
INTRODUCTION
Multiple myeloma (MM) is characterized by the expansion of malignant plasma-cells (PCs) within a permissive bone marrow microenvironment (BMM) that promotes tumor cell survival and proliferation [1]. A preexisting premalignant condition, known as monoclonal gammopathy of undetermined significance (MGUS), is a common step of MM development [2]. The transition from MGUS to MM takes place by progressive mutational events accompanied by changes in the BMM [3, 4]. MM therapy has tremendously improved in the last few years together with the understanding of mechanisms promoting the onset and development of the disease [5]. Most of preclinical findings in MM research that translated into clinics as effective therapies have been developed by the use of a variety of in vivo models, which aim to recapitulate the disease and to provide insights on the interactions between MM cells and the surrounding microenvironment. Such models represent to date the basic tool to investigate and predict the effectiveness of novel therapeutic strategies. In this work, we will review the most relevant in vivo experimental platforms for the study of MM pathogenesis and for drug discovery.
THE 5TMM MODEL: A MURINE BACKGROUND FOR MM DEVELOPMENT
Specific mouse strains may spontaneously develop age-associated malignancies. Among them, C57BL/KalwRij are prone to develop B cell lymphoproliferative disorders, as approximately 80% of these mice carry a monoclonal component (MC), resembling human MGUS [6]. A very small fraction (0.5%) of mice progress to MM and Waldenstrom Macroglobulinemia. Mice spontaneously developing MM disease represent the original 5TMM model. Bone marrow (BM)-MM cells from these mice can be efficiently transplanted into syngeneic mice to easily reproduce the disease. Indeed, from the original 5TMM, several cell lines have been established, such as: 5T2 that reproduces different milestones of advanced disease including serum paraprotein production and lytic bone lesion formation; 5T33, more aggressive, with preferential dissemination to spleen and liver. It is therefore possible to derive mice carrying extensive or very limited skeleton damages, with malignant PC clones confined to the BM and spleen and with different growth patterns. In this model, cytogenetic abnormalities showed hyperdiploid features, with lower frequency of translocations as compared to human disease [7]. MM pathogenesis can be further evaluated by crossing mice with specific genetic backgrounds, such as recombinant activating gene 2 (-/-) (RAG-2 -/-) mice that lack T and B cell proper development [8]. These mice allow to study the interactions between MM cells and the surrounding BMM, including the immune system, and several findings such as the MM cells cross talk with BM stromal cells (BMSCs), the BM homing of malignant PCs and the overwhelming osteoclast (OCL) activity as the promoter of MM related bone disease (MMBD) are among the most relevant achievements of this model (Figure 1). Additionally, this model constitutes a suitable platform for drug discovery and has been largely used for in vivo evaluation of several new compounds, especially due to the significant number of animals bearing the same disease that may be easily produced, thus allowing the conduction of statistically relevant studies. However, the main limitation of this model resides in the exclusive murine genetic background that may limit the study of compounds targeting human specific targets.
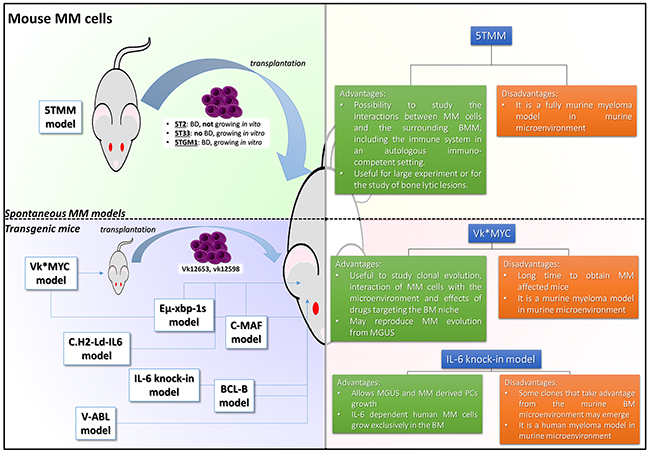
Figure 1: 5TMM and TG mouse models of MM. The pictures illustrate different strategies adopted to recapitulate MM disease. Models are divided according to their main characteristics: the green quadrant includes models of spontaneous mouse model of MM; the blue quadrant includes models of transgenic murine MM; each quadrant is accompanied by a table briefly describing the most relevant advantages and limits of each model. BD: bone disease; MM: multiple myeloma; BMM: bone marrow microenvironment
All these points need to be considered in the translation process to the clinical setting.
THE XENOGRAFT MODELS: PLATFORMS FOR NEW DRUGS SCREENING
Although the 5T model has been a milestone for the comprehension of MM pathogenesis, several investigators have tried to overcome the limit of “genetically murine myeloma”, in the aim of testing innovative drugs against human MM cells and their microenvironment. The earliest methods attempted to engraft human MM cells in an animal recipient relied on the injection either subcutaneously (s.c.) or systemically (intravenously, i.v. or intraperitoneally, i.p.) of MM cell lines in SCID and NOD/SCID mice. In these mice strains, engraftment of tumor cells is allowed by the lack of T and B cells (SCID and NOD/SCID) and reduced NK activity (NOD/SCID). The injection route is relevant in this system as the s.c. route will be followed by the growth of palpable tumors only in the injection site, while the systemic injection will give rise to disseminated involvement of different organs such as spleen, liver, lungs and bone marrow, depending on the cell line used [9–11]. In the s.c. model, the growth of MM cells can be easily measured by a caliper or ultrasound (that further allows the study of angiogenesis) until mouse death or sacrifice. Indeed, tumor volume and mouse survival are the main objective parameters to ascertain the effect of a specific drug. In the i.v. model, MM cell growth is diffuse in different organs and the burden of disease can be evaluated by standard imaging techniques (microCT/MRI/PET) [10]. Alternatively, MM cells are transduced to express a bioluminescent or fluorescent marker, thus allowing the detection of tumor cells by dedicated imaging devices. In both routes models, it is feasible to detect monoclonal component (MC) in the mouse sera as well as other biochemical markers (e.g. beta 2 microglobulin) that represent an indirect measurement of the disease burden. Both models have been developed in the aim to investigate new drugs and still represent the most common methods to study innovative drugs activity in vivo [12, 13].
The s.c. model has the advantage that the tumor can be easily monitored and measured during treatments. However, s.c. growth does not recapitulate the human disease due to several limitations. Specifically, in this model, MM cells proliferate in the absence of the peculiar BMM that sustains the expansion of malignant PCs and drives the development of bone damage and drug resistance. Additionally, MM cell lines used for engraftment are mainly derived from extramedullary sites such as pleural effusions or peripheral blood of advanced MM patients. These cells have lost the dependence from the BMM and have changed significantly their genetic background to a late stage disease- instead of intramedullary MM- type.
When considering the i.v. route, homing to the BM is also found, according to the specific type of MM cell line used. In these cases, the murine BM can be infiltrated with consequent bone damage as in the human MM. However, along the course of the disease, other mice organs host MM cells, reproducing an in vivo scenario commonly not observed in human disease. Thus, both models are very useful and easy-to-handle to quickly assess the activity of new cytotoxic drugs, but do not provide insights on the critical interactions between malignant PCs and their own BMM. This information is presently somewhat relevant to allow a successful translation in clinics of innovative drugs with selective mechanisms of action that in most cases involves the BMM. To overcome the relevant limitations of previous models, some attempts to develop new “humanized” models have been designed and made available (Figure 2). In these models, the basic idea is to reproduce the human BMM by implanting bone recipients in the flank of mice and then seeding them with fresh primary BM explanted cells or IL-6-dependent MM cell lines. The advantage of this approach is that MM cells grow in an orthotopic full human environment, where BMSCs support MM cell proliferation that in turn triggers osteoclasts (OCL) activity and suppresses bone apposition, thus reproducing the development of BD as observed in patients. Several variants of this model have been developed according to the type of the bone chip used (Figure 2).
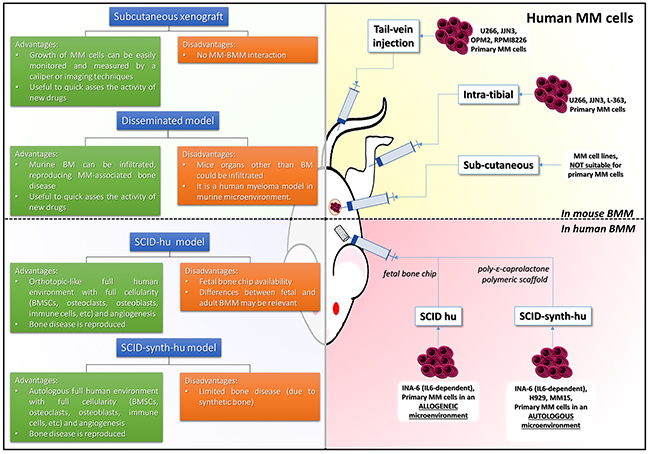
Figure 2: Xenograft and SCID models of MM. The pictures illustrate different strategies to recapitulate human MM disease in mice. The yellow quadrant includes models where human MM cells are grown in murine bone marrow microenvironment; the red quadrant includes models where human MM cells are grown in human autologous or allogenic bone marrow microenvironment; each quadrant is accompanied by a table briefly describing the most relevant advantages and limits of each model. BD: bone disease; MM: multiple myeloma; BMM: bone marrow microenvironment
The SCID-hu model
In the SCID-hu model, a human fetal bone chip (FBC) is implanted in the recipient mouse. In this model [14], MM cell lines home exclusively to the human implant instead of murine bone. Furthermore, when a contralateral FBC was implanted, MM cells migrated from the previously injected FBC to the contralateral one, while sparing the murine bone and obviously all other murine normal tissues. Engrafted MM cells produced MC and IL-6, which correlated with the time from implantation and the tumor burden. Following this initial experience, primary PCs from MM patients were successfully engrafted into SCID-hu models [15]. MC was detected in mouse sera, while human bone sections showed increased OCL activity and neo-angiogenesis, resembling human disease (Figure 3). The FBC has been also demonstrated to be an excellent recipient for engraftment of IL-6 dependent MM cells (INA-6) [16]. These cells, in fact, are fully dependent on human BMSCs and/or human IL-6, which is a well recognized key factor and target for MM [17–28], and cannot be used for production of conventional s.c. xenografts since do not grow under murine IL-6 stimulation. On the other side, administration of human IL-6 induces mice cachexia and therefore these cells cannot be used in vivo without a recipient recapitulating the human bone milieu. In this model, extensive BM infiltration by INA-6 was paralleled by an increased beta 2 microglobulin in mice sera. The advantages of using the INA-6 implanted SCID-hu model are the unlimited high reproducibility of the data with a cell line that recapitulates more accurately the human disease, growing in an orthotopic environment. The latter is a crucial issue to study mechanistic pathways of myeloma-genesis and to ascertain the effects of new drugs against MM cells and BMM [16, 23, 26, 29–33].
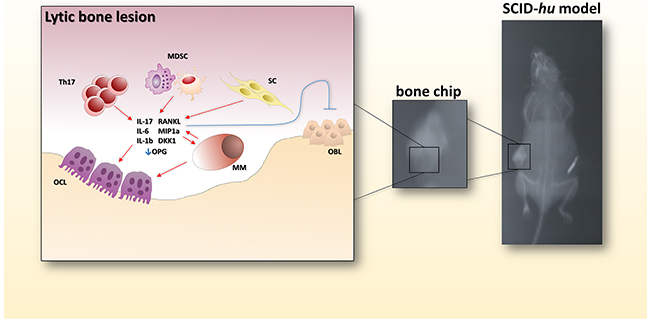
Figure 3: Bone-disease model. The picture shows the radiologic evidence of a bone lytic lesion within a fetal bone chip. In the cartoon on the left, the main actors determining the bone resorption activity are reported. Th17: T helper 17 lymphocytes; MDSC: myeloid-derived suppressor cell; SC: stromal cell; OBL: osteoblast; MM: multiple myeloma cell; OCL: osteoclast.
Indeed, the use of these models allowed investigators to define the role of key signaling pathways or to investigate several new agents. For example, p38 mitogen activated kinase (MAPK) is constitutively activated in MM and contributes to tumor growth [34]. Abrogation of p38 signaling within MM cells attenuates OCL activity and restores bone apposition in a SCID-hu environment. The same model allowed demonstration of the anti-MM effects of thalidomide in vivo [35]. The potency of the model to study new drugs has been further tested by Tai et al., who showed that targeting Bruton tyrosine kinase (BTK) in MM cells and OCLs impairs MM growth and attenuates MMBD [38]. MM-dependent OCL activity is clearly reproducible in the SCID-hu model, where zoledronic acid, the most common bisphosphonate routinely used for MM supportive therapy, efficiently attenuates bone damage [35]. Osteoblasts (OBL) suppression and aberrant OBL/OCL crosstalk through Ephrin B2/B4 (EPHB2/B4) interaction has been also described within FBC [36]. The authors demonstrated that interfering with the EPH axis by EPH B2/B4 -FC molecules determined reversal of OBL suppression and reduced OCL levels in myeloma carrying SCID-hu mice.
Even if the SCID-hu model proved to be effective in studying MM pathogenesis as well as other plasma cell discrasias such as Waldenstrom’s macroglobulinemia [37–39] and the activity of novel drugs, its major limitations rely on FBC availability for many institutions. Furthermore, fetal BMM does not exactly recapitulate adult BMM. Alternative methods have been developed in the aim to overcome these issues.
The SCID-rab model
In the SCID-rab model human fetal bone chips are replaced by rabbit bones. MM cells engraft into the rabbit bone reproducing the human disease [40]. The SCID-rab model has been the backbone for studying the bone-building effects of proteasome inhibitor (PI) bortezomib, which are independent from anti-neoplastic effects of the PI. Indeed, when mice are exposed to PI, bone mineral density (BMD) increases in both myelomatous and non-myelomatous bones. These data represented the basis to further explore bone building activity of novel drugs within SCID-rab. In this model, [41] the effects of anti-DKK-1 mAb that was able to increase BMD in the presence of patient derived malignant PCs, has been evaluated. DKK-1 negatively regulates WNT signaling and OBL differentiation. By neutralizing DKK-1, OBL numbers were restored, while OCLs were significantly reduced. These findings were confirmed and implemented by testing a clinical grade anti DKK-1 Ab in the SCID-hu model [30], thus demonstrating the close relationship between the two models.
The SCID-rab model is a suitable model for preclinical investigation in MM, but it certainly retains its own limits as rabbit bone, although ontogenetically related, cannot be strictly compared to human bone. However, the development of bone chip based models unveiled the potentialities of this approach and prompted investigators to ameliorate this system.
The SCID-synth-hu model
In an effort to reproduce human MM microenvironment in the mouse, synthetic scaffolds, closely resembling the trabecular structure of human bone were developed. A relevant option has been provided with the generation of the SCID-synth-hu model (Figure 2) [42]. It is based on a three-dimensional poly-ε-caprolactone polymeric scaffold (PS), featuring the human trabeculae, wherein MM cells/primary PCs and BMSCs can be successfully loaded. The PS is either cultured in vitro or implanted in the mouse flank as for FBC/rabbit chip. In this latter case, the recipient was then engrafted with cell populations from whole bone marrow aspirates directly in vivo. Treatment with i.p. bortezomib/dexametasone regimen led to dramatic decrease of MC in the mouse sera and clearance of MM cells within the retrieved scaffold. An intriguing preliminary finding was also the development of neo-angiogenesis in the MM coated PS [42]. Following-up this observation, Zhu et al. [43] demonstrated that bone marrow endothelial cells (BMECs) support MM cell growth, migration and BM homing within PS either in vitro and implanted in the mice. BMECs-MM cells interaction is mainly dependent on CD147, which is highly expressed by MM cells as compared to normal PCs and binds cyclophilin A (cyPA). cyPA is secreted by BMECs. CD147 blockade exerted potent anti MM activity as shown in the PS MM model. These data demonstrated that the PS is a powerful platform to recapitulate MM disease and to test a variety of innovative therapeutic approaches [29, 33, 42, 44–46]. Figure 2 summarizes the main characteristics of the most important models discussed in this section.
Table 1: Transgenic murine models of MM
TG Model |
TG |
Genomic aberrations |
Growth Pattern |
C.H2-Ld-IL6 |
Ld-IL-6 |
T(12,15); |
E.M. monoclonal plasmacytomas/B cell lymphomas |
v-ABL |
Eμ-v-abl |
T(12,15); |
E.M. monoclonal plasmacytomas |
Vk*MYC |
V-k exon-c-MYC |
SHM dependent c-MYC expression in CD138+ PCs |
I.M. MGUS/MM |
XBP-1 |
Eμ-xbp1 |
Cyclin D1; MAF upreg. |
I.M. MGUS/MM |
c-MAF |
Eμ-IgH-c-MAF |
X-BP1, Blimp-1 upreg.; PAX-5 dowreg. |
Plasmablastic/plasmacytic lymphomas |
Bcl-B |
Eμ-IgH-Bcl-B |
IGF-1, IL-6, c-MYC, X-BP1, IRF-4 upreg. |
I.M. MM |
IL-6 Knock in |
Rag2 -/- IL2R -/-/hIL-3-MCSF-GMCSF-TPO-IL-6 knock ins |
Parental MM cell aberrations |
Recipient of MGUS/sMM/MM PCs |
Table 1 illustrates the main TG models discussed in the text together with the most relevant genomic aberrations detected in TG mice and the pattern of proliferation of malignant PCs (see text for further details). Abbreviations: rearrang.: genomic rearrangement; upreg.: upregulated gene; downreg: downregulated gene; SHM: somatic hypermutation machinery; I.M.: intramedullary; E.M.: extramedullary.
SPECIAL CONSIDERATION ON THE USE OF NOD SCID GAMMA NULL MOUSE AS XENOGRAFT MODEL OF MM
The recently introduced NOD/SCID gamma null (NSG) mouse strain lacks IL-2 gamma chain, resulting in the absence of B/T/NK activity and severe impairment of antigen presenting cells and complement [47]. Some authors attained successful engraftment of I.V. injected U266 MM cells into NSG recipients following 2.4 gy irradiation (demonstrated to be unnecessary for tumor engraftment in subsequent reports) (Figure 2) [48]. U266 showed specific homing to murine BM, which is observed also with other MM cells such as OPM2, JJN3 and RPMI8226, and in contrast to NOD/SCID IV implants [10, 44, 49, 50]. Homing to the BM is reproducibly attained and classical features of MMBD can be observed with increased OCL activity, OBL reduction and lytic lesions across the skeleton [49]. However, it should be taken into account, that these models may present an extensive burden of extramedullary disease, thus mostly resembling very aggressive or late stage MM disease more than classical indolent myeloma. Again, the major limitation of this model is due to cell lines, that are better representative of a BMM-independent plasma cell leukemia than a MM.
Besides MM cells, patient derived malignant PCs have been engrafted into mice BM reproducing the human disease features. In this model, administration of zoledronic acid is effective in attenuating bone disease, while not affecting tumor burden. PIs such as carfilzomib dramatically reduced tumor burden and showed anabolic effects in MM implanted NSG mice, demonstrating that this model is an ideal platform for drug development and testing [49, 51]. A further refinement of NSG based MM models has been presented by others [52–54], where several MM cell lines, such as U266, L-363 and JJN3, or primary bone marrow cells from MM patients (both CD138 positive and negative fraction) have been directly implanted into NSG mice tibia (Figure 2). The advantages of intratibial implants are due to an accelerated and bone confined growth of MM, whose features are strictly related to the human disease. Interestingly, the disease spread over bone skeleton, and tumor burden could be easily determined by bioluminescent techniques in the presence of luminescent dye tagged MM cells and/or by MRI.
TRANSGENIC MODELS
IL-6 transgenic models
Early studies demonstrated that IL-6 is essential for MM cell growth in vitro and in vivo [55, 56], suggesting that this cytokine is the main supportive factor in mineral oil induced plasmacytomas (MOPCs) [57], which develop within the peritoneal cavity following the generation of inflammatory dependent granulomas [58]. Based on these premises, an IL-6 transgene (Tg) driven by the promoter of Ld gene of the major histocompatibility complex (H2-Ld) was generated in B6 mice and showed the development of massive policlonal plasmacytomas. These tumors were transplanted in mineral oil treated BALB/c mice, where MC component of IgA isotype and clonal plasmacytomas were observed. These plasmacytomas carried t(12;15) and c-myc gene rearrangements. Interestingly, when IL-6 Tg is stabilized in BALB/c background mice through progressive backcrossing until N20, derived c.IL6 mice spontaneously developed monoclonal plasmacytomas or B cell lymphomas within lymphoid tissues. These tumors could be transplanted in mineral oil treated mice arising t(12;15)+ plasmacytomas [59]. These early studies were of relevance as they began to unveil the role of IL-6 dependence in MM growth and the importance of tumor microenvironment in promoting MM development in vivo. C.H2-Ld-IL6 model is still the best reference model to study the pathophysiology of IL-6 in MM [60]. Further evidence of potential microenvironment dependence of MOPC tumors is provided by the derived MOPC315.BM cell line, which is obtained by consecutive i.v. passages of an established MOPC315 MM. Interestingly, these tumors shift from a s.c. growth form to BM selective expansion with extensive osteolytic disease and limited extramedullary involvement, resembling human MM [7].
The v-ABL model
c-myc rearrangement in IL-6 driven plasmacytomas was not an isolated finding in early transgenic studies of MM. In the Eμ-v-abl model [61], the v-abl gene, which encodes for a constitutively active non-receptor tyrosine kinase, is juxtaposed with an IgH enhancer. The resulting Eμ-v-abl strongly promotes plasmacytoma development even in mineral oil free mice. In most cases a t(12;15) or other myc gene rearrangements could be identified, while double Tg Eμ-myc and Eμ-v-abl presented with an accelerated plasmacytoma onset (5-8 wks for double Tg as compared to 11-52 in Eμ-v-abl), thus confirming the putative role of c-MYC in MM pathophysiology. Acceleration of plasmacytoma onset can be attained by crossing the Eμ-v-abl mice with either Bim-/-, BCL2 or MCL-1 Tg mice [62]. The lack of pro- apoptotic signals or the enriched survival signals carried by crossed Tg facilitated MYC driven plasmacytoma-genesis by increasing PC frequency in normal mice and increased cyclin D expression. The synergistic effects of MYC and abl can be due to the activation of relevant pro-survival and proliferation signaling pathways such as RAS and PI3K dependent pathways. This synergy seems to be selective for tumorigenesis in PC lineage rather than lymphocytes, although the mechanisms of such selectivity remain not clarified.
The Vk*MYC model
In the mineral oil/transgene induced plasmacytomas discussed so far, tumors develop in the peritoneum, lymphnodes, spleen and more rarely BM. Therefore, these models resemble at best extramedullary MM cases and do not recapitulate the human disease. As discussed, a common element of these models is the rearrangement of MYC gene that drives tumor onset and development. The Vk*MYC model [63] aims to overcome these limitations and to approximate the generated mouse tumor to human MM (Figure 1). The Vk*MYC is structured with a V-kappa exon sequence splicing in frame with the human MYC locus. The Tg harbors a stop codon within V-kappa exon that generates a DGYW motif. This motif is targeted by somatic hyper-mutation machinery (SHM), which can randomly revert the stop codon and promote MYC expression. In this way, MYC becomes activated along GC reaction, preferentially affecting PC differentiation as light chain regulatory sequences allow for SHM activity. Low proliferating monoclonal PCs accumulate into Vk*MYC mice, along their life, within BM and secondary lymphoid organs, and resemble the feature of MGUS/MM. Antibody production is monoclonal in most cases. Bi- or tri-clonal paraprotein occurs to a lesser extent, indicating the occurrence of different clonal PCs. Clonal PCs are transplanted into syngenic mice recapitulating MGUS with corresponding MC spikes in recipient mice. The range of BM PC infiltration in Vk*MYC mice encompasses true MGUS disease up to overt MM. These mice have on average lower hemoglobin levels, reduced BMD and MM like kidney damage, thus resembling the human disease. The peculiarity of this model is that the tumor recapitulates the slow development and progression of MM with only 1/3 of the mice that evolve towards an extramedullary plasmacytoma as opposite to previously discussed models. The “smoldering” phenotype of the model switches to a more aggressive disease when BM PCs are serially transplanted in congenic mice [64]. The Vk*MYC has been used to evaluate the activity of anti-MM drugs [64]. PIs and ImiDs and innovative drugs such as HDAC inhibitors are effective drugs in the model, while standard chemotherapy lacks relevant activity, resembling the human disease. The model is also suitable to study and develop novel combo therapies. Another peculiarity of the Vk*MYC model is the reproduction of the existing cross talk between clonal PCs and surrounding microenvironment. This distinctive feature proves to be very helpful to study clonal dynamics of the disease. As for other tumor types, the current hypothesis of MM onset and development is based on the existence of competition among different clones at diagnosis and along treatment, that drives towards indolent or more aggressive disease courses [4]. Indeed, the Vk*MYC models allows the study of the evolution of distinct clones in the same mouse along time [65]. These clones are identified after transplant in congenic wild type or Vk*MYC mice to evaluate their behavior within transplanted mice. Interestingly, transplanted clones can compete out the preexisting tumor clones within the recipient mice, coexist with them or even support these clones to proliferate. A similar pattern is observed also in Vk*MYC mice undergoing treatment which by itself creates clonal selection. These different courses resemble the human disease and can be observed also in MM at diagnosis and during subsequent relapses [4]. The model highlights the complex role of MYC in MM onset and development. Several findings indicate that MYC is involved in MGUS to MM transition [66], while MYC inhibitors efficiently promote MM cell death in vitro [67]. If we consider that MYC activation in MM is sustained mainly by secondary translocations not involving Ig Locus and can be present in the early phases of MM development [68], it becomes further evident the relevance of the Vk*MYC model to study MM pathogenesis.
The XBP-1 model
Unfolded protein response (UPR) has a recognized role in PC differentiation and function [69]. Among regulators of UPR, XBP-1 has been investigated [69–71]. XBP-1 is required for PC differentiation as it is rapidly upregulated in B cells upon PC differentiating stimuli, whereas mice lacking XBP-1 retain normal B cell activity but show very low level of antibodies in the absence of PC differentiation. Based on these premises, a Eμ-XBP-1 Tg mouse, with a prominent expression of Tg product in lymphoid organs (spleen, lymphnodes, bone marrow and thymus) has been generated [72]. By 40 wks of age, transgenic mice developed M spike detectable in sera, antibody based cast nephropathy and bone marrow PC infiltrate <10%. At 14-24 months of age, 1/3 of mice developed MM, showing >10% bone marrow PCs and bone lytic lesions. A human MM like gene signature was identified in Eμ-XBP-1 mice including over-expression of Cyclin D1 and MAF. The Eμ-XBP-1 Tg mouse represents a suitable platform to study the consequences of targeting UPR modulators in MM. gp96 is a ER related chaperone, that is strictly related to XBP-1 activity and found upregulated in MM patients [73]. gp96 deletion within Eμ-XBP-1 Tg mice attenuates MM disease, further linking UPR and MM development in this model.
The c-MAF model
c-MAF proto-oncogene is found over-expressed in MM in the presence of t(14;16) [74]. On this basis, a TG mouse carrying the Eμ-IgH-c-MAF vector that allowed expression of the TG in B cell lineage has been generated. The same group previously demonstrated that c-MAF upregulation in T cell compartment led to development of T cell lymphomas [75]. Similarly, B cell compartment restricted over-expression of c-MAF favors B cell lymphomas in aged mice (>50 weeks of age) with typical features: a clonal M spike with hyper-gammaglobulinemia, increased PCs in the bone marrow and cast nephropathy. Interestingly, tumor cells were B220+CD138+CD21+CD23+IgM+IgD-, thus resembling a plasma-cytic or plasma-blastic lymphoma. C-MAF targets such as Ccnd2 and Itgb7 were consistently elevated as expected, while the transcription factors Blimp-1 and XBP-1, which control PC differentiation were increased. In contrast, the main negative regulator of PC development, PAX-5, was downregulated in these mice. Overall, these data confirm the relevance of c-MAF in the development of PC tumors as described in MM patients. However, c-MAF TG mice do not recapitulate MM disease as the behavior of these tumors is more similar to PC lymphomas. Therefore, the mechanism of MM development in C-MAF carrying tumors must be further elucitated.
THE BCL-2 FAMILY PROTEINS TG MODELS
BCL-2 family members exert potent anti-apoptotic activity supporting PC survival upon microenvironment stimuli [76]. Among them, BCL-XL and Bcl-B were studied in TG models of PC tumors. A BCL-XL TG was developed with a K chain enhancer (KE) and promoter (Vk21, KP) [77]. BCL-XL TG developed hyper-gammaglobulinemia, extramedullary PC foci together with a MM like cast nephropathy. However, no M spikes were found. When backcrossed with Eμ-c-MYC mouse, the double TG mice developed aggressive B cell tumors, with severe lymphocytosis, hypergammaglobulinemia and a fatal outcome by 5.5 weeks of age. While multiple extramedullary PC loci are generated, BM PC counts are not increased. However, lytic disease is observed in the long bones of sacrificed mice. Genetic features of malignant PCs of double TG mice were analyzed in a second report [78], showing the presence of several translocations, deletions and insertions. The most frequent aberrant chromosomes were 12 and 16. Among dysregulated genes, cyclin group genes were abnormally expressed as compared to normal PCs.
Bcl-B is a BCL-2 family member, whose role in MM pathogenesis has been previously overlooked [79]. Indeed, Bcl-B expression is increased in MM cell lines and malignant PCs from patients as compared to MGUS and healthy subjects. TG Eμ-IgH-Bcl-B mice showed a significantly shorter survival as compared to WT mice. They developed progressive BM plasmocytosis, anemia and M spike in the sera. Interestingly, IgG2b was the only isotype expressed in TG mice, a feature unexplained by investigators [79]. GEP was carried out on BM PCs from these mice unveiling the expression of typical human MM associated genes, driving PC survival and proliferation, and triggering bone resorption [79]. BM PCs from TG mice were transplantable in syngeneic mice reproducing the disease in the recipient. BM plasmocytosis in TG mice was reduced by treatment with standard anti MM drugs (bortezomib and melphalan). These data indicated that TG Eμ-IgH-Bcl-B mice recapitulate the human MM disease more stringently than BCL-XL TG and can be used as a platform for testing new drugs.
THE “IL-6 KNOCK IN” HUMANIZED MOUSE MODEL
The research of mouse models that could recapitulate human hematopoiesis and hematological malignancies has followed parallel steps along the last 20 years. However, the experiences in SCID-NOD/SCID environment proved to be suboptimal for reproducing human hematopoiesis [80]. On this regard, a major improvement has been achieved with the development of humanized mice where human cytokine genes were knocked in to replace the corresponding mouse cytokine counterpart [80, 81]. This approach allows human hematopoietic cell growth upon more physiological stimuli as the expression of the selected human cytokine is tissue specific and finely regulated as opposite to the insertion of a TG or the administration of an exogenous cytokine. Based on these premises, a Rag2-/- IL2R -/- mouse was generated where human genes for M-CSF, GM-CSF, IL-3 and TPO were knocked in the corresponding mouse loci. This mouse (MITRG) allowed easier engraftment of human hematopoietic cells derived from peripheral blood or BM [81] and development of innate immune system cells (Figure 1). The latter is a very relevant feature of this model as human innate immune system plays a prominent role in the study of tumor growth in the context of chronic inflammatory stimuli. Indeed, Das et al. [82] knocked in IL-6 human gene in the MITRG mouse, followed by injection of INA-6 MM cell line or patient derived malignant PCs into the bone. In this system, MM PCs nicely grew within mouse BM, without showing contralateral bone and splenic proliferation, meaning that tumor cells followed a growth path that was limited to the bone as in the human disease. Interestingly, either CD138+ or CD138- BM cell fraction expanded into MITGR BM. Tumor cell fraction obtained from grafted mice was able to reproduce the disease upon transplantation in tumor free mice. Transplanted tumor showed better engraftment and growth after CD3 T cell depletion from primary tumor to attenuate xenograft versus host disease. MITGR mice proved to be an ideal recipient to allow malignant PC proliferation from MGUS and smoldering MM [82]. Comparison of genomic analysis by whole-exome sequencing between xenograft tumor cells and parental MM demonstrated similar LOH and CNAs patterns. This finding is very relevant when we consider the possibility to evaluate the evolution of distinct sub-clones within the mouse model. Indeed, an ideal model should preserve the clonal distribution observed in the parental tumor cells derived from the patient. However, even in this model, genomic changes could be detected in the xenografted tumors. As authors argued, the changes were likely due to the emergence of minor sub-clonal fractions that take a proliferation advantage in the xenograft BM microenvironment [82]. Table 1 summarizes the different TG models discussed in this section.
IMMUNOTHERAPY: THE NEW CHALLENGE FOR MURINE MODELS
In recent years, we observed a paradigm shift in the treatment of different malignancies with the increasing introduction into the clinical practice of novel agents or innovative strategies aimed to improve patients’ immune response against cancer cells [83–89]. Specifically, the discovery of immune checkpoints and the rapid development of inhibitory monoclonal antibodies to these molecules raised the question on the best suitable model to choose for the study of new immune-modulatory agents. In this view, several immunocompetent mouse models of MM, including the 5TMM and vk*MYC, have been widely used to investigate the role of different immunosuppressive cell populations and the activity of checkpoints inhibitors such as anti-PD1 mAbs [90–95]. However, further efforts are required to generate the “ideal” model for cancer immunotherapy, i.e. mice bearing a fully competent immune system able to mount an effective anti-cancer response [96]. The SCID-hu and the SCID-synth-hu represent a strong improvement in this direction, especially taking into account that immune populations, such as dendritic cells, have been reported to be present and functionally active in the latter model [97].
CONCLUDING REMARKS AND PERSPECTIVES
In vivo models of MM have increased the understanding of the disease complexity. On a historical point of view these models have evolved with the major aim of better defining the complexity of the cross-talk between tumor cells and their BMM and also to provide predictive tools for investigation of new agents. The recent interest in the discovery of agents capable of targeting and modulate regulatory networks driven by non-coding agents [98, 99] has further underscored the need of more reliable models of human MM. On these bases, selecting the right model for each specific pre-clinical research is an essential point for the successful completion of investigation. In general, if we recollect the different systems, it is clear that humanized mouse models likely represent the best platform for future studies. Among others, two major breakthrough findings have recently emerged in the pathogenic and therapeutic scenario of MM: the clonal evolution [4] and the immunotherapy [100, 101]. MM models that allow recapitulation of the clonal competition for investigation of novel biological agents as well as those that allow the design and evaluation of immunotherapeutic strategies within an appropriate microenvironment, will likely provide the most relevant platforms and impact in future translational studies on MM.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FINANCIAL SUPPORT
This work has been supported by the Italian Association for Cancer Research (AIRC), PI: PT. “Special Program Molecular Clinical Oncology - 5 per mille” n. 9980, 2010/15 and its Extension Program 2016/17 n.9980, and “Innovative immunotherapeutic treatments of human cancer”, MultiUnit - Multi Unit Regional n.16695 (co-financed by AIRC and CARICAL foundation).
REFERENCES
1. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016; 91:101–19. https://doi.org/10.1016/j.mayocp.2015.11.007.
2. Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, Melton LJ 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002; 346:564–9. https://doi.org/10.1056/NEJMoa01133202.
3. Botta C, Di Martino MT, Ciliberto D, Cuce M, Correale P, Rossi M, Tagliaferri P, Tassone P. A gene expression inflammatory signature specifically predicts multiple myeloma evolution and patients survival. Blood Cancer J. 2016; 6:e511. https://doi.org/10.1038/bcj.2016.118.
4. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335–48. https://doi.org/10.1038/nrc3257.
5. Botta C, Ciliberto D, Rossi M, Staropoli N, Cucè M, Galeano T, Tagliaferri P, Tassone P. Network meta-analysis of randomized trials in multiple myeloma: efficacy and safety in relapsed/refractory patients. Blood Adv. 2017; 1:455-466.
6. Radl J, De Glopper ED, Schuit HR, Zurcher C. Idiopathic paraproteinemia. II. Transplantation of the paraprotein-producing clone from old to young C57BL/Kalwrij mice. J Immunol. 1979; 122:609–13.
7. van den Akker TW, Radl J, Franken-Postma E, Hagemeijer A. Cytogenetic findings in mouse multiple myeloma and waldenstrom’s macroglobulinemia. Cancer Genet Cytogenet. 1996; 86:156–61.
8. Fowler JA, Mundy GR, Lwin ST, Lynch CC, Edwards CM. A murine model of myeloma that allows genetic manipulation of the host microenvironment. Dis Model Mech. 2009; 2:604–11. https://doi.org/10.1242/dmm.003160.
9. Wu KD, Zhou L, Burtrum D, Ludwig DL, Moore MA. Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis. Cancer Immunol Immunother. 2007; 56:343–57. https://doi.org/10.1007/s00262-006-0196-9.
10. Mitsiades CS, Mitsiades NS, Bronson RT, Chauhan D, Munshi N, Treon SP, Maxwell CA, Pilarski L, Hideshima T, Hoffman RM, Anderson KC. Fluorescence imaging of multiple myeloma cells in a clinically relevant SCID/NOD in vivo model: Biologic and clinical implications. Cancer Res. 2003; 63:6689–96.
11. Huang YW, Richardson JA, Tong AW, Zhang BQ, Stone MJ, Vitetta ES. Disseminated growth of a human multiple myeloma cell line in mice with severe combined immunodeficiency disease. Cancer Res. 1993; 53:1392–6.
12. Di Martino MT, Campani V, Misso G, Gallo Cantafio ME, Gulla A, Foresta U, Guzzi PH, Castellano M, Grimaldi A, Gigantino V, Franco R, Lusa S, Cannataro M, et al. In vivo activity of miR-34a mimics delivered by stable nucleic acid lipid particles (SNALPs) against multiple myeloma. PLoS One. 2014; 9:e90005. https://doi.org/10.1371/journal.pone.0090005.
13. Leotta M, Biamonte L, Raimondi L, Ronchetti D, Di Martino MT, Botta C, Leone E, Pitari MR, Neri A, Giordano A, Tagliaferri P, Tassone P, Amodio N. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J Cell Physiol. 2014; 229:2106–16. https://doi.org/10.1002/jcp.24669.
14. Urashima M, Chen BP, Chen S, Pinkus GS, Bronson RT, Dedera DA, Hoshi Y, Teoh G, Ogata A, Treon SP, Chauhan D, Anderson KC. The development of a model for the homing of multiple myeloma cells to human bone marrow. Blood. 1997; 90:754–65.
15. Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in scid-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998; 92:2908–13.
16. Tassone P, Neri P, Carrasco DR, Burger R, Goldmacher VS, Fram R, Munshi V, Shammas MA, Catley L, Jacob GS, Venuta S, Anderson KC, Munshi NC. A clinically relevant SCID-hu in vivo model of human multiple myeloma. Blood. 2005; 106:713–6. https://doi.org/10.1182/blood-2005-01-0373.
17. Rossi M, Di Martino MT, Morelli E, Leotta M, Rizzo A, Grimaldi A, Misso G, Tassone P, Caraglia M. Molecular targets for the treatment of multiple myeloma. Curr Cancer Drug Targets. 2012; 12:757–67.
18. Neri P, Tagliaferri P, Di Martino MT, Calimeri T, Amodio N, Bulotta A, Ventura M, Eramo PO, Viscomi C, Arbitrio M, Rossi M, Caraglia M, Munshi NC, et al. In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. Br J Haematol. 2008; 143:520–31. https://doi.org/10.1111/j.1365-2141.2008.07387.x.
19. Neri P, Kumar S, Fulciniti MT, Vallet S, Chhetri S, Mukherjee S, Tai Y, Chauhan D, Tassone P, Venuta S, Munshi NC, Hideshima T, Anderson KC, Raje N. Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res. 2007; 13:5903–9. https://doi.org/10.1158/1078-0432.CCR-07-0753.
20. Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gulla A, Rossi M, Neri A, Giordano A, Munshi NC, Anderson KC, Tagliaferri P, Tassone P. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin Cancer Res. 2013; 19:2096–106. https://doi.org/10.1158/1078-0432.CCR-12-3325.
21. Burger R, Le Gouill S, Tai YT, Shringarpure R, Tassone P, Neri P, Podar K, Catley L, Hideshima T, Chauhan D, Caulder E, Neilan CL, Vaddi K, et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol Cancer Ther. 2009; 8:26–35. https://doi.org/10.1158/1535-7163.MCT-08-0149.
22. Pitari MR, Rossi M, Amodio N, Botta C, Morelli E, Federico C, Gulla A, Caracciolo D, Di Martino MT, Arbitrio M, Giordano A, Tagliaferri P, Tassone P. Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget. 2015; 6:27343–58. https://doi.org/10.18632/oncotarget.4398.
23. Tassone P, Neri P, Burger R, Savino R, Shammas M, Catley L, Podar K, Chauhan D, Masciari S, Gozzini A, Tagliaferri P, Venuta S, Munshi NC, Anderson KC. Combination therapy with interleukin-6 receptor superantagonist Sant7 and dexamethasone induces antitumor effects in a novel SCID-hu in vivo model of human multiple myeloma. Clin Cancer Res. 2005; 11:4251–8. https://doi.org/10.1158/1078-0432.CCR-04-2611.
24. Yasui H, Hideshima T, Hamasaki M, Roccaro AM, Shiraishi N, Kumar S, Tassone P, Ishitsuka K, Raje N, Tai YT, Podar K, Chauhan D, Leoni LM, et al. SDX-101, the R-enantiomer of etodolac, induces cytotoxicity, overcomes drug resistance, and enhances the activity of dexamethasone in multiple myeloma. Blood. 2005; 106:706–12. https://doi.org/10.1182/blood-2005-02-0838.
25. Hamasaki M, Hideshima T, Tassone P, Neri P, Ishitsuka K, Yasui H, Shiraishi N, Raje N, Kumar S, Picker DH, Jacob GS, Richardson PG, Munshi NC, Anderson KC. Azaspirane (N-N-diethyl-8,8-dipropyl-2-azaspiro [4.5] decane-2-propanamine) inhibits human multiple myeloma cell growth in the bone marrow milieu in vitro and in vivo. Blood. 2005; 105:4470–6. https://doi.org/10.1182/blood-2004-09-3794.
26. Fulciniti M, Hideshima T, Vermot-Desroches C, Pozzi S, Nanjappa P, Shen Z, Patel N, Smith ES, Wang W, Prabhala R, Tai YT, Tassone P, Anderson KC, Munshi NC. A high-affinity fully human anti-IL-6 mAb, 1339, for the treatment of multiple myeloma. Clin Cancer Res. 2009; 15:7144–52. https://doi.org/10.1158/1078-0432.CCR-09-1483.
27. Tassone P, Galea E, Forciniti S, Tagliaferri P, Venuta S. The IL-6 receptor super-antagonist Sant7 enhances antiproliferative and apoptotic effects induced by dexamethasone and zoledronic acid on multiple myeloma cells. Int J Oncol. 2002; 21:867–73.
28. Tassone P, Forciniti S, Galea E, Savino R, Turco MC, Iacopino P, Tagliaferri P, Morrone G, Ciliberto G, Venuta S. Synergistic induction of growth arrest and apoptosis of human myeloma cells by the IL-6 super-antagonist Sant7 and Dexamethasone. Cell Death Differ. 2000; 7:327–8. https://doi.org/10.1038/sj.cdd.4400635.
29. Tassone P, Neri P, Burger R, Di Martino MT, Leone E, Amodio N, Caraglia M, Tagliaferri P. Mouse models as a translational platform for the development of new therapeutic agents in multiple myeloma. Curr Cancer Drug Targets. 2012; 12:814–22.
30. Fulciniti M, Tassone P, Hideshima T, Vallet S, Nanjappa P, Ettenberg SA, Shen Z, Patel N, Tai YT, Chauhan D, Mitsiades C, Prabhala R, Raje N, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009; 114:371–9. https://doi.org/10.1182/blood-2008-11-191577.
31. Hideshima T, Neri P, Tassone P, Yasui H, Ishitsuka K, Raje N, Chauhan D, Podar K, Mitsiades C, Dang L, Munshi N, Richardson P, Schenkein D, Anderson KC. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin Cancer Res. 2006; 12:5887–94. https://doi.org/10.1158/1078-0432.CCR-05-2501.
32. Neri P, Tassone P, Shammas M, Yasui H, Schipani E, Batchu RB, Blotta S, Prabhala R, Catley L, Hamasaki M, Hideshima T, Chauhan D, Jacob GS, et al. Biological pathways and in vivo antitumor activity induced by atiprimod in myeloma. Leukemia. 2007; 21:2519–26. https://doi.org/10.1038/sj.leu.2404912.
33. Tagliaferri P, Rossi M, Di Martino MT, Amodio N, Leone E, Gulla A, Neri A, Tassone P. Promises and challenges of microrna-based treatment of multiple myeloma. Curr Cancer Drug Targets. 2012; 12:838–46.
34. Yang J, He J, Wang J, Cao Y, Ling J, Qian J, Lu Y, Li H, Zheng Y, Lan Y, Hong S, Matthews J, Starbuck MW, et al. Constitutive activation of p38 MAPK in tumor cells contributes to osteolytic bone lesions in multiple myeloma. Leukemia. 2012; 26:2114–23. https://doi.org/10.1038/leu.2012.71.
35. Yaccoby S, Pearse RN, Johnson CL, Barlogie B, Choi Y, Epstein J. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on osteoclast activity. Br J Haematol. 2002; 116:278–90.
36. Pennisi A, Ling W, Li X, Khan S, Shaughnessy JD Jr, Barlogie B, Yaccoby S. The ephrinB2/EphB4 axis is dysregulated in osteoprogenitors from myeloma patients and its activation affects myeloma bone disease and tumor growth. Blood. 2009; 114:1803–12. https://doi.org/10.1182/blood-2009-01-201954.
37. Ho AW, Hatjiharissi E, Ciccarelli BT, Branagan AR, Hunter ZR, Leleu X, Tournilhac O, Xu L, O’Connor K, Manning RJ, Santos DD, Chemaly M, Patterson CJ, et al. CD27-CD70 interactions in the pathogenesis of waldenstrom macroglobulinemia. Blood. 2008; 112:4683–9. https://doi.org/10.1182/blood-2007-04-084525.
38. Ditzel Santos D, Ho AW, Tournilhac O, Hatjiharissi E, Leleu X, Xu L, Tassone P, Neri P, Hunter ZR, Chemaly MA, Branagan AR, Manning RJ, Patterson CJ, et al. Establishment of BCWM.1 cell line for Waldenström’s macroglobulinemia with productive in vivo engraftment in SCID-hu mice. Exp Hematol. 2007; 35:1366–75. https://doi.org/10.1016/j.exphem.2007.05.022.
39. Tassone P, Neri P, Kutok JL, Tournilhac O, Santos DD, Hatjiharissi E, Munshi V, Venuta S, Anderson KC, Treon SP, Munshi NC. A SCID-hu in vivo model of human waldenstrom macroglobulinemia. Blood. 2005; 106:1341–5. https://doi.org/10.1182/blood-2004-11-4477.
40. Yata K, Yaccoby S. The SCID-rab model: a novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia. 2004; 18:1891–7. https://doi.org/10.1038/sj.leu.2403513.
41. Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD Jr. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007; 109:2106–11. https://doi.org/10.1182/blood-2006-09-047712.
42. Calimeri T, Battista E, Conforti F, Neri P, Di Martino MT, Rossi M, Foresta U, Piro E, Ferrara F, Amorosi A, Bahlis N, Anderson KC, Munshi N, et al. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia. 2011; 25:707–11. https://doi.org/10.1038/leu.2010.300.
43. Zhu D, Wang Z, Zhao JJ, Calimeri T, Meng J, Hideshima T, Fulciniti M, Kang Y, Ficarro SB, Tai YT, Hunter Z, McMilin D, Tong H, et al. The cyclophilin A-CD147 complex promotes the proliferation and homing of multiple myeloma cells. Nat Med. 2015; 21:572–80. https://doi.org/10.1038/nm.3867.
44. Paton-Hough J, Chantry AD, Lawson MA. A review of current murine models of multiple myeloma used to assess the efficacy of therapeutic agents on tumour growth and bone disease. Bone. 2015; 77:57–68. https://doi.org/10.1016/j.bone.2015.04.004.
45. Amodio N, Leotta M, Bellizzi D, Di Martino MT, D’Aquila P, Lionetti M, Fabiani F, Leone E, Gulla AM, Passarino G, Caraglia M, Negrini M, Neri A, et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget. 2012; 3:1246–58. https://doi.org/10.18632/oncotarget.675.
46. Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, Cantafio ME, Gulla A, Conforti F, Morelli E, Tomaino V, Rossi M, Negrini M, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res. 2012; 18:6260–70. https://doi.org/10.1158/1078-0432.CCR-12-1708.
47. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002; 100:3175–82. https://doi.org/10.1182/blood-2001-12-0207.
48. Miyakawa Y, Ohnishi Y, Tomisawa M, Monnai M, Kohmura K, Ueyama Y, Ito M, Ikeda Y, Kizaki M, Nakamura M. Establishment of a new model of human multiple myeloma using NOD/SCID/gammac(null) (NOG) mice. Biochem Biophys Res Commun. 2004; 313:258–62.
49. Lawson MA, Paton-Hough JM, Evans HR, Walker RE, Harris W, Ratnabalan D, Snowden JA, Chantry AD. NOD/SCID-GAMMA mice are an ideal strain to assess the efficacy of therapeutic agents used in the treatment of myeloma bone disease. PLoS One. 2015; 10:e0119546. https://doi.org/10.1371/journal.pone.0119546.
50. Lwin ST, Edwards CM, Silbermann R. Preclinical animal models of multiple myeloma. Bonekey Rep. 2016; 5:772. https://doi.org/10.1038/bonekey.2015.142.
51. Hurchla MA, Garcia-Gomez A, Hornick MC, Ocio EM, Li A, Blanco JF, Collins L, Kirk CJ, Piwnica-Worms D, Vij R, Tomasson MH, Pandiella A, San Miguel JF, et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013; 27:430–40. https://doi.org/10.1038/leu.2012.183.
52. Fryer RA, Graham TJ, Smith EM, Walker-Samuel S, Morgan GJ, Robinson SP, Davies FE. Characterization of a novel mouse model of multiple myeloma and its use in preclinical therapeutic assessment. PLoS One. 2013; 8:e57641. https://doi.org/10.1371/journal.pone.0057641.
53. Hiasa M, Okui T, Allette YM, Ripsch MS, Sun-Wada GH, Wakabayashi H, Roodman GD, White FA, Yoneda T. Bone Pain induced by multiple myeloma is reduced by targeting V-ATPase and ASIC3. Cancer Res. 2017; 77:1283–95. https://doi.org/10.1158/0008-5472.CAN-15-3545.
54. Schueler J, Wider D, Klingner K, Siegers GM, May AM, Wasch R, Fiebig HH, Engelhardt M. Intratibial injection of human multiple myeloma cells in NOD/SCID IL-2Rgamma(null) mice mimics human myeloma and serves as a valuable tool for the development of anticancer strategies. PLoS One. 2013; 8:e79939. https://doi.org/10.1371/journal.pone.0079939.
55. Hilbert DM, Kopf M, Mock BA, Kohler G, Rudikoff S. Interleukin 6 is essential for in vivo development of b lineage neoplasms. J Exp Med. 1995; 182:243–8.
56. Nordan RP, Potter M. A macrophage-derived factor required by plasmacytomas for survival and proliferation in vitro. Science. 1986; 233:566–9.
57. Lattanzio G, Libert C, Aquilina M, Cappelletti M, Ciliberto G, Musiani P, Poli V. Defective development of pristane-oil-induced plasmacytomas in interleukin-6-deficient BALB/c mice. Am J Pathol. 1997; 151:689–96.
58. Potter M. Neoplastic development in plasma cells. Immunol Rev. 2003; 194:177–95.
59. Kovalchuk AL, Kim JS, Park SS, Coleman AE, Ward JM, Morse HC 3rd, Kishimoto T, Potter M, Janz S. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci U S A. 2002; 99:1509–14. https://doi.org/10.1073/pnas.022643999.
60. Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F, Janz S. Preclinical validation of interleukin 6 as a therapeutic target in multiple myeloma. Immunol Res. 2014; 59:188–202. https://doi.org/10.1007/s12026-014-8528-x.
61. Rosenbaum H, Harris AW, Bath ML, McNeall J, Webb E, Adams JM, Cory S. An E mu-v-abl transgene elicits plasmacytomas in concert with an activated myc gene. EMBO J. 1990; 9:897–905.
62. Vandenberg CJ, Waring P, Strasser A, Cory S. Plasmacytomagenesis in Emu-v-abl transgenic mice is accelerated when apoptosis is restrained. Blood. 2014; 124:1099–109. https://doi.org/10.1182/blood-2014-04-570770.
63. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, Kremer R, Cattoretti G, Bergsagel PL. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008; 13:167–80. https://doi.org/10.1016/j.ccr.2008.01.007.
64. Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, Stewart AK, Johnstone RW, Bergsagel PL. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood. 2012; 120:376–85. https://doi.org/10.1182/blood-2012-02-412783.
65. Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, Kumar S, Rajkumar SV, Carpten JD, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012; 120:1067–76. https://doi.org/10.1182/blood-2012-01-405985.
66. Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, Mulligan G, Chesi M, Bergsagel PL, Fonseca R. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011; 25:1026–35. https://doi.org/10.1038/leu.2011.53.
67. Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood. 2012; 120:2450–3. https://doi.org/10.1182/blood-2011-08-371567.
68. Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R, and Intergroupe Francophone du Myélome. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001; 98:3082–6.
69. Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003; 4:321–9. https://doi.org/10.1038/ni907.
70. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004; 21:81–93. https://doi.org/10.1016/j.immuni.2004.06.010.
71. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001; 412:300–7. https://doi.org/10.1038/35085509.
72. Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, Zheng M, Mani M, Henderson J, Pinkus GS, Munshi N, Horner J, Ivanova EV, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007; 11:349–60. https://doi.org/10.1016/j.ccr.2007.02.015.
73. Hua Y, White-Gilbertson S, Kellner J, Rachidi S, Usmani SZ, Chiosis G, Depinho R, Li Z, Liu B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res. 2013; 19:6242–51. https://doi.org/10.1158/1078-0432.CCR-13-2083.
74. Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, Schrock E, Ried T, Kuehl WM. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an ig locus in multiple myeloma. Blood. 1998; 91:4457–63.
75. Morito N, Yoh K, Fujioka Y, Nakano T, Shimohata H, Hashimoto Y, Yamada A, Maeda A, Matsuno F, Hata H, Suzuki A, Imagawa S, Mitsuya H, et al. Overexpression of c-Maf contributes to T-cell lymphoma in both mice and human. Cancer Res. 2006; 66:812–9. https://doi.org/10.1158/0008-5472.CAN-05-2154.
76. Spets H, Stromberg T, Georgii-Hemming P, Siljason J, Nilsson K, Jernberg-Wiklund H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: regulation during interleukin-6(IL-6)-induced growth and survival. Eur J Haematol. 2002; 69:76–89.
77. Linden M, Kirchhof N, Carlson C, Van Ness B. Targeted overexpression of Bcl-xl in B-lymphoid cells results in lymphoproliferative disease and plasma cell malignancies. Blood. 2004; 103:2779–86. https://doi.org/10.1182/blood-2003-10-3399.
78. Boylan KL, Gosse MA, Staggs SE, Janz S, Grindle S, Kansas GS, Van Ness BG. A transgenic mouse model of plasma cell malignancy shows phenotypic, cytogenetic, and gene expression heterogeneity similar to human multiple myeloma. Cancer Res. 2007; 67:4069–78. https://doi.org/10.1158/0008-5472.CAN-06-3699.
79. Hamouda MA, Jacquel A, Robert G, Puissant A, Richez V, Cassel R, Fenouille N, Roulland S, Gilleron J, Griessinger E, Dubois A, Bailly-Maitre B, Goncalves D, et al. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J Exp Med. 2016; 213:1705–22. https://doi.org/10.1084/jem.20150983.
80. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, Manz MG. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013; 31:635–74. https://doi.org/10.1146/annurev-immunol-032712-095921.
81. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, Manz MG, Flavell RA. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014; 32:364–72. https://doi.org/10.1038/nbt.2858.
82. Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, Kocoglu MH, Borsotti C, Zhang L, Branagan A, Eynon E, Manz MG, Flavell RA, Dhodapkar MV. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat Med. 2016; 22:1351–7. https://doi.org/10.1038/nm.4202.
83. Pastina P, Nardone V, Botta C, Croci S, Tini P, Battaglia G, Ricci V, Cusi MG, Gandolfo C, Misso G, Zappavigna S, Caraglia M, Giordano A, et al. Radiotherapy prolongs the survival of advanced non-small-cell lung cancer patients undergone to an immune-modulating treatment with dose-fractioned cisplatin and metronomic etoposide and bevacizumab (mPEBev). Oncotarget. 2017; 8:75904–13. https://doi.org/10.18632/oncotarget.20411.
84. Cusi MG, Botta C, Pastina P, Rossetti MG, Dreassi E, Guidelli GM, Fioravanti A, Martino EC, Gandolfo C, Pagliuchi M, Basile A, Carbone SF, Ricci V, et al. Phase I trial of thymidylate synthase poly-epitope peptide (TSPP) vaccine in advanced cancer patients. Cancer Immunol Immunother. 2015; 64:1159–73. https://doi.org/10.1007/s00262-015-1711-7.
85. Correale P, Botta C, Martino EC, Ulivieri C, Battaglia G, Carfagno T, Rossetti MG, Fioravanti A, Guidelli GM, Cheleschi S, Gandolfo C, Carbone F, Baldari TC, et al. Phase Ib study of poly-epitope peptide vaccination to thymidylate synthase (TSPP) and GOLFIG chemo-immunotherapy for treatment of metastatic colorectal cancer patients. OncoImmunology. 2015; 5:e1101205. https://doi.org/10.1080/2162402X.2015.1101205.
86. Correale P, Botta C, Ciliberto D, Pastina P, Ingargiola R, Zappavigna S, Tassone P, Pirtoli L, Caraglia M, Tagliaferri P. Immunotherapy of colorectal cancer: new perspectives after a long path. Immunotherapy. 2016; 8:1281–92. https://doi.org/10.2217/imt-2016-0089.
87. Botta C, Misso G, Martino EC, Pirtoli L, Cusi MG, Tassone P, Tagliaferri P, Caraglia M, Correale P. The route to solve the interplay between inflammation, angiogenesis and anti-cancer immune response. Cell Death Dis. 2016; 7:e2299. https://doi.org/10.1038/cddis.2016.211.
88. Botta C, Bestoso E, Apollinari S, Cusi MG, Pastina P, Abbruzzese A, Sperlongano P, Misso G, Caraglia M, Tassone P, Tagliaferri P, Correale P. Immune-modulating effects of the newest cetuximab-based chemoimmunotherapy regimen in advanced colorectal cancer patients. J Immunother. 2012; 35:440–7. https://doi.org/10.1097/CJI.0b013e31825943aa.
89. Botta C, Barbieri V, Ciliberto D, Rossi A, Rocco D, Addeo R, Staropoli N, Pastina P, Marvaso G, Martellucci I, Guglielmo A, Pirtoli L, Sperlongano P, et al. Systemic inflammatory status at baseline predicts bevacizumab benefit in advanced non-small cell lung cancer patients. Cancer Biol Ther. 2013; 14:469–75. https://doi.org/10.4161/cbt.24425.
90. Van Valckenborgh E, Schouppe E, Movahedi K, De Bruyne E, Menu E, De Baetselier P, Vanderkerken K, Van Ginderachter JA. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia. 2012; 26:2424–8. https://doi.org/10.1038/leu.2012.113.
91. Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, Teng MW, Colonna M, Ritchie DS, Chesi M, Bergsagel PL, Hill GR, Smyth MJ, Martinet L. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest. 2015; 125:2904. https://doi.org/10.1172/JCI82646.
92. Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL, Keane N, Ahmann GJ, Morrison KM, et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med. 2016; 22:1411–20. https://doi.org/10.1038/nm.4229.
93. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002; 99:12293–7. https://doi.org/10.1073/pnas.192461099.
94. Hong S, Li H, Qian J, Yang J, Lu Y, Yi Q. Optimizing dendritic cell vaccine for immunotherapy in multiple myeloma: tumour lysates are more potent tumour antigens than idiotype protein to promote anti-tumour immunity. Clin Exp Immunol. 2012; 170:167–77. https://doi.org/10.1111/j.1365-2249.2012.04642.x.
95. Botta C, Gulla A, Correale P, Tagliaferri P, Tassone P. Myeloid-derived suppressor cells in multiple myeloma: pre-clinical research and translational opportunities. Front Oncol. 2014; 4:348. https://doi.org/10.3389/fonc.2014.00348.
96. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016; 16:759–73. https://doi.org/10.1038/nrc.2016.91.
97. Botta C, Cuce M, Pitari MR, Caracciolo D, Gulla A, Morelli E, Riillo C, Biamonte L, Cantafio MEG, Prabhala R, Mignogna C, Di Vito A, Altomare E, et al. MiR-29b antagonizes the pro-inflammatory tumor-promoting activity of multiple myeloma-educated dendritic cells. Leukemia. 2017. https://doi.org/10.1038/leu.2017.336.
98. Amodio N, Di Martino MT, Neri A, Tagliaferri P, Tassone P. Non-coding RNA: a novel opportunity for the personalized treatment of multiple myeloma. Expert Opin Biol Ther. 2013; 13:S125–37. https://doi.org/10.1517/14712598.2013.796356.
99. Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, Tassone P. MiR-29s: A family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget. 2015; 6:12837–61. https://doi.org/10.18632/oncotarget.3805.
100. Rossi M, Botta C, Correale P, Tassone P, Tagliaferri P. Immunologic microenvironment and personalized treatment in multiple myeloma. Expert Opin Biol Ther. 2013; 13:S83–93. https://doi.org/10.1517/14712598.2013.799130.
101. Al-Hujaily EM, Oldham RA, Hari P, Medin JA. Development of novel immunotherapies for multiple myeloma. Int J Mol Sci. 2016; 17. https://doi.org/10.3390/ijms17091506.