
INTRODUCTION
Treatment for ovarian cancer remains challenging despite a high initial response rate to first line platinum-taxane treatment. Most patients eventually experience recurrence and require further treatment. However, there is no effective treatment for recurrent drug-resistant ovarian cancer. Despite efforts to overcome resistance using alternate chemotherapy agents, mortality remains high in platinum-resistant patients [1–7]. Therefore, there is a critical need to develop novel strategies to treat advanced and drug resistant ovarian cancer.
STAT3, a promising molecular target for cancer therapies, is a member of the STAT family of transcription factors that mediate cellular responses to cytokines and growth factors. In healthy tissue, STAT3 is predominantly located in the cytoplasm in an inactive form. In response to cytokine stimulation, STAT3 is phosphorylated at Tyr705 by Janus family kinases (JAK) [8, 9]. Phosphorylated STAT3 protein can translocate into the nucleus, bind to DNA, and activate the transcription of various genes that regulate vital cellular functions, including cell survival, proliferation, angiogenesis, and tumor evasion [10]. Normally, the activation of STAT3 by JAK occurs transiently and is tightly regulated. However, in cancer cells STAT3 is constitutively activated [10–14], and its persistent activation is associated with a poor prognosis in cancer patients, including ovarian cancer patients [15, 16].
Several recent studies have demonstrated a critical role of STAT3 in ovarian cancer growth and progression. Inhibition of STAT3 activation has led to reduced tumor growth, decreased peritoneal dissemination, and diminished ascites production in a peritoneal ovarian tumor model [17–19]. In addition, emerging evidence suggests that activation of STAT3 is involved in resistance to both receptor tyrosine kinase -target therapy and conventional chemotherapy [20–26]. In addition, increased STAT3 activation occurs in paclitaxel-resistant ovarian cancer cells, and STAT3 inhibition potently increases anti-tumor activity of paclitaxel [27–29]. Targeting JAK1/STAT3, therefore, could be a potential novel therapeutic approach for treating advanced and chemoresistant ovarian cancer.
Ruxolitinib is a potent and selective oral JAK1 and JAK2 inhibitor that was FDA-approved in 2011 for the treatment of myelofibrosis (MF), post-polycythemia vera myelofibrosis (PPV-MF), and post-essential thrombocythemia myelofibrosis (PET-MF) [30–32]. The therapeutic potential of ruxolitinib in solid tumors is currently undergoing clinical evaluation in ovarian, metastatic breast, and pancreatic cancers [33–35]. However, there is little pre-clinical information available about ruxolitinib in ovarian cancer treatment.
In this study, we investigated the anti-tumor activity of ruxolitinib, either alone or in combination with chemotherapy agents, in human ovarian cancer both in vitro and in vivo.
RESULTS
Effect of ruxolitinib on phosphorylation of STAT3 and cell viability in human ovarian cancer cells
To understand the effect of ruxolitinib on STAT3 phosphorylation, we incubated OVCAR-8, MDAH2774, and SKOV3 human ovarian cancer cells with increasing concentrations of ruxolitinib followed by Western blot analysis. We found that ruxolitinib significantly inhibited phosphorylation of STAT3 in a dose dependent manner in all cells (Figure 1A). To study the anti-tumor activity of ruxolitinib in human ovarian cancer, we first tested the effects of ruxolitinib on the proliferation and viability of OVCAR-8, MDAH2774, and SKOV-3 cells. Cells were incubated with increasing concentrations of ruxolitinib, and cell viability was determined after 72 h. We found that ruxolitinib inhibited cell viability with IC50s ranging from 13.37 μM to 18.53 μM (Figure 1B).
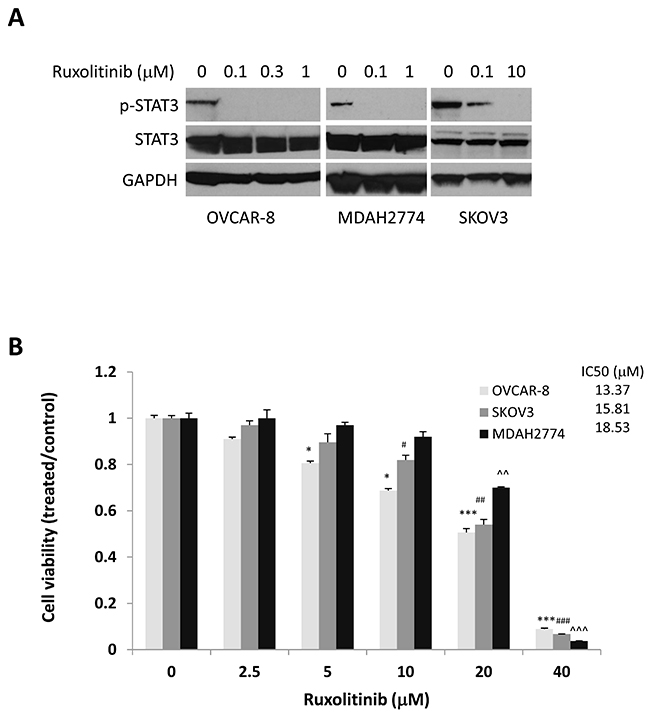
Figure 1: Anti-tumor activity of ruxolitinib in ovarian cancer. (A) Dose-dependent inhibition of STAT3 phosphorylation. Human ovarian cancer cells, OVCAR-8, MDAH2774, and SKOV3, were treated with the indicated concentrations of ruxolitinib for 24 h. Phosphorylation of STAT3 was analyzed by Western blot. (B) Dose dependent inhibition of cell viability. Human ovarian cancer cell lines were treated with the indicated concentrations of ruxolitinib. Cell viability was determined 72 h later. The IC50 was determined by the Chou-Talalay method. *P<0.05; ***P<0.0005, ruxolitinib vs control in OVCAR-8 cells; #P<0.05; ##P<0.005; ###P<0.0005, ruxolitinib vs control in SKOV-3 cells; ^^P<0.005; ^^^P<0.0005, ruxolitinib vs control in MDAH2774 cells.
Next, to investigate the possibility that reduced cell survival by ruxolitinib could be due to the induction of apoptosis, we treated OVCAR-8 and MDAH2774 cells with various concentrations of ruxolitinib for 48 h. The number of apoptotic cells was then determined by annexin V staining (Figure 2A). We found that ruxolitinib induced cell apoptosis in a dose dependent manner in both OVCAR-8 and MDAH2774 cells. Consistent with the annexin V staining results, generation of cleaved poly-ADP ribose polymerase (PARP), a marker for apoptosis, increased in both OVCAR-8 and MDAH2774 cells treated with ruxolitinib for 48 h (Figure 2B). These results indicate that ruxolitinib could inhibit cell viability of human ovarian cancer cells by promoting apoptosis.
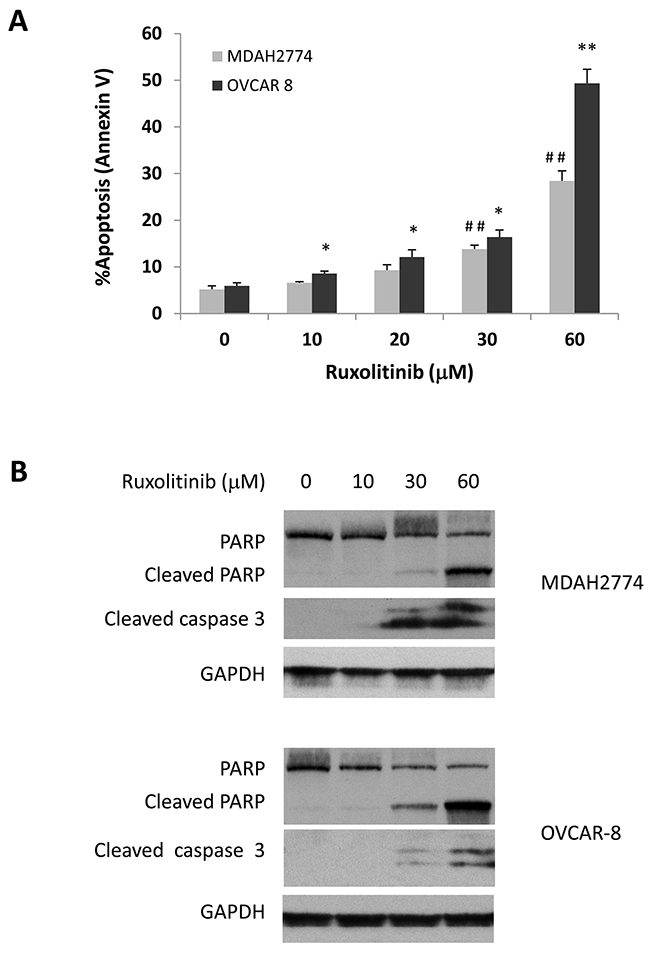
Figure 2: Dose dependent induction of apoptosis. (A) and (B) OVCAR-8 and MDAH 2774 cells were incubated with various concentrations of ruxolitinib for 48 h. Apoptosis was determined by flow cytometry using annexin V and PI staining (A) or using cleaved poly-ADP ribose polymerase (PARP) and cleaved caspase-3 by Western blot (B). *P<0.05; **P<0.005, ruxolitinib vs control in MDAH2774 cells; ##P<0.005, ruxolitinib vs control in OVCAR-8 cells.
Effect of ruxolitinib on cell viability induced by chemotherapy agents
Previous studies suggest that activation of STAT3 may confer cell resistance to chemotherapy reagents in ovarian cancer cells [20–25]. To understand whether inhibition of the STAT3 pathway could enhance the anti-tumor activity of chemotherapy reagents, we incubated human ovarian cancer cells with several chemotherapy agents, either alone or in combination with ruxolitinib. We found that ruxolitinib significantly increased the anti-tumor activity of paclitaxel, cisplatin, and carboplatin – the first line chemotherapy agents in the treatment of ovarian cancer (Figure 3 and 4). The IC50 of paclitaxel was decreased by over two-fold in both OVCAR-8 and MDAH2774 cells (Table 1). Ruxolitinib also increased the anti-tumor activity of doxorubicin and topotecan, commonly used chemotherapy agents for the treatment of relapsed ovarian cancer (Table 1, Figure 3 and 4).
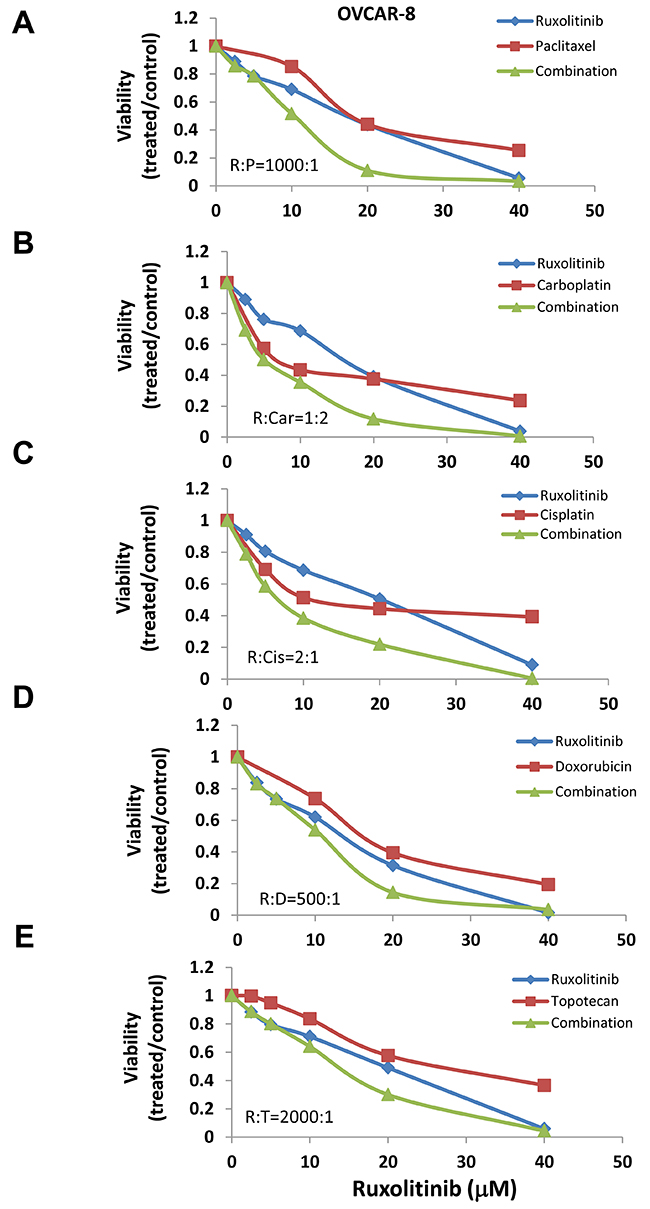
Figure 3: Ruxolitinib enhanced the anti-tumor activity of chemotherapy agents in OVCAR-8 human ovarian cancer cells. OVCAR-8 cells were treated with ruxolitinib either alone or together with chemotherapy agents, paclitaxel (A), carboplatin (B), cisplatin (C), doxorubicin (D), and topotecan (E), at various concentrations in a fixed molar ratio. Cell viability was determined 72 h later.
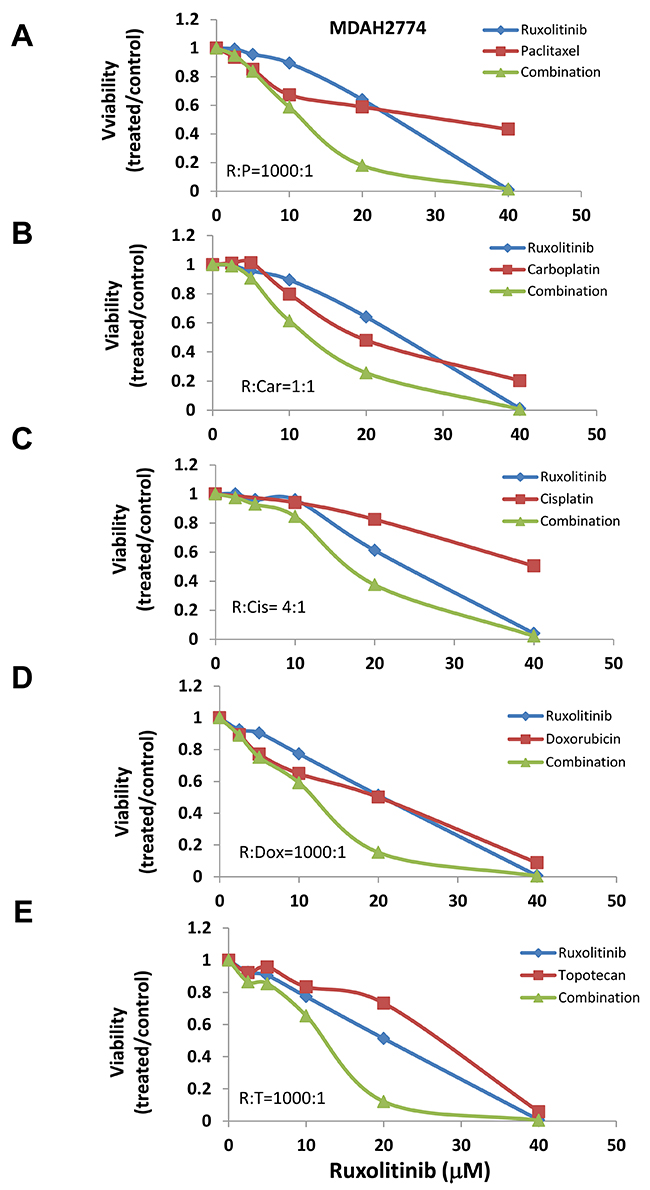
Figure 4: Ruxolitinib enhanced the anti-tumor activity of chemotherapy agents in MDAH2774 human ovarian cancer cells. MDAH2774 cells were treated with ruxolitinib either alone or together with chemotherapy agents, paclitaxel (A), carboplatin (B), cisplatin (C), doxorubicin (D), and topotecan (E), at various concentrations in a fixed molar ratio. Cell viability was determined 72 h later.
Table 1: Ruxolitinib enhanced anti-tumor activity of chemotherapy reagents in human ovarian cancer cells
Cells |
Fold reduction |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Paclitaxel |
Carboplatin |
Cisplatin |
Doxorubicin |
Topotecan |
|||||||||||
IC50 |
IC75 |
IC95 |
IC50 |
IC75 |
IC95 |
IC50 |
IC75 |
IC95 |
IC50 |
IC75 |
IC95 |
IC50 |
IC75 |
IC95 |
|
OVCAR-8 |
2.75 |
2.90 |
8.29 |
1.72 |
2.51 |
4.71 |
2.52 |
9.49 |
88.06 |
2.20 |
4.13 |
11.97 |
1.54 |
1.63 |
1.79 |
MDAH2774 |
2.89 |
5.36 |
14.97 |
1.94 |
2.23 |
2.81 |
2.32 |
3.3 |
5.93 |
1.59 |
2.26 |
3.96 |
2.31 |
2.83 |
3.95 |
To understand whether the increased activity was additive or synergistic, the combination index (CI) was determined using the Chou-Talalay method (CI=1, additive effect; CI<1, synergism; CI>1, antagonism) [36]. We found that ruxolitinib can synergistically increase the anti-tumor activity of paclitaxel in both OVCAR-8 and MDAH2774 cells (Table 2, Figure 3 and 4). The effect of ruxolitinib on the anti-tumor activity of other chemotherapy agents was dependent on the cell-type. For example, the combination of ruxolitinib and ciaplatin or carboplatin is synergistic in OVCAR-8 cells, but not in MDAH2774. The combination of ruxolitinib and doxorubicin or topotecan is not synergistic in both OVCAR-8 and MDAH 2774 cells. To determine the optimal ruxolitinib:paclitaxel molar ratio, OVCAR-8 cells were incubated with ruxolitinib (fixed at 40 μM) and paclitaxel (20 nM to 160 nM) at various paclitaxel:ruxolitinib molar ratios (1:250, 1:500, 1:1000 and 1:2000) (Figure 5A). The combination treatment produced a synergism at each molar ratio (Table 3); however, the 1:1000 molar ratio produced stronger synergy and a lower IC50 for both agents in OVCAR-8 cells. To understand whether anti-tumor effect of paclitaxel can also be enhanced by other JAK/STAT3 inhibitors, AZD1480, another JAK/STAT3 inhibitor, was combined with paclitaxel in MDAH2774 cells. The combined treatment is much more effective than either alone with CI<1 (Figure 5B and 5C). Taken together, our results demonstrated that blocking JAK/STAT3 pathway can synergistically enhance anti-tumor activity of paclitaxel in ovarian cancer cells.
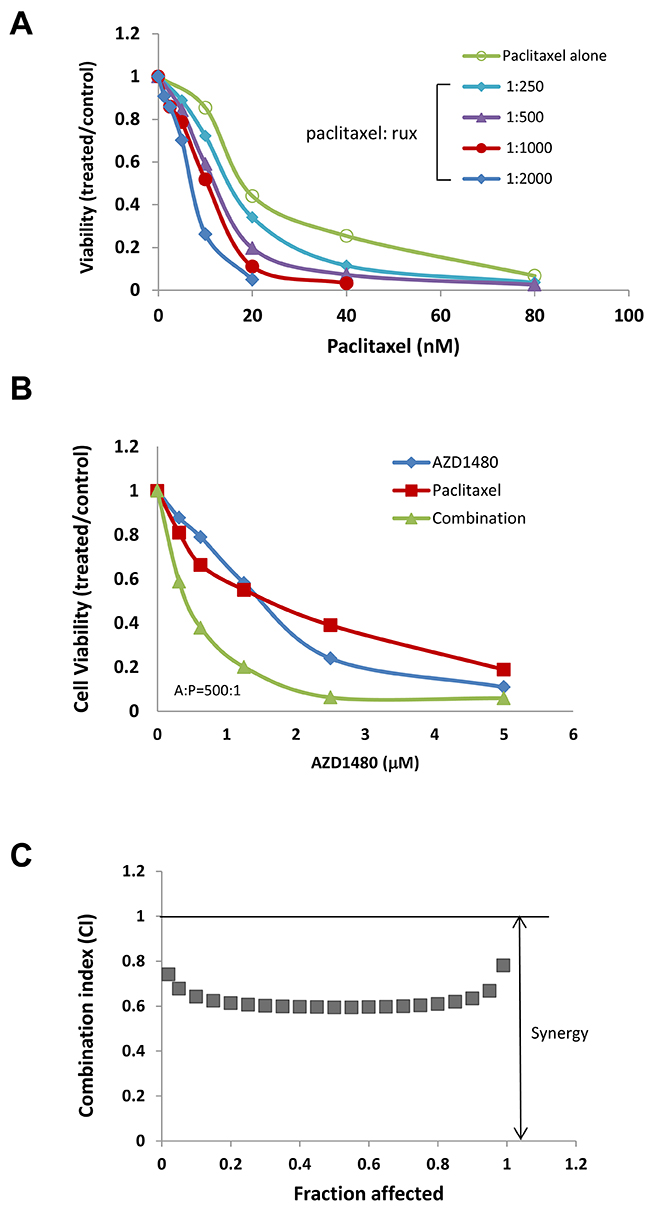
Figure 5: JAK/STAT3 inhibitors enhanced the anti-tumor activity of paclitaxel in human ovarian cancer cells. (A) OVCAR-8 cells were treated with ruxolitinib either alone or together with paclitaxel in variety of molar ratios. Cell viability was determined 72 h later. (B) and (C) MDAH2774 cells were treated with AZD1480 (another JAK/STAT3 inhibitor) and paclitaxel, either alone or together. Cell viability was determined 72 h later. CIs (combination index) were determined by the Chou-Talalay method.
Table 2: Interaction between ruxolitinib and chemotherapy agents in human ovarian cancer cells
Cells |
Combination index (CI) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Paclitaxel |
Carboplatin |
Cisplatin |
Doxorubicin |
Topotecan |
|||||||||||
ED50 |
ED75 |
ED90 |
ED50 |
ED75 |
ED90 |
ED50 |
ED75 |
ED90 |
ED50 |
ED75 |
ED90 |
ED50 |
ED75 |
ED90 |
|
OVCAR-8 |
1.01 |
0.93 |
0.87 |
1.06 |
0.85 |
0.69 |
0.85 |
0.48 |
0.33 |
1.38 |
1.21 |
1.13 |
1.26 |
1.22 |
1.19 |
MDAH2774 |
0.96 |
0.86 |
0.83 |
1.24 |
1.15 |
1.08 |
1.08 |
1.01 |
0.99 |
1.30 |
1.12 |
0.99 |
1.15 |
1.05 |
0.96 |
Table 3: Synergistic interaction between ruxolitinib and paclitaxel in variety of molar ratios in OVCAR-8 cells
Paclitaxel: Ruxolitinib |
Combination index (CI) |
IC50 |
|||
|---|---|---|---|---|---|
ED50 |
ED75 |
ED90 |
Ruxolitinib (μM) |
Paclitaxel (nM) |
|
1:250 |
1.00 |
0.93 |
0.86 |
3.70 |
14.8 |
1:500 |
1.01 |
0.93 |
0.87 |
5.66 |
11.32 |
1:1000 |
1.06 |
0.96 |
0.87 |
7.93 |
7.93 |
1:2000 |
1.12 |
1.07 |
1.01 |
11.02 |
5.51 |
Effects of combination treatment with ruxolitinib and paclitaxel on apoptosis
Next, we determined whether the synergistic effect of ruxolitinib and paclitaxel is due to induction of apoptosis. OVCAR-8 and MDAH2774 cells were treated with ruxolitinib and paclitaxel either alone or in combination for 48 h, and the number of apoptotic cells was determined by annexin V staining. Paclitaxel-induced apoptosis increased from 23.48% to 51.33% in OVCAR-8 cells and from 6.91% to 23.92% in MDAH2774 cells when combined with ruxolitinib (Figure 6A). Consistent with the annexin V staining results, more cleaved caspase 3 and cleaved poly-ADP ribose polymerase (PARP) were generated in cells that were treated with both ruxolitinib and paclitaxel (Figure 6B). These results indicate that inhibition of the JAK/STAT3 pathway could enhance the sensitivity of these human ovarian cancer cells to paclitaxel by promoting apoptosis.
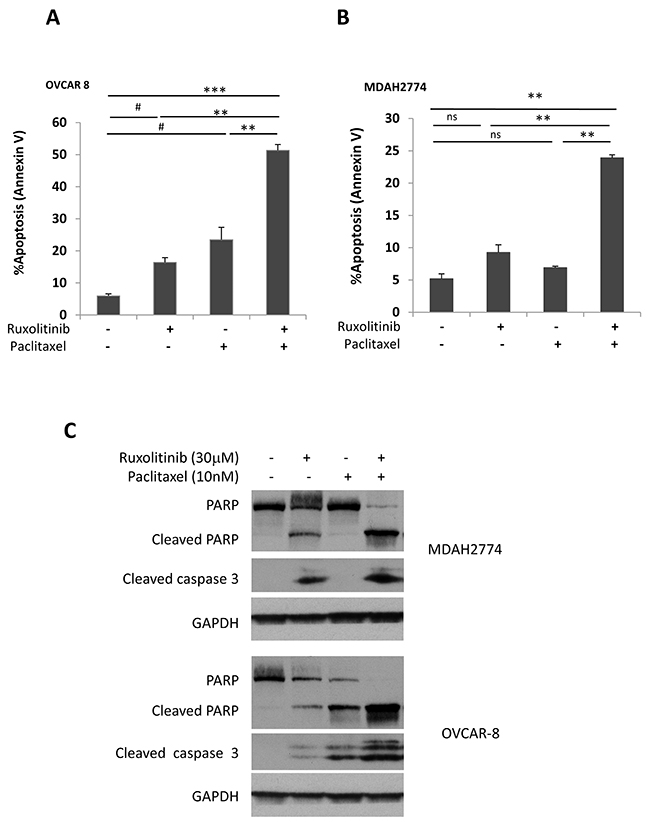
Figure 6: Ruxolitinib enhanced paclitaxel-induced apoptosis in human ovarian cancer cells. (A) OVCAR-8 and (B) MDAH2774 cells were treated with ruxolitinib (30μM), paclitaxel (10nM) either alone or together, for 48 h. Apoptosis was determined by flow cytometry using annexin V and PI staining (A&B) or by cleaved poly-ADP ribose polymerase (PARP) and cleaved caspase-3 by Western blot (C). ns: not significant; #P<0.05, ruxolitinib alone or paclitaxel alone vs control; **P<0.005; ***P<0.0005, ruxolitinib alone, paclitaxel alone or control vs combination.
Effect of combination treatment on ovarian cancer growth in mice
Next, we investigated whether the combination treatment could suppress tumor growth more effectively than either treatment alone in a mouse tumor model that represents late stage ovarian cancer with peritoneal metastasis and ascites formation. OVCAR-8-ip-Luc cells were generated and used for this model. OVCAR-8-ip-Luc is a highly metastatic human ovarian cancer cell derived from OVCAR-8 cells by selecting for a peritoneal metastatic phenotype in the mice (Materials and Methods). One week following i.p. injection of OVCAR-8-ip-Luc, mice were randomized into four groups and treated with vehicle control, ruxolitinib, paclitaxel, or paclitaxel plus ruxolitinib. Ruxolitinib was given orally in chow formulation (2g ruxolitinib in 1kg chow), which has been successfully used in a number of studies [37–41]. The serum level of ruxolitinib was shown to fall within the range achieved in humans. Both food consumption and body weight were monitored over the course of treatment and they were comparable between mice with ruxolitinib chow and mice with control chow. Treatment with ruxolitinib chow alone decreased tumor weight from 1.724g to 1.276g, and treatment with paclitaxel alone decreased tumor weight from 1.724g to 0.348g. However, the combination treatment further decreased the tumor weight to 0.142g (Figure 7A), suggesting that the combination treatment was more effective than any single treatment.
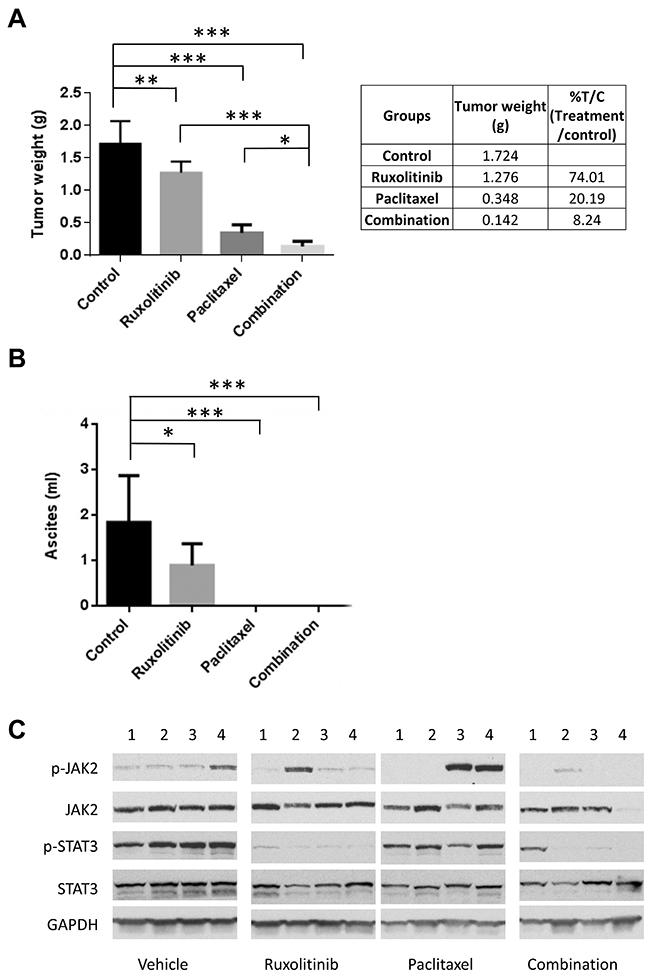
Figure 7: Ruxolitinib enhanced anti-tumor activity of paclitaxel in mice. OVCAR-8-ip-luc cells were implanted into peritoneal cavity of NSG mice. Mice were randomized into 4 groups and treated with control chow, chow supplemented with ruxolitinib (2000mg/kg), paclitaxel (10mg/kg; via i.p. injection every 4 days for 3 times), or the combination of both. Mice were euthanized four weeks later. (A) Large tumor and small tumor nodules throughout the peritoneal cavity were excised and weighed. (B) Ascites were collected, and their volumes were measured. n=4-9, *P<0.05; **P<0.005; ***P<0.0005. (C) Ruxolitinib inhibited the activation of STAT3 in the tumors. Whole tumor lysates were prepared and analyzed for expression of p-STAT3, p-JAK2, total STAT3, and total JAK2 by Western blot.
To investigate the molecular changes in the tumors upon treatment, tumor tissue lysates were analyzed for the expression of p-STAT3 and p-JAK2 by Western blot. Phosphorylation of STAT3 was blocked in the presence of ruxolitinib, either alone or in combination with paclitaxel (Figure 7C). Overall, these results support a synergistic effect of ruxolitinib and paclitaxel on ovarian cancer cell growth and survival both in vitro and in vivo.
Dual treatment of ruxolitinib and paclitaxel led to the reduction of MCL-1 expression in ovarian cancer cells
To understand the molecular mechanism underlying this synergistic effect, we investigated the effect of combined treatment on the expression of proteins involved in cell signaling and cell survival. A number of signaling pathways, including MAPK/ERK, PI3K/AKT and JAK/STAT3 pathways, are constitutively activated and play important roles in the growth and progression of ovarian cancer. To study the effect of ruxolitinib and paclitaxel on these signaling pathways, OVCAR-8 and MDAH2774 cells were treated with ruxolitinib and paclitaxel either alone or in combination for 24 hours, and tested for the expression of p-STAT3, p-AKT, p-ERK, as well as BCL-XL and MCL-1, two important STAT3 downstream proteins, by Western blot. As shown in Figure 8A, the combination of ruxolitinib and paclitaxel caused a significantly reduction of MCL-1, an important pro-survival protein. Consistent with this result, MCL-1 expression was also reduced in the tumor tissues that were treated with both ruxolitinib and paclitaxel (Figure 8B). Taken together, our results suggested that the synergistic effect by combining ruxolitinib and paclitaxel might be mediated via reducing pro-survival proteins, such as MCL-1 and promoting apoptosis. However, additional study is needed to further understand the mechanism underlying the synergy.
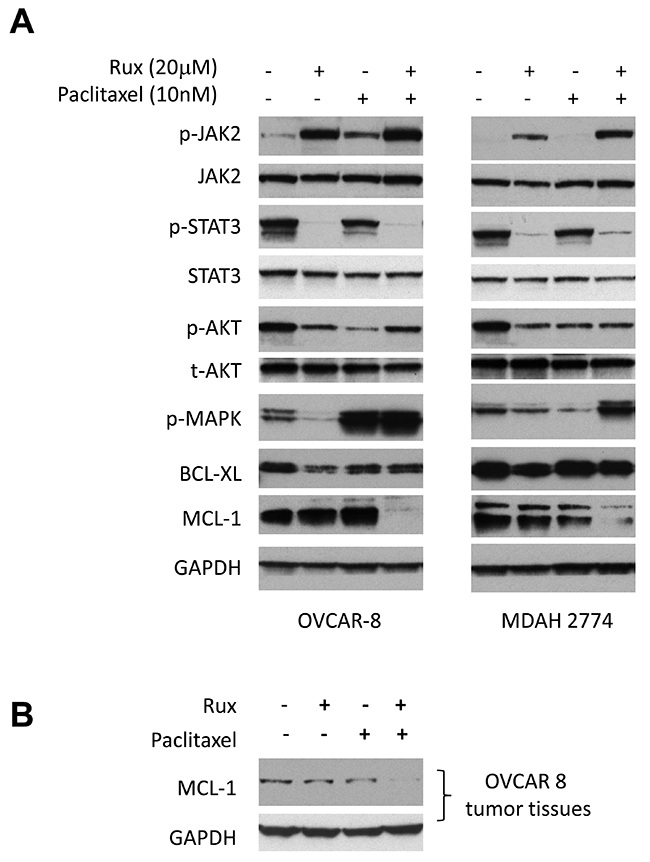
Figure 8: Combined treatment of ruxolitinib and paclitaxel led to the reduction of MCL-1 expression. (A) OVCAR-8 and MDAH2774 cells were treated with ruxolitinib (20μM), paclitaxel (10nM) or the combination for 24 h. Whole cells were collected and determined for the change of STAT3, AKT and ERK pathways and expression of BCL-XL and MCL-1 by Western blot. (B) Whole tumor lysates from the respective groups (4-8) were pooled and analyzed for expression of MCL-1 by Western blot.
DISCUSSION
The JAK/STAT3 pathway has emerged as an attractive target for cancer treatment [42, 43]. In this study, we investigated the therapeutic potential of the JAK1/JAK2 inhibitor ruxolitinib in ovarian cancer treatment, either alone or in combination with conventional chemotherapy agents. Ruxolitinib has been previously FDA-approved for the treatment of MF [30]. Our results demonstrate that ruxolitinib, either alone or in combination with paclitaxel, can significantly inhibit STAT3 activation and ovarian tumor growth both in cells and in a mouse model.
Drug resistance remains one of the major challenges in the treatment of ovarian cancer [24, 44, 45]. Most patients eventually develop resistance to treatment despite high initial response rates to first line platinum-taxane treatment. Currently available agents used to treat drug resistant patients include paclitaxel, pegylated liposomal doxorubicin, and topotecan [44]. However, the response rate is in the 10-15% range, and the overall survival rate is only about 12 months. To improve the treatment for drug resistant ovarian cancer, alternative approaches have been developed, including the use of novel cytotoxic reagents and the combination of chemotherapy with targeted agents. Previous studies have shown that constitutive activation of STAT3 confers resistance to platinum and paclitaxel induced apoptosis in ovarian cancer [20, 27]. Our study demonstrates that ruxolitinib, an FDA-approved JAK inhibitor, can potently enhance the anti-tumor activity of paclitaxel in ovarian cancer both in vitro and in vivo. Although the mechanism of this synergistic interaction is currently unclear, our preliminary results suggest that suppression of STAT3 activation may involve down-regulation of anti-apoptosis proteins, such as MCL-1.
To disrupt STAT3 signaling and activity, various approaches have been explored to identify small molecule inhibitors targeting members of the STAT3 signaling pathway [46]. Although a number of inhibitors have been developed to directly inhibit STAT3 activity in preclinical studies, none of these direct inhibitors are currently in clinical studies for cancer treatment. In contrast, targeting the upstream kinase activity of STAT3 has led to the discovery of several inhibitors in clinical studies, including ruxolitinib (INCB018424), tofacitinib (CP-690550), fedratinin (TG101348), pacritinib (NCT01773187, SB1518), momelotinib (CYT387, NCT00935987), and baricitinib (INCB 028050) [46–53]. However, off-target toxicity is still a concern due to the conserved ATP binding site in the kinase family and the ability of kinase to activate more than one target. Ruxolitinib is the only JAK1/JAK2 inhibitor FDA-approved for the treatment of MF [30]. The therapeutic application of ruxolitinib in solid tumors is currently in clinical study in several solid tumors including ovarian, metastatic breast, and pancreatic cancers [34, 54]. The combined treatment of ruxolitinib with paclitaxel and carboplatin is currently under clinical evaluation in human ovarian cancer. However, little preclinical information is available about the effect of ruxolitinib in ovarian cancer growth. Our results demonstrated for the first time that the combination of ruxolitinib with paclitaxel is more effective against ovarian cancer growth than either agent alone, suggesting that ruxolitinib may be used to improve drug sensitivity and reduce undesirable side effects associated with conventional chemotherapy.
Taken together, our findings provide a valuable preclinical foundation for clinical trials with ruxolitinib either as a single agent or in combination with paclitaxel for the treatment of recurrent and advanced ovarian cancer.
MATERIALS AND METHODS
Reagents
Ruxolitinib was kindly provided by Incyte Inc. AZD1480 was kindly provided by AstraZeneca. Antibodies against p-STAT3 (Y705), STAT3, p-JAK2 (Y1007/1008), JAK2, p-AKT, p-ERK, BCL-XL, PARP, Caspase 3, and GAPDH were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against AKT and MCL-1 were from Santa Cruz Biotechnology (Dallas, TX). The antibody against actin was obtained from Sigma (St. Louis, MO).
Cell culture
SKOV3 and MDAH2774 cells were obtained from ATCC. The OVCAR-8 cells were obtained from the National Cancer Institute. SKOV3 and MDAH2774 cells were cultured in DMEM medium. OVCAR-8 cells were cultured in RPMI1640 medium. Culture media were supplemented with 10% FBS and 1% penicillin/streptomycin (P/S). All cells were grown in 5% (v/v) CO2 at 37°C.
To minimize mouse-to-mouse variation in tumor formation of OVCAR-8 cells, a subline of OVCAR-8, OVCAR-8-ip, was generated. To do this, parental OVCAR-8 cells were inoculated into the peritoneal cavity of NOD/SCID/IL2R gamma null (NSG) mice and allowed to form tumor. The mouse that had the most ascites and peritoneal metastasis at earliest time point was used to isolate tumor cells from the ascites. The isolated tumor cells, called OVCAR-8-ip, produced ascites and formed peritoneal tumors with much less variation between mice when these cells were implanted into mice. To monitor peritoneal tumor growth using imaging, OVCAR-8-ip cells were stably transfected with CMV-p:EGFP-ffluc pHIV7 (a gift from Christine Brown at City of Hope) as previously described to create OVCAR-8-ip-Luc cells [55].
Cell viability assays
Cells were plated in 96-well plate format in 100 μl growth medium. To ensure that all cells had similar cell confluence when placed, we placed MDAH2774 at 7000 cells per well and other cells at 4000 per well. Cells were treated with DMSO or drugs the next day at the indicated concentrations and incubated for an additional 1 to 3 days. Viable cells were determined either by MTS assay (Promega, Madison, WI, USA) or acid phosphatase assay as described previously [56]. The IC50 was determined using Calcusyn (Biosoft, Ferguson, MO).
Determination of combination index (CI)
The combination index (CI) was determined using the Chou-Talalay method [36] using Calcusyn (Biosoft, MO).
Annexin V staining
Apoptosis was measured using an Annexin V Apoptosis Detection Kit (BD bioscience). Briefly, ovarian cancer cells were treated with ruxolitinib, paclitaxel, or both. After 48 h, floating and attached cells were collected and stained with FITC-Annexin V and PI (propidium iodide). The staining intensity was then quantified using fluorescence-activated cell sorting (FACS).
Western blot analysis
Western blots were performed as described previously [57]. Cells were grown in complete medium overnight and treated with DMSO or drugs at various concentrations for 24 h. Cells were washed in cold PBS and lysed in RIPA lysis buffer (Thermo Scientific) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Proteins were quantified using BCA protein assay reagent (Thermo Scientific). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and incubated with total and phosphorylated protein-specific antibodies. Binding of the primary antibody was detected using a horseradish peroxidase (HRP)-conjugated secondary antibody and chemiluminescent substrates (Thermo Scientific).
Animal models
All animal studies were carried out under protocols approved by the City of Hope Institutional Animal Care and Use Committee (IACUC) in accordance with guidelines of the association for Assessment and Accreditation of Laboratory Animal Care.
OVCAR-8-ip-Luc cells (5×106 in 100 μl) were inoculated into the peritoneal cavity of 6- to 8- week-old female NSG mice. Starting one week after inoculation, mice were treated with control, ruxolitinib, paclitaxel (10mg/kg via i.p. injection, every 4 days for total 3 times), or combination of both. Ruxolitinib was given orally in chow formulation (2g ruxolitinib in 1kg chow) as described previously (kindly provided by Incyte) [37–41]. We monitor food consumption during period of treatment to ensure comparable amount of food was taken between mice with ruxolitinib chow and mice with control chow. Body weight was monitored weekly as an indicator of drug-induced toxicity and overall health of the mice. The mice were monitored for ascites production and any adverse effects. Mice were euthanized 25 days after cell inoculation. Visible tumor nodules were excised and weighed, and the ascites fluid was collected and measured for the volume.
Statistical analysis
Data are presented as mean ± S.D. Student’s t-test was used to compare the means of two groups. Experiments were carried out in triplicate or more. P < 0.05 was considered statistically significant.
Author contributions
Conception and design of study: ESH, WW, RJ, JHY; In vitro and in vivo experiments: WW, JW, SAL; Technical and material support: WW, THD; Analysis and interpretation of data: ESH, WW, THD, RJ, JHY; Writing, review and/or revision of the manuscript: ESH, WW, THD and JHY. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We thank Dr. Holly Koblish at Incyte for providing ruxolitinib and her voluble suggestions, AstraZeneca for providing AZD1480, Dr. Chris Gandhi for critical reading of this manuscript, Lucy Brown and the Analytical Cytometry Core, the Animal Tumor Models Program, Small Animal Image Core, as well as animal facility for their technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING SUPPORT
Research reported in this publication included work performed in the Analytical Cytometry, Animal Tumor Models Program and Small Animal Image Cores supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
1. Vaughan S, Coward JI, Bast RC Jr, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011; 11:719-25.
2. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004; 351:2519-29.
3. Cristea M, Han E, Salmon L, Morgan RJ. Practical considerations in ovarian cancer chemotherapy. Ther Adv Med Oncol. 2010; 2:175-87.
4. Burger RA. Advances in ovarian cancer disease control. Gynecol Oncol. 2012; 124:5-9.
5. Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE, Boente M, Birrer MJ, Liang SX; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011; 365:2473-83.
6. Mor G, Yin G, Chefetz I, Yang Y, Alvero A. Ovarian cancer stem cells and inflammation. Cancer Biol Ther. 2011; 11:708-13.
7. Kimball KJ, Numnum TM, Kirby TO, Zamboni WC, Estes JM, Barnes MN, Matei DE, Koch KM, Alvarez RD. A phase i study of lapatinib in combination with carboplatin in women with platinum sensitive recurrent ovarian carcinoma. Gynecol Oncol. 2008; 111:95-101.
8. Darnell JE Jr, Kerr IM, Stark GR. Jak-stat pathways and transcriptional activation in response to ifns and other extracellular signaling proteins. Science. 1994; 264:1415-21.
9. Darnell JE Jr. Stats and gene regulation. Science. 1997; 277:1630-5.
10. Yu H, Pardoll D, Jove R. Stats in cancer inflammation and immunity: a leading role for stat3. Nat Rev Cancer. 2009; 9:798-809.
11. Yu H, Jove R. The stats of cancer—new molecular targets come of age. Nat Rev Cancer. 2004; 4:97-105.
12. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr. Stat3 as an oncogene. Cell. 1999; 98:295-303.
13. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernández-Luna JL, Nuñez G, Dalton WS, Jove R. Constitutive activation of stat3 signaling confers resistance to apoptosis in human u266 myeloma cells. Immunity. 1999; 10:105-15.
14. Darnell JE. Validating stat3 in cancer therapy. Nat Med. 2005; 11:595-6.
15. Rosen DG, Mercado-Uribe I, Yang G, Bast RC Jr, Amin HM, Lai R, Liu J. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer. 2006; 107:2730-40.
16. Anglesio MS, George J, Kulbe H, Friedlander M, Rischin D, Lemech C, Power J, Coward J, Cowin PA, House CM, Chakravarty P, Gorringe KL, Campbell IG, et al. Il6-stat3-hif signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res. 2011; 17:2538-48.
17. Ma Y, Zhang X, Xu X, Shen L, Yao Y, Yang Z, Liu P. Stat3 decoy oligodeoxynucleotides-loaded solid lipid nanoparticles induce cell death and inhibit invasion in ovarian cancer cells. PLoS One. 2015; 10:e0124924.
18. Wen W, Liang W, Wu J, Kowolik CM, Buettner R, Scuto A, Hsieh MY, Hong H, Brown CE, Forman SJ, Horne D, Morgan R, Wakabayashi M, et al. Targeting jak1/stat3 signaling suppresses tumor progression and metastasis in a peritoneal model of human ovarian cancer. Mol Cancer Ther. 2014; 13:3037-48.
19. Gritsina G, Xiao F, O’Brien SW, Gabbasov R, Maglaty MA, Xu RH, Thapa RJ, Zhou Y, Nicolas E, Litwin S, Balachandran S, Sigal LJ, Huszar D, Connolly DC. Targeted blockade of jak/stat3 signaling inhibits ovarian carcinoma growth. Mol Cancer Ther. 2015; 14:1035-47.
20. Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, Hampel C, Lee H, Seiden MV. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin Cancer Res. 2006; 12:5055-63.
21. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014; 26:207-21.
22. Eichten A, Su J, Adler AP, Zhang L, Ioffe E, Parveen AA, Yancopoulos GD, Rudge J, Lowy I, Lin HC, MacDonald D, Daly C, Duan X, Thurston G. Resistance to anti-vegf therapy mediated by autocrine il6/stat3 signaling and overcome by il6 blockade. Cancer Res. 2016; 76:2327-39.
23. Kandala PK, Srivastava SK. Diindolylmethane suppresses ovarian cancer growth and potentiates the effect of cisplatin in tumor mouse model by targeting signal transducer and activator of transcription 3 (stat3). BMC Med. 2012; 10:9.
24. Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback activation of stat3 as a cancer drug-resistance mechanism. Trends Pharmacol Sci. 2016; 37:47-61.
25. Wen W, Wu J, Liu L, Tian Y, Buettner R, Hsieh MY, Horne D, Dellinger TH, Han ES, Jove R, Yim JH. Synergistic anti-tumor effect of combined inhibition of egfr and jak/stat3 pathways in human ovarian cancer. Mol Cancer. 2015; 14:100.
26. Jung JG, Shih IM, Park JT, Gerry E, Kim TH, Ayhan A, Handschuh K, Davidson B, Fader AN, Selleri L, Wang TL. Ovarian cancer chemoresistance relies on the stem cell reprogramming factor pbx1. Cancer Res. 2016; 76:6351-61.
27. Ji T, Gong D, Han Z, Wei X, Yan Y, Ye F, Ding W, Wang J, Xia X, Li F, Hu W, Lu Y, Wang S, et al. Abrogation of constitutive stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013; 341:231-39.
28. Selvendiran K, Ahmed S, Dayton A, Kuppusamy ML, Rivera BK, Kálai T, Hideg K, Kuppusamy P. Ho-3867, a curcumin analog, sensitizes cisplatin-resistant ovarian carcinoma, leading to therapeutic synergy through stat3 inhibition. Cancer Biol Ther. 2011; 12:837-45.
29. Hu Y, Hong Y, Xu Y, Liu P, Guo DH, Chen Y. Inhibition of the jak/stat pathway with ruxolitinib overcomes cisplatin resistance in non-small-cell lung cancer nsclc. Apoptosis. 2014; 19:1627-36.
30. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, Dipersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366:799-807.
31. Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/stat pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013; 19:1933-40.
32. Saenz DT, Fiskus W, Manshouri T, Rajapakshe K, Krieger S, Sun B, Mill CP, DiNardo C, Pemmaraju N, Kadia T, Parmar S, Sharma S, Coarfa C, et al. Bet protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary aml cells. Leukemia. 2017; 31:678-87.
33. Yu HA, Perez L, Chang Q, Gao SP, Kris MG, Riely GJ, Bromberg J. A phase 1/2 trial of ruxolitinib and erlotinib in patients with egfr-mutant lung adenocarcinomas with acquired resistance to erlotinib. J Thorac Oncol. 2017; 12:102-9.
34. Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM 3rd, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA, Manges R, Garrett WM, Hunter DS, et al. Randomized, double-blind, phase ii study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015; 33:4039-47.
35. Ju W, Zhang M, Wilson KM, Petrus MN, Bamford RN, Zhang X, Guha R, Ferrer M, Thomas CJ, Waldmann TA. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or navitoclax in a murine model of human hodgkin’s lymphoma. Proc Natl Acad Sci U S A. 2016; 113:1624-29.
36. Chou TC. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010; 70:440-46.
37. Iacobucci I, Li Y, Roberts KG, Dobson SM, Kim JC, Payne-Turner D, Harvey RC, Valentine M, McCastlain K, Easton J, Yergeau D, Janke LJ, Shao Y, et al. Truncating erythropoietin receptor rearrangements in acute lymphoblastic leukemia. Cancer Cell. 2016; 29:186-200.
38. Taniguchi K, Moroishi T, De Jong PR, Krawczyk M, Grebbin BM, Luo H, Xu RH, Golob-Schwarzl N, Schweiger C, Wang K, Di Caro G, Feng Y, Fearon ER, et al. Yap–il-6st autoregulatory loop activated on apc loss controls colonic tumorigenesis. Proc Natl Acad Sci U S A. 2017; 114:1643-48.
39. Maude SL, Dolai S, Delgado-Martin C, Vincent T, Robbins A, Selvanathan A, Ryan T, Hall J, Wood AC, Tasian SK, Hunger SP, Loh ML, Mullighan CG, et al. Efficacy of jak/stat pathway inhibition in murine xenograft models of early t-cell precursor (etp) acute lymphoblastic leukemia. Blood. 2015; 125:1759-67.
40. Dolai S, Sia KC, Robbins AK, Zhong L, Heatley SL, Vincent TL, Hochgräfe F, Sutton R, Kurmasheva RT, Revesz T, White DL, Houghton PJ, Smith MA, et al. Quantitative phosphotyrosine profiling of patient-derived xenografts identifies therapeutic targets in pediatric leukemia. Cancer Res. 2016; 76:2766-77.
41. Ollila S, Domènech-Moreno E, Laajanen K, Wong IP, Tripathi S, Pentinmikko N, Gao Y, Yan Y, Niemelä EH, Wang TC, Viollet B, Leone G, Katajisto P, et al. Stromal lkb1 deficiency leads to gastrointestinal tumorigenesis involving the il-11–jak/stat3 pathway. J Clin Invest. 2017; 128:402-14.
42. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting stat3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014; 14:736-46.
43. Sansone P, Bromberg J. Targeting the interleukin-6/jak/stat pathway in human malignancies. J Clin Oncol. 2012; 30:1005-14.
44. Giornelli GH. Management of relapsed ovarian cancer: a review. Springerplus. 2016; 5:1197.
45. Brasseur K, Gévry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget. 2017; 8:4008-42. https://doi.org/10.18632/oncotarget.14021.
46. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting stat3 for cancer treatment. ACS Chem Biol. 2016; 11:308-18.
47. Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, et al. Efficacy of tg101348, a selective jak2 inhibitor, in treatment of a murine model of jak2v617f-induced polycythemia vera. Cancer Cell. 2008; 13:311-20.
48. Geron I, Abrahamsson AE, Barroga CF, Kavalerchik E, Gotlib J, Hood JD, Durocher J, Mak CC, Noronha G, Soll RM, Tefferi A, Kaushansky K, Jamieson CH. Selective inhibition of jak2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008; 13:321-30.
49. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, et al. Preclinical characterization of the selective jak1/2 inhibitor incb018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115:3109-17.
50. Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, William AD, Lee A, Poulsen A, et al. Sb1518, a novel macrocyclic pyrimidine-based jak2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011; 25:1751-59.
51. Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. Cyt387, a selective jak1/jak2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009; 23:1441-45.
52. Flanagan ME, Blumenkopf TA, Brissette WH, Brown MF, Casavant JM, Shang-Poa C, Doty JL, Elliott EA, Fisher MB, Hines M, Kent C, Kudlacz EM, Lillie BM, et al. Discovery of cp-690,550: a potent and selective janus kinase (jak) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem. 2010; 53:8468-84.
53. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, et al. Selective inhibition of jak1 and jak2 is efficacious in rodent models of arthritis: preclinical characterization of incb028050. J Immunol. 2010; 184:5298-307.
54. Tavallai M, Booth L, Roberts JL, Poklepovic A, Dent P. Rationally repurposing ruxolitinib (jakafi ((r))) as a solid tumor therapeutic. Front Oncol. 2016; 6:142.
55. Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, Shih CC, Badie B, Hudecek M, Riddell SR, Jensen MC. Recognition and killing of brain tumor stem-like initiating cells by cd8+ cytolytic t cells. Cancer Res. 2009; 69:8886-93.
56. Yang TT, Sinai P, Kain SR. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Anal Biochem. 1996; 241:103-8.
57. Lu J, Zhang K, Nam S, Anderson RA, Jove R, Wen W. Novel angiogenesis inhibitory activity in cinnamon extract blocks vegfr2 kinase and downstream signaling. Carcinogenesis. 2010; 31:481-88.