
INTRODUCTION
The Sonic Hedgehog (Shh) signaling pathway is physiologically activated during embryogenesis and development [1]. The hedgehog gene was originally discovered in Drosophila in 1980 and described by Nüsslein-Volhard and Wieschaus [2]. The name “hedgehog” comes from the spiky aspect of the embryonic cuticle observed in the mutant for this gene. In mammals, the Shh pathway plays a major role in development of the brain, the limbs, and pulmonary organogenesis. The Shh pathway is also reactivated in various solid cancers, such as thoracic cancers including small-cell lung cancer (SCLC) [3], non-small cell lung cancer (NSCLC) [4–6] and malignant pleural mesothelioma (MPM) [7]. Shh activation is associated with the maintenance of cancer stem cells (CSCs) and is involved in early stages of carcinogenesis [8, 9]. In addition, the Shh pathway is associated with resistance to radiotherapy and chemotherapy in several models of cancer. Until recently, however, the role of the Shh pathway in thoracic cancers was poorly described. Recent studies have expanded our comprehension of the role of Shh pathway activation and its interaction with CSCs in thoracic cancers.
In this review, we aim to describe the current knowledge of CSCs in lung cancer and detail the mechanisms of activation of the Shh pathway in thoracic malignancies (SCLC, NSCLC, and MPM), with a focus on recent discoveries of the correlation between Shh expression and CSCs in lung cancer.
GENERALITIES OF CSCS IN LUNG CANCER
CSCs represent a very small proportion of tumor cells and are undifferentiated, with high oncogenesis, proliferation and differentiation potential. These cells are involved at the very early steps of oncogenesis. CSCs are defined by three essential properties: self-renewal, production of differentiated “daughter” cell lines and in vivo oncogenic capacity (xenograft formation) [10]. Several key genes are expressed in CSCs, such as SOX2, NANOG and OCT4/POU5F1. CSCs have been found in many solid tumors, including lung cancer. Carney et al. showed that some NSCLC and SCLC cells, representing less than 2% of tumor cells, were able to produce new tumor cell colonies. These colonies could induce tumors in vivo similar to the initial tumor in nude mice [11].
Several different biomarkers have been described so far for the detection and isolation of CSCs. Membranous markers are the most commonly used for CSCs detection, such as CD133, CD44, and breast cancer resistance protein (BCRP). CD133 is a transmembrane receptor with unknown function. CD133+ cells represent between 0.30% and 6% of tumor cells in NSCLC and exhibit the ability to grow as spheroids in agarose culture and to differentiate to CD133- tumor cells, and show high in vivo oncogenic potential in NOD/SCID mice [12, 13]. CD44 is a membranous protein involved in cell adhesion, migration and interaction with the extracellular matrix [14]. Notably, CD44 interacts with EGFR and Met [15, 16]. In NSCLC, CSC genes (NANOG, OCT4/POU5F1, and SOX2) are overexpressed in CD44+ cells [17]. BCRP is a member of the ABC transporter family that is also overexpressed in CSCs and involved in chemoresistance [18, 19]. The phenomenon of Hoechst 33342 efflux (“side population” (SP)), i.e. the active transport of Hoechst 33342 from the intracellular compartment to the extracellular space through membranous ABC transporters, has also been used as a marker of CSCs [18]. More recently, several studies have validated ALDH (aldehyde dehydrogenase) activity as a marker of CSC, especially in NSCLC [20, 21]. ALDH represents a group of enzymes responsible for the oxidation of aldehydes. Their activity appears to be increased in stem cells. ALDH activity is involved in alcohol metabolism, vitamin A metabolism, resistance to certain chemotherapy agents (such as cyclophosphamide) and early differentiation of stem cells [22]. In NSCLC, ALDH+ cells display a CSC phenotype, both in vitro and in vivo [20, 21]. Specific techniques using ALDH activity that can be used in NSCLC have been developed [20, 21], such as ALDEFLUOR™ (StemCell Technology) [23].
Despite the identification of CSC markers, several issues remain, limiting the potential use of such CSC markers in lung cancer. Meng et al. showed that CD133 was not specific enough to isolate CSCs in vitro in NSCLC cell lines, as some CD133- cells had CSC features (colony formation, self-renewal, proliferation, differentiation, and chemoresistance) [24]. Moreover, PCR and flow cytometry analyses in lung cancer cell lines showed that CD133 was much more specific to CSCs in SCLC than in NSCLC [25]. Finally, the interpretation of ALDH activity can be problematic in lung tumors, as it can also be increased in normal pneumocytes from smokers [26]. The use of several markers to reach good specificity for CSC isolation in flow cytometry has also been described [27], but this increases the complexity of the assays and requires the use of several lasers with strict rules of compensation, inducing a possible higher rate of false-positives and false-negatives. New biomarkers for CSCs detection and isolation are therefore needed.
THE SHH PATHWAY
The Shh protein
Three different types of hedgehog proteins have been described: Indian, Desert and Sonic Hedghehog (Shh). In mammals, the Shh protein is the main expressed protein. In physiological conditions, the SHH gene, located in q736, produces a pre-protein of 45 kDa. The pre-protein contains an auto-cleavage site, and cleavage results in the production of a 20 kDa N-terminal protein (Shh-N) and a 25 kDa C-terminal protein (Shh-C) [28]. The Shh-C protein has cholesterol transferase activity and catalyzes the addition of cholesterol to the C-terminus part of the Shh-N protein [29, 30]. In addition, palmitoyl residues are also covalently attached to the Shh-N protein (palmitoylation). These lipidic modifications enable the Shh-N protein to anchor to the cell membrane before being secreted into the extracellular space [31]. All physiological functions during development are linked to the Shh-N protein, and the palmitoylated Shh-N protein is approximately 30 times more active than its non-palmitoylated form [32]. The Shh-C protein has no known physiological role outside its cholesterol transferase activity and is also freely secreted into the extracellular space.
Activation of the Shh pathway
The receptor for Shh is Patched (Ptch), a 12-transmembrane domain receptor, with two isoforms (Ptch1 and Ptch2). In the absence of ligand (Figure 1A), Ptch inhibits the migration of Smoothened (Smo) to the membrane, keeping Smo inactive inside the cell. The Gli transcription factors (Gli1, Gli2 and Gli3) are bound to the protein SUFU (suppressor of fused homolog) and remain inactive in the cytoplasm [33]. Gli3 is also phosphorylated by PKA (protein kinase A), which induces the formation of a GLIR inhibitor protein that represses the transcription of the Shh-target genes.
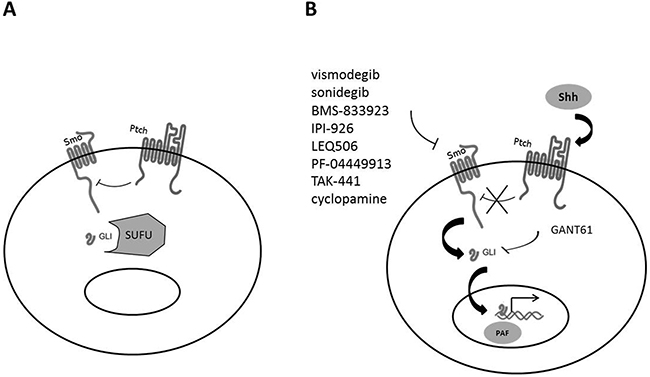
Figure 1: Shh signaling pathway. (A) In the absence of Shh binding to Ptch, Ptch exerts an inhibitory action on Smo. Gli is associated with SUFU in the cytoplasm and is inactive. (B) When Shh binds to Ptch, Smo is activated and induces the migration of Gli into the nucleus with subsequent activation of the transcription of target genes. Main inhibitors of the Shh pathway are also shown.
When Shh binds to its receptor Ptch (Figure 1B), Smo migrates to the cell membrane. Gli proteins then dissociate from SUFU and translocate into the nucleus to activate the transcription of target genes.
Several mechanisms that negatively regulate the Shh pathway under physiological conditions have been described. HHIP (hedgehog interacting protein) binds to Shh on the cell membrane and thus competes with Ptch [34]. Upon Shh binding to Ptch, the Shh-Ptch complex is internalized in the cell before its degradation by the lysosome, stopping the activation of Smo [35]. The ZIC transcription factors interact with Gli proteins through their zinc finger domain [36] and either activate or inhibit Gli, depending on the cell type and physiological conditions [37].
SHH PATHWAY AND SOLID CANCERS
Mechanisms of activation of the Shh pathway in solid cancers
Several mechanisms have been described for the reactivation of the Shh pathway in solid cancers. The Shh pathway can be activated by somatic mutations. For example, Gorlin syndrome, which induces the formation of multiple basal cell carcinomas, rhabdomyosarcomas and medulloblastomas, is characterized by the presence of inactivating mutations of PTCH that result in constant activation of Smo [38, 39]. SUFU mutations have also been described in sporadic cases of medulloblastomas [40]. In sporadic basal cell carcinomas, approximately 70% of cases present a mutation of Shh pathway-related genes (inactivating mutation of PTCH or activating mutation of SMO) [41]. The importance of the activation of the Shh pathway in this type of cancer has led to the development of a Smo inhibitor (vismodegib, Genentech, USA) for the treatment of basal cell carcinoma [42].
In several other cancers, an activation of the Shh pathway was described without evidence of mutation of Shh pathway-related genes. The mechanism of activation is autocrine and paracrine activation through the secretion of the Shh protein. This mechanism of activation has been described in breast [43–45], hepatic [46, 47], pancreatic [48], gallbladder [49], gastric [50] and prostatic cancers [51, 52]. A recent meta-analysis on 39 studies (4496 cases) showed a prognostic role for Gli1 expression in most solid tumors, with worse 3-year, 5-year, and 10-year overall survival and disease-free survival in case of Shh pathway activation [53].
In cancer cells, various genes are activated by Gli proteins, depending on the context and the type of cell [54]. Most of the Gli target genes are involved in proliferation, cell survival, epithelial-mesenchymal transition (EMT) and CSC phenotype. Some genes are also involved in negative control of the Shh pathway. Table 1 summarizes the main target genes of the Shh pathway in cancer.
Table 1: Main cellular processes and genes activated by the Shh pathway
Cellular processes |
Gene |
|---|---|
FOXC2 |
|
SNAI1 |
|
EMT |
TWIST2 |
ZEB1 |
|
ZEB2 |
|
JAG2 |
|
FST |
|
GREM1 |
|
BMP4 |
|
WNT2B |
|
CSCs |
WNT5A |
PDGFRA |
|
BMI1 |
|
LGR5 |
|
CD44 |
|
CD133 |
|
Positive feedback of the Shh pathway |
GLI1 |
Negative feedback of the Shh pathway |
PTCH1 |
PTCH2 |
|
HHIP1 |
|
Cell proliferation |
MYCN |
CCND1 |
|
CCND2 |
|
NEAC |
|
FOXM1 |
|
CCNB1 |
|
CDC25B |
|
Cell survival |
BCL2 |
CFLAR |
|
Other processes |
FOXF1 |
FOXL1 |
|
PRDM1 |
|
PTHLH |
Adapted from [55].
EMT: epithelial-mesenchymal transition; CSCs: cancer stem cells.
The Shh pathway and CSCs
The relationship between CSCs and the Shh pathway has been well documented. Li et al. isolated cells with a CSC phenotype (CD44 (+) CD24 (+) ESA (+)) from primary cultures of pancreatic adenocarcinomas [8]. These cells had a 100-fold greater tumorigenesis potential than other cells, as measured by the formation of xenografts in immunocompromised mice. These cells also showed overexpression of Shh pathway compared with other tumor cells. In another work on lung cancer cell lines (HCC and H1339), inhibition of the Shh pathway by a pharmacological Smo-inhibitor (vismodegib) resulted in a decrease of the population of CSCs measured by the ability of efflux of Hoechst 33342 (SP+ cells) using flow cytometry analysis [55]. The population of SP+ cells decreased from 0.45% (HCC) and 0.75% (H13339) to 0.24% (HCC) and 0.18% (H1339) after treatment with vismodegib. Similar results were shown in PTEN-dependent glioblastoma models [56]. In gallbladder cancer, CSCs isolated by Fluorescence-activated cell sorting (FACS) (through CD44 and aberrantly glycosylated integrin α3β1 staining) showed high level of Shh pathway activation, which was required for CSC renewal [57]. Finally, Lemjabbar-Alaoui et al. used a model of lung carcinogenesis induced by cigarette smoke in which human bronchial cells from primary cultures were exposed to tobacco smoke for 8 days [9]. After 8 days of tobacco exposure, bronchial cells had acquired a tumor phenotype, with an increase of proliferation rate, capacity of growth on agarose gel, and ability to form tumor xenografts in immunocompromised mice. The authors also found an activation of the Wnt and Shh signaling pathways in these cells. Inhibition of the Shh pathway prevented the tobacco-induced tumor phenotype. Interestingly, nicotine seems to induce the proliferation of CSCs in pancreatic cancer through the activation of the Shh pathway [58].
Several factors of cell proliferation are overexpressed in CSCs, such as Myc proteins [59]. A correlation between the Shh pathway and Myc activation has been shown in several studies [60–62]. Gli inhibition by GANT-61 inhibits CSC proliferation in vitro and in vivo and the expression of c-Myc and CSC genes (NANOG, OCT4, and SOX2) [63]. In NSCLC, c-Myc activation is closely associated with hPAF1C, a transcription regulator activated in CSCs [64, 65]. hPAF1C expression has prognostic impact in early-stage NSCLC and showed a correlation with c-Myc expression in tumor samples and in NSCLC cell lines [66]. Interestingly, the inhibition of hPAF1C also inhibits Shh expression [65].
Activation of the Shh pathway in NSCLC
Several studies have demonstrated that the Shh pathway is activated in NSCLC. Yuan et al. showed an activation of the Shh pathway in a number of NSCLC cell lines [4]. The authors also analyzed the expression of Gli1 by immunohistochemistry in a panel of 120 samples of NSCLC in a tissue microarray and found that 87% of adenocarcinomas and 93% of squamous cell carcinoma demonstrated Shh pathway activation. Another study performed immunohistochemistry analyses of several components of the Shh pathway on 80 surgical samples of NSCLC (stage I to III); a large majority of the samples were positive for Shh (97.5%), Ptch1 (78.7%), Smo (72.5%), Gli1 (98.7%) and Gli2 (87.5%) [5]. The expression of Shh pathway factors was present only in tumor cells and not in normal tissues. In another study of 248 early-stage NSCLC, the expression of Shh pathway factors was not correlated to recurrence-free survival or overall survival [6].
The pro-oncogenic role of the Shh pathway in NSCLC was confirmed by Huang et al. [67]. The authors analyzed the expression of Shh pathway genes by PCR in two different cohorts of patients with lung squamous cell carcinoma (n = 178 and n = 56), as well as in four squamous cell carcinoma cell lines (H520, H2170, H226, and SK-MES-1). The results showed that the Shh pathway was activated both in the cell lines and in the two cohorts. In vitro, inhibition of the Shh pathway by Gli2 siRNA or a Gli-inhibitor molecule (GANT61) decreased cell proliferation and activated apoptosis. The use of GANT61 in xenograft models in vivo also showed antitumor activity.
Shh and chemoresistance in NSCLC
Concerning the role of the Shh pathway in chemoresistance, a correlation has been shown between the activation of the Shh pathway in cancer cells and the expression of ABC (ATP-binding cassette) transporters, such as MDR1 and ABCG2, which have demonstrated involvement in chemoresistance [68]. We showed that activation of the Shh pathway was correlated with the acquisition of a mesenchymal phenotype (EMT) by tumor cells, a known factor of chemoresistance, with an inverse correlation between the expression of Gli and epithelial markers such as E-cadherin and a positive correlation with migration and invasion [69, 70]. Moreover, vismodegib exhibits anti-tumor activity in cisplatin-resistant cancer cell lines [71]. Ahmad et al. performed analyses using the A549M NSCLC cell line, in which A549 cells were made resistant to cisplatin and erlotinib by treatment with TGFβ and acquisition of a mesenchymal phenotype [72]. The IC50 values of cisplatin and erlotinib in A549M cells were significantly lower with pretreatment by vismodegib or anti-Shh siRNA. Moreover, the administration of vismodegib decreased the expression of CSC-related genes, such as SOX2 and NANOG. Finally, we demonstrated that chemorefractory NSCLC (i.e. NSCLC with early tumor progression with first-line platinum-based chemotherapy) showed overexpression of Gli2 compared with chemosensitive NSCLC and that inhibition of Shh pathway had a synergistic effect with cisplatin in the most chemoresistant cell lines in vitro [73].
Shh and resistance to EGFR tyrosine kinase inhibitors in EGFR-mutated NSCLC
During embryogenesis, there is a cross-talk between the Shh and EGFR pathways [74]. Kim et al. showed a correlation between the expression of Shh-related proteins and the presence of EGFR mutation in surgically resected lung adenocarcinoma [75]. In EGFR-mutated NSCLC, pre-clinical models have suggested a role of the Shh pathway activation in tumor progression with EGFR tyrosine kinase inhibitors (TKIs). The Shh pathway is closely associated with EMT, as discussed above, and EMT is one of the mechanisms associated with acquired resistance to EGFR TKIs [76]. In addition, EGFR-mutated lung cancer cells resistant to EGFR TKIs also harbor SMO amplification along with Met activation [77]. In these cells, combined Shh and Met inhibition had significant antitumor activity. The potentiation of the activity of EGFR TKIs by the inhibition of the Shh pathway has been proven in several other pre-clinical studies, with an inhibition of CSC renewal and activity [78–80]. The identification of drugs targeting both Met and Smo could lead to the development of new treatments for EGFR-mutated NSCLC [81].
Shh and resistance to radiotherapy
Activation of the Shh pathway has also been correlated with resistance to radiotherapy. Radiation treatment in hepatocellular carcinoma, colorectal and pancreatic carcinoma induced activation of the Shh pathway in vitro [47, 82]. Shh pathway activation is also associated with the risk of local tumor relapse in cervical carcinoma after radiotherapy [83]. In vitro irradiation of NSCLC cell lines induced overexpression of CSC markers, as CD44 and CD166, and also EMT [84].
Activation of the Shh pathway in SCLC
Watkins et al. demonstrated that the Shh pathway was activated in SCLC, with an autocrine and juxtacrine activation mechanism [3]. Immunohistochemistry and western blot analysis of SCLC human samples and cell lines showed an overexpression of Shh and Gli1 in tumor and cancer cells. Inhibition of the Shh pathway by cyclopamine (a Smo inhibitor) blocked cell growth in vitro and in vivo in SCLC xenografts. These results were confirmed in transgenic mouse models with deletion of RB1 (retinoblastoma 1) and TRP53 (transformation related protein 53) [85]. Tumor growth was inhibited by deletion of Smo in these models and treatment with pharmacological inhibitors of the Shh pathway.
Smo inhibitors have been tested in clinical trials in SCLC. A phase II trial (Eastern Cooperative Oncology Group ECOG-1508 trial) evaluated the benefit of adding vismodegib to cisplatin and etoposide chemotherapy in advanced SCLC as first-line treatment [86]. No difference in terms of progression-free survival or overall survival was shown between patients who received only chemotherapy and those treated by vismodegib and chemotherapy. Several hypotheses could explain these negative results: no selection of a specific patient phenotype (inhibition of the Shh pathway benefits only non-responder tumors?), the absence of a predefined biomarker to identify patients who will likely benefit from vismodegib, or the treatment strategy used in this trial (concomitant treatment). A recent phase I trial testing another Smo antagonist (sonidegib) in addition to cisplatin-etoposide for advanced SCLC (n=15) showed a 79% response rate [87]. Interestingly, one patient experienced a prolonged progression free survival (27 months), with a tumor showing amplification of SOX2, a CSC-related gene. Finally, recent data suggested a complex mechanism of activation of the Shh pathway in SCLC. As the canonical Shh pathway (through Ptch-Smo activation) seems to be strongly involved in SCLC cancer cells, a non-canonical Shh pathway involving a Smo-independent upregulation of cyclin B1 induced chromosomal instability in cells lacking both p53 and Rb1 [88]. These Smo-independent mechanisms in SCLC could partially explain the negative results of Smo inhibitors in SCLC.
Activation of the Shh pathway in MPM
Similar to lung cancer, previous studies have shown an overexpression of Shh pathway proteins in MPM, while normal pleural tissue does not express these proteins [7]. Activation of the Shh pathway in MPM seems to be regulated by the protein kinase CK2α, which is overexpressed in MPM [89]. Moreover, in models of primary cultures of MPM, Smo inhibition induced a significant decrease in tumor growth through Gli1 inhibition. The inhibition of Smo also had an anti-tumor action in animal xenograft models in vivo. Similar results were obtained by direct inhibition of Gli by siRNA or a small inhibitory molecule of Gli (Gli-I) [90, 91] or GANT61 [92]. Interestingly, inhibition of Gli with a Gli-I molecule had a synergistic cytotoxic effect with pemetrexed in MPM lines [90].
FULL-LENGTH SHH PROTEIN AS A NEW CSC MARKER AND FUTURE THERAPEUTIC STRATEGIES
While the Shh pathway is closely associated with CSCs, which represent less than 1% of tumor cells, the majority of NSCLC cells show positive Gli1 and Gli2 expression by immunohistochemistry [5, 69, 70], suggesting a complex mechanism of Shh pathway activation in NSCLC. The main hypothesis is that the CSCs are the source of Shh production in the tumor, with paracrine activity and activation of downstream Shh pathway proteins (Figure 2). We confirmed this hypothesis in a recent work [93]. We used FACS to identify a small proportion of Shh-positive (Shh+) cells. When analyzing SHH gene expression by digital droplet PCR on sorted cells, we found that only Shh+ cells expressed SHH, whereas Shh-negative (Shh-) cells had no significant SHH expression. These Shh+ cells had paracrine activity on Shh- cells in term of proliferation and migration, with activation of Ptch, Smo and Gli in Shh- cells. Moreover, the Shh+ cells harbored CSC features, with spheroid formation in serum-free medium conditions, chemoresistance, and tumor initiation in nude mice, even with inoculation with as low as 1,500 cells. Surprisingly, while the full-length Shh protein is normally cleaved in the cytosol to produce functional N-terminal peptide, we found that Shh+ cells produced a full-length Shh protein that migrated to the membrane before its secretion. The underlying mechanisms of the production of unprocessed Shh protein are not yet clearly understood, and further studies are in process to clarify these mechanisms. Interestingly, the production of full-length Shh protein by CSCs does not seem to be restricted to NSCLC, as Shh+ can also be found in pancreatic adenocarcinoma, MPM and melanoma [93].
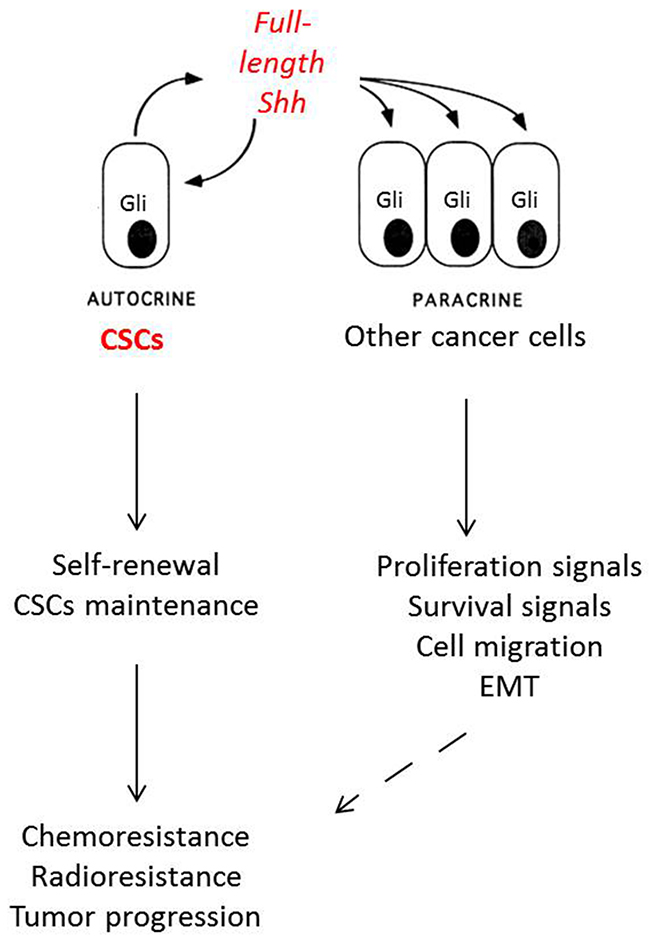
Figure 2: Mechanisms of Shh activation in non-small cell lung cancer. Cancer stem cells (CSCs) are Shh+ cells that secrete Shh full-length protein and induce autocrine (self-renewal and CSC stock maintenance) and paracrine activity (proliferation and survival signals, cell migration and epithelial-mesenchymal transition). All these signals are responsible for resistance to chemotherapy, radiotherapy and tumor proliferation in non-small cell lung cancer.
These findings of production of full-length Shh protein by CSCs represent a milestone in the understanding of CSC biology in lung cancer and implicate full-length Shh protein as a potential CSC marker. This new CSC marker, unlike many other markers used so far (CD133, CD44), has a functional role, and Shh-targeted therapies are already available. Treatment with vismodegib is able to induce a complete extinction of the Shh+ signal in FACS, both in vitro and in vivo [93]. Pre-treatment of NSCLC cells by vismodegib prevented tumor initiation in nude mice [93]. Targeting the Shh pathway should therefore be encouraged in association with chemotherapy to specifically inhibit CSCs and therefore enhance the efficacy of chemotherapy. One of the main challenges in targeting the Shh pathway is that all of the drugs currently available in clinical trials are Smo-inhibitors (shown in Table 2). But this strategy targets only the canonical Shh pathway, and non-canonical Shh pathway activation has been shown in various malignancies, such as in SCLC [88] or in MPM [91]. In these situations, targeting Gli could be more efficient, and the development of specific Gli-inhibitors is in process [91]. The other therapeutic option could be directly targeting the full-length Shh protein. We recently showed that treatment with an antibody specifically designed to target the C-terminal peptide of Shh protein, and therefore also the full-length Shh protein, showed encouraging anti-tumor activity in vivo [94], and could be a promising treatment in the future.
Table 2: Main Smo-inhibitors developed in clinical trials in thoracic oncology
Inhibitor |
Company |
Clinical development |
Clinicaltrial ID |
Type of thoracic malignancy |
|---|---|---|---|---|
vismodegib (GDC-0449) |
Genentech-Roche |
Phase II |
NCT02465060 |
SCLC |
sonidegib (LDE225) |
Novartis |
Phase I |
NCT01579929 |
SCLC |
BMS-833923 |
Bristol-Meyers-Squibb |
Phase I |
NCT00927875 |
SCLC |
SCLC: small cell lung cancer.
CONCLUSIONS
The presence of Shh activation in thoracic cancers has been established for several years. However, the recent discoveries on the critical role of Shh as a CSC marker with a functional role and its impact on the global resistance to anti-cancer treatment and tumor proliferation have highlighted the pivotal role of this pathway in solid cancers, especially in NSCLC. Shh+ cells are the source of the full-length Shh protein production and exhibit CSC features, with a strong impact on oncogenesis and chemoresistance. A new avenue for the development of therapies targeting CSCs through Shh pathway inhibition is beginning, with expected improvements in terms of clinical benefits for patients with thoracic cancers.
Author contributions
EGL wrote the manuscript.
BH and DJ reviewed the manuscript.
ACKNOWLEDGMENTS
We thank Gabrielle White Wolf, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
CONFLICTS OF INTEREST
None.
REFERENCES
1. McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003; 53:1–114. https://doi.org/10.1016/S0070-2153(03)53002-2
2. Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980; 287:795–801. https://doi.org/10.1038/287795a0
3. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003; 422:313–17. https://doi.org/10.1038/nature01493
4. Yuan Z, Goetz JA, Singh S, Ogden SK, Petty WJ, Black CC, Memoli VA, Dmitrovsky E, Robbins DJ. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007; 26:1046–55. https://doi.org/10.1038/sj.onc.1209860
5. Gialmanidis IP, Bravou V, Amanetopoulou SG, Varakis J, Kourea H, Papadaki H. Overexpression of hedgehog pathway molecules and FOXM1 in non-small cell lung carcinomas. Lung Cancer. 2009; 66:64–74. https://doi.org/10.1016/j.lungcan.2009.01.007
6. Raz G, Allen KE, Kingsley C, Cherni I, Arora S, Watanabe A, Lorenzo CD, Edwards V DK, Sridhar S, Hostetter G, Weiss GJ. Hedgehog signaling pathway molecules and ALDH1A1 expression in early-stage non-small cell lung cancer. Lung Cancer. 2012; 76:191–96. https://doi.org/10.1016/j.lungcan.2011.10.015
7. Shi Y, Moura U, Opitz I, Soltermann A, Rehrauer H, Thies S, Weder W, Stahel RA, Felley-Bosco E. Role of hedgehog signaling in malignant pleural mesothelioma. Clin Cancer Res. 2012; 18:4646–56. https://doi.org/10.1158/1078-0432.CCR-12-0599
8. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007; 67:1030–37. https://doi.org/10.1158/0008-5472.CAN-06-2030
9. Lemjabbar-Alaoui H, Dasari V, Sidhu SS, Mengistab A, Finkbeiner W, Gallup M, Basbaum C. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS One. 2006; 1:e93. https://doi.org/10.1371/journal.pone.0000093
10. Rivera C, Rivera S, Loriot Y, Vozenin MC, Deutsch E. Lung cancer stem cell: new insights on experimental models and preclinical data. J Oncol. 2011; 2011:549181. https://doi.org/10.1155/2011/549181
11. Carney DN, Gazdar AF, Bunn PA Jr, Guccion JG. Demonstration of the stem cell nature of clonogenic tumor cells from lung cancer patients. Stem Cells. 1982; 1:149–64.
12. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008; 15:504–14. https://doi.org/10.1038/sj.cdd.4402283
13. Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, Rocco G, Pirozzi G. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009; 36:446–53. https://doi.org/10.1016/j.ejcts.2009.03.063
14. Fasano M, Sabatini MT, Wieczorek R, Sidhu G, Goswami S, Jagirdar J. CD44 and its v6 spliced variant in lung tumors: a role in histogenesis? Cancer. 1997; 80:34–41. https://doi.org/10.1002/(SICI)1097-0142(19970701)80:1<34::AID-CNCR5>3.0.CO;2-F
15. Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002; 16:3074–86. https://doi.org/10.1101/gad.242602
16. Bourguignon LY, Peyrollier K, Gilad E, Brightman A. Hyaluronan-CD44 interaction with neural Wiskott-Aldrich syndrome protein (N-WASP) promotes actin polymerization and ErbB2 activation leading to beta-catenin nuclear translocation, transcriptional up-regulation, and cell migration in ovarian tumor cells. J Biol Chem. 2007; 282:1265–80. https://doi.org/10.1074/jbc.M604672200
17. Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y, Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010; 5:e14062. https://doi.org/10.1371/journal.pone.0014062
18. Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007; 67:4827–33. https://doi.org/10.1158/0008-5472.CAN-06-3557
19. Sung JM, Cho HJ, Yi H, Lee CH, Kim HS, Kim DK, Abd El-Aty AM, Kim JS, Landowski CP, Hediger MA, Shin HC. Characterization of a stem cell population in lung cancer A549 cells. Biochem Biophys Res Commun. 2008; 371:163–67. https://doi.org/10.1016/j.bbrc.2008.04.038
20. Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, Wang H, Liu Z, Su Y, Stass SA, Katz RL. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009; 7:330–38. https://doi.org/10.1158/1541-7786.MCR-08-0393
21. Liang D, Shi Y. Aldehyde dehydrogenase-1 is a specific marker for stem cells in human lung adenocarcinoma. Med Oncol. 2012; 29:633–39. https://doi.org/10.1007/s12032-011-9933-9
22. Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci U S A. 1999; 96:9118–23. https://doi.org/10.1073/pnas.96.16.9118
23. Moreb JS, Zucali JR, Ostmark B, Benson NA. Heterogeneity of aldehyde dehydrogenase expression in lung cancer cell lines is revealed by Aldefluor flow cytometry-based assay. Cytometry B Clin Cytom. 2007; 72:281–89. https://doi.org/10.1002/cyto.b.20161
24. Meng X, Li M, Wang X, Wang Y, Ma D. Both CD133+ and CD133- subpopulations of A549 and H446 cells contain cancer-initiating cells. Cancer Sci. 2009; 100:1040–46. https://doi.org/10.1111/j.1349-7006.2009.01144.x
25. Cui F, Wang J, Chen D, Chen YJ. CD133 is a temporary marker of cancer stem cells in small cell lung cancer, but not in non-small cell lung cancer. Oncol Rep. 2011; 25:701–08. https://doi.org/10.3892/or.2010.1115
26. Patel M, Lu L, Zander DS, Sreerama L, Coco D, Moreb JS. ALDH1A1 and ALDH3A1 expression in lung cancers: correlation with histologic type and potential precursors. Lung Cancer. 2008; 59:340–49. https://doi.org/10.1016/j.lungcan.2007.08.033
27. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009; 106:16281–86. https://doi.org/10.1073/pnas.0905653106
28. Lee JJ, Ekker SC, von Kessler DP, Porter JA, Sun BI, Beachy PA. Autoproteolysis in hedgehog protein biogenesis. Science. 1994; 266:1528–37. https://doi.org/10.1126/science.7985023
29. Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998; 273:14037–45. https://doi.org/10.1074/jbc.273.22.14037
30. Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996; 274:255–59. https://doi.org/10.1126/science.274.5285.255
31. Robbins DJ, Hebrok M. Hedgehogs: la dolce vita. Workshop on Hedgehog-Gli signaling in cancer and stem cells. EMBO Rep. 2007; 8:451–55. https://doi.org/10.1038/sj.embor.7400959
32. Wendler F, Franch-Marro X, Vincent JP. How does cholesterol affect the way Hedgehog works? Development. 2006; 133:3055–61. https://doi.org/10.1242/dev.02472
33. Kogerman P, Grimm T, Kogerman L, Krause D, Undén AB, Sandstedt B, Toftgård R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999; 1:312–19. https://doi.org/10.1038/13031
34. Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999; 397:617–21. https://doi.org/10.1038/17611
35. Torroja C, Gorfinkiel N, Guerrero I. Patched controls the Hedgehog gradient by endocytosis in a dynamin-dependent manner, but this internalization does not play a major role in signal transduction. Development. 2004; 131:2395–408. https://doi.org/10.1242/dev.01102
36. Koyabu Y, Nakata K, Mizugishi K, Aruga J, Mikoshiba K. Physical and functional interactions between Zic and Gli proteins. J Biol Chem. 2001; 276:6889–92. https://doi.org/10.1074/jbc.C000773200
37. Mizugishi K, Aruga J, Nakata K, Mikoshiba K. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J Biol Chem. 2001; 276:2180–88. https://doi.org/10.1074/jbc.M004430200
38. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996; 85:841–51. https://doi.org/10.1016/S0092-8674(00)81268-4
39. Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996; 272:1668–71. https://doi.org/10.1126/science.272.5268.1668
40. Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997; 57:842–45.
41. Tang JY, So PL, Epstein EH Jr. Novel Hedgehog pathway targets against basal cell carcinoma. Toxicol Appl Pharmacol. 2007; 224:257–64. https://doi.org/10.1016/j.taap.2006.12.011
42. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012; 366:2171–79. https://doi.org/10.1056/NEJMoa1113713
43. Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, Kuroki S, Katano M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004; 64:6071–74. https://doi.org/10.1158/0008-5472.CAN-04-0416
44. Song L, Wang W, Liu D, Zhao Y, He J, Wang X, Dai Z, Zhang H, Li X. Targeting of sonic hedgehog-Gli signaling: A potential therapeutic target for patients with breast cancer. Oncol Lett. 2016; 12:1027–33. https://doi.org/10.3892/ol.2016.4722
45. Noman AS, Uddin M, Rahman MZ, Nayeem MJ, Alam SS, Khatun Z, Wahiduzzaman M, Sultana A, Rahman ML, Ali MY, Barua D, Ahmed I, Islam MS, et al. Overexpression of sonic hedgehog in the triple negative breast cancer: clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci Rep. 2016; 6:18830. https://doi.org/10.1038/srep18830
46. Li W, Miao S, Miao M, Li R, Cao X, Zhang K, Huang G, Fu B. Hedgehog signaling activation in hepatic stellate cells promotes angiogenesis and vascular mimicry in hepatocellular carcinoma. Cancer Invest. 2016; 34:424–30. https://doi.org/10.1080/07357907.2016.1227442
47. Chen YJ, Lin CP, Hsu ML, Shieh HR, Chao NK, Chao KS. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. Int J Radiat Oncol Biol Phys. 2011; 80:851–59. https://doi.org/10.1016/j.ijrobp.2011.01.003
48. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003; 425:851–56. https://doi.org/10.1038/nature02009
49. Matsushita S, Onishi H, Nakano K, Nagamatsu I, Imaizumi A, Hattori M, Oda Y, Tanaka M, Katano M. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014; 105:272–80. https://doi.org/10.1111/cas.12354
50. Ertao Z, Jianhui C, Chuangqi C, Changjiang Q, Sile C, Yulong H, Hui W, Shirong C. Autocrine Sonic hedgehog signaling promotes gastric cancer proliferation through induction of phospholipase Cγ1 and the ERK1/2 pathway. J Exp Clin Cancer Res. 2016; 35:63. https://doi.org/10.1186/s13046-016-0336-9
51. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004; 431:707–12. https://doi.org/10.1038/nature02962
52. Lubik AA, Nouri M, Truong S, Ghaffari M, Adomat HH, Corey E, Cox ME, Li N, Guns ES, Yenki P, Pham S, Buttyan R. Paracrine sonic hedgehog signaling contributes significantly to acquired steroidogenesis in the prostate tumor microenvironment. Int J Cancer. 2017; 140:358–69. https://doi.org/10.1002/ijc.30450
53. Cheng J, Gao J, Tao K, Yu P. Prognostic role of Gli1 expression in solid malignancies: a meta-analysis. Sci Rep. 2016; 6:22184. https://doi.org/10.1038/srep22184
54. Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009; 9:873–86. https://doi.org/10.2174/156652409789105570
55. Tian F, Mysliwietz J, Ellwart J, Gamarra F, Huber RM, Bergner A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin Exp Med. 2012; 12:25–30. https://doi.org/10.1007/s10238-011-0135-8
56. Xu Q, Yuan X, Liu G, Black KL, Yu JS. Hedgehog signaling regulates brain tumor-initiating cell proliferation and portends shorter survival for patients with PTEN-coexpressing glioblastomas. Stem Cells. 2008; 26:3018–26. https://doi.org/10.1634/stemcells.2008-0459
57. Li C, Du Y, Yang Z, He L, Wang Y, Hao L, Ding M, Yan R, Wang J, Fan Z. GALNT1-mediated glycosylation and activation of sonic hedgehog signaling maintains the self-renewal and tumor-initiating capacity of bladder cancer stem cells. Cancer Res. 2016; 76:1273–83. https://doi.org/10.1158/0008-5472.CAN-15-2309
58. Al-Wadei MH, Banerjee J, Al-Wadei HA, Schuller HM. Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur J Cancer. 2016; 52:188–96. https://doi.org/10.1016/j.ejca.2015.10.003
59. Mimeault M, Batra SK. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol Aspects Med. 2014; 39:3–32. https://doi.org/10.1016/j.mam.2013.08.001
60. Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003; 130:15–28. https://doi.org/10.1242/dev.00182
61. Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004; 131:217–28. https://doi.org/10.1242/dev.00891
62. Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006; 124:47–59. https://doi.org/10.1016/j.cell.2005.10.042
63. Fu J, Rodova M, Roy SK, Sharma J, Singh KP, Srivastava RK, Shankar S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013; 330:22–32. https://doi.org/10.1016/j.canlet.2012.11.018
64. Moniaux N, Nemos C, Schmied BM, Chauhan SC, Deb S, Morikane K, Choudhury A, Vanlith M, Sutherlin M, Sikela JM, Hollingsworth MA, Batra SK. The human homologue of the RNA polymerase II-associated factor 1 (hPaf1), localized on the 19q13 amplicon, is associated with tumorigenesis. Oncogene. 2006; 25:3247–57. https://doi.org/10.1038/sj.onc.1209353
65. Ponnusamy MP, Deb S, Dey P, Chakraborty S, Rachagani S, Senapati S, Batra SK. RNA polymerase II associated factor 1/PD2 maintains self-renewal by its interaction with Oct3/4 in mouse embryonic stem cells. Stem Cells. 2009; 27:3001–11. https://doi.org/10.1002/stem.237
66. Zhi X, Giroux-Leprieur E, Wislez M, Hu M, Zhang Y, Shi H, Du K, Wang L. Human RNA polymerase II associated factor 1 complex promotes tumorigenesis by activating c-MYC transcription in non-small cell lung cancer. Biochem Biophys Res Commun. 2015; 465:685–90. https://doi.org/10.1016/j.bbrc.2015.08.017. Erratum in: Biochem Biophys Res Commun. 2015; 468:934.
67. Huang L, Walter V, Hayes DN, Onaitis M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014; 20:1566–75. https://doi.org/10.1158/1078-0432.CCR-13-2195
68. Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007; 26:5674–79. https://doi.org/10.1038/sj.onc.1210356
69. Yue D, Li H, Che J, Zhang Y, Tseng HH, Jin JQ, Luh TM, Giroux-Leprieur E, Mo M, Zheng Q, Shi H, Zhang H, Hao X, et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res. 2014; 33:34. https://doi.org/10.1186/1756-9966-33-34
70. Li H, Yue D, Jin JQ, Woodard GA, Tolani B, Luh TM, Giroux-Leprieur E, Mo M, Chen Z, Che J, Zhang Z, Zhou Y, Wang L, et al. Gli promotes epithelial-mesenchymal transition in human lung adenocarcinomas. Oncotarget. 2016; 7:80415–25. https://doi.org/10.18632/oncotarget.11246
71. Tian F, Schrödl K, Kiefl R, Huber RM, Bergner A. The hedgehog pathway inhibitor GDC-0449 alters intracellular Ca2+ homeostasis and inhibits cell growth in cisplatin-resistant lung cancer cells. Anticancer Res. 2012; 32:89–94.
72. Ahmad A, Maitah MY, Ginnebaugh KR, Li Y, Bao B, Gadgeel SM, Sarkar FH. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J Hematol Oncol. 2013; 6:77. https://doi.org/10.1186/1756-8722-6-77
73. Giroux Leprieur E, Vieira T, Antoine M, Rozensztajn N, Rabbe N, Ruppert AM, Lavole A, Cadranel J, Wislez M. Sonic Hedgehog pathway activation is associated with resistance to platinum-based chemotherapy in advanced non-small-cell lung carcinoma. Clin Lung Cancer. 2016; 17:301–08. https://doi.org/10.1016/j.cllc.2015.12.007
74. Reinchisi G, Parada M, Lois P, Oyanadel C, Shaughnessy R, Gonzalez A, Palma V. Sonic Hedgehog modulates EGFR dependent proliferation of neural stem cells during late mouse embryogenesis through EGFR transactivation. Front Cell Neurosci. 2013; 7:166. https://doi.org/10.3389/fncel.2013.00166
75. Kim JE, Kim H, Choe JY, Sun P, Jheon S, Chung JH. High expression of Sonic hedgehog signaling proteins is related to the favorable outcome, EGFR mutation, and lepidic predominant subtype in primary lung adenocarcinoma. Ann Surg Oncol. 2013 (Suppl 3); 20:S570–76. https://doi.org/10.1245/s10434-013-3022-6
76. Califano R, Morgillo F, De Mello RA, Mountzios G. Role of mesenchymal-epithelial transition amplification in resistance to anti-epidermal growth factor receptor agents. Ann Transl Med. 2015; 3:81. https://doi.org/10.3978/j.issn.2305-5839.2015.03.44
77. Della Corte CM, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, Accardo M, Fabozzi A, Fiorelli A, Fasano M, Papaccio F, Martinelli E, Troiani T, Troncone G, et al. SMO gene amplification and activation of the Hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin Cancer Res. 2015; 21:4686–97. https://doi.org/10.1158/1078-0432.CCR-14-3319
78. Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-mediated regulation of Sox2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non-small cell lung cancer. Neoplasia. 2015; 17:538–51. https://doi.org/10.1016/j.neo.2015.07.001
79. Bai XY, Zhang XC, Yang SQ, An SJ, Chen ZH, Su J, Xie Z, Gou LY, Wu YL. Blockade of Hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One. 2016; 11:e0149370. https://doi.org/10.1371/journal.pone.0149370
80. Lin EH, Kao YR, Lin CA, Kuo TY, Yang SP, Hsu CF, Chou TY, Ho CC, Wu CW. Hedgehog pathway maintains cell survival under stress conditions, and drives drug resistance in lung adenocarcinoma. Oncotarget. 2016; 7:24179–93. https://doi.org/10.18632/oncotarget.8253
81. Morgillo F, Amendola G, Della Corte CM, Giacomelli C, Botta L, Di Maro S, Messere A, Ciaramella V, Taliani S, Marinelli L, Trincavelli ML, Martini C, Novellino E, et al. Dual MET and SMO negative modulators overcome resistance to EGFR inhibitors in human nonsmall cell lung cancer. J Med Chem. 2017; 60:7447–58. https://doi.org/10.1021/acs.jmedchem.7b00794
82. Ma J, Tian L, Cheng J, Chen Z, Xu B, Wang L, Li C, Huang Q. Sonic hedgehog signaling pathway supports cancer cell growth during cancer radiotherapy. PLoS One. 2013; 8:e65032. https://doi.org/10.1371/journal.pone.0065032
83. Chaudary N, Pintilie M, Hedley D, Fyles AW, Milosevic M, Clarke B, Hill RP, Mackay H. Hedgehog pathway signaling in cervical carcinoma and outcome after chemoradiation. Cancer. 2012; 118:3105–15. https://doi.org/10.1002/cncr.26635
84. Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, Epperly M, Levina V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer. 2013; 12:94. https://doi.org/10.1186/1476-4598-12-94
85. Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, Bernard K, Conklin JF, Szczepny A, Yuan J, Guo R, Ospina B, Falzon J, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011; 17:1504–08. https://doi.org/10.1038/nm.2473
86. Belani CP, Dahlberg SE, Rudin CM, Fleisher M, Chen HX, Takebe N, Velasco MR Jr, Tester WJ, Sturtz K, Hann CL, Shanks JC, Monga M, Ramalingam SS, Schiller JH. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer. 2016; 122:2371–78. https://doi.org/10.1002/cncr.30062
87. Pietanza MC, Litvak AM, Varghese AM, Krug LM, Fleisher M, Teitcher JB, Holodny AI, Sima CS, Woo KM, Ng KK, Won HH, Berger MF, Kris MG, Rudin CM. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer. 2016; 99:23–30. https://doi.org/10.1016/j.lungcan.2016.04.014
88. Szczepny A, Rogers S, Jayasekara WS, Park K, McCloy RA, Cochrane CR, Ganju V, Cooper WA, Sage J, Peacock CD, Cain JE, Burgess A, Watkins DN. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene. 2017; 36:5544–50. https://doi.org/10.1038/onc.2017.173
89. Zhang S, Yang YL, Wang Y, You B, Dai Y, Chan G, Hsieh D, Kim IJ, Fang LT, Au A, Stoppler HJ, Xu Z, Jablons DM, You L. CK2α, over-expressed in human malignant pleural mesothelioma, regulates the Hedgehog signaling pathway in mesothelioma cells. J Exp Clin Cancer Res. 2014; 33:93. https://doi.org/10.1186/s13046-014-0093-6
90. Li H, Lui N, Cheng T, Tseng HH, Yue D, Giroux-Leprieur E, Do HT, Sheng Q, Jin JQ, Luh TW, Jablons DM, He B. Gli as a novel therapeutic target in malignant pleural mesothelioma. PLoS One. 2013; 8:e57346. https://doi.org/10.1371/journal.pone.0057346
91. Bosco-Clément G, Zhang F, Chen Z, Zhou HM, Li H, Mikami I, Hirata T, Yagui-Beltran A, Lui N, Do HT, Cheng T, Tseng HH, Choi H, et al. Targeting Gli transcription activation by small molecule suppresses tumor growth. Oncogene. 2014; 33:2087–97. https://doi.org/10.1038/onc.2013.164
92. You M, Varona-Santos J, Singh S, Robbins DJ, Savaraj N, Nguyen DM. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg. 2014; 147:508–16. https://doi.org/10.1016/j.jtcvs.2013.08.035
93. Leprieur EG, Tolani B, Li H, Leguay F, Hoang NT, Acevedo LA, Jin JQ, Tseng HH, Yue D, Kim IJ, Wislez M, Wang C, Jablons DM, He B. Membrane-bound full-length Sonic Hedgehog identifies cancer stem cells in human non-small cell lung cancer. Oncotarget. 2017; 8:103744–57. https://doi.org/10.18632/oncotarget.21781
94. Tolani B, Hoang NT, Acevedo LA, Giroux Leprieur E, Li H, He B, Jablons D. Preclinical characterization of therapeutic antibodies targeted at the carboxy-terminus of Sonic Hedgehog. Oncotarget. 2018 Feb 15. https://doi.org/10.18632/oncotarget.24510. [Epub ahead of print].