
INTRODUCTION
Glioblastoma multiforme (GBM) represents the most common and most malignant primary brain tumor in adults with an incidence of 3-4/100,000/year [1]. The prognosis of patients diagnosed with GBM remains extraordinary poor with the median survival only 12.1–14.6 months [2, 3]. Moreover, no more than 3–5% of patients survive longer than 3 years [1, 4]. Genomics and epigenomics of GBM, together with other glial tumors, were extensively researched during the last decade that led into the identification of several abnormalities in major cellular signaling pathways with a diversity of mutated genes in gliomas [5–7]. The essential role among them represents the metabolic enzyme isocitrate dehydrogenase (IDH) [5, 8]. Isocitrate dehydrogenases (three isoforms IDH1, IDH2, and IDH3) catalyze the oxidative carboxylation of isocitrate to alpha-ketoglutarate and reduce nicotinamide adenine dinucleotide phosphate (NADP) to NADPH, which is necessary for the regeneration of reduced glutathione that serves as the main cellular antioxidant [9]. The mutated IDH acquires aberrant and oncogenic function, namely the conversion of alpha-ketoglutarate to the oncometabolite 2-hydroxyglutarate (2-HG), which subsequently leads to genome-wide epigenetic changes in human gliomas and their progression [10, 11]. The mutations in IDH1/2 represent also an independent and important GBM prognostic factor [12–16] and their routine assessment should be the standard in the clinical management of patients with gliomas (including GBM) according to the recently updated World Health Organization (WHO) 2016 classification of CNS tumors and European Association for Neuro-Oncology (EANO) guidelines on the diagnosis and treatment of glial tumors [17, 18].
The GBM microenvironment and its involvement in cancer development and progression was extensively studied, especially tumor angiogenesis and neovascularization [19, 20]. GBM is highly vascularized tumor with substantial microvascular proliferation surrounding necrotic areas [17, 21, 22]. There is still a relative lack of studies, above that with inconclusive results, which assess the relation of GBM genetics and especially IDH mutation status with tumor angiogenesis and microvessel formation. Originally, it was hypothesized that the aberrantly produced 2-HG by mutated IDH may compete with alpha-ketoglutarate and inhibit the prolyl hydroxylase (PHD), which in turn can lead to the pathological stabilization of hypoxia inducible factor (HIF) and induction of the expression of the major proangiogenic vascular endothelial growth factor (VEGF) and thus initiation of tumor angiogenesis [23, 24]. However, subsequent studies did not prove this hypothesis [25, 26]. Above that, more recent studies in human astrocytes, colorectal and erythroleukemia cell lines found the negative association between IDH mutations and HIF and VEGF expression [27, 28]. The conclusive information about the association of IDH mutations and tumor microvascular proliferation would be of a great clinical importance in relation to the development of targeted anticancer and antiangiogenic therapy for malignant gliomas [29]. Moreover, hypoxia may play the key role in the development and progression of the malignancies in CNS as has been recently presented in the context of the aggressive metastatic disease triggered by certain subtypes of breast cancer [30–33].
The aim of this study was to examine the hypoxia-related angiogenesis and tumor-specific microcirculation assessed by immunohistochemistry in the tissue samples obtained during surgery from patients with newly-diagnosed GBM, and to find the relationship between tumor microcirculation and IDH1 mutation status in the same patients. GBM microcirculation was assessed by histological quantification of expression of pan-endothelial markers (CD31, CD34, and von Willebrand factor – vWF) as well as the marker of newly-formed microvessels (CD105 - endoglin) together with the expression of the major proangiogenic vascular endothelial growth factor (VEGF) and the brain-specific activator of angiogenesis in the hypoxic condition – hypoxia inducible factor 2 alpha (HIF2α). The proliferative activity (Ki-67 antigen expression) was also assessed and correlated with GBM microcirculation. The prognostic significance of these markers and IDH1 mutation status in relation to patient’s progression-free survival (PFS) and overall survival (OS) was examined as well.
RESULTS
The occurrence of IDH1 R132H mutation and its relation to patients’ survival
The IDH1 R132H mutation was detected in the tissue samples from 20 GBM patients (38.5%), whereas the IDH1 wild-type tumor was observed in remaining 32 patients (61.5%). Because of the relatively high portion of IDH1 mutated tumors, the separation of primary and secondary GBM (progressing from the low-grade gliomas) was performed on the basis of clinical information from the patient history, where possible. The diagnosis of secondary GBM was established if there was a history of previously assessed low grade glioma, or if the patient suffered from presumably tumor corresponding neurological symptomatology (epileptic seizures, focal neurological deficit) at least 6 months before the final GBM diagnosis. Based on these criteria, IDH1 R132H mutation was detected in 4 from 34 primary GBMs (11.8%), whereas the majority of secondary GBMs comprised the mutation (16 of 18, 88.9%) (Table 1).
Table 1: The distribution of IDH1 R132H mutated tumors among primary and secondary glioblastomas
Mutation status |
Primary glioblastoma (n=34) |
Secondary glioblastoma (n=18) |
|---|---|---|
IDH1 R132H |
4 (11.8 %) |
16 (88.9 %) |
IDH1 wild type |
30 (88.2 %) |
2 (11.1 %) |
The detection of IDH1 R123H mutation in the tumor tissue represented a significant positive prognostic factor for both PFS and OS of GBM patients (Table 2). Subjects with GBM positive for IDH1 R132H mutation experienced longer median PFS than patients with wild-type tumors (136 vs. 44 days; P = 0.037, log-rank test) (Figure 1). Likewise, patients with IDH1 R132H positive tumors had significantly longer median OS than those without the mutation (270 vs. 124 days; P = 0.035, log-rank test) (Figure 2). In the multivariate analysis, the IDH1 R132H mutation remained positive prognostic factor for OS (HR = 0.433, P = 0.011) but not PFS (HR = 0.560, P = 0.092) of GBM patients. The other significant prognostic factors in multivariate analysis were age at diagnosis (P = 0.002) and treatment after surgery (P = 0.003) (Table 3).
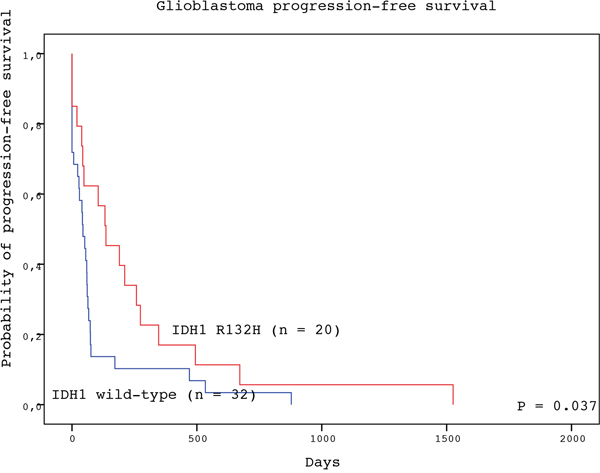
Figure 1: Progression-free survival of GBM patients with IDH1 R132H-mutated (red line) and IDH1 wild type (blue line) tumors (log-rank test).
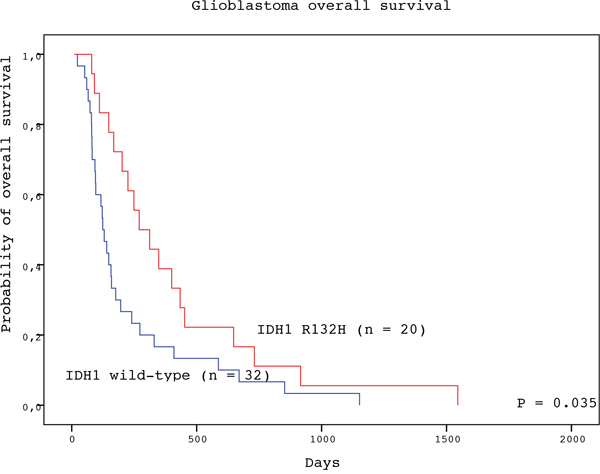
Figure 2: Overall survival of GBM patients with IDH1 R132H-mutated (red line) and IDH1 wild type (blue line) tumors (log-rank test).
Table 2: Progression-free survival and overall survival of GBM patients in relation to the IDH1 mutation status
Survival analysis |
Number of patients |
Median [days] (95% Cl) |
P value (log-rank) |
|---|---|---|---|
Progression-free Survival |
|
|
|
IDH1 R132H |
20 |
136 (22-249) |
|
IDH1 wild type |
32 |
44 (17-71) |
0.037 |
Overall Survival |
|
|
|
IDH1 R132H |
20 |
270 (139-400) |
|
IDH1 wild type |
32 |
124 (93-155) |
0.035 |
Table 3: Multivariate Cox proportional hazards regression for progression free survival (PFS) and overall survival (OS) of patients with GBM
Characteristics |
PFS |
P value |
OS |
P value |
|---|---|---|---|---|
Age |
|
|
|
|
< 60 |
1.000 |
0.088 |
1.000 |
0.002 |
≥ 60 |
1.945 (0.907 – 4.172) |
|
3.691 (1.606 – 8.484) |
|
Gender |
|
|
|
|
Female |
1.000 |
0.936 |
1.000 |
0.162 |
Male |
0.974 (0.513 – 1.851) |
|
0.632 (0.332 – 1.203) |
|
KPS |
|
|
|
|
< 90 |
1.000 |
0.607 |
1.000 |
0.319 |
≥ 90 |
0.85 (0.458 – 1.578) |
|
0.726 (0.387 – 1.363) |
|
Treatment |
|
|
|
|
Surgery alone |
1.000 |
<0.001 |
1.000 |
0.003 |
Surgery + RT/CT |
0.241 (0.112 – 0.519) |
|
0.343 (0.168 – 0.699) |
|
IDH1 mutation status |
|
|
|
|
IDH1 wild type |
1.000 |
0.092 |
1.000 |
0.011 |
IDH1 R132H |
0.560 (0.285 – 1.1) |
|
0.433 (0.227 – 0.823) |
|
PFS – progression free survival; OS – overall survival; HR – hazard ratio; CI - confidence interval; KPS - Karnofsky performance score; RT - radiotherapy; CT – chemotherapy.
Mutual correlations among microvascular, angiogenesis, hypoxia, and proliferation markers and other clinicopathological characteristics
The histologically quantified expression of microvascular endothelial markers (CD31, CD34, vWF) together with markers of active endothelia (CD105), proportions of newly-formed microvessels (ratios CD105/CD31, CD105/CD34 and CD105/vWF), intratumor hypoxia (HIF2α), angiogenesis (VEGF), and proliferation (Ki-67) were correlated each other and also with patient’s clinicopathological characteristics such as the age at diagnosis, pre-operative Karnofsky Performance Status (KPS), OS and PFS. The Spearman’s rank correlation coefficients are presented in Table 4. Statistically significant results (P < 0.05) are highlighted.
Table 4: Mutual correlations among the expression of hypoxia, angiogenesis, microvascular, proliferation markers, and clinical-pathological characteristics (Spearman’s rank-order correlation coefficients)
|
KPS |
PFS |
OS |
CD31 |
CD34 |
vWF |
CD105 |
VEGF |
HIF2 |
CD105/CD31 |
CD105/CD34 |
CD105/vWF |
Ki-67 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Age |
-0.076 |
-0.380** |
-0.583** |
-0.119 |
-0.105 |
0.020 |
0.203 |
-0.009 |
-0.278* |
0.306* |
0.358** |
0.158 |
0.004 |
KPS |
- |
0.207 |
0.082 |
0.054 |
0.070 |
0.028 |
0.025 |
0.037 |
-0.087 |
-0.094 |
-0.149 |
-0.107 |
0.097 |
PFS |
- |
- |
0.781** |
0.229 |
0.181 |
0.082 |
-0.043 |
-0.078 |
0.179 |
-0.318* |
-0.288* |
-0.156 |
-0.118 |
OS |
- |
- |
- |
0.206 |
0.210 |
0.089 |
0.023 |
-0.124 |
0.259 |
-0.242 |
-0.232 |
-0.048 |
-0.073 |
CD31 |
- |
- |
- |
- |
0.845** |
0.760** |
0.482** |
0.163 |
0.189 |
-0.492** |
-0.405** |
-0.298* |
-0.193 |
CD34 |
- |
- |
- |
- |
- |
0.859** |
0.590** |
0.270 |
0.285* |
-0.300* |
-0.475** |
-0.321* |
-0.143 |
vWF |
- |
- |
- |
- |
- |
- |
0.662** |
0.189 |
0.102 |
-0.164 |
-0.258 |
-0.389** |
-0.195 |
CD105 |
- |
- |
- |
- |
- |
- |
- |
0.199 |
0.072 |
0.440** |
0.368** |
0.374** |
-0.143 |
VEGF |
- |
- |
- |
- |
- |
- |
- |
- |
0.330* |
0.004 |
-0.132 |
-0.049 |
0.228 |
HIF2 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
-0.155 |
-0.317* |
-0.050 |
0.194 |
CD105/CD31 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
0.854** |
0.768** |
0.112 |
CD105/CD34 |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
0.817** |
0.005 |
CD105/vWF |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
0.049 |
** Correlation is significant at the 0.01 level (2-tailed)
* Correlation is significant at the 0.05 level (2-tailed)
Relation between microvascular, angiogenesis, hypoxia, and proliferation markers and IDH1 R132H mutation status
The differences in expression of endothelial markers (CD31, CD34, vWF) together with markers of active endothelia (CD105), proportions of newly-formed microvessels (ratios CD105/CD31, CD105/CD34 and CD105/vWF), intratumor hypoxia (HIF2α), angiogenesis (VEGF), and proliferative activity (Ki-67) were examined between GBM samples with IDH1 R132H mutation by comparison with IDH1 wild-type tumors.
The only statistically significant result was observed for VEGF with higher expression in IDH1 wild-type than IDH1 R132H mutated tumors (median 0.02285 vs. 0.017706, respectively; P = 0.01, Mann-Whitney U test).
Relation between microvascular, angiogenesis, and hypoxia markers and patients’ survival
Patients with GBM expressing endothelial marker CD31 above the median (60.4 mm-2) experienced significantly longer estimated median PFS than patients with CD31 bellow this value (64 vs. 43 days, respectively; P = 0.041, long-rank test). Similarly, expression of endothelial marker CD34 above the median (63.4 mm-2) was associated with significantly longer estimated median PFS than CD31 bellow this value (61 vs. 48 days, respectively; P = 0.042, long-rank test).
On the contrary, the higher expression of markers assessing the newly-formed microvessels (ratio CD105/CD31) over the median (0.547) was associated with shorter estimated median PFS compared with expression bellow this value (48 vs. 74 days, respectively; P = 0.026, long-rank test).
DISCUSSION
Recurrent IDH mutations and their role for oncogenesis and tumor progression were described for the first time in GBM [5, 34]. This observation led to new insights into GBM biology with the major role of alterations in cancer cell metabolism for some portion of these tumors [35]. Predominantly secondary GBM that progressed from the low grade tumors were identified to harbor the IDH mutations [36]. The IDH1 and IDH2 mutations were subsequently indentified in the majority of lower grade gliomas, mostly in diffuse astrocytomas (62%), anaplastic astrocytomas (46%), oligodendrogliomas (77%), and anaplastic oligodendrogliomas (73%) [37].
The main oncogenic role of IDH mutations are likely caused by the change in the metabolic activity of this enzyme. Instead of the production of alpha-ketoglutarate, mutated IDH produced oncometabolite 2-hydroxyglutarate (2-HG) that was highly accumulated in cancer cells [11]. Subsequently, 2-HG inhibits the functions of the alpha-ketoglutarate dependent superfamily of dioxygenases that have diverse cellular functions including histone demethylation and demethylation of hypermethylated DNA [38, 39]. IDH mutations and 2-HG production were identified to be the sufficient steps in the process leading to glioma hypermethylator phenotype [40]. This finding was important for understanding of glioma oncogenesis and highlighted the interplay between genomic and epigenomic changes in cancers [41].
The assessment of IDH mutations, predominantly IDH1 codon 132 and IDH2 codon 172 missense mutations, was recently incorporated into the updated 2016 WHO classification of CNS tumors [17]. The so-called “integrated diagnosis” combining the histological tumor typing, tumor grading using the four-tiered WHO grading scheme, and the tissue-based molecular analyses of IDH mutations and other molecular characteristics represents now the standard in the diagnostic process of CNS astrocytic and oligodendroglial tumors, including GBM [17, 18].
From the clinical point of view, the assessment of IDH mutations in gliomas is crucial for the prediction of patient prognosis, which is especially emphasized for GBM. Across a number of studies, the substantial differences in OS between IDH-mutant and IDH wildtype (IDH-WT) GBM were identified such as 3.8 versus 1.1 years [5], 2.6 versus 1.3 years [8], 2.3 versus 1.2 years [9], and 3 years versus 1 year [13]. Similarly, OS differences were observed in patients with IDH-mutant versus IDH-WT anaplastic astrocytomas - 5.4 versus 1.7 years [8], 6.8 versus 1.6 years [9], and 7 versus 2 years [13]. IDH mutations keep their important diagnostic and prognostic role also for patients with low-grade gliomas [7, 42–44]. Recent meta-analysis confirmed the prognostic role of IDH1/2 mutations in gliomas as well [45].
In our study we also proved the presence of IDH1 R132H mutation in the tumor tissue to serve as the positive prognostic factor for patients with GBM in relation to PFS as well as OS. The IDH1 mutation remained positive prognostic factor for OS (HR = 0.433, P = 0.011) but not PFS (HR = 0.560, P = 0.092) in the multivariate analysis as well. However, the differences in median PFS (136 vs. 44 days) and OS (270 vs. 124 days) between patients with and without IDH1 mutated tumors are not as meaningful as in other studies. The reason for this could be a big heterogeneity of treatments applied to our patients in the real clinical practice. The recommended standard treatment protocol with neurosurgery followed by concomitant chemo-radiotherapy with temozolomide [46] was implemented in 15 patients. The radiotherapy only after surgery was provided to other 15 patients. Importantly, 22 patients were treated neither by radiotherapy nor by chemotherapy after the surgery. The treatment after surgery (radiotherapy or radiotherapy plus chemotherapy) compared with the neurosurgery alone served as a strong positive prognostic factor for patients PFS (HR = 0.241, P<0.001) and OS (HR = 0.343, P=0.003) in multivariate analysis as well. Moreover, older patients (age ≥ 60 years) had significantly worse prognosis for OS than younger patients (HR = 3.691, P = 0.002). Both the age at diagnosis and postsurgical treatment with either radiotherapy or radiotherapy plus chemotherapy are known strong prognostic factors for patients with GBM [47–50].
A limited number of patients included into the study can impinge the survival analysis as well, which is caused mainly by low incidence of GBM among other tumor types. Because of a relatively high number of IDH1 R132H mutated tumors (20 from 52 patients, 38.5%) identified in this study, distinction between primary and secondary GBM was done based on clinical information from each patient history, where possible. After that the majority of IDH1 mutated tumors (16 from 18 patients, 88.9%) were classified as secondary GBM that is in accordance with similar studies [8, 36, 51].
GBM is highly vascularized tumor with substantial microvascular proliferation predominantly surrounding necrotic areas [17, 21, 22, 29]. The process of angiogenesis is turned on by the presence of hypoxia that results in upregulation of hypoxia inducible factors (HIF1α and HIF2α), which subsequently leads to the upregulation of vascular growth factors such as VEGF and microvascular proliferation [52–54]. In addition to the essential role of VEGF also other growth factors were identified to play an important role in GBM angiogenesis such as hepatocyte growth factor (HGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), or angiopoietins and interleukin-8 [55–58]. The pathological angiogenesis can be also constitutively stimulated in non-hypoxia dependent manner by the aberrant activation of the main cellular pathways in GBM such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) signalizations [21, 54, 59].
Abundant GBM microvasculature as a response to aberrant angiogenesis was observed in a number of studies, even if the selection of appropriate endothelial marker is an essential factor for the microvessels assessment. The pan-endothelial marker CD34 was expressed in the significant portion of giant cell GBM (73%) as well as non-giant cell GBM (56%) with the strong widespread staining in 55% and 25% of tumors, respectively [60]. The significant relationship between the expression of pan-endothelial marker CD31 and tumor grade was found in the study with 45 astrocytomas where the highest expression was observed in GBM samples (P = 0.001) [61]. Another study examined the degree of angiogenesis in GBM measured by microvessel density (MVD) using both pan-endothelial marker CD31 and marker CD105/endoglin, which preferentially react with active endothelial cells in angiogenic tissue [62]. MVD was correlated with the expression of VEGF and patient prognosis. MVD was increasing with the higher grades of gliomas and the GBM was the most vascularized tumor. MVD assessed by CD105 was more closely correlated with VEGF expression than by CD31 endothelial marker. Moreover, GBM patients showing higher CD105 MVD had a significantly shorter survival than those with lower CD105 MVD tumors (P = 0.0131), which was not the case for CD31 [62]. The study concluded that CD105 may be a better marker than CD31 for evaluation of angiogenesis and prediction of prognosis in astrocytic tumors. The comparison between CD105 and CD31 MVD in the assessment of GBM angiogenesis was examined in another study with 46 tumor samples as well [63]. CD105 MVD was significantly higher than CD31 MVD (median 49 vs. 37 microvessels per field, respectively, P<0.001) and CD105 MVD was more closely correlated with VEGF (R = 0.421, P = 0.003) than CD31 MVD (R = 0.330, P = 0.024). Moreover, patients with lower CD105 MVD had increased survival compared with those with higher CD105 MVD (P = 0.045) that was not the case for CD31 (P = 0.340). More recently, the angiogenesis was examined by the assessment of CD105 and CD31 MVD in addition to VEGF expression in 50 adult GBM patients [63]. CD105 MVD was significantly higher in GBM compared with peritumoral tissue samples (P=0.012), that was not observed for CD 31. However, inside the tumor tissue the positive correlation was found between MVD assessed by CD 105 and CD 31 endothelial markers (R = 0.630, P<0.001). Both CD105 and CD31 MVD significantly correlated with VEGF expression (P<0.001). The large study including 208 GBM samples evaluated tumor angiogenesis by the expression of CD34, PDGF-C, VEGF and CD105 markers and their relation to HIF1α expression [64]. The expression of HIF1α positively correlated with VEGF and PDGF-C expression in tumor samples (P < 0.001). Moreover, endothelial cells expressing PDGF-C and VEGF were also positive for CD105 confirming the proangiogenic effect of these factors in GBM. Very recently, the immunohistochemical expression of CD34 and CD105 was examined in 43 GBM patients of which 20 experienced tumor hemorrhage [65]. Interestingly, tumors that hemorrhaged had higher expression of CD34 (P = 0.0583) and CD105 (P = 0.0467) as well as HIF1α mRNA (P = 0.0073) by comparison with non-hemorrhaged tumors. Therefore increased hypoxia-induced angiogenesis and microvascular formation may play a role in glioblastoma hemorrhage as well. The relationship between the vascularization assessed by histopathology from tumor samples and radiological methods before the surgery was also observed in recent prospective study with 25 newly diagnosed GBM patients [66]. CD105 MVD expression significantly positively correlated with biomarkers related to tumor perfusion assessed by perfusion computed tomography (permeability surface-area product) and dynamic contrast-enhanced magnetic resonance imaging (volume transfer constant) in this study (R = 0.644, P < 0.001 and R = 0.683, P < 0.001, respectively).
In addition to other studies we evaluated multiple pan-endothelial markers (CD31, CD34, and vWF) together with the marker of active endothelia and newly-formed microvessels (CD105/endoglin) in the assessment of GBM microcirculation and tried to find their mutual relationships. Strong correlations were observed among the pan-endothelial markers CD31, CD34, and vWF. The marker of active endothelia CD105 also correlated with CD31, CD34, as well as vWF, however the relationships were weaker (R = 0.482, 0.59, and 0.662 respectively). Therefore the pan-endothelial markers are commutable with one another, whereas the combination of one pan-endothelial marker with marker of active endothelia provides additional information to the analyses of GBM microvascularity. Only a portion of microvessels detected in the tumor tissue are in fact formed by mitotically active endothelial cells (assessed by CD105 expression). On the other hand, the relationship between the tumor microvascularity (assessed by expression of pan-endothelial as well as CD105 markers) and the leading proangiogenic factor VEGF was not observed in our study. The tumor hypoxia was examined by the assessment of expression of hypoxia inducible factor HIF2α, which is more specific than HIF1α for brain tissue and is widely expressed in high grade gliomas including GBM [67, 68]. Even if the VEGF moderately correlated with HIF2α (R = 0.33), there was no relationship between the tumor microcrovascularity and the hypoxia inducible factor HIF2α, with the only exception of a weak positive correlation between HIF2α and CD34 expression (R = 0.285). We observed no relationships among tumor proliferative activity (Ki-67) and tumor microvascularity, angiogenesis, or hypoxia. We also examined the portion of GBM-associated newly-formed microvessels by the assessment of the ratios between the expression of CD105 and pan-endothelial markers (CD105/CD31, CD105/CD34, and CD105/vWF) and their relation to VEGF and HIF2α. Although there were substantial mutual correlations among these ratios, no association to VEGF was found and only moderate negative correlation of CD105/CD34 with HIF2α was observed (R = 0.317). Therefore the process of angiogenesis in our GBM samples could be only very little dependent on the HIF/VEGF axis. It could rather go through the alternative pathways such as the aberrant activation of MAPK, PI3K, or Notch signaling that were also described in GBM [54, 59, 69]. Interestingly, the CD105/CD31 and CD105/CD34 ratios were positively associated with the patient age (R = 0.319 and R = 368, respectively). This indicated that older compared with younger patients had more newly-formed microvessels in their tumors, even though the correlations were relatively weak.
We observed that the expression of CD31 and CD34 above the median levels served as the positive prognostic factor for patient PFS. On the contrary, patients with higher portion of newly-formed microvessels (assessed by the ratio CD105/CD31 above the median level) experienced significantly shorter PFS. Therefore in addition to other studies, we explored that just the portion of newly-formed microvessels in tumor tissue can be a negative prognostic factor for survival of patients with GBM.
The relationship between the presence of IDH mutations and tumor angiogenesis has been also studied, though with inconclusive results. As discussed above, HIF1α and HIF2α have the central role in cellular response to hypoxia and initiating of VEGF-mediated angiogenesis. The activity of hypoxia inducible factors is negatively regulated by the prolyl hydroxylase (PHD) that promote HIF degradation by the hydroxylation of two proline residues in its oxygen-dependent degradation domain, which in turns leads to association of HIF with the pVHL E3 ligase complex and its degradation via the ubiquitin-proteasome pathway [70]. PHD belongs to the alpha-ketoglutarate dependent dioxigenases. Therefore it was hypothesized that the aberrantly produced 2-HG by mutated IDH may compete with alpha-ketoglutarate and inhibit PHD, which in turn can lead to the pathological stabilization of HIF, induction of VEGF expression and thus initiation of tumor angiogenesis [23, 24]. However, subsequent studies did not prove this hypothesis when no significant correlation was observed between IDH mutations and HIF1α expression in glioma samples [25, 26]. Above that, more recent studies in human astrocytes, colorectal and erythroleukemia cell lines found the negative association between IDH mutations and HIF1α expression caused by the 2-HG-dependent stimulation rather than inhibition of PHD [27, 28].
We also examined if there was a difference in expression of hypoxia inducible factor (HIF2α) as well as its downstream pro-angiogenetic factor VEGF in accordance to IDH1 R123H mutation status in our GBM samples. In addition to studies discussed above, we examined whether there was a difference in microvessels formation between the IDH1 mutated and IDH1 wild-type tumors as well. We found that HIF2α expression and tumor microvessels formation were independent of the IDH1 mutation status. Moreover, IDH1 mutated tumors experienced in fact decreased expression of VEGF by comparison to IDH1 wild-type GBM. Finally, the tumor proliferative activity assessed by the expression of Ki-67 antigen did not differ with the IDH1 mutation status.
The main limitation of the presented study is a relatively low number of included patients as well as the substantial heterogeneity in applied postsurgical treatment that can impinge the survival analysis. Also the mechanism study is needed to elucidate the role of IDH1 mutation in GBM angiogenesis and neovascularization, which is considered for designing the follow-up projects.
In summary, our results did not prove the hypothesis that angiogenesis in IDH-mutated GBM is caused mainly by IDH mutation-dependent expression of HIF and downstream overexpression of VEGF. Rather than VEGF-mediated angiogenesis, the alternative mechanisms of microvessels formation such as the aberrant activation of MAPK, PI3K, or Notch signaling can play a significant role in our GBM tumors. We also showed that the combination of pan-endothelial marker together with the marker of active endothelia CD105/endoglin can improve the histological assessment of GBM microvasculature and better predict of patient’s prognosis.
MATERIALS AND METHODS
Patients
52 patients with a diagnosis of GBM (n = 52; 28 males and 24 females; median age 64.3 years) who were treated (tumor resection, radiotherapy, and chemotherapy with temozolomide) in the Faculty Hospital in Pilsen between the years 2009 and 2014 were included into the retrospective study (Table 5). The study protocol was approved by the ethics committee.
Table 5: Characteristics of glioblastoma patients included in the study
Patient characteristics |
|
|---|---|
Sex |
|
Male |
28 |
Female |
24 |
Age, years |
|
Median |
67 |
Range |
35 - 87 |
KPS |
|
Median |
80 |
Range |
30 - 100 |
Postoperative treatment |
|
RT + CT |
15 |
RT alone |
15 |
None |
22 |
KPS - Karnofsky performance score; RT - radiotherapy; CT - chemotherapy
DNA isolation
DNA was extracted from 10 μm sections of formalin-fixed, paraffin-embedded (FFPE) tissue samples following macro dissection of tumor tissue and normal brain tissue using the QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany). The 10 μm sections corresponded to haematoxylin and eosin representative with tumor tissue verified by a pathologist specialized in brain tumors.
Mutation detection
For detection of mutant allele IDH1 c.395G>A (p.R132H, COSMIC ID 28746), TaqMan Mutation Detection Assays (assay name: IDH1 28746mu and IDH1 rf) with the TaqMan Mutation Detection IPC Reagent Kit (Life Technologies, Carlsbad, California, USA) were used. Mutant allele detection was performed according to recommended procedure and reaction conditions in the manual. For the amplification, the Stratagene Mx3000P realtime PCR system instrument (Agilent Technologies, Inc., Santa Clara, CA, USA) was used. Detection of mutant alleles was performed in duplicate in a reaction volume of 20 μL. Detection of reference gene was also performed in duplicate. The analysis of positive samples was repeated. Before analysis of collection of tumor samples, the samples of normal brain tissue were analyzed for detection of cut-off amplification curve. No amplification of mutant allele was present in normal brain tissue. On the basis of these results and the shape of amplification curve of positive tumor samples, the 25 ΔCt cut-off value was determined.
Histological processing and quantification
Tissue FFPE samples for light microscopy were cut into 5 μm sections and stained with hematoxylin and eosin. Adjacent sections were processed immunohistochemically using Ventana Benchmark XT automated stainer (Ventana Medical System, Inc., Tucson, AZ, USA) with diaminobenzidine visualization and counterstaining with Mayer’s haematoxylin. The details on monoclonal primary antibodies employed together with appropriate positive controls are listed in Table 6. Briefly, three markers were used to detect the endothelial cells lining the tumorous blood vessels (CD34, CD31, and vWF) and another marker (CD105) was used for newly-formed proliferation-associated endothelial cells. Stimulation of angiogenesis was assessed using the anti-VEGF-A (vascular endothelial growth factor A - VEGF) antibody. Tissue hypoxia and possible regulation of the VEGF expression was assessed using the anti HIF2α (type 2 hypoxia inducible factor) antibody. The proliferative activity of the tumor was assessed by the expression of Ki-67 antigen.
Table 6: Immunohistochemical reagents for the histological analysis
Primary antibody, clone and dilution |
Manufacturer |
Used for detection of |
|---|---|---|
Monoclonal Mouse Anti-Human CD34 Class II, Clone QBEnd 10 (Dako), 1:30 |
DakoCytomation, Glostrup, Denmark |
endothelial cells |
Monoclonal Mouse Anti-Human CD31 , Endothelial cell, Clone JC70A, 1:40 |
DakoCytomation, Glostrup, Denmark |
endothelial cells |
Polyclonal Rabbit Anti-Human Von Willebrand Factor (vWF), 1:200 |
DakoCytomation, Glostrup, Denmark |
endothelial cells |
Monoclonal Mouse Anti-Human CD105, Endoglin, Clone SN6h, 1:5 |
DakoCytomation, Glostrup, Denmark |
proliferation-associated endothelial cells |
Polyclonal Rabbit Anti-Human VEGF-A (vascular endothelial growth factor), 1:100 |
Zytomed Systems, Berlin, Germany |
up-regulation of angiogenesis |
Monoclonal Mouse Anti-Human HIF2α (hypoxia inducible factor), Clone ep190b, 1:30 |
Abcam, Cambridge, UK |
tissue hypoxia and regulation of VEFG expression |
Monoclonal Mouse Anti-Human Ki67, Clone MIB-1, 1:400 |
Dako, Glostrup, Denmark |
proliferation-associated antigen |
All quantitative estimates were done using stereological methods and the Ellipse software (ViDiTo, Kosice, Slovakia). Stereological methods used in the study are based on counting intersections of the structures of interest with stereological grids [71] or counting frames [72] randomly superposed on the micrographs. During the histological evaluation, the observers had no knowledge of the biological status of the samples or the patient histories. Magnification and sampling of microscopic image fields per section used for estimating all the quantitative parameters are summarized in Table 7. The borders of the glioblastoma tumor were outlined in sections stained with haematoxylin-eosin and the borders were respected in all the consecutive sections stained by immunohistochemistry. Non-tumorous brain tissue and necrotic areas were excluded from the quantification. The microvascular bed was quantified as the number of CD31, CD34, vWF, of CD105-positive microvessel profiles per section area QA of the vascular wall [73, 74]. Unbiased counting frames with known area were projected on micrographs and profiles of microvessels inside the frames or touching the admittance borders but not touching the forbidden lines were counted (Figure 3).
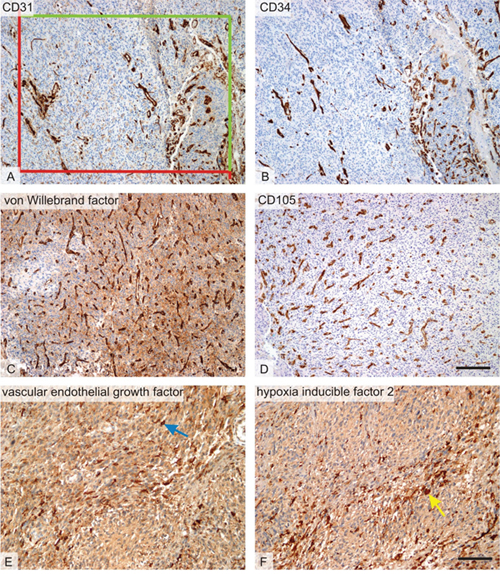
Figure 3: Histological markers of angiogenesis. Micrographs representing the six methods used for quantitative assessment of microvascular bed and its regulation within the glioblastoma tumors. (A) Microvessels with CD31-positive endothelium. In all endothelial markers, the number of microvessel profiles per section area was counted using a projection of unbiased counting frame consisting of two admittance (green) and two forbidden (red) borders. (B) Microvessels with CD34-positive endothelium on a micrograph representing an area on a serial section corresponding to figure A. (C) Microvessels with endothelium positive to von Willebrand factor (vWF). In most glioblastoma samples, von Willebrand factor was less readable due to a slightly positive background when compared to CD31 of CD34 immunohistochemistry. (D) Microvessels with proliferation-associated endothelium positive to von CD105. The micrograph was taken from an area corresponding to that on the B. (E) Vascular endothelial growth factor (VEGF)-positive cells (blue arrow) and (F) hypoxia inducible factor 2 (HIF-2)-positive cells (yellow arrow) were frequently found close to the necrosis (necrotic areas were excluded from the quantification). Positive cells were quantified using their area fraction within the tumor. Immunohistochemical detection of the CD31 (A), CD34 (B), von Willebrand factor (C), CD105 (D), VEGF(E), and HIF-2 (F), visualization of the immunoreaction with diaminobenzidine (dark brown), counterstaining with hematoxylin. Scale bar 200 μm (A-D) and 100 μm (E-F).
Table 7: Sampling of histological sections and microscopic image fields for quantification
Quantitative parameter (component, reference space) |
Microscope objective used |
Image fields sampled per patient |
Number of counting events per patient |
|---|---|---|---|
QA(CD34): number of CD34-positive microvessel profiles per section area |
10× |
4 |
307 vascular profiles counted on average |
QA(CD31): number of CD31-positive microvessel profiles per section area |
10× |
4 |
331 vascular profiles counted on average |
QA(vWF): number of vWF-positive microvessel profiles per section area |
10× |
4 |
289 vascular profiles counted on average |
QA(CD105): number of CD105-positive microvessel profiles per section area |
10× |
4 |
162 vascular profiles counted on average |
AA(VEGF, tumor): area fraction of VEGF-immunopositive cell profiles |
20× |
8 |
>150 intersections with VEGF-positive areas, 204 on average |
AA(HIF2α, tumor): area fraction of HIF2α -immunopositive cell profiles |
20× |
8 |
>150 intersections HIF2α -positive areas, 237 on average |
AA(component, space) – area fractions of the respective components within their reference spaces; QA – number of microvessel profiles per section area. The microscope objective and magnification used for the quantitative assessment of each of the parameters was at the lowest setting that permitted an exact and unambiguous identification of the counting events with respect to histological staining methods. The number of counting events per sample is provided, and the resulting data are presented as arithmetic means of all image fields representing each patient.
The presence of cells expressing VEGF-A and HIF2α was quantified as area fractions AA of their positive immunostaining within the histological section of the glioblastoma tissue. When estimating the area fraction of the detected structures, points of a stereological point grid were superimposed on the micrographs. The area fraction was calculated as a ratio between the number of points hitting the immunopositive cell profiles and all the points hitting the reference area of the glioblastoma tissue. The proliferative activity of the tumor tissue was determined as percentage of MIB-1 stained nuclei per total number of nuclei in four representative cross-sections in each tumor sample.
Statistical analysis
Statistical analyses were carried out using the SPSS 22 software package (IBM, Armonk, NY). Spearman’s rank-order correlation method was used for mutual correlations of expression of hypoxia, angiogenesis, and microvascular markers with clinical-pathological characteristics. The Mann-Whitney U test was used for the determination of differences between markers in IDH1 mutated compared with IDH1 wild type tumors. Overall survival (OS) was defined as the time between the diagnosis and death or last follow-up. Progression-free survival (PFS) was defined as the time between the diagnosis and recurrence or last follow-up. Survival time was estimated by Kaplan-Meier analyses and compared among patient subsets using log-rank test. Multivariate analysis was performed with the Cox regression model to test independent significance while adjusting for covariates, data are presented as hazard ratios (HR) and 95% confidence intervals (95%CI). Reported P-values were two-sided. P-values of less than 0.05 were considered to indicate statistical significance.
Abbreviations
2-HG, 2-hydroxyglutarate; CD, cluster of differentiation; EANO, European Association for Neuro-Oncology; FFPE, formalin-fixed, paraffin-embedded; FGF, fibroblast growth factor; GBM, glioblastoma multiforme; HGF, hepatocyte growth factor; HIF1α, hypoxia inducible factor 1 alpha; HIF2α, hypoxia inducible factor 2 alpha; IDH, isocitrate dehydrogenase; IDH-WT, IDH wildtype; IHC, immunohistochemistry; KPS, Karnofsky performance status; MAPK, mitogen-activated protein kinase; mRNA, messenger RNA; MVD, microvessel density; NADP, nicotinamide adenine dinucleotide phosphate; OS, overall survival; PDGF, platelet-derived growth factor; PFS, progression-free survival; PHD, prolyl hydroxylase; PI3K, phosphatidylinositol 3-kinase; pVHL, von Hippel-Lindau protein; RT-PCR, real-time polymerase chain reaction; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor; WHO, World Health Organization.
Author contributions
JP jr. is the project coordinator who has created the main scientific ideas and hypotheses presented in the manuscript, has performed the literature search, analysed the data and drafted the manuscript. ZT conducted the histological analyses. MP and PP conducted the IDH mutation analysis. OH confirmed pathological diagnosis of glioblastoma in all patients. JP jr., ZT and LH have designed the final version of the manuscript. JP has contributed to the manuscript by his expertise in glioma research and treatment. ZT and TK have provided expertise in the quantitative histology. All the authors have read and approved the final manuscript.
CONFLICTS OF INTEREST
Authors declare that they have no conflicts of interests regarding the publication of this paper.
FUNDING
This study was supported by the Charles University Research Fund (Progres Q39). This work was supported by MH CZ - DRO (Faculty Hospital Plzen - FNPl, 00669806), and by the National Sustainability Program I (NPU I) Nr. LO1503 provided by the Ministry of Education Youth and Sports of the Czech Republic, and by the grant SVV 2017 No 260 390.
REFERENCES
1. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro-oncol. 2016; 18:v1–75.
2. Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G, and German Glioma Network. Long-term survival with glioblastoma multiforme. Brain. 2007; 130:2596–606.
3. Polivka J Jr, Polivka J, Holubec L, Kubikova T, Priban V, Hes O, Pivovarcikova K, Treskova I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017; 37:21–33.
4. Polivka J Jr, Polivka J, Rohan V, Topolcan O, Ferda J. New molecularly targeted therapies for glioblastoma multiforme. Anticancer Res. 2012; 32:2935–46.
5. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807–12.
6. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–68.
7. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, et al, and Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015; 372:2481–98.
8. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–73.
9. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009; 27:4150–54.
10. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012; 483:479–83.
11. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010; 465:966.
12. Xia L, Wu B, Fu Z, Feng F, Qiao E, Li Q, Sun C, Ge M. Prognostic role of IDH mutations in gliomas: a meta-analysis of 55 observational studies. Oncotarget. 2015; 6:17354–65. https://doi.org/10.18632/oncotarget.4008.
13. Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, Reifenberger G, Weller M, Loeffler M, von Deimling A. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010; 120:707–18.
14. Polivka J, Polivka J Jr, Rohan V, Pesta M, Repik T, Pitule P, Topolcan O. Isocitrate dehydrogenase-1 mutations as prognostic biomarker in glioblastoma multiforme patients in West Bohemia. BioMed Res Int. 2014; 2014:735659.
15. Polivka J, Polivka J Jr, Rohan V, Topolcan O. Glioblastoma Multiforme - a Review of Pathogenesis, Biomarkers and Therapeutic Perspectives. Cesk Slov Neurol N. 2013; 76/109:575–83.
16. Polivka J, Polivka J Jr, Krakorova K, Peterka M, Topolcan O. Current status of biomarker research in neurology. EPMA J. 2016; 7:14.
17. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803–20.
18. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Le Rhun E, Balana C, Chinot O, Bendszus M, Reijneveld JC, Dhermain F, et al, and European Association for Neuro-Oncology (EANO) Task Force on Gliomas. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017; 18:e315–29.
19. Popescu AM, Purcaru SO, Alexandru O, Dricu A. New perspectives in glioblastoma antiangiogenic therapy. Contemp Oncol (Pozn). 2016; 20:109–18.
20. Zhang M, Ye G, Li J, Wang Y. Recent advance in molecular angiogenesis in glioblastoma: the challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015; 32:229–36.
21. Rodriguez FJ, Orr BA, Ligon KL, Eberhart CG. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget. 2012; 3:98–106. https://doi.org/10.18632/oncotarget.427.
22. Chi A, Norden AD, Wen PY. Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther. 2007; 7:1537–60.
23. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009; 324:261–65.
24. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011; 19:17–30.
25. Metellus P, Colin C, Taieb D, Guedj E, Nanni-Metellus I, de Paula AM, Colavolpe C, Fuentes S, Dufour H, Barrie M, Chinot O, Ouafik L, Figarella-Branger D. IDH mutation status impact on in vivo hypoxia biomarkers expression: new insights from a clinical, nuclear imaging and immunohistochemical study in 33 glioma patients. J Neurooncol. 2011; 105:591–600.
26. Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, von Deimling A, Zagzag D. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1α upregulation in adult glioma. Acta Neuropathol. 2011; 121:279–81.
27. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012; 483:484–88.
28. Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013; 339:1621–25.
29. Lombardi G, Pambuku A, Bellu L, Farina M, Della Puppa A, Denaro L, Zagonel V. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2017; 111:94–102.
30. Polivka J Jr, Kralickova M, Polivka J, Kaiser C, Kuhn W, Golubnitschaja O. Mystery of the brain metastatic disease in breast cancer patients: improved patient stratification, disease prediction and targeted prevention on the horizon? EPMA J. 2017; 8:119–27.
31. Golubnitschaja O. Feeling cold and other underestimated symptoms in breast cancer: anecdotes or individual profiles for advanced patient stratification? EPMA J. 2017; 8:17–22.
32. Bubnov R, Polivka J Jr, Zubor P, Konieczka K, Golubnitschaja O. “Pre-metastatic niches” in breast cancer: are they created by or prior to the tumour onset? “Flammer Syndrome” relevance to address the question. EPMA J. 2017; 8:141–57.
33. Zubor P, Gondova A, Polivka J Jr, Kasajova P, Konieczka K, Danko J, Golubnitschaja O. Breast cancer and Flammer syndrome: any symptoms in common for prediction, prevention and personalised medical approach? EPMA J. 2017; 8:129–40.
34. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
35. Zhang C, Moore LM, Li X, Yung WK, Zhang W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-oncol. 2013; 15:1114–26.
36. Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncol. 2009; 11:341–47.
37. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39:D945–50.
38. Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008; 4:152–56.
39. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012; 483:474–78.
40. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012; 483:479–83.
41. Ye D, Ma S, Xiong Y, Guan KL. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell. 2013; 23:274–76.
42. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, Sarkar G, Caron AA, Kollmeyer TM, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015; 372:2499–508.
43. Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, Yamamoto T, Tanahashi K, Ranjit M, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015; 47:458–68.
44. Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V, Mittelbronn M, Schittenhelm J, Herold-Mende C, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015; 129:133–46.
45. Zou P, Xu H, Chen P, Yan Q, Zhao L, Zhao P, Gu A. IDH1/IDH2 mutations define the prognosis and molecular profiles of patients with gliomas: a meta-analysis. PLoS One. 2013; 8:e68782.
46. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al, and European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, and National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–96.
47. Gittleman H, Lim D, Kattan MW, Chakravarti A, Gilbert MR, Lassman AB, Lo SS, Machtay M, Sloan AE, Sulman EP, Tian D, Vogelbaum MA, Wang TJ, et al. An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG Oncology RTOG 0525 and 0825. Neuro Oncol. 2017; 19:669–77.
48. Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, Lacombe D, Stupp R. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008; 9:29–38.
49. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al, and European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups, and National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459–66.
50. Pichlmeier U, Bink A, Schackert G, Stummer W, and ALA Glioma Study Group. Resection and survival in glioblastoma multiforme: an RTOG recursive partitioning analysis of ALA study patients. Neuro-oncol. 2008; 10:1025–34.
51. Li R, Li H, Yan W, Yang P, Bao Z, Zhang C, Jiang T, You Y. Genetic and clinical characteristics of primary and secondary glioblastoma is associated with differential molecular subtype distribution. Oncotarget. 2015; 6:7318–24. https://doi.org/10.18632/oncotarget.3440.
52. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008; 13:206–20.
53. Wang N, Jain RK, Batchelor TT. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics. 2017; 14:321–32.
54. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016; 388:518–29.
55. Schmidt NO, Westphal M, Hagel C, Ergün S, Stavrou D, Rosen EM, Lamszus K. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999; 84:10–18.
56. Shih AH, Holland EC. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006; 232:139–47.
57. Reiss Y, Machein MR, Plate KH. The role of angiopoietins during angiogenesis in gliomas. Brain Pathol. 2005; 15:311–17.
58. Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-oncol. 2005; 7:122–33.
59. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014; 142:164–75.
60. Galloway M. CD34 expression in glioblastoma and giant cell glioblastoma. Clin Neuropathol. 2010; 29:89–93.
61. Khattab AZ, Ahmed MI, Fouad MA, Essa WA. Significance of p53 and CD31 in astrogliomas. Med Oncol. 2009; 26:86–92.
62. Yao Y, Kubota T, Takeuchi H, Sato K. Prognostic significance of microvessel density determined by an anti-CD105/endoglin monoclonal antibody in astrocytic tumors: comparison with an anti-CD31 monoclonal antibody. Neuropathology. 2005; 25:201–06.
63. Behrem S, Zarkovic K, Eskinja N, Jonjic N. Endoglin is a better marker than CD31 in evaluation of angiogenesis in glioblastoma. Croat Med J. 2005; 46:417–22.
64. Clara CA, Marie SK, de Almeida JR, Wakamatsu A, Oba-Shinjo SM, Uno M, Neville M, Rosemberg S. Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1α in human glioblastoma. Neuropathology. 2014; 34:343–52.
65. McGahan BG, Neilsen BK, Kelly DL, McComb RD, Kazmi SA, White ML, Zhang Y, Aizenberg MR. Assessment of vascularity in glioblastoma and its implications on patient outcomes. J Neurooncol. 2017; 132:35–44.
66. Jia ZZ, Shi W, Shi JL, Shen DD, Gu HM, Zhou XJ. Comparison between perfusion computed tomography and dynamic contrast-enhanced magnetic resonance imaging in assessing glioblastoma microvasculature. Eur J Radiol. 2017; 87:120–24.
67. Renfrow J, Mott R, Huang Y, Wong T, Lesser G, Debinski W, Strowd RE. In situ expression of hypoxia inducible factor 2-alpha in gliomas. J Clin Oncol. 2017; 35 Suppl; abstr e13534.
68. Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003; 17:271–73.
69. Teodorczyk M, Schmidt MH. Notching on Cancer’s Door: Notch Signaling in Brain Tumors. Front Oncol. 2015; 4:341.
70. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010; 40:294–309.
71. Howard V, Reed M. Unbiased Stereology: Three-Dimensional Measurement in Microscopy. Garland Science. 2004.
72. Gundersen HJ. Notes on the estimation of the numerical density of arbitrary profiles: the edge effect. J Microsc. 1977; 111:219–23.
73. Tonar Z, Egger GF, Witter K, Wolfesberger B. Quantification of microvessels in canine lymph nodes. Microsc Res Tech. 2008; 71:760–72.
74. Veselá P, Tonar Z, Sálek D, Vokurka S, Trněný M, Kodet R, Moulis M, Kašparová P, Vernerová Z, Velenská Z, Stříteský J, Michal M, Boudová L. Microvessel density of mantle cell lymphoma. A retrospective study of its prognostic role and the correlation with the Ki-67 and the mantle cell lymphoma international prognostic index in 177 cases. Virchows Arch. 2014; 465:587–97.