, Bromhead CJ. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009; 25:1961–2. https://doi.org/10.1093/bioinformatics/btp313.
40. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002; 30:97–101. https://doi.org/10.1038/ng786.
41. Lunter G, Goodson M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Research. 2011; 21: 936–9. https://doi.org/10.1101/gr.111120.110.
42. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
43. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303. https://doi.org/10.1101/gr.107524.110.
44. NHLBI GO Exome Sequencing Project (ESP). (Seattle, WA, USA: Exome Variant Server).
45. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, and 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015; 526:68–74. https://doi.org/10.1038/nature15393.
46. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–11.
47. Wellcome Trust Case Control Consortium. Nature. 2007; 447:661–78. https://doi.org/http://www.nature.com/nature/journal/v447/n7145/suppinfo/nature05911_S1.html.
48. Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, Casey G, Hunter DJ, Sellers TA, Gruber SB, Dunning AM, Michailidou K, Fachal L, et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol Biomarkers Prev. 2017; 26:126–35. https://doi.org/10.1158/1055-9965.epi-16-0106.
49. Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009; 5: e1000529. https://doi.org/10.1371/journal.pgen.1000529.
50. Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, Danecek P, Malerba G, Trabetti E, Zheng HF, Gambaro G, Richards JB, Durbin R, et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nature Communications. 2015; 6: 8111. https://doi.org/10.1038/ncomms9111.
51. Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007; 39:906–13. https://doi.org/http://www.nature.com/ng/journal/v39/n7/suppinfo/ng2088_S1.html.
52. Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, Berrettini W, Knouff CW, Yuan X, Waeber G, Vollenweider P, Preisig M, Wareham NJ, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010; 42:436–40. https://doi.org/http://www.nature.com/ng/journal/v42/n5/suppinfo/ng.572_S1.html.