
Introduction
Lung cancer is the most frequent cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers [1]. Only a minority of NSCLC patients is suitable for radical treatment as curative care. Most patients have advanced disease at diagnosis and palliative therapy is the mainstay of management. Conventional chemotherapy of NSCLC has apparently reached a plateau of effectiveness in improving survival of lung cancer patients, and treatment outcomes must still be considered disappointing [2]. Based on a better understanding of the biology of lung cancer, targeted therapies are also available to treat patients with NSCLC. Predictive markers of response to these agents are undergoing prospective validation and promising results have been reported.
The epidermal growth factor receptor (EGFR) signaling pathway is importantly implicated in tumor cell growth, local invasion, angiogenesis, metastasis, protein translation and cell metabolism. It activates two major pathways in solid tumors, RAS/RAF/MAPK/ERK and PI3K/AKT/mTOR, as seen in Figure 1 [3]. Molecular aberrations on the EGFR pathway are the most commonly studied predictive biomarkers of response/ resistance to targeted agents in lung cancer. This review delineates the current role of EGFR inhibitors in the treatment of advanced NSCLC according to EGFR and KRAS status of the tumor, strategies to overcome resistance to agents targeting EGFR and also discusses other recently discovered molecular aberration in lung cancer, ALK rearrangement, which is being efficiently targeted with ALK inhibitors.
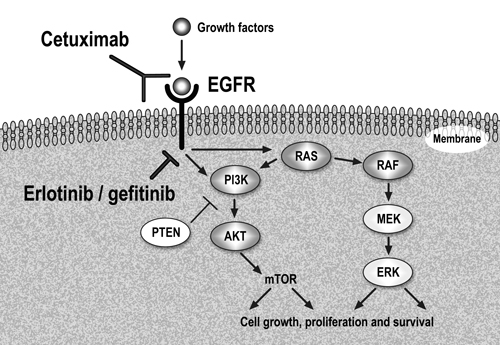
Figure 1: Epidermal Growth Factor Receptor (EGFR) pathway and anti-EGFR therapy in clinical use.
EGFR pathway: molecular aberrations, anti-EGFR therapy and predictive markers of response
NSCLC is associated with EGFR overexpression in up to 80% of the patients and a high EGFR gene copy number is found in nearly 60% of the cases [4-6]. Mutation of the EGFR proto-oncogene is found in 10% to 20% of lung carcinomas (mostly adenocarcinomas) and nearly 90% of lung cancer-specific EGFR mutations comprise a leucine-to-arginine substitution at position 858 (L858R) and deletion mutations in exon 19 (delE746-A750) [7-10]. These mutations cause constitutive activation of the tyrosine kinase of the EGFR [11]. DNA sequencing is the most accurate method for identification of EGFR mutations in tissue samples. Using polymerase-chain reaction (PCR) amplification, deletions in exon 19 and exon 21 point mutations in codon 858 can be detected by length analysis and specific probes for wild-type and mutant sequences [12]. In addition, the Scorpion Amplified Refractory Mutation System (SARMS) technology can be used to detect EGFR mutations in serum genomic DNA or circulating lung-cancer cells [13, 14].
The EGFR kinase domain can be targeted with tyrosine kinase inhibitors (TKIs), such as erlotinib and gefitinib. In addition, another strategy to inhibit EGFR activity is with monoclonal antibodies such as cetuximab, a human-mouse chimeric IgG1 agent. It has been demonstrated that a subgroup of NSCLC patients achieves impressive response rates (RR), symptomatic improvement and long-term progression-free survival (PFS) with these agents. Since the first reports of an association between somatic mutations in EGFR exons 19 and 21 and response to gefitinib, treatment of NSCLC has changed dramatically [7, 8]. It has been shown that exon 19 deletions are more sensitive to erlotinib inhibition than the L858R mutation, a finding demonstrated by kinetic analysis [15] and also confirmed in clinical studies [16-18]. On the other hand, cetuximab is not as potent as EGFR TKIs in tumors with exon 19 deletion or L858R EGFR mutations [19].
Differences in the design of the clinical studies and technical approaches have led to some confusion about the role of molecular diagnostics in guiding the use of EGFR-targeted therapy in NSCLC. Most information regarding clinical benefit with these agents comes from retrospective analysis of large studies. At the present time, prospective clinical data confirming the predictive value of receptor mutations for response to EGFR TKIs is available [20, 21]. Apart from mutation analysis, EGFR protein expression determined by immunohistochemistry (IHC) and EGFR gene copy number determined by fluorescent in situ hybridization (FISH) have been evaluated as markers for clinical decision making regarding EGFR TKI therapy. Technical considerations are important in assessing IHC, which suffers from the lack of a standard methodology and inconsistencies among testing centers [6]. In addition, gene copy number evaluation by FISH may be affected by tumor heterogeneity within analyzed specimens. Therefore, a detailed review of the clinical trials evaluating molecular markers of response to anti-EGFR agents is warranted.
Review of clinical trials with anti-EGFR agents in NSCLC
Gefitinib and Erlotinib
Phase I studies of gefitinib defined dose-limiting toxicities at 700 to 1000 mg/day [22, 23]. However, pharmacodynamic data showed that a dose of 150 mg/day was sufficient to suppress EGFR signaling in skin biopsy specimens [24]. As chronic daily doses higher than 500 mg/day were relatively not well tolerated, further studies evaluated 250 and 500 mg/day doses. Gefitinib was examined as monotherapy in two phase II studies called IDEAL trials [25, 26]. Response rates with doses of 250 and 500 mg/day were similar, ranging from 10% to 18%. Notably, responses were more likely in patients with specific characteristics: Asians, females, non-smoking and those with adenocarcinoma tumors. Posterior analysis demonstrated that patients with and EGFR mutation had an improved RR to gefitinib compared to wild-type patients (46% versus 10%), as shown in Table 1 [27].
Table 1: Response rate to EGFR tyrosine kinase inhibitors according to EGFR mutation status. NR: not reported.
|
EGFR mutation (+) |
EGFR mutation (-) |
Reference |
% (patients) |
% (patients) |
||
Gefitinib |
|||
Retrospective evaluation |
|||
IDEAL trial |
46% (13) |
10% (56) |
27 |
ISEL trial |
37% (16) |
2% (116) |
30 |
INTEREST trial |
42% (19) |
NR |
32 |
Prospective evaluation |
|||
Inoue et al. |
75% (16) |
- |
43 |
Sequist et al. |
55% (31) |
- |
45 |
Kim et al. |
53% (45) |
- |
18 |
Erlotinib |
|||
Retrospective evaluation |
|||
BR.21 |
27% (15) |
7% (101) |
36 |
Janne et al. |
66% (32) |
8% (48) |
47 |
Prospective evaluation |
|||
Spanish Lung Cancer trial |
70% (217) |
- |
16 |
The early trials that evaluated EGFR TKIs for the second- and third-line settings of advanced NSCLC did not select patients on the basis of any EGFR marker [28, 29]. The ISEL trial evaluated the role of second-line gefitinib 250 mg/day in 1,692 patients with advanced NSCLC [29]. Median overall survival (OS) was 5.6 months, compared to 5.1 months in the placebo group, difference not statistically significant. High EGFR gene copy number (30.8% of patients) was associated with a nonsignificant trend toward improved OS with gefitinib treatment and patients with EGFR mutations had higher RR than patients without (37.5% versus 2.6%) [30]. In the INTEREST trial, 1,466 pretreated patients with advanced NSCLC were randomly assigned to receive gefitinib or docetaxel in the second-line setting [31]. Non-inferiority of gefitinib compared to docetaxel was confirmed for OS (7.6 versus 8.0 months). EGFR mutation-positive patients had significantly longer PFS (HR – Hazard Ratio = 0.16) and higher objective RR (42.1% versus 21.1%) with gefitinib compared to docetaxel. Patients with high EGFR copy number also had higher RR with gefitinib (13% versus 7.4%) [32].
In contrast to gefitinib, dose of erlotinib studied in phase II and III trials is the maximum-tolerated dose defined in phase I trials (150 mg/ day) [33]. A phase II trial in patients with refractory NSCLC confirmed RR of approximately 12% and survival outcomes similar to gefitinib [34]. The BR.21 study (erlotinib in second- and third-line settings of advanced NSCLC) found superiority in survival with erlotinib group as compared to placebo (median OS of 6.7 versus 4.7 months) [28]. Molecular analysis of BR.21 trial showed that FISH positivity (45% of tumors) was predictive of improved survival with erlotinib (HR = 0.44) [35]. EGFR mutations were confirmed in 17% of tumors (34 patients had classical EGFR activating mutations). Response rate was significantly higher in patients with these mutations (27% versus 7%) compared to patients with wild-type or other indeterminate mutations [36]. This trial also confirmed symptomatic and survival benefit of erlotinib in patients without initial clinical predictors of response to EGFR TKIs [37]. One possible explanation to this finding is that erlotinib was administered at its maximum-tolerated dose, which could have different effects in the population of patients with wild-type EGFR, not hypersensitive to EGFR TKIs. This correlates with the higher prevalence of rash, a known marker of efficacy with these agents, in the erlotinib trial (76%, as compared to 37% in the gefitinib trial) [38].
Another study with relevant molecular data is the SATURN trial, which randomly assigned 889 patients with advanced NSCLC who had response or stable disease after four cycles of platinum-based chemotherapy to erlotinib or placebo as maintenance treatment [39]. Progression-free survival was significantly improved in the erlotinib maintenance arm (HR = 0.71). Tumor biomarker analysis of EGFR mutation status was available for 437 patients: those with EGFR mutation had median PFS of 44.6 weeks on erlotinib compared to 13 weeks with placebo (HR = 0.1), a remarkable benefit for this subgroup of patients. EGFR mutation was the only marker significantly predictive of differential erlotinib effect [39].
Cetuximab
In the FLEX trial, 1,125 patients with advanced NSCLC and EGFR-positive tumors by IHC were randomly assigned to chemotherapy plus cetuximab or chemotherapy alone [40]. Patients given chemotherapy plus cetuximab survived longer than those in the chemotherapy-alone group (median OS of 11.3 versus 10.1 months; HR for death 0.87, p = 0.044). Molecular biomarker study showed that no significant differences in survival were detected between patients with FISH positive or negative tumors in either treatment arm [41]. As anticipated by preclinical data, EGFR mutation status (positive in 17% of cases) was not predictive of benefit with cetuximab. However, the prognostic value of mutation was confirmed, with longer overall survival in patients with EGFR mutation tumors compared to wild-type tumors in both treatment arms [41].
A similar trial was carried out in the USA in 676 nonselected patients with advanced NSCLC (BMS099) [42]. There was no survival benefit with the addition of cetuximab to paclitaxel-carboplatin combination chemotherapy. Tissue samples for biomarker subanalysis were available for one-third of the patients. EGFR expression, EGFR copy number and EGFR mutations were not associated with treatment outcomes [42].
Prospective studies with EGFR-TKIs in selected patient populations
Studies with first-line EGFR TKIs in advanced lung cancer have been recently reported. The first study was conducted in Asian patients and examined EGFR status in 75 patients [43]. Sixteen patients with EGFR mutation were enrolled onto the study and received gefitinib 250 mg/day. Overall RR was 75% and median PFS was 9.7 months. As seen in Table 1, a confirmatory trial with gefitinib as first-line treatment of advanced EGFR mutant NSCLC reported RR of 53% in 45 patients [18]. Interesting data came from another prospective trial in Japan that enrolled 30 patients with EGFR mutations and poor Performance Status (PS) without indication for palliative chemotherapy [44]. The overall RR was 66% and the disease control rate was 90%. Of note, 15 of 22 patients with baseline PS 3 improved to PS 1. Median PFS and OS were 6.5 months and 17.8 months, respectively [44].
Prospective screening studies in non-Asian populations have also been reported. The first study screened chemotherapy-naïve patients with advanced NSCLC and clinical characteristic associated with EGFR mutations [45]. Response rate was 55% in 31 patients who received gefitinib and the median PFS was 9.2 months. The authors also evaluated EGFR gene copy number by FISH and concluded that it did not provide additional predictive information. This was because most patients who harbor EGFR mutations also had high gene copy numbers [46]. The Spanish Lung Cancer Group trial observed a prevalence of EGFR mutations in patients with lung cancer in Spain around 16% (350 of 2,105 cases) [16]. In 60% of the patients, EGFR mutations were also detected in the serum. Median PFS and OS for 217 patients who received erlotinib were 14 months and 27 months, respectively, and radiologic RR was around 70%. Duration of response was similar for patients receiving first-line therapy (14 months) or second-line therapy (13 months) [16].
Recently, results of the phase II randomized trial of erlotinib compared to erlotinib in combination with paclitaxel-carboplatin chemotherapy in treatment-naïve, never or light former smokers with advanced lung adenocarcinoma [47]. Of the 182 patients randomized, 67 (39%) had mutant and 105 (61%) wild-type EGFR. In the overall patient population, addition of chemotherapy did not significantly increase response rate or PFS. EGFR mutant patients had median PFS of 15.7 months with single-agent erlotinib as compared to 17.2 months with combination therapy. On the other hand, PFS in patients with tumors harboring wild-type EGFR was only 2.4 months with single-agent erlotinib and 4.8 months with the addition of chemotherapy [47].
Based on these findings, several phase III trials are comparing first-line treatment of advanced NSCLC with chemotherapy or EGFR TKI in specific populations of patients: (a) in a clinically-enriched subgroup; or (b) in a biomarker-selected subgroup - those with documented EGFR exon 19 or 21 mutations. The first published trial, named IPASS, selected more than 1,200 patients to receive gefitinib 250 mg/day or standard paclitaxel-carboplatin chemotherapy [17]. Only patients with clinical characteristics predictive of response to EGFR TKIs were enrolled (adenocarcinoma histology; nonsmokers, defined as patients who had smoked <100 cigarettes in their lifetime; or former light smokers, defined as those who had stopped smoking at least 15 years previously and had a total of ≤10 pack-years of smoking). The study met its primary objective of showing the noninferiority of gefitinib and also showed its superiority, as compared to chemotherapy, with regards to PFS (HR for progression or death = 0.74). The 12-months rates of PFS were 24.9% with gefitinib and 6.7% with paclitaxel-carboplatin. However, the overall population result is clearly of less relevance than the outcome in subgroups of patients. As shown in Table 2, in the 261 patients who were positive for the EGFR mutation, PFS was significantly longer among those who received gefitinib than among those who received paclitaxel-carboplatin (HR = 0.48), whereas in the subgroup of 176 patients who were negative for the mutation, PFS was significantly longer among those who received chemotherapy (HR for progression or death with gefitinib = 2.85). In the mutation-negative population, objective RR was only 1.1% with gefitinib versus 23.5% with chemotherapy. High EGFR copy number by FISH was predictive for efficacy only when accompanied by the presence of concomitant EGFR mutation. Patients with a high EGFR copy number and wild-type EGFR did not benefit from gefitinib, but did benefit from chemotherapy [17]. No difference in OS was seen in patients with EGFR mutation regardless of the treatment assigned [48]. The major reason for the lack of survival benefit is the cross-over effect, as half of the patients in the chemotherapy arm received EGFR TKIs at disease progression [48]. The second trial assigned 309 chemotherapy-naïve patients with adenocarcinomas who had never smoked to receive 250 mg gefitinib daily or gemcitabine plus cisplatin at standard doses [49]. Progression-free survival was similar in both arms (6.1 months for gefitinib, 6.6 months for chemotherapy) and in the subgroup of patients with known EGFR mutations (44%), gefitinib produced a higher RR, as shown in Table 2 [49]. Both studies emphasize the importance of molecular selection of patients for first-line treatment with an EGFR TKI.
Table 2: Prospective randomized studies of first-line EGFR TKIs versus standard chemotherapy in clinically-enriched and biomarker-selected patient populations. HR: Hazard Ratio, NS: non significant.
Study |
Population |
Treatment Arms |
Response Rate |
Progression-free survival (favoring EGFR TKI) |
Overall survival |
Reference |
IPASS |
Asians, never/ former light smokers, adenocarcinoma (216 patients with EGFR mutation) |
Gefitinib x Paclitaxel/ Carboplatin |
71% x 47% |
HR 0.48 (0.36 - 0.64) |
HR 1.0 (0.76-1.33) |
17 |
First-SIGNAL |
Asians, never smokers, adenocarcinoma (26 patients with EGFR mutation received gefitinib) |
Gefitinib x Gemcitabine/ Cisplatin |
85% x 37% |
8.4 x 6.7 months (NS) |
Not available |
49 |
North-East Japan |
EGFR mutation, 200 patients |
Gefitinib x Paclitaxel/ Carboplatin |
74% x 31% |
10.8 x 5.4 months |
30.5 x 23.6 months (NS) |
20 |
WJTOG3405 |
EGFR mutation, 177 patients |
Gefitinib x Docetaxel/ Cisplatin |
62% x 32% |
9.2 x 6.3 months |
Not available |
21 |
OPTIMAL |
EGFR mutation, 165 patients |
Erlotinib x Gemcitabine/ Carboplatin |
83% x 36% |
13.1 x 4.6 months |
Not available |
50 |
EURTAC |
EGFR mutation, approximately 170 patients |
Erlotinib x Platinum doublet |
Pending |
Pending |
Pending |
|
LUX-Lung 3/ LUX-Lung 6 |
EGFR mutation, approximately 330 patients each study |
BIBW2992 x Pemetrexed/ Cisplatin or Gemcitabine/ Cisplatin |
Pending |
Pending |
Pending |
|
Preliminary results of other phase III trials that randomized patients with metastatic NSCLC and EGFR mutations at baseline to receive EGFR TKIs or standard first-line platinum-based regimens are presented in Table 2. The North-East Japan trial (prematurely interrupted after interim analysis) and the WJTOG3405 trial demonstrated that treatment with gefitinib doubles the RR and significantly improves PFS as compared to chemotherapy [20, 21]. The first study with erlotinib as comparator was recently presented. In the OPTIMAL trial, Asian patients with chemonaïve NSCLC with EGFR mutations were randomized to erlotinib 150 mg/day or gemcitabine plus carboplatin [50]. Both RR and PFS were higher in the EGFR TKI subgroup [50]. Additional prospective studies are underway, as seen in Table 2.
Overcoming resistance to EGFR TKIs
Tumors become resistant when they reactivate downstream signaling despite the presence of the EGFR inhibitor. Zhang et al. summarize the current understanding of the functional role of activating EGFR mutations in addition to the pivotal primary and acquired resistance mechanisms to EGFR inhibitors [51]. Resistance is typically caused by mutations in the EGFR gene that are not associated with sensitivity to EGFR TKIs, such as insertion mutations in exon 20 [52], or by other somatic mutations in genes that have an impact on the EGFR signaling pathway, such as KRAS [53]. Acquired resistance may be caused by additional mutations in the EGFR gene obtained during the course of treatment that change the protein-coding sequence or by amplification of another oncogene signaling pathway [54].
The most commonly identified mechanism of resistance is an EGFR mutation at position 790 (T790M), resulting in substitution of a threonine residue with methionine, which abrogates the ability of gefitinib or erlotinib to inhibit EGFR [55, 56]. This mutation can be found in 50% of the tissue samples from patients with acquired gefitinib resistance [57]. Another acquired mutation in EGFR, which leads to substitution of alanine for threonine at position 854 (T854A) and hinders the inhibition of tyrosine phosphorylation by erlotinib, has also been reported [58]. Various irreversible EGFR inhibitors, such as BIBW2992/ afatinib (targeting EGFR and HER2) and PF00299804 (targeting EGFR, HER2 and HER4) are undergoing clinical development. These agents may prevent and overcome primary and acquired resistance to first-generation reversible EGFR TKIs. In the LUX-Lung 2 study, 129 patients with activating EGFR mutations and no previous EGFR TKI therapy received BIBW2992 as single agent [59]. Overall RR was 60%, with a promising PFS of 14 months. This drug has also shown activity in patients whose tumors harbored less common EGFR mutations [60]. Its efficacy was also evaluated as a rescue treatment after failure to erlotinib or gefitinib in a randomized phase III trial [61]. At primary analysis (358 events in 585 patients), median OS was 10.8 months in the BIBW2992 group and 11.9 months in the placebo group. A significant PFS increase was seen in patients that received the study drug (3.3 versus 1.1 months) [61]. Regarding the PF00029804 compound, preliminary data from a phase II randomized trial showed that PFS was superior when compared to erlotinib in the general population of patients with chemotherapy-refractory NSCLC, not selected according to EGFR mutation status (12.4 weeks versus 8.3 weeks) [62]. As first-line treatment of patients with known EGFR mutation or clinically selected (Asians with adenocarcinoma and non-or light smoking history), PF00029804 showed encouraging efficacy, with 6 month-PFS rate of 67% (85% in those with EGFR mutation) [63].
Activation of downstream signaling via alternative mechanisms that stimulate the RAS/RAF/MAPK/ERK and PI3K/AKT/mTOR pathways is another mechanism of resistance to EGFR TKIs. This occurs with activation of the IGF-1R pathway (Insulin-like Growth Factor 1 Receptor) [64], amplification/ mutations of MET (the receptor for hepatocyte growth factor receptor, identified in 10-20% of NSCLC) [65-67], and PIK3CA amplification/ mutations (identified in up to 17% of NSCLC) [68]. In this situation, there appears to be dual input to signaling and combined inhibition of EGFR and the alternative pathway may be necessary to kill tumor cells. A combination approach may prevent the emergence of resistance that eventually occurs following initial response to EGFR TKIs and may increase the proportion of EGFR wild-type patients that respond. Preliminary data of a phase II randomized trial of erlotinib and placebo or in combination with a non-ATP competitive receptor TKI of c-MET (ARQ 197) were recently presented [69]. Progression-free survival was significantly higher for the dual inhibition approach (HR = 0.68, p < 0.05) [69]. A monoclonal antibody targeting MET (MetMab) is also under clinical development. In patients with advanced NSCLC and MET expression by IHC analysis, the combination of erlotinib and MetMab significantly increased PFS (HR = 0.56, p = 0.05) as compared to erlotinib and placebo in a phase II randomized trial [70]. Multiple phase I and II trials are underway to evaluate the additive benefit of other targeted agents, such as anti-IGF1R monoclonal antibodies, PI3K/mTOR and MEK inhibitiors.
KRAS status and response to anti-EGFR agents
KRAS mutations were identified in NSCLC tumors more than 20 years ago [71]. KRAS encodes a GTPase downstream of EGFR and mutations (most frequent in exons 12, 13 and 61) lead to stimulus-independent, persistent activation of downstream effectors of the RAF/MAPK/MEK/ERK cascade [72]. They are associated with significant tobacco exposure and worse prognosis, although contradictory data have been reported [73, 74]. Prevalence of mutation is about 20% in the overall lung cancer patient population, 5% to 15% in never-smokers patients with adenocarcinoma and approximately 5% in the squamous cell carcinoma subtype [75-77].
KRAS mutations are associated with primary resistance to EGFR TKIs [78]. Phase II trials have shown very small or absent response rates to erlotinib in patients with KRAS mutations [79]. A recent meta-analysis demonstrated 3% rate of objective tumor response to EGFR TKIs in patients with KRAS mutations, as compared to 26% in those with wild-type KRAS [80]. Data from the TRIBUTE trial (chemotherapy with or without erlotinib for previously untreated patients with NSCLC) suggest that, in KRAS mutated patients, OS and RR may be worse with the addition of EGFR TKI [81]. KRAS mutant patients (20% of available tumor samples) showed a RR of 8% with paclitaxel-carboplatin plus erlotinib, compared with 23% for patients that received the chemotherapy doublet alone. Of note, the RR for patients treated with chemotherapy alone did not differ significantly by KRAS mutation status (26% in those without mutation versus 23%) [81]. In the INTEREST trial, KRAS mutation was not a predictive factor for a differential survival effect between gefitinib and docetaxel [32]. In the BR.21 trial, significant survival benefit from second- or third-line erlotinib therapy was observed for patients with wild-type KRAS (HR = 0.69, p=0.03) but not for patients with mutant KRAS (HR = 1.67, p = 0.31) [36]. Molecular data from the maintenance SATURN trial suggested that KRAS mutation was a negative prognostic factor for patients receiving placebo, with significantly shorter PFS (HR = 1.5, p = 0.017) [39]. Patients with wild-type KRAS had statistically significant benefit with erlotinib in terms of PFS (HR = 0.7, p = 0.0009). The hazard ratio for PFS was similar in the KRAS mutant population, but the benefit was not statistically significant (HR = 0.77, p = 0.22) [39].
Based on data from KRAS mutation status and benefit of cetuximab in advanced colon cancer, molecular analysis of FLEX trial was conducted. Of the 1,125 patients enrolled, 395 had tumor samples available and KRAS mutation was detected in 19%. The comparison of the cetuximab treatment effects in patients with KRAS wild-type tumors and those with KRAS mutant tumors showed no marked differences with regard to PFS or OS [41]. In this trial, the benefit of cetuximab was observed regardless of KRAS mutational status. The same results were obtained with retrospective analysis of the BMS099 trial [42]. Therefore, KRAS status does not predict sensitivity to cetuximab in NSCLC.
ALK rearrangement and targeted therapy
A fusion gene between echinoderm microtubule-associated protein like 4 (EML4) and the anaplastic lymphoma kinase (ALK) has recently been identified in NSCLC [82]. Although more frequent in hematological malignancies, recurrent chromosome translocations may play a role in the molecular pathogenesis of solid tumors. The fusion gene EML4-ALK, inv(2)(p21p23), becomes activated to exert a marked oncogenicity both in vitro and in vivo, possibly by switching on the RAS/RAF signaling pathway [83]. ALK rearrangement can be identified by FISH analysis using break-apart probes to ALK, which detects disruption of the ALK locus but does not confirm EML4 as the partner fusion gene. Recently, a novel highly sensitive antibody allowed for the routine detection of ALK-rearranged lung carcinomas by standard IHC [84].
Among 266 resected NSCLCs in an East Asian population, the EML4-ALK fusion gene was found in about 5% of cases, as assessed by reverse transcriptase-PCR and posterior sequencing [85]. EML4-ALK was associated with younger age of cancer onset and with never-smoking status. EML4-ALK, EGFR, and KRAS mutations were all mutually exclusive, suggesting that ALK mutation may be an important oncogenic factor, and a potential therapeutic target in EGFR wild-type and KRAS wild-type lung cancer. When patients are selected for genetic screening on the basis of two or more of clinical characteristics (female sex, Asian ethnicity, never/light smoking history, and adenocarcinoma histology), a recent study demonstrated that among 141 tumors evaluated, 19 (13%) were EML4-ALK mutant and 31 (22%) were EGFR mutant [86]. Considering only never/ light smokers without EGFR mutation, the frequency of EML4-ALK was 33%. EML4-ALK positivity was related with resistance to EGFR TKIs but similar RR to platinum-based combination chemotherapy. In addition, presence of EML4-ALK mutation was not associated differences in OS, as compared to those patients with wild type EML4-ALK and EGFR [86].
Impressive clinical activity was demonstrated in a phase I trial of an oral ATP- competitive TKI of ALK and c-MET, crizotinib (PF-02341066), in patients with advanced NSCLC and whose tumors harbored ALK rearrangement by FISH analysis [87]. Patients enrolled shared several key clinical features with EGFR-mutated patients (adenocarcinoma histology and nonsmoking history). Among 82 patients treated in an expanded cohort of the dose-escalation study (250 mg twice daily), 57% had an objective RR and 72% were progression-free at 6 months [87]. All patients tested negative for EGFR mutation and amplification of MET, another target for crizotinib, which suggested that the therapeutic effect is through inhibition of ALK. Further confirmatory studies are underway, along with a phase III trial comparing treatment of ALK rearrangement positive patients with crizotinib or standard chemotherapy in the second-line setting.
Conclusions
Identifying the patients who are most likely to obtain clinical benefit from targeted therapies in NSCLC is of paramount importance. To make rational clinical decisions, in addition to understanding the biology of the disease, oncologists need to rely on standardized and validated methods of molecular assessment. EGFR amplification by FISH and protein expression measured by IHC are not informative for personalized therapy in advanced NSCLC but further analysis of studies that combine chemotherapy with monoclonal antibodies targeting EGFR is indicated. In addition, the value of determining KRAS mutation status to select EGFR TKI therapy is not clear. On the other hand, it is now well established that specific genetic lesions that drive the proliferation of cancer cells render some tumors very sensitive to therapeutic inhibitors targeting the mutated pathway. The most useful biomarker is EGFR mutation status and its determination is mandatory for proper therapeutic decisions. It is a good prognostic factor and has also predictive value for selecting treatment with EGFR TKIs. Different from the initially unsuccessful trials of EGFR inhibitors in nongenotyped patients, data from multiple phase III trials show superiority of gefitinib/ erlotinib over standard chemotherapy for advanced NSCLC in terms of RR and PFS in the biomarker-selected EGFR mutation positive subgroup of patients. Additionally, ALK rearrangement is a promising biomarker of benefit with ALK inhibitors and its detection resulted in prompt translation of preclinical data to patient care. Only 3 years after the initiation of the phase I trial, a phase III registration study of crizotinib in ALK-positive patients started enrollment. Participation in a clinical trial is the best alternative for this subgroup of patients. Importantly, although EGFR and ALK mutations are found mostly in patients with history of no smoking or light smoking who have adenocarcinoma, genotyping should be offered to all patients with advanced NSCLC if treatment with specific TKIs is available.
In conclusion, EGFR TKI therapy should be recommended to patients with activating EGFR mutations in the course of the disease. However, the lack of OS advantage with early treatment raises the question of whether we should give these drugs up-front, as maintenance therapy or as second-/ third-line options. Clinical practice in the treatment of advanced NSCLC patients has shown that progression and symptomatic deterioration can occur very short after treatment discontinuation. EGFR TKI therapy may be started once the mutation status is known: (a) as first-line therapy in all patients, especially those unfit or who do not agree with chemotherapy; and (b) as maintenance/ “early second-line” therapy for patients that received previous chemotherapy. Erlotinib may be considered a salvage treatment in unselected patient populations of chemotherapy-refractory NSCLC, as disease stabilization and symptomatic improvement was observed independent of molecular or clinical predictors of benefit. In addition, more clinical and translational data on irreversible EGFR inhibitors and dual targeted therapy in molecularly-selected subgroups of patients will help oncologists to personalize therapy of advanced NSCLC even further.
Acknowledgements
The authors wish to thank Dr. Josep Tabernero for critical review and Vall d’Hebron Institute of Oncology for financial support of graphic design (Figure 1).
References
1. Herbst RS, Heymach JV, Lippman SM. Molecular origins of cancer: Lung cancer. New Engl J Med. 2008; 359: 1367-80.
2. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH; Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small cell lung cancer. New Engl J Med. 2002; 346: 92-8.
3. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nature Rev Cancer. 2005; 5: 341-4.
4. Rusch V, Baselga J, Cordon-Cardo CV, Orazem J, Zaman M, Hoda S, McIntosh J, Kurie J, Dmitrovsky E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1003; 53: 2379-85.
5. Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J, Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clin Cancer Res. 1997; 3: 515-22.
6. Hirsch FR, Dziadziuszko R, Thatcher N, Mann H, Watkins C, Parums DV, Speake G, Holloway B, Bunn PA Jr, Franklin WA. Epidermal growth factor receptor immunohistochemistry: comparison of antibodies and cutoff points to predict benefit from gefitinib in a phase 3 placebo-controlled study in advanced non small-cell lung cancer. Cancer. 2008; 112: 1114-21.
7. Lynch TJ, Bell DW, Sordell R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New Eng J Med. 2004; 350: 2129-39.
8. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304: 1497-500.
9. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancer from never smokers and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004; 101: 13306-11.
10. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004; 64: 8919-23.
11. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung-cancer-derived EGFR mutants and inhibitors complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007; 11: 217-27.
12. Molina-Vila MA, Bertran-Alamillo J, Reguart N, Taron M, Castellà E, Llatjós M, Costa C, Mayo C, Pradas A, Queralt C, Botia M, Pérez-Cano M, Carrasco E, Tomàs M, Mate JL, Moran T, Rosell R. A sensitive method for detecting EGFR mutations in non-small cell lung cancer samples with few tumor cells. J Thorac Oncol. 2008; 3: 1224-35.
13. Kimura H, Kasahara K, Kawaishi M, Kunitoh H, Tamura T, Holloway B, Nishio K. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res. 2006; 12: 3915-21.
14. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, Settleman J, Tompkins RG, Lynch TJ, Toner M, Haber DA. Detection of mutations in EGFR in circulating lung-cancer cells. New Engl J Med. 2008; 359: 366-77.
15. Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006; 66: 8163-71.
16. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, et al. Screening for epidermal growth factor receptor mutations in lung cancer. New Engl J Med. 2009; 361: 958-67.
17. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or paclitaxel-carboplatin in pulmonary adenocarcinoma. New Engl J Med. 2009; 361: 947-57.
18. Kim DW, Lee SH, Lee JS, Lee MA, Kang JH, Kim SY, Shin SW, Kim HK, Heo DS. A multicenter phase II study to evaluate the efficacy and safety of gefitinib as first-line treatment for Korean patients with advanced pulmonary adenocarcinoma harboring EGFR mutations. Lung Cancer. 2011; 71: 65-9.
19. Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, Tenen DG, Johnson BE, Jänne PA. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005; 27: 1185-94.
20. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. New Engl J Med. 2010; 362: 2380-8.
21. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11: 121-8.
22. Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, Miller V, Averbuch S, Ochs J, Morris C, Feyereislova A, Swaisland H, Rowinsky EK. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results from a phase I trial. J Clin Oncol. 2002; 20: 2240-50.
23. Herbst RS, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Baselga J, Rojo F, Hong WK, Swaisland H, Averbuch SD, Ochs J, LoRusso PM. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD 1839 is generally well tolerated and has activity in non-small cell lung cancer and other solid tumors. Results from a phase I trial. J Clin Oncol. 2002; 20: 3815-25.
24. Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, Kaye SB, Gianni L, Harris A, Bjork T, Averbuch SD, Feyereislova A, Swaisland H, Rojo F, Albanell J. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumors. J Clin Oncol. 2002; 20: 4292-302.
25. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The Ideal I Trial). J Clin Oncol. 2003; 21: 2237-46.
26. Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC. Efficacy of gefitinib, an epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003; 290: 2149-58.
27. Bell DW, Lynch TJ, Haseriat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB, Krebs AD, Johnson DH, Giaccone G, Herbst RS, Manegold C, Fukuoka M, et al. Epidermal growth factor receptor mutations and gene amplification in non-small cell lung cancer: molecular analysis of IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005; 23: 8081-92.
28. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, et al. Erlotinib in previously treated non-small cell lung cancer. New Engl J Med. 2005; 353: 123-32.
29. Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small cell lung cancer: results from a randomized, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung cancer). Lancet. 2005; 366: 1527-37.
30. Hirsch FR, Varella-Garcia M, Bunn PA Jr, Franklin WA, Dziadziuszko R, Thatcher N, Chang A, Parikh P, Pereira JR, Ciuleanu T, von Pawel J, Watkins C, Flannery A, Ellison G, Donald E, Knight L, et al. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol. 2006; 24: 5034-42.
31. Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, Li LY, Watkins CL, Sellers MV, Lowe ES, Sun Y, Liao ML, Osterlind K, Reck M, Armour AA, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): A randomised phase III trial. Lancet. 2008; 372: 1809-18.
32. Douillard JY, Shepherd F, Hirsh V, Mok T, Socinski MA, Gervais R, Liao ML, Bischoff H, Reck M, Sellers MV, Watkins CL, Speake G, Armour AA, Kim ES. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non–small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol. 2010; 28: 744-52.
33. Hidalgo M, Siu LL, Nemunaitis J, Rizzo J, Hammond LA, Takimoto C, Eckhardt SG, Tolcher A, Britten CD, Denis L, Ferrante K, Von Hoff DD, Silberman S, Rowinsky EK. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor-tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001; 19: 3267-79.
34. Perez-Soler R, Chachoua A, Hammond LA, Rowinsky EK, Huberman M, Karp D, Rigas J, Clark GM, Santabárbara P, Bonomi P. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2004; 22: 3238-47.
35. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, et al. Erlotinib in lung cancer: molecular and clinical predictors of outcome. New Engl J Med. 2005; 353: 133-44.
36. Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire JA, Kamel-Reid S, Seymour L, Shepherd FA, Tsao MS; National Cancer Institute of Canada Clinical Trials Group Study BR.21. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008; 26: 4268-75.
37. Bezjak A, Tu D, Seymour L, Clark G, Trajkovic A, Zukin M, Ayoub J, Lago S, de Albuquerque Ribeiro R, Gerogianni A, Cyjon A, Noble J, Laberge F, Chan RT, Fenton D, von Pawel J, et al. Symptom improvement in lung cancer patients treated with erlotinib: quality of life analysis of the National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2006; 24: 3831-7.
38. Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res. 2007; 13: 3913-21.
39. Cappuzzo F, Ciuleanu T, Stelmarkh L, Cicenas S, Szczésna A, Juhász E, Esteban E, Molinier O, Brugger W, Melezínek I, Klingelschmitt G, Klughammer B, Giaccone G; SATURN investigators. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010; 11: 521-29.
40. Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT, Ganul V, Roh JK, Bajetta E, O’Byrne K, de Marinis F, Eberhardt W, Goddemeier T, Emig M, Gatzemeier U; FLEX Study Team. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009; 373: 1525-31.
41. O’Byrne K, Rodrigues Pereira J, von Pawel J, et al. Molecular and clinical biomarkers of outcome with cetuximab: Data from the phase III FLEX study in non-small cell lung cancer. Eur J Cancer 2009; 7 (suppl.): 553 [abstract 9160].
42. Khambata-Ford S, Harbison CT, Hart LL, Awad M, Xu LA, Horak CE, Dakhil S, Hermann RC, Lynch TJ, Weber MR. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol. 2010; 28: 918-27.
43. Inoue A, Suzuki T, Fukuhara T, Maemondo M, Kimura Y, Morikawa N, Watanabe H, Saijo Y, Nukiwa T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol. 2006; 24: 3340-6.
44. Inoue A, Kobayashi K, Usui K, Maemondo M, Okinaga S, Mikami I, Ando M, Yamazaki K, Saijo Y, Gemma A, Miyazawa H, Tanaka T, Ikebuchi K, Nukiwa T, Morita S, Hagiwara K; North East Japan Gefitinib Study Group. First-line gefitinib for patients with advanced non–small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J Clin Oncol. 2009; 27: 1394-400.
45. Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, Kuhlmann GL, Han M, Goldberg JS, Settleman J, Iafrate AJ, Engelman JA, et al. First-line gefitinib in patients with advanced non-small cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008; 26: 2442-9.
46. Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Shibata T, Sakiyama T, Yoshida T, Tamura T. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005; 23: 6829-37.
47. Janne PA, Wang XF, Socinski MA, Crawford J, Capelletti M, Edelman MJ, Villalona-Calero MA, Kratzke RA, Vokes EE, Miller VA. Randomized phase II trial of erlotinib alone or in combination with carboplatin/paclitaxel in never or light former smokers with advanced lung adenocarcinoma: CALGB 30406. J Clin Oncol. 2010; 15s (suppl.): [abstract 7503].
48. Yang CH, Fukuoka M, Mok TS, Wu Y-L, Thongprasert S, Saijo N, Chu DT, Jiang H, Duffield EL, Ichinose Y. Final overall survival results from a phase III, randomised, open label, first-line study of gefitinib v. carboplatin/paclitaxel in clinically selected patients with advanced non-small cell lung cancer in Asia (IPASS). Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract LBA2].
49. Lee JS, Park K, Kim SM, et al. A randomized phase III study of gefitinib (Iressa) versus standard chemotherapy (gemcitabine plus cisplatin) as a first-line treatment for never-smokers with advanced or metastatic adenocarcinoma of the lung. J Thorac Oncol. 2009; 4 (suppl. 1): [abstract PRS.4].
50. Zhou C, Wu Y-L, Chen G, Feng J, Liu X, Wang C, Zhang S, Wang J, Zhou S, Ren S. Efficacy results from the randomised phase III OPTIMAL (CTONG 0802) study comparing first-line erlotinib versus carboplatin plus gemcitabine, in Chinese advanced non-small-cell lung cancer patients with EGFR activating mutations. Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract LBA13].
51. Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget. 2010 Nov;1(7):497-514.
52. Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors and non-small-cell lung cancer: Current knowledge and future directions. J Clin Oncol. 2005; 23: 2556-68.
53. Raponi M, Winkler H, Dracopoli NC. KRAS mutations predict response to EGFR inhibitors. Curr Opin Pharmacol. 2008; 8: 41-8.
54. Hammerman PS, Jänne PA, Johnson E. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2009; 15: 7502-9.
55. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New Engl J Med. 2005; 352: 786-92.
56. Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008; 14: 2895-99.
57. Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006; 12: 5764-9.
58. Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA, Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008; 14: 7519-25.
59. Yang C, Shih J, Su W, et al. A phase II study of BIBW 2992 in patients with adenocarcinoma of the lung and activating EGFR/HER1 mutations (LUX-LUNG 2). Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract 367PD].
60. Shih J, Yu C, Su W, et al. Activity of BIBW 2992, an irreversible EGFR/HER1 and HER2 TKI, in lung adenocarcinoma patients harbouring less common EGFR mutations. Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract 415P].
61. Miller VA, Hirsh V, Cadranel J, Chen Y-M, Park K, Kim SW, Caicun Z, Oberdick M, Shahidi M, Yang CH. Phase IIB/ III double-blind randomized trial of afatinib (BIBW 2992, an irreversible inhibitor of the EGFR/HER1 and HER2) + best supportive care (BSC) versus placebo + BSC in patients with NSCLC failing 1-2 lines of chemotherapy and erlotinib or gefitinib (LUX-LUNG 1). Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract LBA1].
62. Ramalingam SS, Boyer MJ, Park K, et al. Randomized phase 2 study of PF299804, an irreversible human epidermal growth factor receptor (EGFR) inhibitor, versus erlotinib in patients with advanced non-small cell lung cancer after chemotherapy failure: quantitative and qualitative benefits. Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract 365PD].
63. Mok T, Spigel DR, Park K, Socinski MA, Tung SY, Kim D-W, Ou S-H I, Zhang H, O’Connell JP, Jänne P. Efficacy and safety of PF299804 as first-line treatment of patients with advanced NSCLC selected for activating mutation of epidermal growth factor receptor (EGFR). Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract LBA18].
64. Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Investig. 2008; 18: 2609-19.
65. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007; 104: 20932-7.
66. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316: 1039-43.
67. Cipriani NA, Abidoye OO, Vokes E, Salgia R. MET as a target for treatment of chest tumors. Lung Cancer. 2009; 63: 169-79.
68. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Date H, Lam WL, Minna JD, Gazdar AF. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008; 68: 6913-21.
69. Sequist LV, Akerley WL, Brugger W, et al. Final results from ARQ197-209: a global randomized placebo-controlled phase 2 clinical trial of erlotinib plus ARQ197 versus erlotinib plus placebo in previously treated EGFR-inhibitor naïve patients with advanced non-small cell lung cancer. Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract 3630].
70. Spigel D, Ervin T, Ramlau R, Daniel D, Goldschmidt J, Krzakowski M, Godbert B, Yu W, Patel PH, Petersonet A. Randomized multicenter double-blind placebo controlled phase II study evaluating METMAB, an antibody to MET receptor, in combination with erlotinib, in patients with non-small cell lung cancer. Annals Oncol. 2010; 21 (suppl. 8): viii122–viii161 [abstract LBA15].
71. Santos E, Martin-Zanca D, Reddy E, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-RAS oncogene in lung carcinoma but not in the normal tissue of the same patient. Science. 1984; 223: 661-4.
72. Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for Epidermal Growth Factor Receptor-targeted therapy? J Clin Oncol. 2010; 28: 4769-77.
73. Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006; 8: 30-8.
74. Marks JL, Broderick S, Zhou Q, Chitale D, Li AR, Zakowski MF, Kris MG, Rusch VW, Azzoli CG, Seshan VE, Ladanyi M, Pao W. Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol. 2008; 3: 111-6.
75. Graziano SL, Gamble GP, Newman NB, Abbott LZ, Rooney M, Mookherjee S, Lamb ML, Kohman LJ, Poiesz BJ. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol. 1999; 17: 668-75.
76. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W, Miller VA, Ladanyi M. Frequency and distinctive spectrum of KRAS mutations in never-smokers with lung adenocarcinoma. Clin Cancer Res. 2008; 14: 5731-4.
77. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009; 6: 201-5.
78. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib and erlotinib. PLoS Med. 2005; 2: e17.
79. Jackman DM, Yeap BY, Lindeman NI, Fidias P, Rabin MS, Temel J, Skarin AT, Meyerson M, Holmes AJ, Borras AM, Freidlin B, Ostler PA, Lucca J, Lynch TJ, Johnson BE, Jänne PA. Phase II clinical trial of chemotherapy-naïve patients > or = 70 years of age treated with erlotinib for advanced non–small-cell lung cancer. J Clin Oncol. 2007; 25: 760-6.
80. Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, Li J, Chen Q. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010; 69: 272-8.
81. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, Ramies DA, Sebisanovic D, Stinson JA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005; 23: 5900-9.
82. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448: 561-6.
83. Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Science. 2008; 99: 2349-55.
84. Mino-Kenudson M, Chirieac LR, Law K, Hornick JL, Lindeman N, Mark EJ, Cohen DW, Johnson BE, Jänne PA, Iafrate AJ, Rodig SJ. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010; 16: 1561-71.
85. Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M; University of Hong Kong Lung Cancer Study Group. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009; 115: 1723-33.
86. Shaw TA, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark EJ, Rodig SJ, Chirieac LR, Kwak EL. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009; 27: 4247-53.
87. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. New Engl J Med. 2010; 363: 1693-703.