
INTRODUCTION
Today the risk to develop cancer is quite high and the number of tumor patients unfortunately is still even increasing [1, 2]. If not slowed down, it is expected that the global annual new cases will reach 15 million by 2020 [3]. As a form of cancer treatment, the emergence of chimeric antigen receptors (CARs) T cell therapy brings hope to tumor patients. The CAR-T cells can target tumor antigens independent of MHC restriction [4, 5], which include an extracellular antigen binding domain, a trans-membrane portion and an intracellular signalling domain (Figure 1) that is very important to the complete activation of CAR-T cells [5, 6]. Upon the recognition of specific antigens, CAR-T cells are activated to proliferate and secrete cytokines. CAR-T cells can promote cancer killing and has shown promise for the immunotherapy of some human malignancies [7, 8]. But, the treatment efficacy of solid tumors utilizing CAR-T cells is unsatisfactory and a set of challenges still are not solved, such as antigen specificity, mechanisms of exhaustion and safety issues [9, 10].
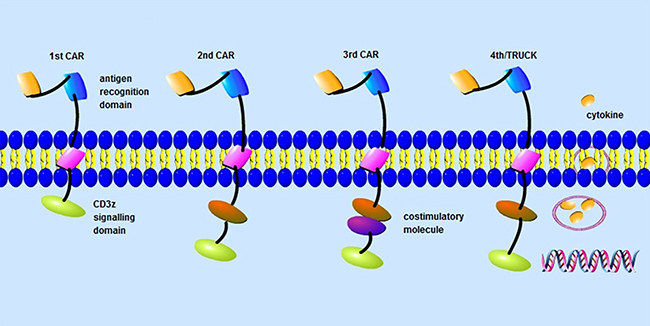
Figure 1: CAR-T-cell design. Chimeric antigen receptors (CARs) are composed of an extracellular domain, a transmembrane domain and an intracellular signaling domain. First generation CARs only have a CD3z signalling domain. By contrast, second generation CARs have a costimulatory signalling domain to enhance the signal function of the CD3z signalling domain. In third generation CARs, two costimulatory signalling domains are added to amplify anti-tumor effect of secondgeneration CARs. While in the fourth generation CARs (TRUCKs), cytokine genes are added.
EXTRACELLULAR ANTIGENIC TARGETS
The identification of targets happens before and is a prerequisite of CAR-T cell therapy. In order to avoid damage to healthy tissues caused by CAR-T cell therapy, the targets must be restricted on tumor cells [11, 12]. So far, a variety of tumor associated antigens (TAAs) have been targeted to achieve ideal therapeutic effect [13, 14].
Hematological malignancies
Within the past few years, clinical trials of CAR-T cell therapy were tested in hematological malignancies. For example, CD19-targeted CARs to treat B cell cancers are up to 70%–90% response rate in acute and chronic leukemias [15–18]. Although the great success with CD19 specific CARs, CD19 escape variants have been confirmed after therapy and responding patients with subsequent target loss have a recurrence of the disease [19–21]. To overcome such antigen target issues about escape variants, one method is to investigate other tumor antigen targets, such as CD22 [22], CD20 [23], CD138 [24], CD33 [25], CD123 [26], inactive tyrosine-protein kinase transmembrane receptor ROR1 (ROR1) [27], immunoglobulin kappa chain (Igκ) [28], B-cell maturation antigen (BCMA) [29], and Lewis Y antigen (LeY) [30, 31] (Table 1). Another way is to develop new strategies to design CAR-T cells, such as bi-specific chimeric antigen receptors. For example, the design of CD19/CD20 tandem CAR-T cells that can kill tumor cells efficiently when encountering either of the antigens [32, 33]. In addition to the methods above, the concepts of double CARs or dual receptors within one T cell, switchable CARs mentioned below also can be used here to prevent the development of antigen escape variants (Figure 2). Besides CD19 escape variants, the missing of HVEM (Herpes Virus Entry Mediator) also is reported, which caused lymphomas in vivo due to the destruction of inhibitory interactions between the HVEM and BTLA (B and T lymphocyte attenuator) receptors. CAR-T cells that secrete HVEM have shown great therapeutic efficacy against xenografted lymphomas in vivo [34].
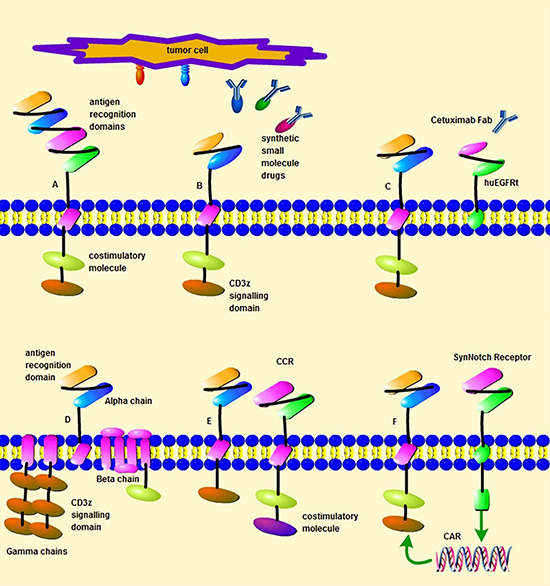
Figure 2: Different design strategies of CAR T cells. (A) A bi-specific CAR targeting two different antigens. (B) A CAR that target tumor antigens through synthetic small molecule drugs, such as the avidin-CAR, sCAR or UniCAR. (C) A CAR designed with huEGFRt. (D) The design of multi-chain CAR based on FcεRI receptor scaffold (E) A suboptimal CAR and a chimeric co-stimulatory receptor (CCR) expressed by one T cell. (F) The expression of a CAR induced by a synNotch receptor within one T cell.
Table 1: CAR-T-cell targets for the treatment of hematological tumors
Target |
CAR structure |
Malignancy |
Reference |
|---|---|---|---|
BCMA |
CD3ζ and 41BB |
MM |
NCT02215967 [29] |
CD19 |
CD3ζ and CD28; CD3ζ and 41BB KIR2DS2 and DAP12- |
Lymphoma; Leukemia |
NCT01044069 [17] NCT01626495 [18] NCT02685670 [105] [114] |
CD22 |
CD3ζ and CD28 |
FL; NHL; DLBCL; ALL |
NCT02315612 [22] |
CD20 |
CD3ζ; CD3ζ and 41BB- |
CD20positive malignancies |
NCT01735604 [23] |
CD138 |
CD3ζ and 41BB |
MM |
NCT01886976 [24] |
CD33 |
CD3ζ and 41BB |
AML |
NCT01864902 [25] |
CD123 |
CD3ζ and CD28 |
AML |
NCT02159495 [26] |
CD19 CD20 |
CD3ζ and 41BB |
Leukemia; Lymphoma |
NCT03097770 [32] |
CD19 PSMA |
CD3ζ and CD28 PD-1 or CTLA4 |
Leukemias |
[67] |
FITC-CD19 Ab |
CD3ζ and CD28 |
CD19 positive cancers |
[126] |
Igκ |
CD3ζ and CD28 |
CLL |
NCT00881920 [28] |
LeY |
CD3ζ and CD28 |
AML |
NCT01716364 [30] |
ROR1 |
CD3ζ and 41BB |
CLL; SLL |
NCT02194374 [27] |
AML, acute myeloid leukaemia; ALL, acute lymphoblastic leukaemia; BCMA, Bcell maturation antigen; CLL, chronic lymphocytic leukaemia; CTLA4, Cytotoxic T lymphocyte associated antigen 4; DLBCL, diffuse large Bcell lymphoma; DAP12, DNAX-activating protein of 12 kDa; FL, follicular lymphoma; FITC, fluoresceine isothiocyanate; Igκ, immunoglobulin kappa chain; KIR2DS2, stimulatory killer immunoglobulin-like receptor 2DS2; LeY, Lewis Y antigen; MM, multiple myeloma; NHL, nonHodgkin lymphoma; PSMA, prostatespecific membrane antigen; PSMA-CAR (iCAR) [67], inhibitory chimeric antigen receptor; PD-1, programmed death 1; ROR1, inactive tyrosineprotein kinase transmembrane receptor ROR1; SLL, small lymphocytic lymphoma.
Solid tumors
With the comforting results of CD19 CARs, much attention has been paid to the development of CARs to look for effective methods to treat solid tumors successfully. About solid tumors, many tumor-associated antigens are also targeted to get the optimal efficacy (Table 2).
Table 2: CAR-T-cell targets for the treatment of solid tumors
Target |
CAR structure |
Malignancy |
Reference |
|---|---|---|---|
Biotin |
CD3ζ, CD28 and 41BB |
EGFRvIII positive cancer |
[125] |
CD171 |
CD3ζ and 4-1BB; CD3ζ, CD28 and 4-1BB |
Neuroblastoma |
NCT02311621 [70] |
EGFRvIII |
CD3ζ and 41BB CD3ζ and ICOS- |
Glioma |
NCT02209376 [40] [107] |
FAP |
CD3ζ and CD28 KIR2DS2 and DAP12- |
Mesothelioma; Lung cancer |
[114] |
FR |
CD3ζ and CD27 |
Ovarian cancer; Breast cancer |
[98] |
Glypican-3 |
CD3ζ, CD28 and 41BB |
Hepatocellular carcinoma |
NCT02395250 [112] |
HER2 |
CD3ζ and CD28 |
HER2 positive cancer; Sarcoma |
NCT02713984 [61] NCT00902044 [60] |
HER2 MUC1 |
CD3ζ and CD28 |
Breast cancer |
[62] |
HER2 IL13Rα2 |
CD3ζ and CD28 |
Glioblastoma |
[47] |
IL13Rα2 |
CD3ζ; CD3ζ and 41BB CD3ζ and CD28 CD3ζ, CD28 and 41BB CD3ζ, CD28 and OX40- |
Glioma |
NCT02208362 [45] [46] |
Mesothelin |
CD3ζ; CD3ζ and CD28 CD3ζand 41BB CD3ζ and ICOS KIR2DS2 and DAP12- |
Mesothelioma; Pancreatic cancer; Non-small cell lung cancer |
NCT01355965 [49] NCT02465983 [50] [106] [114] |
Mesothelin CD19 |
CD3ζand 41BB |
Pancreatic cancer |
NCT02465983[50] |
MUC1 |
CD3ζ and 41BB |
MUC1 positive solid tumor |
NCT02587689 [54] |
NKG2D |
CD3ζ; CD3ζ and DAP10 CD3ζ and 41BB CD3ζ and CD28 |
Ovarian cancer Ewing sarcoma |
|
PSMA |
CD3ζ and CD28 |
Prostate cancer |
NCT01140373 [64] NCT00664196 [65] |
PD1 and CD19; PD1 and Mesothelin; |
CD3ζ and CD28 CD3ζ, CD28 and 41BB |
PD-L1 positive cells |
[87] |
DAP12, DNAX-activating protein of 12 kDa; DAP10, DNAX-activating protein of 10 kDa; EGFRVIII, epidermal growth factor receptor variant III; FAP, fibroblast activation protein; FR, folate receptor; FL, follicular lymphoma; HER2, human epidermal growth factor receptor 2; IL13Rα2, interleukin 13 receptor α2; KIR2DS2, stimulatory killer immunoglobulin-like receptor 2DS2; MUC1, mucin 1; NKG2D, Natural Killer Group 2D; PSMA, prostatespecific membrane antigen; PD-1, programmed death 1; PD-L1, programmed death ligand 1.
Epidermal growth factor receptor (EGFR) and EGFR variant (EGFRvIII) are over-expressed in many cancer types and are commonly associated with the malignancy of glioblastoma [35, 36]. The expression of EGFRvIII within a cell is often associated with survival, invasion, angiogenesis and resistance against radiation and chemotherapy [37, 38]. EGFRvIII specific CAR-T cells have shown great antitumor efficacy in preclinical studies and now they have been being evaluated in clinical trials [39, 40]. EGFR also could be modified as a useful tool, which retains a cetuximab binding site and lacks domains I and II and its cytoplasmic tail. Cetuximab can recognize the truncated EGFR (huEGFRt) so that CAR-T cells expressing the truncated EGFR can be selected, tracked and ablated in vivo after administration of cetuximab [41].
Interleukin 13 receptor α2 (IL13Rα2) is a glioma-associated antigen and also is associated with a reduced survival rate of patients [42, 43]. In a study, after CAR-T cell treatment, regression of tumors along with corresponding increases of cytokines and immune cells was observed [44, 45]. However, the IL13Rα2 specific CARs can also recognize interleukin 13 receptor α1 (IL13Rα1). To solve the problem, IL13Rα2 specific single-chain variable fragment (scFv) 47 is used as an antigen binding domain and the specificity indeed has been enhanced [46]. Specificity could also be improved by a CAR targeting two or more antigens. IL13Rα2 and human epidermal growth factor receptor-2 (HER2) specific CARs are designed with CD3z and CD28 domains to make tandem CARs (TanCARs) [47]. These CAR-T cells can distinctively and effectively recognize tumors, mitigate antigen escape and have also shown enhanced persistence in the presence of the both targets.
Mesothelin is a TAA expressed by many malignant cancers [48]. CARs specific for mesothelin have been investigated in clinical trials to treat patients with pancreatic cancer and malignant pleural mesothelioma [49, 50]. In the terms of persistence, patients with pancreatic cancer have been treated with T cells that simultaneously express two CARs targeting mesothelin and CD19 in clinical trials [50]. Thus, the influence of antibodies on the mesothelin directed CARs can be blocked due to the eradication of B cells by the CD19-specific CARs and then these CAR-T cells can function for a long time.
Aberrant expression of glycoform has been found on the cell membrane mucin-1 (MUC1), a large protein carrying O-glycan over-expressed by most adenocarcinomas [51, 52]. CARs targeting the MUC1 glycopeptide epitope are designed based on a monoclonal antibody (5E5) and these CAR-T cells have shown ability to eliminate pancreatic tumors [53–55]. Interleukin-4 (IL-4) has several pathophysiologic and therapeutic links to cancers and can promote the function of CAR-T cells. MUC1 specific CAR-T cells engineered with IL-4 receptor ectodomain have shown enhanced resistance to immunosuppressive cytokines and improved anti-tumor efficacy [56, 57].
HER2 is a member of receptor tyrosine-protein kinase family, which is over-expressed by many tumor cells and also is expressed by some epithelial cells [58, 59]. In clinical trials, patients with tumors expressing HER2 have been treated with second generation HER2 targeted CARs (CD28/CD3z) [60, 61]. Several research groups are attempting to design two CARs in a single T cell, which can specially recognize tumor cells. In a trial, HER2 and MUC1 specific CARs with CD3z and co-stimulatory molecule respectively within one T cell have been designed, which can eliminate tumor cells efficiently and offset tumor antigen escape variants when encountering target cells co-expressing MUC1 and HER2 [62].
Most prostate-cancer cells and many tumor-associated neo-vasculatures express prostate specific membrane antigen (PSMA) [63, 64]. Thus anti-angiogenic effects together with direct anti-tumor effects might be able to get by PSMA specific CARs [65]. To improve the specificity of CAR-T cells, PSMA expressed by normal tissues is targeted to provide negative signaling to the PSMA specific dual targets CAR-T cells with the co-stimulatory molecule of programmed death-1 (PD1) or cytotoxic T lymphocyte associated antigen 4 (CTLA4) [66, 67]. The strategy of inhibitory chimeric antigen receptors (iCARs) above can be used not only to enhance antigen recognition, but also to increase safety.
Neural cell adhesion molecule L1, also named CD171, is expressed on many tumors, but also on normal tissues [68]. However, the expression pattern of CD171 by cancers is glycosylated, different from that of normal cells. CARs have been developed to target the glycosylated CD171 expressed on malignant cells and these T cells have been demonstrated safety without on-target off-tumor toxicity [69]. Moreover, clinical trials using CD171 specific CARs are also in progress [70]. In addition to targeting tumor antigens, receptors or ligands can also be targeted to enhance specificity of CARs, such as the use of Natural Killer Group 2D (NKG2D) [71, 72].
Tumor microenvironment
Infiltration and trafficking
T cells must be able to traffic to tumor sites in order to exert their effector functions in vivo. Extracellular matrix (ECM) is the main barrier of transport, which contains the heparan sulfate proteoglycans (HSPGs), the main component of ECM [73]. T cells must get rid of obstacle of HSPGs in stroma-rich tumor microenvironment to reach tumor sites. However, T cells have no ability to express the enzyme heparanase (HPSE) to degrade heparan sulfate proteoglycans. Therefore, CAR-T cells that can secrete heparanase are engineered, which can promote infiltration and anti-tumor activity [74]. Chemokine receptors can also be used to enhance traffic. For example, CD30-directed CAR-T cells engineered with CC-chemokine receptor 4 (CCR4) have enhanced migratory capacity in murine Hodgkin’s lymphoma xenograft models [75]. CAR-T cells expressing CC chemokine receptor 2b (CCR2b) also have improved migration in mesothelioma and neuroblastoma that naturally secrete large quantities of CC chemokine ligand 2 (CCL2) [76, 77].
Target tumor vasculatures
Abundant blood vessels in tumor tissues can express immunosuppressive molecules and promote the growth of tumors. Thus, targeting tumor vasculature is a strategy to improve cell immunotherapy with CARs. Studies have confirmed that poor prognosis and metastasis of tumors is due to the over-expression of vascular endothelial growth factor (VEGF) and their receptors (VEGFR) in the tumor microenvironment [78]. Now vascular endothelial growth factor receptor-2 (VEGFR2) has been targeted to treat patients with metastatic tumors and enhanced efficacy has been achieved by these T cells [79]. Anti-angiogenic therapy also can lead to increased expression of adhesion molecules and chemokines that can enhance infiltration [80].
Target immunosuppressive cells and factors
At present, the main targets of CAR-T cell therapy for tumors are PSMA, Mesothelin, HER2, EGFR, and so on. To date, it lacks methods to overcome the inhibitory effect of tumor microenvironment on CAR-T cells. Therefore, novel CARs need to be developed to improve inhibition of tumor microenvironment and enhance anti-tumor abilities. It has been pointed that PD1 or CTLA4 can inhibit the function of T cells in the tumor microenvironment [81, 82]. However, checkpoint inhibiting antibodies can block the inhibitory signal to the T cells and have achieved successful results for the treatment of different tumor types [83–86]. Now, CARs that can secrete PD1 and/or CTLA4 antibodies have been designed to improve immunosuppression and enhance anti-tumor effect in clinical trials and it has been demonstrated that tumor volume can be decreased by PD1 specific CARs [87–89]. Our research group is designing CTLA4-specific CAR-T cells to study the effects of improving immunosuppressive microenvironment and enhancing anti-tumor cytotoxicity. Adenosine, as a potent immunosuppressive factor, is regarded as a potential target [90]. It is reported that adenosine A2A receptors (A2ARs) can be up-regulated by CAR-T cells to exert a negative immune reaction when combining adenosine. Blockade of A2ARs has achieved great responses significantly, particularly in the help of PD-1 blockade [91]. Thus, new CARs may be designed to secrete adenosine antagonists or adenosine antagonists and PD1 antibodies to enhance anti-tumor efficacy.
SPATIAL DISTANCE AND SPACER REGION
Some studies have reported that the distance between antigen recognition domains and the specific antigen targets can affect the function of CARs. It has been demonstrated that the epitope near a more-proximal position on the membrane can activate CAR-T cells more efficiently [92–94]. In a trial, CARs can greatly recognize and attack tumor cells in vitro through targeting an epitope in a distal position on the membrane with a shortened extracellular spacer region compared with a longer one [46, 95]. It is a question whether the CAR-T cells without a hinge domain can enhance tumor killing. Therefore, two kinds of CARs, with or without a hinge domain, have been designed to study the problem. Finally, it is concluded that a hinge can enhance the expansion and anti-tumor efficacy for some specific CAR-T cells [96, 97]. Thus, the antigen target location and hinge length should be taken into account when designing CARs, which are critical for the activity of CAR-T cells.
INTRACELLULAR SIGNALLING DOMAINS
Many co-stimulatory molecules have been investigated, including CD28, 4-1BB (CD137), CD27 and OX40 (CD134), which have been incorporated into CARs to further enhance therapeutic effect [98, 99] (Table 2). With the development of co-stimulatory molecules, CAR-T cells have experienced four generations of development (Figure 1). The first generation only utilizes CD3z chain typically to provide an activation signal. Early studies show that the persistence of the first generation CARs is superior, but the expansion ability and anti-tumor efficacy are unsatisfactory [99]. Subsequently, a co-stimulatory molecule is added into the structures of CARs to augment the proliferation and responses, which is so-called second generation CARs [100]. It has been pointed that CD28 can enhance the telomere length, which can affect the persistence and anti-tumor efficacy of T cells [101, 102]. Thus, senescent T cells can be regenerated by restoration of CD28 expression [103]. CAR-T cells with CD28 or 4-1BB signaling domain have shown potent anti-tumor efficacy in vivo for B cell malignancies [104, 105]. ICOS co-stimulatory domain also has been used and CARs with ICOS tend to have enhanced survival time than CARs with CD28 or 4-1BB co-stimulatory domain [106, 107]. In order to further strengthen the function of the second generation CARs, the third generation has been designed that has two co-stimulatory molecules [108]. CAR-T cells with CD28 and 4-1BB domains have shown enhanced functionality and increased persistence [109–112]. In addition to these co-stimulatory molecules mentioned above, some other molecules are also being studied, such as CTLA-4 or PD-1. Antigen specific suppression of CAR-T cells with CTLA-4 or PD-1 can be achieved to prevent the damage of inadequate T cell specificity to normal tissues [67, 113]. Stimulatory killer immunoglobulin-like receptor (KIR) KIR2DS2 and DNAX-activating protein of 12 kDa (DAP12) also are used to replace CD3z and co-stimulatory molecule to enhance the proliferation and function of CAR-T cells, which can destroy immunotherapy-resistant solid tumors efficiently [114]. Different from the first three generation CARs, cytokine genes that can improve the activation and expansion of T cells and promote the resistance to immunosuppression have been introduced into the fourth generation CARs (TRUCKs) and these CAR-T cells modified with cytokine genes can use some valid components of the tumor microenvironment to amplify anti-tumor efficacy [115–117].
SAFETY
Although remarkable clinical efficacy, it is still difficult to apply the current CAR-T therapy generally due to the restriction of serious treatment-related toxicities [118]. Bi-specific chimeric antigen receptors and dual CARs strategies mentioned above can help to reduce the risk of development of side effects. Besides, dual receptors in one T cell also can prevent the development of side effects and increase specificity. For instance, a CAR and a chimeric co-stimulatory receptor (CCR) or a CAR and a synthetic Notch receptor are designed in one T cell to target two different antigens. These T cells only eliminate tumors with both antigens but do not destroy cells with either antigen alone [119, 120]. Multi-chain CARs based on FcεRI receptor scaffold also have been investigated to increase safety. FcεRI receptor scaffold has three different polypeptide chains (alpha, beta and gamma) and these polypeptide chains are substituted by an antigen recognition domain, a co-stimulatory molecule and CD3z respectively. Between the antigen binding domain and a hinge domain, FKBP domains and/or FRB are incorporated, which have a high affinity to the rapamycin and FKBP-rapamycin complex respectively. With the application of a small molecule of rapamycin or analog of rapamycin, antitumor cytotoxicity and advantage for safety are shown by these designed CAR-T cells [121]. A split-receptor design has been used to engineer CAR-T cells, which have antigen binding and intracellular signaling domains on separate polypeptides. These T cells can be activated only on the presence of the heterodimerizing small molecule and tumor antigens. Moreover, the activity of these T cells is titratable by the dose of the small molecules, which increase the safety of CAR-T cells application [122] (Figure 2).
It is highly desirable to design universal CARs that have the ability to recognize multiple TAAs and minimize the risk of treatment-related toxicities. A study reported a novel and universal CAR strategy that can extend the specificity and safety potential of CAR-T cells by using a biotin-avidin system [123, 124]. EGFRvIII+ gliomas were targeted by biotinylated monoclonal antibody (biotin-4G1) and then avidin-CARs were used against the biotin-4G1. This therapeutic strategy is proved valid by EGFRvIII+ glioma-bearing mice [125]. Other analogous “switch” molecules also have been explored to regulate CAR-T cells activity in vivo to minimize toxicities, while maintaining potent anti-tumor activity, such as the switch molecules modified with fluoresceine isothiocyanate (FITC) or peptide neo-epitopes (PNE). FITC or PNE -specific CAR-T cells kill tumors dependent on the presence of switch molecule, which can enhance the activity of the CAR-T cells by dose titration [126–129]. To reduce the risk of side effects and broaden the range of application of CAR-T cells, a modular CAR platform (UniCAR) was developed to target different tumor antigens through different specific targeting modules (TMs) that have incorporated a peptide epitope E5B9. The function of E5B9 specific CAR-T cells is completely dependent on the presence of specific TMs and specific targets. Moreover, the activity of these cells can be turned on and off by the TMs [130–132]. However, it is a problem whether these synthetic small molecules are absolutely safe after long-term application. We need to think about the problems that whether similar elements to these molecules will be produced in the body and whether the body will be resistance to these molecules.
The development of gene editing technology has also helped to improve safety. The activity of CAR-T cells can be eliminated by activating the suicide gene caspase-9 (iCasp9) that can effectively induce apoptosis of CAR-T cells to overcome side effects [133]. Another elimination gene is the truncated EGFR mentioned above. The activity of CAR-T cells can be rapidly eliminated with administration of cetuximab to prevent the events of serious toxicities [41]. However, suicide gene strategies can result in terminating therapeutic responses because of eliminating T cells indiscriminately. Moreover, gene editing also is capable of producing CAR-T cells that have ability to avoid graft versus host disease (GvHD) induced by allogeneic CAR-T cells through eliminating the expression of the endogenous T cell receptor (TCR) to enhance safety [134, 135].
CONCLUSIONS
In recent years, CAR-T cell immunotherapy has achieved highly effective results in treating hematological malignancies and achieved much progress on the aspects of antigen targets, intracellular signal domains and the combined application of immune cells and synthetic small molecule drugs. Despite significant progress, some major challenges still have not been solved in engineered T cells to treat solid tumors and have remain significant barriers to its broader clinical application, especially in terms of specificity, persistence, safety, and immunosuppressive microenvironment [136]. We expect the reliable, safe, and effective CAR-T cells and extend it toward the treatment of a broad range of tumors in the future.
ACKNOWLEDGMENTS AND FUNDING
This study is supported by Jiangsu Planned Projects for Postdoctoral Research Funds, No. 1501060A; Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents.
CONFLICTS OF INTEREST
The authors have no competing interests to declare.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66:115–32. https://doi.org/10.3322/caac.21338.
3. Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001; 2:533–43. https://doi.org/10.1016/S1470-2045(01)00486-7.
4. Cheadle EJ, Sheard V, Hombach AA, Chmielewski M, Riet T, Berrevoets C, Schooten E, Lamers C, Abken H, Debets R, Gilham DE. Chimeric antigen receptors for T-cell based therapy. Methods Mol Biol. 2012; 907:645–66. https://doi.org/10.1007/978-1-61779-974-7_36.
5. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013; 3:388–98. https://doi.org/10.1158/2159-8290.CD-12-0548.
6. Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009; 183:5563–74. https://doi.org/10.4049/jimmunol.0900447.
7. Gonzalez S, Naranjo A, Serrano LM, Chang WC, Wright CL, Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J Gene Med. 2004; 6:704–11. https://doi.org/10.1002/jgm.489.
8. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011; 118:6050–6. https://doi.org/10.1182/blood-2011-05-354449.
9. Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer J. 2014; 20:151–5. https://doi.org/10.1097/PPO.0000000000000032.
10. Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015; 27:466–74. https://doi.org/10.1097/CCO.0000000000000232.
11. Kalos M. Tumor antigen-specific T cells and cancer immunotherapy: current issues and future prospects. Vaccine. 2003; 21:781–6.
12. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009; 21:215–23. https://doi.org/10.1016/j.coi.2009.02.009.
13. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA, Rosenberg SA. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012; 23:1043–53. https://doi.org/10.1089/hum.2012.041.
14. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, Wang LL, Han WD. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015; 23:184–91. https://doi.org/10.1038/mt.2014.164.
15. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371:1507–17. https://doi.org/10.1056/NEJMoa1407222.
16. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016; 126:2123–38. https://doi.org/10.1172/JCI85309.
17. US National Library of Science. Precursor B Cell Acute Lymphoblastic Leukemia (B-ALL) Treated With Autologous T Cells Genetically Targeted to the B Cell Specific Antigen CD19. 2010. https://clinicaltrials.gov/ct2/show/NCT01044069.
18. US National Library of Science. Phase I/IIA Study of CART19 Cells for Patients With Chemotherapy Resistant or Refractory CD19+ Leukemia and Lymphoma (Pedi CART19). 2012. https://clinicaltrials.gov/ct2/show/NCT01626495.
19. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013; 368:1509–18. https://doi.org/10.1056/NEJMoa1215134.
20. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015; 385:517–28. https://doi.org/10.1016/S0140-6736(14)61403-3.
21. Yu H, Sotillo E, Harrington C, Wertheim G, Paessler M, Maude SL, Rheingold SR, Grupp SA, Thomas-Tikhonenko A, Pillai V. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am J Hematol. 2017; 92:E11-E3. https://doi.org/10.1002/ajh.24594.
22. US National Library of Science. Anti-CD22 Chimeric Receptor T Cells in Pediatric and Young Adults With Recurrent or Refractory CD22-expressing B Cell Malignancies. 2014. https://clinicaltrials.gov/ct2/show/NCT02315612.
23. US National Library of Science. Genetically Engineered Lymphocyte Therapy in Treating Patients With Lymphoma That is Resistant or Refractory to Chemotherapy. 2012. https://clinicaltrials.gov/ct2/show/NCT01735604.
24. US National Library of Science. Treatment of Chemotherapy Refractory Multiple Myeloma by CART-138 (CART-138). 2013. https://clinicaltrials.gov/ct2/show/NCT01886976.
25. US National Library of Science. Treatment of Relapsed and/or Chemotherapy Refractory CD33 Positive Acute Myeloid Leukemia by CART-33 (CART33). 2013. https://clinicaltrials.gov/ct2/show/NCT01864902.
26. US National Library of Science. Genetically Modified T-cell Immunotherapy in Treating Patients With Relapsed/Refractory Acute Myeloid Leukemia and Persistent/Recurrent Blastic Plasmacytoid Dendritic Cell Neoplasm. 2014. https://clinicaltrials.gov/ct2/show/NCT02159495.
27. US National Library of Science.Autologous ROR1R-CAR-T Cells for Chronic Lymphocytic Leukemia (CLL). 2014. https://clinicaltrials.gov/ct2/show/NCT02194374.
28. US National Library of Science. Kappa-CD28 T Lymphocytes, Chronic Lymphocytic Leukemia, B-cell Lymphoma or Multiple Myeloma, CHARKALL (CHARKALL). 2009. https://clinicaltrials.gov/ct2/show/NCT00881920.
29. US National Library of Science. Study of T Cells Targeting B-Cell Maturation Antigen for Previously Treated Multiple Myeloma. 2014. https://clinicaltrials.gov/ct2/show/NCT02215967.
30. US National Library of Science. Safety Study of Anti LewisY Chimeric Antigen Receptor in Myeloma, Acute Myeloid Leukemia or Myelodysplastic Syndrome. 2012. https://clinicaltrials.gov/ct2/show/NCT01716364.
31. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol. 2016; 13:370–83. https://doi.org/10.1038/nrclinonc.2016.36.
32. US National Library of Science. Treatment of Relapsed and/or Chemotherapy Refractory B-cell Malignancy by Tandem CAR T Cells Targeting CD19 and CD20. 2017. https://clinicaltrials.gov/ct2/show/NCT03097770.
33. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016; 4:498–508. https://doi.org/10.1158/2326-6066.CIR-15-0231.
34. Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, Tam W, Teruya-Feldstein J, de Stanchina E, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016; 167:405–18 e13. https://doi.org/10.1016/j.cell.2016.08.032.
35. Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist. 2002; 7:31–9.
36. Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000; 60:3081–7.
37. Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res. 2004; 10:3216–24.
38. Liu X, Liu K, Qin J, Hao L, Li X, Liu Y, Zhang X, Liu X, Li P, Han S, Mao Z, Shen L. C/EBPbeta promotes angiogenesis through secretion of IL-6, which is inhibited by genistein, in EGFRvIII-positive glioblastoma. Int J Cancer. 2015; 136:2524–34. https://doi.org/10.1002/ijc.29319.
39. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015; 7:275ra22. https://doi.org/10.1126/scitranslmed.aaa4963.
40. US National Library of Science. Autologous T Cells Redirected to EGFRVIII-With a Chimeric Antigen Receptor in Patients With EGFRVIII+ Glioblastoma. 2014. https://clinicaltrials.gov/ct2/show/NCT02209376.
41. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011; 118:1255–63. https://doi.org/10.1182/blood-2011-02-337360.
42. Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, Barish ME, Forman SJ, Jensen MC. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012; 18:2199–209. https://doi.org/10.1158/1078-0432.CCR-11-1669.
43. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004; 64:9160–6. https://doi.org/10.1158/0008-5472.CAN-04-0454.
44. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016; 375:2561–9. https://doi.org/10.1056/NEJMoa1610497.
45. US National Library of Science. Genetically Modified T-cells in Treating Patients With Recurrent or Refractory Malignant Glioma. 2014. https://clinicaltrials.gov/ct2/show/NCT02208362.
46. Krenciute G, Krebs S, Torres D, Wu MF, Liu H, Dotti G, Li XN, Lesniak MS, Balyasnikova IV, Gottschalk S. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Mol Ther. 2016; 24:354–63. https://doi.org/10.1038/mt.2015.199.
47. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK, Brawley VS, Byrd TT, Krebs S, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. 2016; 126:3036–52. https://doi.org/10.1172/JCI83416.
48. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008; 44:46–53. https://doi.org/10.1016/j.ejca.2007.08.028.
49. US National Library of Science. Autologous Redirected RNA Meso-CIR T Cells. 2011. https://clinicaltrials.gov/ct2/show/NCT01355965.
50. US National Library of Science. Pilot Study of Autologous T-cells in Patients With Metastatic Pancreatic Cancer. 2015. https://clinicaltrials.gov/ct2/show/NCT02465983.
51. Finn OJ, Gantt KR, Lepisto AJ, Pejawar-Gaddy S, Xue J, Beatty PL. Importance of MUC1 and spontaneous mouse tumor models for understanding the immunobiology of human adenocarcinomas. Immunol Res. 2011; 50:261–8. https://doi.org/10.1007/s12026-011-8214-1.
52. Tarp MA, Clausen H. Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim Biophys Acta. 2008; 1780:546–63. https://doi.org/10.1016/j.bbagen.2007.09.010.
53. Tarp MA, Sorensen AL, Mandel U, Paulsen H, Burchell J, Taylor-Papadimitriou J, Clausen H. Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology. 2007; 17:197–209. https://doi.org/10.1093/glycob/cwl061.
54. US National Library of Science. Phase I/II Study of Anti-Mucin1 (MUC1) CAR T Cells for Patients With MUC1+ Advanced Refractory Solid Tumor. 2015. https://clinicaltrials.gov/ct2/show/NCT02587689.
55. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, Cogdill AP, Chen TJ, Song D, et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity. 2016; 44:1444–54. https://doi.org/10.1016/j.immuni.2016.05.014.
56. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, Spicer JF, Davies DM, Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010; 285:25538–44. https://doi.org/10.1074/jbc.M110.127951.
57. Ramachandran M, Dimberg A, Essand M. The cancer-immunity cycle as rational design for synthetic cancer drugs: novel DC vaccines and CAR T-cells. Semin Cancer Biol. 2017; 45:23–35. https://doi.org/10.1016/j.semcancer.2017.02.010.
58. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989; 244:707–12.
59. Tong ZJ, Shi NY, Zhang ZJ, Yuan XD, Hong XM. Expression and prognostic value of HER-2/neu in primary breast cancer with sentinel lymph node metastasis. Biosci Rep. 2017; 37:BSR20170121. https://doi.org/10.1042/BSR20170121.
60. US National Library of Science. Her2 Chimeric Antigen Receptor Expressing T Cells in Advanced Sarcoma. 2009. https://clinicaltrials.gov/ct2/show/NCT00902044.
61. US National Library of Science. A Clinical Research of CAR T Cells Targeting HER2 Positive Cancer. 2016. https://clinicaltrials.gov/ct2/show/NCT02713984.
62. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012; 32:1059–70. https://doi.org/10.1007/s10875-012-9689-9.
63. Haffner MC, Kronberger IE, Ross JS, Sheehan CE, Zitt M, Muhlmann G, Ofner D, Zelger B, Ensinger C, Yang XJ, Geley S, Margreiter R, Bander NH. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum Pathol. 2009; 40:1754–61. https://doi.org/10.1016/j.humpath.2009.06.003.
64. US National Library of Science. Adoptive Transfer of Autologous T Cells Targeted to Prostate Specific Membrane Antigen (PSMA) for the Treatment of Castrate Metastatic Prostate Cancer (CMPC). 2010. https://clinicaltrials.gov/ct2/show/NCT01140373.
65. US National Library of Science. Trial of Anti-PSMA Designer T Cells in Advanced Prostate Cancer After Non-Myeloablative Conditioning. 2008. https://clinicaltrials.gov/ct2/show/NCT00664196.
66. Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015; 36:494–502. https://doi.org/10.1016/j.it.2015.06.004.
67. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013; 5:215ra172. https://doi.org/10.1126/scitranslmed.3006597.
68. Huszar M, Moldenhauer G, Gschwend V, Ben-Arie A, Altevogt P, Fogel M. Expression profile analysis in multiple human tumors identifies L1 (CD171) as a molecular marker for differential diagnosis and targeted therapy. Hum Pathol. 2006; 37:1000–8. https://doi.org/10.1016/j.humpath.2006.03.014.
69. Kunkele A, Taraseviciute A, Finn LS, Johnson AJ, Berger C, Finney O, Chang CA, Rolczynski LS, Brown C, Mgebroff S, Berger M, Park JR, Jensen MC. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin Cancer Res. 2017; 23:466–77. https://doi.org/10.1158/1078-0432.CCR-16-0354.
70. US National Library of Science. Engineered Neuroblastoma Cellular Immunotherapy (ENCIT)-01. 2014. https://clinicaltrials.gov/ct2/show/NCT02311621.
71. Demoulin B, Cook WJ, Murad J, Graber DJ, Sentman ML, Lonez C, Gilham DE, Sentman CL, Agaugue S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017; 13:1593–605. https://doi.org/10.2217/fon-2017-0102.
72. Gilham DE, Maher J. ‘Atypical’ CAR T cells: NKG2D and Erb-B as examples of natural receptor/ligands to target recalcitrant solid tumors. Immunotherapy. 2017; 9:723–33. https://doi.org/10.2217/imt-2017-0045.
73. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999; 68:729–77. https://doi.org/10.1146/annurev.biochem.68.1.729.
74. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015; 21:524–9. https://doi.org/10.1038/nm.3833.
75. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009; 113:6392–402. https://doi.org/10.1182/blood-2009-03-209650.
76. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ Jr, Riley JL, June CH, Albelda SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011; 17:4719–30. https://doi.org/10.1158/1078-0432.CCR-11-0351.
77. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010; 33:780–8. https://doi.org/10.1097/CJI.0b013e3181ee6675.
78. Chen JC, Chang YW, Hong CC, Yu YH, Su JL. The role of the VEGF-C/VEGFRs axis in tumor progression and therapy. Int J Mol Sci. 2012; 14:88–107. https://doi.org/10.3390/ijms14010088.
79. US National Library of Science. CAR T Cell Receptor Immunotherapy Targeting VEGFR2 for Patients With Metastatic Cancer. 2010. https://clinicaltrials.gov/ct2/show/NCT01218867.
80. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, Kreuger J, Dimberg A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015; 29:227–38. https://doi.org/10.1096/fj.14-250985.
81. Chen X, Shao Q, Hao S, Zhao Z, Wang Y, Guo X, He Y, Gao W, Mao H. CTLA-4 positive breast cancer cells suppress dendritic cells maturation and function. Oncotarget. 2017; 8:13703–15. https://doi.org/10.18632/oncotarget.14626.
82. Tanvetyanon T, Gray JE, Antonia SJ. PD-1 checkpoint blockade alone or combined PD-1 and CTLA-4 blockade as immunotherapy for lung cancer? Expert Opin Biol Ther. 2017; 17:305–12. https://doi.org/10.1080/14712598.2017.1280454.
83. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–64. https://doi.org/10.1038/nrc3239.
84. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017; 27:38–58. https://doi.org/10.1038/cr.2016.154.
85. Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, Van Allen E, Merghoub T, Wolchok JD, Snyder A, Hammerbacher J. Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer Immunol Res. 2017; 5:84–91. https://doi.org/10.1158/2326-6066.CIR-16-0019.
86. Parra K, Valenzuela P, Lerma N, Gallegos A, Reza LC, Rodriguez G, Emmenegger U, Di Desidero T, Bocci G, Felder MS, Manciu M, Kirken RA, Francia G. Impact of CTLA-4 blockade in conjunction with metronomic chemotherapy on preclinical breast cancer growth. Br J Cancer. 2017; 116:324–34. https://doi.org/10.1038/bjc.2016.429.
87. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016; 76:1578–90. https://doi.org/10.1158/0008-5472.CAN-15-2524.
88. US National Library of Science. PD-1 Antibody Expressing CAR-T Cells for EGFR Family Member Positive Advanced Solid Tumor. 2016. https://clinicaltrials.gov/ct2/show/NCT02873390.
89. US National Library of Science. CTLA-4 and PD-1 Antibodies Expressing MUC1-CAR-T Cells for MUC1 Positive Advanced Solid Tumor. 2017. https://clinicaltrials.gov/ct2/show/NCT03179007.
90. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004; 22:657–82. https://doi.org/10.1146/annurev.immunol.22.012703.104731.
91. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, Johnstone RW, Trapani JA, Stagg J, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017; 127:929–41. https://doi.org/10.1172/JCI89455.
92. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ, Press OW. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008; 180:7028–38.
93. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012; 119:3940–50. https://doi.org/10.1182/blood-2011-10-387969.
94. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013; 121:1165–74. https://doi.org/10.1182/blood-2012-06-438002.
95. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013; 19:3153–64. https://doi.org/10.1158/1078-0432.CCR-13-0330.
96. Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, Li B, Lin S, Wang S, Wu Q, Liang Q, Li Y, Zhang X, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017; 10:68. https://doi.org/10.1186/s13045-017-0437-8.
97. Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, Andersen JT, Wang MY, Kvalheim G, Gaudernack G, Kyte JA. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015; 22:391–403. https://doi.org/10.1038/gt.2015.4.
98. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012; 119:696–706. https://doi.org/10.1182/blood-2011-03-344275.
99. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011; 121:1822–6. https://doi.org/10.1172/JCI46110.
100. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009; 17:1453–64. https://doi.org/10.1038/mt.2009.83.
101. Barrett DM, Singh N, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy. 2014; 16:619–30. https://doi.org/10.1016/j.jcyt.2013.10.013.
102. Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005; 175:7046–52.
103. Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med. 2003; 198:947–55. https://doi.org/10.1084/jem.20021288.
104. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015; 33:540–9. https://doi.org/10.1200/JCO.2014.56.2025.
105. US National Library of Science. Competitive Transfer of αCD19-TCRz-CD28 and αCD19-TCRz-CD137 CAR-T Cells for B-cell Leukemia/Lymphoma (MatchCART). 2016. https://clinicaltrials.gov/ct2/show/NCT02685670.
106. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD Jr, Scholler J, Scholler N, Bonneau R, June CH. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014; 124:1070–80. https://doi.org/10.1182/blood-2013-10-535245.
107. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, Zhao YY, Zhao LM, Cui J, Gupta P, Wong AJ, Han SY. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. 2013; 6:33. https://doi.org/10.1186/1756-8722-6-33.
108. Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011; 129:2935–44. https://doi.org/10.1002/ijc.25960.
109. Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015; 28:415–28. https://doi.org/10.1016/j.ccell.2015.09.004.
110. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010; 18:413–20. https://doi.org/10.1038/mt.2009.210.
111. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009; 106:3360–5. https://doi.org/10.1073/pnas.0813101106.
112. US National Library of Science. Anti-GPC3 CAR T for Treating Patients With Advanced HCC. 2015. https://clinicaltrials.gov/ct2/show/NCT02395250.
113. Kobold S, Grassmann S, Chaloupka M, Lampert C, Wenk S, Kraus F, Rapp M, Duwell P, Zeng Y, Schmollinger JC, Schnurr M, Endres S, Rothenfusser S. Impact of a New Fusion Receptor on PD-1-Mediated Immunosuppression in Adoptive T Cell Therapy. J Natl Cancer Inst. 2015; 107:djv146. https://doi.org/10.1093/jnci/djv146.
114. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, Newick K, Sun J, Lo A, Baradet T, Feldman MD, Barrett D, Pure E, et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res. 2015; 3:815–26. https://doi.org/10.1158/2326-6066.CIR-15-0054.
115. Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev. 2014; 257:91–106. https://doi.org/10.1111/imr.12126.
116. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015; 15:1145–54. https://doi.org/10.1517/14712598.2015.1046430.
117. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012; 119:4133–41. https://doi.org/10.1182/blood-2011-12-400044.
118. Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016; 44:51–60. https://doi.org/10.1016/j.ctrv.2016.02.001.
119. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013; 31:71–5. https://doi.org/10.1038/nbt.2459.
120. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016; 164:770–9. https://doi.org/10.1016/j.cell.2016.01.011.
121. Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep. 2016; 6:18950. https://doi.org/10.1038/srep18950.
122. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015; 350:aab4077. https://doi.org/10.1126/science.aab4077.
123. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012; 72:1844–52. https://doi.org/10.1158/0008-5472.CAN-11-3890.
124. Urbanska K, Powell DJ. Development of a novel universal immune receptor for antigen targeting: To Infinity and beyond. Oncoimmunology. 2012; 1:777–9. https://doi.org/10.4161/onci.19730.
125. Liu K, Liu X, Peng Z, Sun H, Zhang M, Zhang J, Liu S, Hao L, Lu G, Zheng K, Gong X, Wu D, Wang F, et al. Retargeted human avidin-CAR T cells for adoptive immunotherapy of EGFRvIII expressing gliomas and their evaluation via optical imaging. Oncotarget. 2015; 6:23735–47. https://doi.org/10.18632/oncotarget.4362.
126. Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016; 113:E450–8. https://doi.org/10.1073/pnas.1524193113.
127. Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, Gonzalez J, Schultz PG, Young TS, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015; 137:2832–5. https://doi.org/10.1021/jacs.5b00106.
128. Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS, Mazagova M, Choi SH, Yun HY, Xiao H, Yang P, Luo X, Lim RK, et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew Chem Int Ed Engl. 2016; 55:7520–4. https://doi.org/10.1002/anie.201601902.
129. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, Ma J, Nunez V, Shen J, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016; 113:E459–68. https://doi.org/10.1073/pnas.1524155113.
130. Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016; 6:e458. https://doi.org/10.1038/bcj.2016.61.
131. Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, Ziller-Walter P, Kegler A, Koristka S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017; 8:31368–85. https://doi.org/10.18632/oncotarget.15572.
132. Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gartner S, Schmitz M, Ehninger A, Cartellieri M, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017; 6:e1287246. https://doi.org/10.1080/2162402X.2017.1287246.
133. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014; 5:235. https://doi.org/10.3389/fphar.2014.00235.
134. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, Lebuhotel C, Eyquem J, Cheung GW, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015; 75:3853–64. https://doi.org/10.1158/0008-5472.CAN-14-3321.
135. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, Brown AE, Triggiano MA, Hux JA, Pham CD, Bartsevich VV, Turner CA, Lape J, et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol Ther. 2017; 25:949–61. https://doi.org/10.1016/j.ymthe.2017.02.005.
136. Lipowska-Bhalla G, Gilham DE, Hawkins RE, Rothwell DG. Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer Immunol Immunother. 2012; 61:953–62. https://doi.org/10.1007/s00262-012-1254-0.