
INTRODUCTION
Androgen receptor (AR) signaling is crucial for the growth and development of normal and malignant prostate cells. In fact, androgen deprivation therapy (ADT) with GnRH agonists and antagonists is an essential step in prostate cancer treatment [1]. However, ADT eventually fails twelve to eighteen months after treatment initiation leading to the development of castration-resistant prostate cancer (CRPC) [1]. Second generation AR-targeting drugs enzalutamide and abiraterone are approved for the treatment of metastatic CRPC before and after chemotherapy with docetaxel. While enzalutamide blocks the ligand-binding domain of AR and thus prevents the binding of its natural androgen ligands [2], abiraterone inhibits cytochrome P450 17A1, which is required for adrenal and intratumoral androgen production, and therefore suppresses the androgen level in the tumor [2]. However, primary and secondary resistance to these drugs have been observed [3]. Indeed, response to the second AR-targeted drug in CRPC is decreased and a decline of progression-free survival has been reported with each additional treatment line [4].
A potential mechanism of resistance to enzalutamide and abiraterone is the expression of alternative mRNA splicing variants of the AR [5], e.g. AR-V7 [6, 7]. In contrast to native full-length AR (AR-FL), the AR-V7 lacks the C-terminal ligand binding-domain, which prevents the binding of androgens. At the same time, AR-V7 exerts its transcriptional functions without androgen binding resulting in a constitutive activation of the AR transcriptional program. This ultimately results in enhanced prostate cancer cell proliferation and survival despite enzalutamide and abiraterone treatment [2, 6].
Pro-survival autophagy is another mechanism of drug resistance in human prostate cancer cells [8, 9]. It helps cancer cells to overcome stress conditions, such as chemo- and radiotherapy [10]. In contrast, inhibitors of autophagy may increase anticancer therapy outcome [10].
Recently, we reported on the activity and the mechanism of action of semi-synthetic marine compound rhizochalinin (1) in human CRPC cells (Figure 1). Rhizochalinin decreases AR-V7 expression accompanied by a resensitization of CRPC cells to enzalutamide [11]. Additionally, the marine compound inhibits pro-survival autophagy [11, 12]. In the current study, we chemically modified the structure of rhizochalin in order to evaluate its structure-activity relationships and optimize anticancer properties. Therefore, the following features of rhizochalinin and its derivates were determined: i) main mechanisms of drug-induced tumor suppression, such as cytotoxic action, antiproliferative activity, and apoptosis induction; ii) inhibition of pro-survival autophagy and iii) inhibition of AR signaling [8, 9].
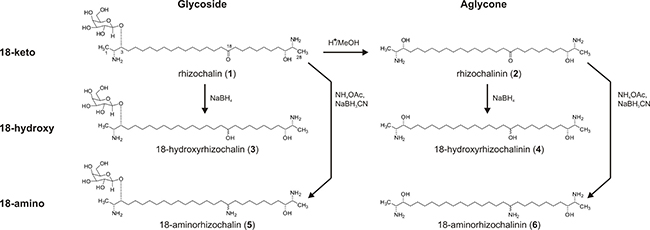
Figure 1: Schema of synthesis of the derivatives (2)–(6) from rhizochalin (1).
RESULTS AND DISCUSSION
Chemistry
Analyses of structure-activity relationships and further structure optimization are essential steps in the drug development process. For rhizochalin (1) (Figure 1) [13] and similar compounds it has been described that sugar elimination may lead to the increased pro-apoptotic activity [14–16]. The change in the molecule polarity due to an additional reduction of the keto group, or the introduction of nitrogen in the molecule may also affect the biological activity, including pro-apoptotic activity of the drug, as well its effect on autophagy [17]. Thus, taking the chemical properties and stability of the rhizochalin molecule into account we applied the following approaches in order to modify its structure: i) elimination of the sugar residue; ii) reduction of 18-keto group to hydroxyl group; or iii) consecutive reduction and amination of 18-keto group (Figure 1). Rhizochalinin (2) (rhizochalin aglycon) [11, 18] was synthesized from rhizochalin (1) [13] via hydrolysis as reported previously [18] (Figure 1). The four new derivatives (3)–(6) were synthesized from rhizochalin (1) and rhizochalinin (2) via hydrogenation (compounds (3) and (4)) and reductive amination (compounds (5) and (6)). All compounds were purified by reversed-phase flash column chromatography or reversed-phase HPLC and elucidated by spectroscopic methods as follows (for detailed 1H NMR data see Supplementary Figure 1):
18-Hydroxyrhizochalin (3). Amorphous solid (61%); 1H NMR (CD3OD, 500 MHz) δ 4.32 (d, J = 7.4 Hz, 1H), 3.79 (d, J = 3.1 Hz, 1H), 3.74 (dd, J = 8.0, 11.7 Hz, 1H), 3.71 (d, J = 4.1, 11.8 Hz, 1H), 3.66 (ddd, J = 3.3, 7.3, 9.8 Hz, 1H), 3.53 (dd, J = 6.5, 14.0 Hz, 1H), 3.51 (dd, J = 7.3, 9.7 Hz, 1H), 3.49 (m, 1H), 3.46 (dd, J = 3.1, 9.7 Hz, 1H), 3.39 (m, 1H), 3.15 (p, J = 6.7 Hz, 1H), 3.01 (p, J = 6.7 Hz, 1H), 1.68 (m, 1H), 1.54 (m, 1H), 1.53 (m, 1H), 1.42 (m, 2H), 1.39 (m, 1H), 1.37 (m, 2H), 1.27–1.30 (br.s, 17H), 1.26 (d, J = 6.6 Hz, 3H), 1.22 (d, J = 6.6 Hz, 3H); 13C NMR (CD3OD, 125MHz): δ 104.6 (C-1’), 81.4 (C-3), 77.6 (C-5’), 75.2 (C-3’), 74.5 (C-26), 73.3 (C-2’), 73.1 (C-18), 71.1 (C-4’), 63.6 (C-6’), 53.9 (C-27), 52.5 (C-2), 39.1 (C-17), 39.0 (C-19), 35.3 (C-25), 33.3 (C-4), 31.3-31.5 (C-5-C-16, C-20-C-24), 17.2 (C-28), 16.3 (C-1); HRESIMS: m/z 635.52065 [M+H]+ (calcd for C34H71N2O8, 635.52049).
18-Hydroxyrhizochalinin (4). Amorphous solid (97%); 1H NMR (CD3OD, 500 MHz) δ 3.50 (m, 1H), 3.43 (m, 2H), 3.08 (sept, J = 6.7 Hz, 2H), 1.55 (m, 1H), 1.53 (m, 1H), 1.42 (m, 5H), 1.41 (m, 1H), 1.32 (m, 3H), 1.29 (br.s, 15H), 1.26 (d, J = 6.8 Hz, 6H); 13C NMR (CD3OD, 125MHz): δ 73.8 (C-3, C-26), 73.1 (C-18), 54.1 (C-2, C-27), 39.1 (C-17, C-19), 35.3 (C-25), 26.9 (C-4), 31.2-31.5 (C-6-C-16, C-20-C-23), 27.4 (C-5, C-24), 16.6 (C-1, C-28); HRESIMS: m/z 473.4687 [M+H]+ (calcd for C28H61N2O2, 473.4678).
18-Aminorhizochalin (5). Amorphous solid (79%); 1H NMR (CD3OD, 500 MHz) δ 4.32 (d, J = 7.4 Hz, 1H), 3.79 (d, J = 3.1 Hz, 1H), 3.74 (dd, J = 8.0, 11.7 Hz, 1H), 3.71 (d, J = 4.1, 11.8 Hz, 1H), 3.66 (ddd, J = 3.3, 7.3, 9.8 Hz, 1H), 3.54 (m, 1H), 3.51 (dd, J = 7.4, 9.8 Hz, 1H), 3.46 (dd, J = 3.1, 9.7 Hz, 1H), 3.39 (m, 1H), 3.15 (p, J = 6.5 Hz, 1H), 3.07 (p, J = 6.5 Hz, 1H), 3.00 (p, J = 6.5 Hz, 1H), 1.68 (m, 1H), 1.60 (m, 2H), 1.55 (m, 1H), 1.53 (m, 3H), 1.39 (m, 1H), 1.37 (m, 4H), 1.27–1.30 (br.s, 15H), 1.26 (d, J = 6.6 Hz, 3H), 1.21 (d, J = 6.8 Hz, 3H); 13C NMR (CD3OD, 125MHz): δ 104.6 (C-1’), 81.4 (C-3), 77.6 (C-5’), 75.2 (C-3’), 74.6 (C-26), 73.3 (C-2’), 71.1 (C-4’), 63.6 (C-6’), 53.9 (C-27), 53.6 (C-18), 52.6 (C-2), 35.0 (C-17), 35.3 (C-19, C-25), 33.5 (C-4), 31.3-31.5 (C-5-C-15, C-21-C-24), 26.9 (C-16, C-20), 17.3 (C-28), 16.3 (C-1); HRESIMS: m/z 634.5357 [M+H]+ (calcd for C34H72N3O7, 634.5365).
18-Aminorhizochalinin (6). Amorphous solid (97%); 1H NMR (CD3OD, 500 MHz) δ 3.39 (m, 2H), 3.08 (p, J = 6.4 Hz, 1H), 3.01 (m, 2H), 1.60 (m, 2H), 1.55 (m, 2H), 1.54 (m, 2H), 1.39 (m, 2H), 1.27–1.30 (br.s, 15H), 1.22 (d, J = 6.8 Hz, 6H); 13C NMR (CD3OD, 125 MHz): δ 74.5 (C-3, C-26), 53.9 (C-2, C-27), 53.6 (C-18), 35.3 (C-4, C-25), 35.0 (C-17, C-19), 31.1-31.4 (C-6-C-16, C-20-C-23), 17.3 (C-1), 17.2 (C-28); HRESIMS: m/z 472.4828 [M+H]+ (calcd for C28H62N3O2, 472.4837).
Biology
Cytotoxicity
First, we evaluated the cytotoxic, antiproliferative and pro-apoptotic effects of the substances (1)–(6). The synthesized compounds were tested in human prostate cancer cell lines PC-3, DU145, LNCaP, 22Rv1, and VCaP. All cell lines except LNCaP cells are abiraterone/enzalutamide-resistant due to the absence of AR (PC-3 and DU145) or the presence of AR-V7 (22Rv1 and VCaP). Additionally, PC-3 cells have previously been reported to be docetaxel-resistant [19]. Remarkably, the compounds exhibited cytotoxic activity in all cells lines at micro- or nanomolar concentrations [20, 21] (Table 1). The aglycons (2), (4) and (6) possessed ~10-fold stronger in vitro activity when compared to glycosides (1), (3) and (5) (Figure 2A). Additionally, an increase of cytotoxicity was observed in the row 18-amino < 18-hydroxy < 18-keto derivatives (Table 1, Figure 2A).
Table 1: Cytotoxic activity of compounds (1)–(6) in human prostate cancer cells after 48 h of treatment
Compound |
IC50, μM |
||||
|---|---|---|---|---|---|
PC-3 |
DU145 |
LNCaP |
22Rv1 |
VCaP |
|
rhizochalin (1) |
16.55 ± 1.37 |
10.75 ± 1.48 |
7.88 ± 2.4 |
7.37 ± 0.69 |
5.81 ± 0.23 |
rhizochalinin (2) |
1.14 ± 0.04 |
1.05 ± 0.02 |
1.69 ± 0.38 |
0.87 ± 0.33 |
0.42 ± 0.11 |
18-hydroxyrhizochalin (3) |
22.62 ± 0.3 |
24.38 ± 0.38 |
9.34 ± 0.57 |
11 ± 1.14 |
15.89 ± 5.23 |
18-hydroxyrhizochalinin (4) |
2.72 ± 0.13 |
2.13 ± 0.19 |
3.55 ± 0.45 |
1.77 ± 0.99 |
0.61 ± 0.08 |
18-aminorhizochalin (5) |
46.57 ± 13.78 |
19.29 ± 13.08 |
8.97 ± 2.47 |
14.21 ± 5.09 |
18.59 ± 3.46 |
18-aminorhizochalinin (6) |
3.39 ± 0.30 |
7.82 ± 1.12 |
9.31 ± 2.12 |
3.46 ± 1.2 |
2.67 ± 0.52 |
Data are presented as mean ± SEM.
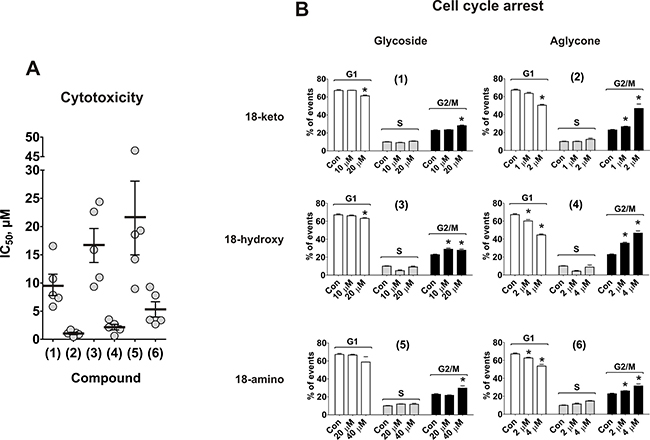
Figure 2: Effect on viability and cell cycle progression of human prostate cancer cells. (A) MTT assay: each dot represents IC50 value (μM) of the compound against certain cancer cell line. The values are equal to those represented in the Table 1. (B) PC-3 cells were treated with compounds (1)–(6) for 48 h and the cell cycle phase distribution was quantified using the Cell Quest Pro software. *p < 0.05 (Student’s t-test).
Cell cycle progression
To investigate the antiproliferative activity of the compounds we examined their effects on cell cycle progression. Mild, but statistically significant G2/M arrest was observed with a 48 h treatment of all compounds in human cancer PC-3 cells. The effect was most pronounced for aglycones (2) and (4) (Figure 2B). However, in contrast to PC-3 cells, no pronounced cell cycle arrest was observed in 22Rv1 cells (data now shown). This may result from a different genetic background of the cells. Thus, rhizochalin and its derivatives exhibit mainly cytotoxic, rather than antiproliferative activity.
Induction of apoptosis
Next, effects on induction of apoptosis were examined. Treatment with rhizochalinin and its derivatives increased the sub-G1 population of PC-3 cells, indicating DNA fragmentation (Figure 3A). Additionally, caspase-3/7 activation was detected in cells treated for 48 h (Figure 3B).
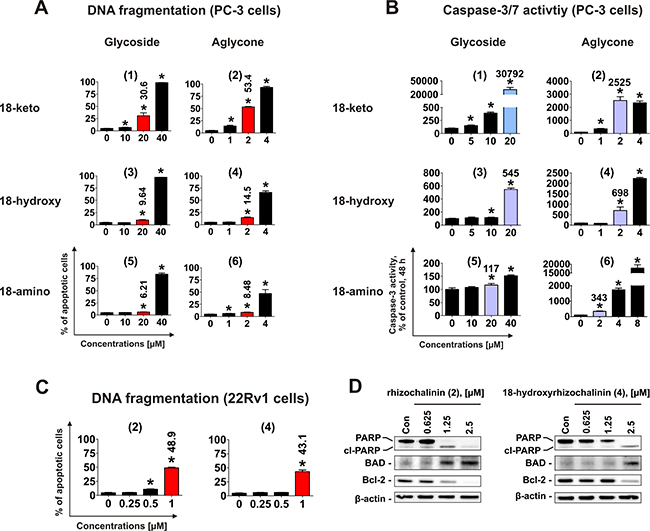
Figure 3: Pro-apoptotic activity of the compounds (1)–(6) in human cancer cells. (A) Analysis of DNA fragmentation in PC-3 cells treated with compounds (1)–(6) for 48 h at different concentrations. The number of cells with fragmented DNA was assessed with flow cytometry and assumed as the sub-G1 population in cycle analysis. The effects on DNA fragmentation were compared at either 20 μM or 2 μM (red bars on the graphs). (B) Analysis of caspase-3/7 activity in PC-3 cells treated with the investigated compounds at different concentrations for 48 h. The effects on caspase-3/7 activity were compared at either 20 μM or 2 μM (blue bars on the graphs). (C) FACS analysis of DNA fragmentation in 22Rv1 cells treated with compounds (2) and (4) for 48 h at different concentrations. The effects on DNA fragmentation were compared at 1 μM (red bars on the graphs). (D) Analysis of several pro- and anti-apoptotic protein expression in 22Rv1 cells treated with compounds (2) and (4) for 48 h. *p < 0.05 (Student’s t-test).
To identify the most active derivative, we examined the ability of the different compounds to induce the DNA fragmentation and caspase-3/7 activation. The glucose-containing compounds (1), (3), and (5) are less cytotoxic (i.e. having higher IC50s) in comparison with the correspondent aglycons (2), (4), and (6) (Figure 2A, Table 1). Therefore for the further experiments, 20 μM for glycoside derivatives (1), (3) and (5); and 2 μM for aglycons (2), (4) and (6) have been used (Figure 3A, 3B). In accordance with the results of cytotoxicity assay (Figure 2A), 18-keto derivatives exhibited the most pronounced pro-apoptotic activity within the same glycoside or aglycone family, whereas 18-amino derivatives showed the weakest activity. Pro-apoptotic activity of aglycon derivatives (2), (4) and (6) was distinctly more pronounced when compared with glycosides (1), (3) and (5) (Figure 3A, 3B).
Rhizochalinin (2) and 18-hydroxyrhizochalinin (4) were identified to have the strongest cytotoxic effect exerted through the apoptotic mechanism (Figure 3A, 3B). Therefore, we additionally examined the pro-apoptotic effects of these derivatives in castration-resistant AR-V7-positive 22Rv1 cells. Similar to PC-3 cell line, we both compounds induced DNA fragmentation in this prostate cancer cell model (Figure 3C). Interestingly, the pro-apoptotic activity of the compounds (2) and (4) was stronger in 22Rv1 cells when compared to PC-3 cells (Figure 3C). We also examined the effects of the compounds on the expressional levels of several apoptosis-related Bcl-2 family proteins (Figure 3D). Up-regulation of the pro-apoptotic BAD and down-regulation of anti-apoptotic Bcl-2 were detected (Figure 3D), while Bax and Bcl-xL expression were not influenced significantly (data not shown). Finally, another apoptotic marker – PARP cleavage – was detected (Figure 3D). Taken together these results indicate apoptotic character of the drug-induced cells death, which is in line with the caspase-dependent apoptosis induced by rhizochalinin (2) in cancer cells, recently reported by us [11].
Effect on autophagy
In human cancer the role of autophagy is discussed controversially [22]. However, it is believed that autophagy is cytotoxic in very early stages of tumor development, but cytoprotective in late stages [22]. Indeed, autophagy leads to selective or non-selective bulk degradation of cellular proteins and organelles. This results in extra nutrients supply and helps cancer cells to overcome stressful conditions (lack of nutrients, chemo- and radiotherapy, etc.), ultimately leading to drug resistance [23, 24]. Thus, autophagy has been identified to be an important mediator of drug-resistance in human CRPC [23, 24]. Rhizochalinin (2) inhibits cytoprotective autophagy in human CRPC cells and thus may be capable to overcome drug resistance [11]. Consequently, we examined the effect of the novel derivatives on autophagy in prostate cancer PC-3 cells [25]. The expression of LC3B-I/II proteins is a well-established marker of autophagy [26]. In this model, an increased LC3B-II level after 48 h-treatment indicates autophagy inhibition, whereas LC3B-II decrease reflects induction of autophagy [27]. All compounds caused an increase of LC3B-II in PC-3 cells (Figure 4). Unlike to the cytotoxic and pro-apoptotic activity, the strongest autophagy inhibitory effect was observed for 18-hydroxy derivatives (3) and (4), and the weakest for 18-amino derivatives (5) and (6) (Figure 4).
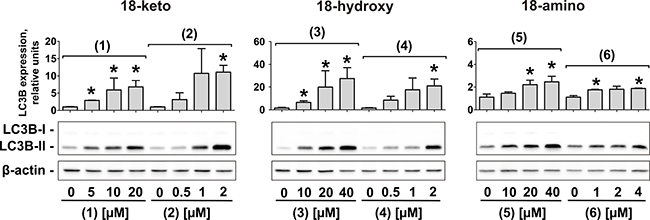
Figure 4: Inhibition of autophagy in human prostate cancer cells under treatment with the compounds (1)–(6). Western blotting analysis of LC3B-I/II proteins in PC-3 cells treated with different concentrations of the compounds (1) – (6) for 48 h. The signal intensity was quantified with Quantity One 4.6 software and normalized to the signal of β-actin. *p < 0.05 (Student’s t-test).
Effect on androgen receptor signaling
AR-V7 expression is accompanied by autoactivation of AR-signaling in prostate cancer cells, mediating resistance to AR-targeting drugs enzalutamide and abiraterone in vitro and in vivo [6, 20, 21]. In patients, AR-V7 expression is associated with drug resistance and poor prognosis [6]. Thus, treatment approaches which are capable of suppressing AR-V7 expression are of high interest.
Recently, we demonstrated that rhizochalinin (2) decreases the basal PSA expression, which is produced in non-DHT-stimulated 22Rv1 cells due to AR-V7 expression [11]. Therefore, we postulated that rhizochalinin suppresses AR-signaling [11]. Consequently, we examined the effect of the newly synthesized derivatives on the same biological target in 22Rv1 cells. The aglycon derivatives (2), (4) and (6) expectably decreased the basal PSA expression, while in contrast the glycosides (1), (3) and (5) surprisingly appeared to increase the PSA production (Figure 5). This finding suggests the particular importance of the sugar elimination for the AR-signaling suppressive properties of the synthesized derivatives.
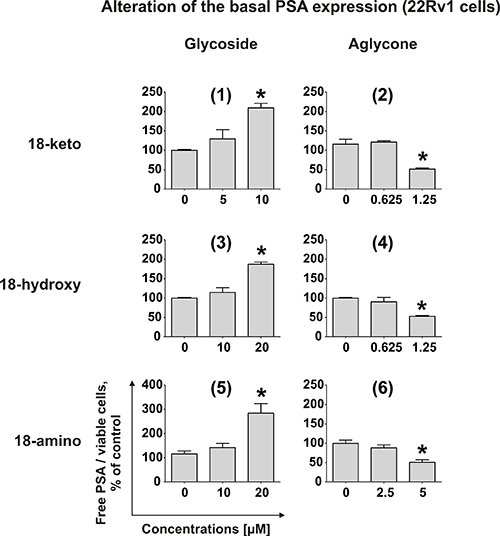
Figure 5: Effect on PSA protein expression. Effect of compounds (1)–(6) on PSA protein expression in 22Rv1 cells. The concentration of PSA in the culture supernatant was analyzed using ELISA and normalized to the number of viable cells. *p < 0.05 (Student’s t-test).
Taking together the results of the cytotoxic, antiproliferative, pro-apoptotic, autophagy inhibitory, and AR-targeting effects examination, rhizochalinin (2) and 18-hydroxyrhizochalinin (4) have been identified as the most promising derivatives in terms of anticancer activity in human CRPC cells (Table 2). Thus, these two molecules were chosen to further determine their ability to suppress AR-signaling in 22Rv1 cells. 22Rv1 cells are capable of both AR-FL- and AR-V7 expression (Figure 6A) and therefore are resistant to ADT and to the second generation AR-targeted therapies [20, 21]. We used the qPCR technique to examine the effects of derivatives (2) and (4) on the expression of mRNA levels of specific endogenous target genes of AR-V7 (genes AKT1 and UBE2C) (Figure 6B) and AR-FL (genes PSA, TMPRSS2, and FKBP5) (Figure 6C). The AR-FL pathway and the correspondent gene expression were activated by DHT treatment. Similar to the results reported in DHT non-stimulated 22Rv1 cells (see Figure 5), both compounds (2) and (4) significantly suppressed the DHT-stimulated expression of PSA mRNA (Figure 6C), as well as two other AR-FL-controlled genes – TMPRSS2 and FKBP5 (Figure 6C). Even more important, AR-V7-controlled AKT1 and UBE2C were also suppressed in 22Rv1 cells (Figure 6B). Remarkably, (2) and (4) down-regulated AR-V7 protein expression (Figure 6D). This may explain the suppression of AR-V7-dependent signaling (Figure 6D). In contrast, AR-V7 and AR-FL mRNA expression remained unaffected by the investigated drugs and DHT (Figure 6B, 6C). Taken together, rhizochalinin (2) and 18-hydroxyrhizochalinin (4) suppress both, AR-V7- and AR-FL-dependent signaling.
Table 2: Summary of biological activities of the compounds (1)–(6) in human prostate cancer cells
Actitiy* |
Compound |
|||||
|---|---|---|---|---|---|---|
(1) |
(2) |
(3) |
(4) |
(5) |
(6) |
|
Cytotoxicity |
++ |
+++ |
+ |
+++ |
+ |
++ |
Anti-proliferative activity |
+ |
++ |
+ |
++ |
+ |
+ |
Pro-apoptotic activity |
++ |
+++ |
+ |
+++ |
+ |
++ |
Autophagy inhibition |
++ |
++ |
+++ |
+++ |
+ |
+ |
AR signaling inhibition |
– |
+++ |
– |
+++ |
- |
++ |
*“+++” – high activity; “++” – moderate activity; “+” – low activity; “–“ – no activity.
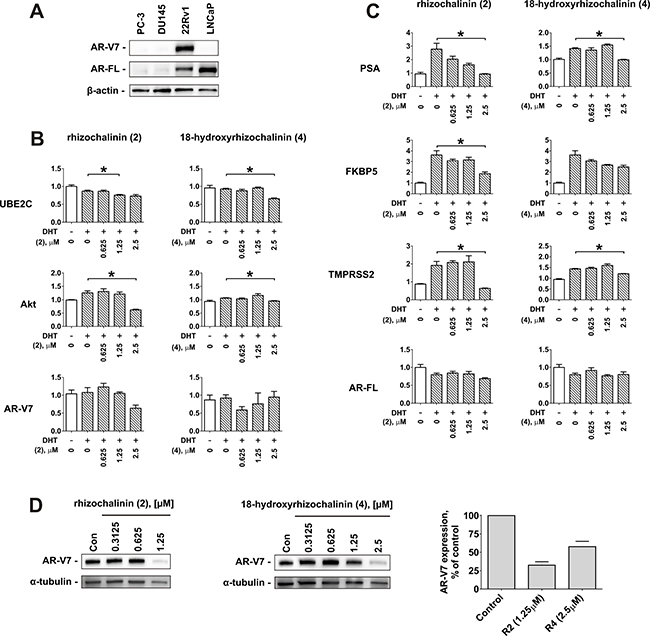
Figure 6: Effect of rhizochalinin (2) and 18-hydroxyrhizochalinin (4) on the expression of different genes, controlled by AR-FL and AR-V7. (A) Analysis of AR-V7 and AR-FL protein expression in different human prostate cancer cells lines. (B, C) mRNA expression levels of the genes controlled by AR-V7 (B) or AR-FL (C). 22Rv1 cells were pre-treated with the indicated concentrations of investigated compounds in 0.1% FBS/RPMI media for 30 min followed by co-treatment with 20 nM DHT for another 24 h. (D) Analysis of AR-V7 expression in 22Rv1 cells lines treated with compounds (2) and (4) for 48 h. Signal intensity was quantified with Quantity One 4.6 software and normalized to the signal of α-tubulin. Protein expression was analyzed by Western blotting. mRNA expression was analysed by qPCR. *p < 0.05 (Student’s t-test).
MATERIALS AND METHODS
Chemistry
The 1H and 13C NMR spectra were obtained using Bruker Avance III HD-500 spectrometer. Chemical shifts were referenced to the corresponding residual solvent signal (δH 3.30/δC 49.60 for CD3OD). ESI mass spectra were obtained on an Agilent 6510 Q-TOF LC-MS spectrometer by direct injection in MeOH. HPLC was performed using a Shimadzu Instrument equipped with a differential refractometer RID-10A and YMC-ODS-A (250 × 10 mm) column. TLC was performed on Silica plates (5–17 μm, Sorbfil, Russia), and visualization was accomplished by charring at 150° C with 10% (v/v) H2SO4 in EtOH.
General procedure for the isolation of rhizochalin (1) and synthesis of rhizochalinin (2)
Rhizochalin ((1), Figure 1) was isolated from the marine sponge Rhizochalina incrustata as described before [13]. Rhizochalinin (rhizochalin aglycon, (2), Figure 1), was synthesized from rhizochalin (1) via hydrolysis as reported previously [18].
General procedure for the synthesis of hydroxyderivatives (3) and (4)
An excess of NaBH4 was added to solution of compound (1) (69 mg) or (2) (31 mg) in 2 mL methanol and the reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was neutralized with acetic acid and the solvent was then evaporated under reduced pressure. The product was purified by YMC gel column flash chromatography using ethanol/water (0 → 60%) mixture as eluent and monitored by thin layer chromatography. The fractions that were eluted with 60% ethanol after reversed phase HPLC (70% ethanol/0.1% trifluoroacetic acid) yielded 18-hydroxyrhizochalin ((3), 42 mg) and 18-hydroxyrhizochalinin ((4), 30 mg) (Figure 1).
General procedure for the synthesis of aminoderivatives (5) and (6)
An excess of ammonium acetate and solution of (1) (73.6 mg) or (2) (31.0 mg) in 2 mL methanol were mixed. Then, an excess of sodium cyanoborohydride was added and the reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was neutralized with acetic acid and the solvent was evaporated under reduced pressure. The resulting product was further purified either by reversed phase HPLC using 65% ethanol/0.1% trifluoroacetic acid solvent system to afford 18-aminorhizochalin ((5), 58.2 mg) or by YMC gel ODS-A column flash chromatography using ethanol/water (0 → 60%) mixture as eluent to afford 18-aminorhizochalinin ((6), 30 mg).
Reagents and antibodies
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent, propidium iodide (PI) and dihydrotestosterone (DHT) were purchased from Sigma (Taufkirchen, Germany). For the protein detection the following antibodies were used: anti-AR-FL (Santa Cruz, sc-816, 1:200), anti-AR-V7 (abcam, #198394, 1:1000), anti-BAD (Cell Signaling, #9239, 1:1000), anti-Bax (Cell Signaling, #5023, 1:1000), anti-Bcl-2 (Cell Signaling, #2876, 1:1000), anti-Bcl-xL (Cell Signaling, #2764, 1:1000), anti-LC3B-I/II (Cell Signaling, #2775, 1:1000), anti-rabbit IgG-HRP (Cell Signaling, #7074, 1:5000), anti-mouse IgG-HRP (GE Healthcare, NXA931, 1:10000), anti-β-Actin-HRP (Santa Cruz, sc-1616, 1:200), anti-α-Tubulin (Sigma-Aldrich, T5168, 1:5000).
Cell lines and culture conditions
The human prostate cancer cells PC-3, DU145, LNCaP, 22Rv1, and VCaP were purchased from ATCC (Manassas, VA, USA). Cell lines were cultured according to the manufacturers’ protocols in the correspondent media as has been described before [11]. Cells were continuously kept in culture for a maximum of 3 months, and were routinely checked for contamination with mycoplasma and inspected microscopically for stable phenotype. The previous pharmacokinetics study revealed the effect of rhizochalinin (2) to be maximal after 48 h of treatment [11], therefore for most of experiments 48 h incubation was chosen. The current research was performed according to the Good Laboratory Practice regulations (GLPs).
In vitro MTT-based drug sensitivity assay
The in vitro cytotoxicity of the investigated compounds was evaluated using the MTT assay, which was performed as previously described [27, 28]. Cells were incubated with the drugs for 48 h.
Cell cycle and DNA fragmentation analysis
The cell cycle distribution was analyzed by flow cytometry using PI staining as described before [29]. Cells were pre-incubated overnight in six-well plates (0.2 × 106 cells/well) and then incubated with rhizochalin and its derivates for 48 h. Afterwards cells were trypsinized, fixed, stained with propidium iodide / RNase containing buffer, and analyzed with a BD Bioscience FACS Calibur analyzer (BD Bioscience, Bedford, MA, USA) and BD Bioscience Cell Quest Pro v.5.2.1. software (BD Bioscience, Bedford, MA, USA). Apoptotic cells containing fragmented DNA were detected as a sub-G1 population.
Caspase-3/7 activity assay
The enzymatic activities of caspase-3 and -7 were measured using Caspase-Glo® 3/7 Assay Kit (Promega) as described before [30]. In brief, 6000 cells per well were seeded in a 96-well white flat-bottom sterile plate, incubated overnight, and treated with the investigated substances at various concentrations for 48 h. Then the Caspase-Glo® 3/7 reagent was added to the plates and luminescence was measured using an Infinite F200PRO reader (TECAN, Männedorf, Switzerland). Cell viability was measured using the modified MTT assay as described before [30]: the treated cells were incubated with the MTT reagent for 2 h, the media was removed and the plates were dried, then the formazan crystals were dissolved in DMSO, the solution was transferred to new transparent plates and optical density was measured. The caspase-3/7 activity was normalized to the cell viability at the correspondent drug concentration.
Analysis of PSA expression
The analysis of free PSA expression was determined as described before [11]. 22Rv1 cells (0.4 × 106 cells/well) were seeded in 6-well plates, incubated overnight and the media was replaced with fresh drug-containing media. After 48 h incubation the aliquots were collected and extracellular human prostate-specific antigen (PSA) was measured in the supernatant by ELISA using the ProStatus™ PSA Free-/Total DELFIA® Kit (PerkinElmer, Turku, Finland). PSA concentrations were normalized to the number of viable cells in the correspondent wells, which was measured by trypan blue-based viability assays as described before [28].
Quantitative real-time PCR (qPCR)
Cells were seeded in Petri dishes (2 × 106 cells per ø 6 cm dish in 5 mL of media for 22Rv1) in 10% FBS/RPMI media and incubated overnight. Then the media was replaced with 5 mL of fresh 0.1% FBS/RPMI media. After 24 h of incubation the cells were treated with the investigated compounds for 30 min in 0.1% FBS/RPMI media followed by co-treatment with 20 nM DHT for another 24 h. Cells were harvested by scratching, pelleted, homogenized using QIAshredder (Cat. # 79654, QIAGEN, Hilden, Germany). Total RNA was isolated using PureLink® RNA Mini Kit (Cat. # 12183018A, Invitrogen, Carlsbad, CA, USA) with the on-column DNA digestion using PureLink™ DNase (Cat. # 12185-010, Invitrogen). RNA was diluted up to 30 μL and concentrations were measured. RNA was transcribed into cDNA using Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Cat. # K1642, Thermo Scientific, Vilnius, Lithuania). qPCR was performed using 2X KAPA SYBR FAST qPCR Master Mix Optimized for Roche LightCycler 480 (Cat. # KK4609, KAPA biosystems, Worburn, MA, USA) according to the manufacturer’s instructions. 2 pmol of primers and 20 ng of template cDNA were used per one reaction. Expressions of human AR-FL, AR-V7, PSA, AKT, FKBP5, TMPRSS2, UBE2C, and GAPDH genes were analyzed using the specific primers, synthesized by Eurofins MWG-Biotech AG (Ebersberg, Germany). Primer sequences and melting temperatures (Tm) are presented in Supplementary information, Supplementary Table 1. The PCR conditions were 30 sec 95° C, followed by 40 cycles of 15 sec 95° C, 5 sec Tm, and 26 sec 72° C (measurement of fluorescence). Melting curve analysis (10 sec 95° C, 60 sec 65° C and 1 sec 97° C) was performed directly after each PCR run. Relative expression was calculated using the 2–ΔΔCT method. To test statistical significance, data were analyzed by unpaired Student’s t-tests.
Protein preparation and western blotting
Preparation of protein extracts and Western blotting were performed as described previously with slight modifications [28]. In brief, 1 × 106 cells/well were seeded in Petri dishes (ø 6 cm, 5 mL/dish) and incubated overnight. Then, the media was substituted with drug-containing media and cells were incubated for 48 h. Cells were harvested, the proteins were extracted using the lysis buffer (1% NP-40 [v/v], 50 mM Tris-HCl (pH 7.6), 0.88% [w/v] NaCl, 0.25% [w/v] sodium cholate, 1 mM Na3VO4, 0.1 mM PMSF, 1 tablet/10 mL cOmplete Mini EDTA-free EASYpacks protease inhibitors cocktail (Roche, Mannheim, Germany)), subjected to electrophoresis and transferred to PVDF membrane. The membrane was incubated with the primary and secondary antibody. The signal was detected as described before [31]. The signal intensity was quantified with Quantity One 4.6 software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
The GraphPad Prism software v. 5.01 (GraphPad Prism software Inc., La Jolla, CA, USA) was used to perform the statistical analyses. Data are presented as mean ± SEM (standard error of the mean). The experiments were performed in triplicates and repeated at least three times. The unpaired Student’s t-test was used to compare the control group and the drug-treated group. Differences were considered to be statistically significant and marked with an asterisk (*) if p < 0.05.
CONCLUSIONS
In conclusion, the newly synthesized derivatives of marine natural compound rhizochalin exhibit structure-activity relationships with an increase of cytotoxic properties in the row 18-amino < 18-hydroxy < 18-keto derivatives. In general, aglycones were distinctly more active when compared with glycosides. In fact, sugar elimination was critical for the ability to suppress AR-signaling while all compounds inhibited cytoprotective autophagy.
Based on these results, rhizochalinin (2) and 18-hydroxyrhizochalinin (4) were identified as the most promising derivatives (Table 2). Both compounds suppressed the AR-FL- and AR-V7-depending signaling in human CRPC cells and induced cancer cell apoptosis, and therefore are promising new treatment options for castration-resistant, AR-V7-positive prostate cancer. Remarkably, we have recently shown the efficacy and low toxicity of rhizochalinin (2) in vivo in human castration-resistant subcutaneously xenotransplanted prostate cancer cells, whereas 18-hydroxyrhizochalinin (4) still awaits for the in vivo examinations [11]. The further structure optimization of the rhizochalin derivatives as well as development of its chemical synthesis from commercially available reagents is currently in progress. The new structures with improved biological and chemical properties as well as the synthesis are to be patented in order to promote further drug development.
Abbreviations
22Rv1: ADT-resistant, AR/AR-V7 positive prostate cancer cell line [21]; AR: Androgen receptor; AR-FL: Full-length androgen receptor; AR-V7: Androgen receptor splice variant 7; ADT: Androgen deprivation therapy; ATCC: American type culture collection; DU145: ADT-resistant, AR-negative prostate cancer cell line [32]; LC3B: Microtubule-associated protein light chain 3; LNCaP: ADT-sensitive, AR-positive prostate cancer cell line [32]; mCRPC: Metastatic castration resistant prostate cancer; PC-3: ADT-resistant, AR-negative prostate cancer cell line [32]; PSA: Prostate specific antigen; VCaP: ADT-resistant, AR/AR-V7 positive prostate cancer cell line [21].
CONFLICTS OF INTEREST
Authors disclose no conflicts of interest.
FUNDING
The research was partially supported by the Grant from RFBR #16-03-00553a.
REFERENCES
1. Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. 2010; 17:S72–S9.
2. Nelson PS. Targeting the androgen receptor in prostate cancer—a resilient foe. N Engl J Med. 2014; 371:1067–9. https://doi.org/doi:10.1056/NEJMe1409306.
3. Armstrong CM, Gao AC. Drug resistance in castration resistant prostate cancer: resistance mechanisms and emerging treatment strategies. American Journal of Clinical and Experimental Urology. 2015; 3:64–76.
4. Caffo O, De Giorgi U, Fratino L, Alesini D, Zagonel V, Facchini G, Gasparro D, Ortega C, Tucci M, Verderame F. Clinical outcomes of castration-resistant prostate cancer treatments administered as third or fourth line following failure of docetaxel and other second-line treatment: results of an Italian multicentre study. European Urology. 2015; 68:147–53. https://doi.org/10.1016/j.eururo.2014.10.014.
5. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008; 68:5469–77. https://doi.org/10.1158/0008-5472.can-08-0594.
6. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371:1028–38. https://doi.org/10.1056/NEJMoa1315815.
7. Boudadi K, Antonarakis ES. Resistance to novel antiandrogen therapies in metastatic castration-resistant prostate cancer. Clin Med Insights Oncol. 2016; 10:1–9. https://doi.org/10.4137/cmo.s34534.
8. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death & Disease. 2013; 4:e838.
9. Kung HJ, Changou C, Nguyen HG, Yang JC, Evans CP, Bold RJ, Chuang F. Autophagy and prostate cancer therapeutics. Prostate Cancer: Springer. 2013; 497–518.
10. Nguyen H, Yang J, Kung H, Shi X, Tilki D, Lara P, White RD, Gao A, Evans C. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014; 33:4521–30. https://doi.org/10.1038/onc.2014.25.
11. Dyshlovoy SA, Otte K, Alsdorf WH, Hauschild J, Lange T, Venz S, Bauer CK, Bähring R, Amann K, Mandanchi R, Schumacher U,Schröder-Schwarz J, Makarieva TN, et al. Marine compound rhizochalinin shows high in vitro and in vivo efficacy in castration resistant prostate cancer. Oncotarget. 2016; 7:69703–17. https://doi.org/10.18632/oncotarget.11941.
12. Dyshlovoy SA, Otte K, Venz S, Hauschild J, Junker H, Makarieva TN, Balabanov S, Alsdorf WH, Madanchi R, Honecker F, Bokemeyer C, Stonik VA, von Amsberg G. Proteomic-based investigations on the mode of action of the marine anticancer compound rhizochalinin. Proteomics. 2017. 17:1700048 [1-11]. https://doi.org/10.1002/pmic.201700048.
13. Makarieva TN, Denisenko VA, Stonik VA, Milgrom YM, Rashkes YV. Rhizochalin, a novel secondary metabolite of mixed biosynthesis from the sponge Rhizochalina incrustata. Tetrahedron Letters. 1989; 30:6581–4. https://doi.org/10.1016/s0040-4039(01)89027-4.
14. Khanal P, Kang BS, Yun HJ, Cho HG, Makarieva TN, Choi HS. Aglycon of rhizochalin from the Rhizochalina incrustata induces apoptosis via activation of AMP-activated protein kinase in HT-29 colon cancer cells. Biological & Pharmaceutical Bulletin. 2011; 34:1553–8.
15. Jin JO, Shastina V, Park JI, Han JY, Makarieva T, Fedorov S, Rasskazov V, Stonik V, Kwak JY. Differential induction of apoptosis of leukemic cells by rhizochalin, two headed sphingolipids from sponge and its derivatives. Biological & Pharmaceutical Bulletin. 2009; 32:955–62.
16. Fedorov SN, Makarieva TN, Guzii AG, Shubina LK, Kwak JY, Stonik VA. Marine two-headed sphingolipid-like compound rhizochalin inhibits EGF-induced transformation of JB6 P+ Cl41 cells. Lipids. 2009; 44:777–85. https://doi.org/10.1007/s11745-009-3322-6.
17. Manallack DT, Prankerd RJ, Yuriev E, Oprea TI, Chalmers DK. The significance of acid/base properties in drug discovery. Chemical Society Reviews. 2013; 42:485–96. https://doi.org/10.1039/c2cs35348b.
18. Molinski TF, Makarieva TN, Stonik VA. (-)-Rhizochalin is a dimeric enantiomorphic (2R)-sphingolipid: absolute configuration of pseudo-C(2v)-symmetric bis-2-amino-3-alkanols by CD. Angew Chem Int Ed Engl. 2000; 39:4076–9. https://doi/org/10.1002/1521-3757(20001117)112:22<4242::AID-ANGE4242>3.0.CO;2-2.
19. Tamaki H, Harashima N, Hiraki M, Arichi N, Nishimura N, Shiina H, Naora K, Harada M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget. 2014; 5:11399–412. https://doi.org/10.18632/oncotarget.2550.
20. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM. Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Res. 2013; 73:483–9. https://doi.org/10.1158/0008-5472.can-12-3630.
21. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014; 20:3198–210. https://doi.org/10.1158/1078-0432.ccr-13-3296.
22. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007; 7:961–7. https://doi.org/10.1038/nrc2254.
23. Farrow JM, Yang JC, Evans CP. Autophagy as a modulator and target in prostate cancer. Nat Rev Urol. 2014; 11:508–16. https://doi.org/10.1038/nrurol.2014.196.
24. Kung HJ, Changou C, Nguyen HG, Yang JC, Evans CP, Bold RJ, Chuang F. Autophagy and prostate cancer therapeutics. In: Tindal DJ, ed. Prostate cancer: Springer. 2013; 497–518.
25. Chlenski A, Nakashiro KI, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. The Prostate. 2001; 47:66–75. https://doi.org/10.1002/pros.1048.
26. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1–222. https://doi.org/10.1080/15548627.2015.1100356.
27. Dyshlovoy SA, Menchinskaya ES, Venz S, Rast S, Amann K, Hauschild J, Otte K, Kalinin VI, Silchenko AS, Avilov SA, Alsdorf W, Madanchi R, Bokemeyer C, et al. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Int J Cancer. 2016; 138:2450–65. https://doi.org/10.1002/ijc.29977.
28. Dyshlovoy SA, Naeth I, Venz S, Preukschas M, Sievert H, Jacobsen C, Shubina LK, Gesell Salazar M, Scharf C, Walther R, Krepstakies M, Priyadarshini P, Hauber J, et al. Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with antiproliferative activity. Journal of Proteome Research. 2012; 11:2316–30. https://doi.org/10.1021/pr300170p.
29. Pelageev DN, Dyshlovoy SA, Pokhilo ND, Denisenko VA, Borisova KL, Keller-von Amsberg G, Bokemeyer C, Fedorov SN, Honecker F, Anufriev VP. Quinone-carbohydrate nonglucoside conjugates as a new type of cytotoxic agents: synthesis and determination of in vitro activity. Eur J Med Chem. 2014; 77:139–44. https://doi.org/10.1016/j.ejmech.2014.03.006.
30. Dyshlovoy SA, Tabakmakher KM, Hauschild J, Shchekaleva RK, Otte K, Guzii AG, Makarieva TN, Kudryashova EK, Fedorov SN, Shubina LK, Bokemeyer C, Honecker F, Stonik VA, et al. Guanidine alkaloids from the marine sponge Monanchora pulchra show cytotoxic properties and prevent EGF-induced neoplastic transformation in vitro. Marine Drugs. 2016; 14:133 [1–17]. https://doi.org/10.3390/md14070133.
31. Dyshlovoy SA, Fedorov SN, Kalinovsky AI, Shubina LK, Bokemeyer C, Stonik VA, Honecker F. Mycalamide A shows cytotoxic properties and prevents EGF-induced neoplastic transformation through inhibition of nuclear factors. Marine Drugs. 2012; 10:1212–24. https://doi.org/10.3390/md10061212.
32. Russell PJ, Kingsley EA. Human prostate cancer cell lines. Prostate Cancer Methods and Protocols: Springer. 2003; 21–39.