
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common malignant brain tumor in adults, with a median survival time of 12–15 months [1]. The current clinical standard of care includes surgical resection, followed by a combination of radiation and chemotherapy [2–4]. Nevertheless, these treatments failed to provide significant benefit for GBM patients.
The ERM (Ezrin/Radixin/Moesin) protein family, including ezrin, radixin, moesin and NF2 (neurofibromatosis type 2), links the plasma membrane with actin filaments. The activation of ERM proteins is the concomitant carboxy-terminal threonine phosphorylation after the recruitment to the plasma membrane (ezrin-567, radixin-564, moesin-558) [5]. The physiological role of moesin is organizing and maintaining cell cortex which is crucial for cell division, morphology, differentiation and regulating downstream signaling pathways [6, 7]. Moesin transcript is found among the list of 884 genes in the cancer genome atlas (TCGA) with worst prognosis signature [8]. Similarly, CD44 is associated with poorer prognosis in a subset of glioblastomas [9, 10]. Importantly, CD44 and moesin expression patterns is closely related to poor prognosis [11]. Additional, in comparison with a normal human astrocyte (NHA) cell line, increased moesin levels and higher CD44 levels were detected in 10 glioblastoma cell lines [12]. Recently, moesin was regard as a glioma progression marker with higher expression in higher grade of glioma in human specimen. Moesin phosphorylation induced by CD44/moesin combination promoted cell migration in glioblastoma and CD44/moesin/β-catenin signaling served as a potential target to inhibit cell proliferation [12]. β-catenin activation played a crucial role in glioblastoma cell differentiation, while CD44, a hyaluronic acid (HA) receptor, was involved in several physiological processes including cell migration, wound healing, leukocyte homing, tumor cell migration and metastasis, and in the meantime referred as a cell surface marker of the cancer stem cells. Therefore, we assume that CD44/moesin may act as a therapeutic target of glioblastoma. It was found that CD44-HA interaction potentially mediated invasion, migration, and resistance to chemotherapy in several malignancies, and HA promoted moesin activation by enhancing the combination of CD44 and moesin. HA-CD44-moesin aggregation induced tumor epithelial-mesenchymal transition and high amounts of HA in gliomas have been associated with poor prognosis in GBM patients [13].
Consumption of phytoestrogen-rich foods was demonstrated to protect against several kinds of cancers, including breast cancer and prostate cancer [14]. Daidzein (daid) was found to inhibit the proliferation of breast cancer cell lines, including T-47D, MCF-7 and MDA-MB-231 cells with low toxicity [15, 16]. Subtoxic doses of daid combined with TRAIL induced rapid apoptosis in glioma cells [17]. Consumption of foods rich in antioxidants and certain phytoestrogens, especially daid, suggested a protective role against gliomagenesis [18]. In this study, we demonstrated that daid002 (Figure 1), a novel daid derivative, inhibited the proliferation of GBM in vivo and vitro through suppressing the phosphorylation of moesin via disturbing the combination of CD44 and moesin, which resulted in decreased β-catenin nuclear translocation-mediated G0/G1 cell cycle arrest. These results showed that daid002 could be an effective candidate for glioblastoma therapy and biological research via regulating CD44/moesin pathway. Moreover, moesin would be a potential target for GBM therapeutics.
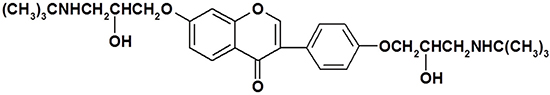
Figure 1: The chemical structure of daid002.
RESULTS
Daid002 inhibited the glioblastoma cell proliferation
To explore the inhibitory effects of daid002 (Figure 1) on cancer cell proliferation in general, we examined the effects of daid002 on lung carcinoma cells (A549), colon cancer cells (HCT116), hepatoma carcinoma cells (HepG-2), gastric carcinoma cells (MKN-28), and glioblastoma cells (U87MG) (Figure 2A). Daid002 substantially inhibited U87MG cell proliferation (Figure 2A). Therefore, the anti-proliferation effect of daid002 (0, 5, 10, 20, 40, 80 μM) was examined on four GBM cell lines (U87MG, U251, U-118MG and A-172) using a 24 h methyl thiazolyl tetrazolium (MTT) assay. Daid002 inhibited proliferation of all GBM cells in a dose-dependent manner (Figure 2B–2E). The half maximal inhibitory concentration (IC50) of daid002 for U87MG, U251, U-118MG, and A-172 were 6.308 ± 2.001, 5.613 ± 2.098, 16.66 ± 5.892 and 8.319 ± 1.057 μM, respectively (Table 1). We also used trypan blue assay to look at if daid002 induced cell death at 24 h. Daid002 increased the number of dead cells in a dose-dependent manner (Figure 2F). However, daid, the parent compound of daid002, showed a much weaker inhibitory effect on those four GBM cell lines (Supplementary Figure 1), and the IC50 of daid for U87MG, U251, U-118MG, and A-172 were 615.60 ± 56.95, 716.27 ± 144.25, 603.33 ± 100.01 and 810.10 ± 130.95 μM (Supplementary Table 1).
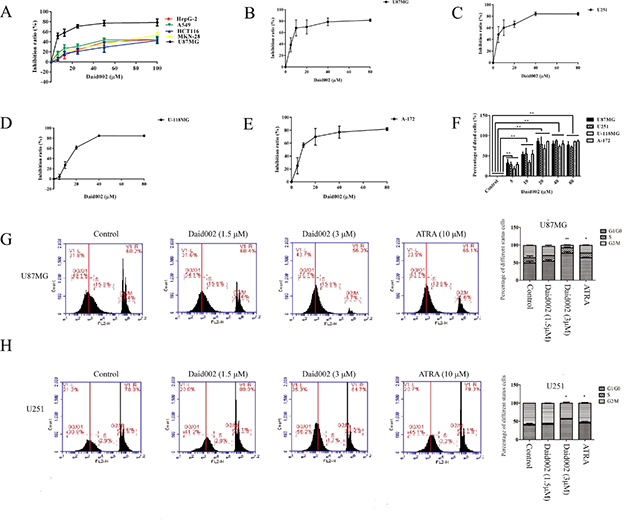
Figure 2: The effect of daid002 on cancer cell proliferation and cell cycle. (A) U87MG, A549, HepG-2, HCT116 and MKN-28 Cells were treated with different concentrations of daid002 for 24 h (0, 5, 10, 20, 40, 80 μM). Cell viability was determined by the MTT assay. GBM cells were treated with different concentrations of daid002 for 24 h (0, 5, 10, 20, 40, 80 μM). Cell viability was determined by the MTT assay. ((B): U87MG, (C): U251, (D): U-118MG, (E): A-172). Data was expressed as means ± SD of three independent experiments. (F) GBM cells were treated with different concentrations of daid002 for 24 h (0, 5, 10, 20, 40, 80 μM) in 6 wells plates, then cells were stained with trypan blue and counted. (G and H) Cell cycle was detected by flow cytometry following PI staining. Cells (U87MG/U251) were also treated with different concentrations of daid002 for 24 h. Different percentages of three cell phases (G0/G1, S and G2/M) were shown. All the control cells (U87MG/U251) were set to 100%. Results were presented as means ± SD (n = 3), and *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group.
Table 1: The IC50 of daid002 on GBM cell lines
GBM cell lines |
IC50(μM) |
|---|---|
U251 |
5.613 ± 2.098 |
U87MG |
6.308 ± 2.001 |
U-118MG |
16.66 ± 5.892 |
A-172 |
8.319 ± 1.057 |
Daid002 exhibited the most marked growth inhibitory effect on U87MG and U251 cells. Next, to explore the effect of daid002 on cell cycle arrest, U87MG and U251 cells were treated with daid002 for 24 h, followed by cell cycle analysis. Daid002 (3 μM) significantly increased the G0/G1 population in U87MG and U251 cells, while at lower concentration (1.5 μM) it did not have the same effect (Figure 2G, 2H). All-trans retinoic acid (ATRA), a differentiation therapy agent, was used as a positive control. These data suggest that daid002 might serve as an anti-proliferative agent, with marked effects on the cell cycle in GBM cells.
Daid002 reduced the expression of p-moesin in GBM cell lines
Previous studies indicated that CD44 and moesin expression patterns strong correlated with poor prognosis in GBM. To further determined CD44 and moesin expression were related in high grade GBM samples, we analyzed data in the Cancer Genome Atlas data (TCGA). mRNA expression of CD44 correlated with expression of MSN (i.e., moesin, Pearson: 0.64, Spearman: 0.69) in 580 GBM tissues based on the TCGA database (Cell, 2013, http://www.cbioportal.org/study?id=gbm_tcga_pub2013#summary. Supplementary Figure 2), suggesting that moesin and CD44 were glioma progression markers, with highly correlated expression levels. To study the effect of daid002 on moesin phosphorylation, we treated all GBM cell lines with daid002 for 24 h and measured expression levels of CD44, moesin and p-moesin by western blot. Expression of p-moesin was reduced by daid002 treatment in a dose-dependent manner (Figure 3A–3D). Daid002 had no effect on CD44 and moesin protein levels. After stimulation of daid, protein levels of CD44, moesin and p-moesin remained unchanged with a much weaker anti-proliferation effect on GBM cells. These results suggest that daid002 reduces phosphorylation of moesin in GBM cells.
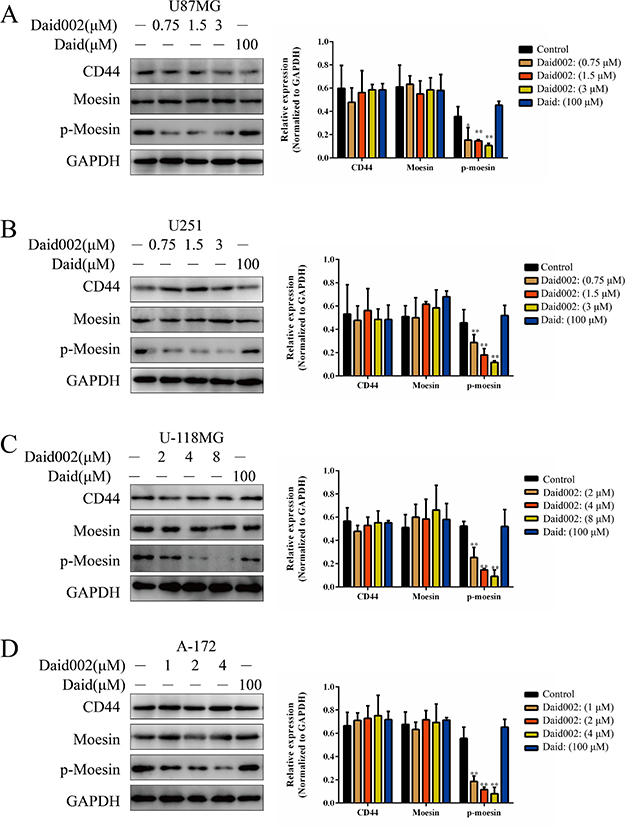
Figure 3: Daid002 reduced the expression of p-moesin in GBM cell lines. (A, B, C and D) All GBM cells were treated with different concentrations of daid002 for 24 h, the expression of p-moesin was reduced in a dose-dependent manner (A: U87MG, B: U251, C: U-118MG, D: A-172). Results were presented as means ± SD (n = 3), and *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group.
Daid002 inhibited proliferation in U87MG and U251 cells via regulating moesin
Overexpression of moesin substantially shortened the survival time in an orthotopic mouse model of glioblastoma [12]. To determine whether defects in proliferation were due to daid002-mediated effects on moesin, we knocked-down (KD) moesin expression in the U87MG and U251 cells (Figure 4A). Depletion of moesin resulted in strong inhibition of proliferation (Figure 4B, 4C). Transfection of moesin-siRNA or stimulation of daid002 led to proliferation inhibition effect at day 3 after transfection or after treatment. Nevertheless, after genetic depletion, cell proliferation recovered with moesin expression at day 5 (Figure 4D, 4E, 4F). To reverse this proliferative effect via moesin expression recuperation, daid002 was used to treat moesin-siRNA transfected cells for 5 days. Proliferation defects were detected in U87MG and U251 cells (Figure 4D, 4E, 4F).
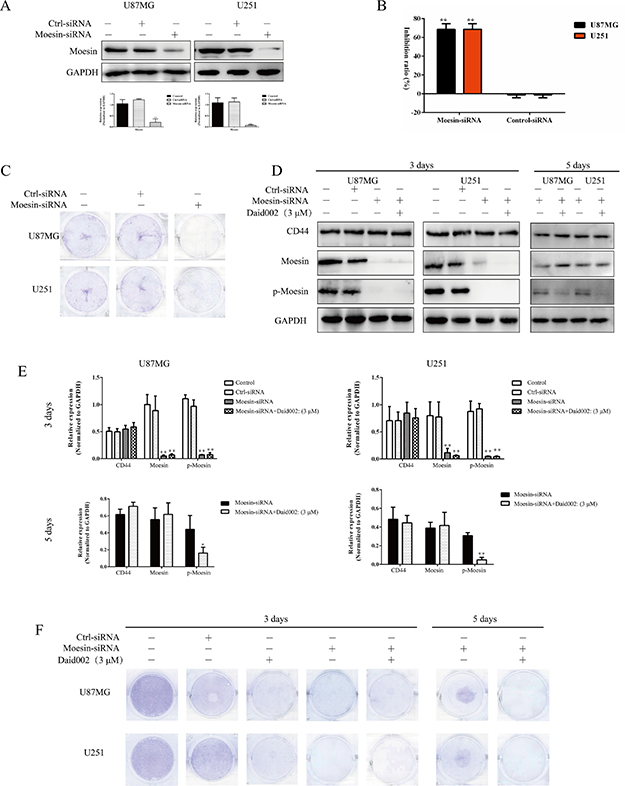
Figure 4: Daid002 inhibited proliferation in U87MG and U251 cells via moesin. (A) U87MG and U251 cells were transfected with moesin-siRNA, expression of moesin was detected. GAPDH as a loading control. (B) U87MG and U251 cells were transfected with moesin-siRNA for 48 h and cell viability was determined by the MTT assay. (C) U87MG and U251 cells were transfected with moesin-siRNA for 48 h, cells were stained with crystal violet. (D and E) U87MG and U251 cells were transfected with moesin-siRNA or treatment with daid002 (3 μM), then daid002 (3 μM) were simulated with moesin KD, CD44, moesin, p-moesin were detected by western blot (D) and quantitated (E). GAPDH as a loading control. (F) U87MG and U251 cells were transfected with moesin-siRNA or treatment with daid002 (3 μM), then daid002 (3 μM) were simulated with moesin KD, cells were stained with crystal violet. Results were presented as means ± SD (n = 3), and *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group.
The interaction between HA and CD44 promoted moesin activation by increasing the combination of CD44 and moesin [7, 19]. Upregulation of p-moesin was detected by western blot (Figure 5A and 5B), and stimulation with HA (100 μg/mL) led to over-proliferation in U87MG and U251 cells, and daid002 reversed this over-proliferation induced by HA (Figure 5C). Immunofluorescence (IF) results showed that level of p-moesin was increased and daid002 reduced the upregulation of p-moesin (Figure 5D). As shown Figure 5F, moesin siRNA significantly reversed HA-induced over-proliferation at day 3 (Figure 5F), and cell proliferation resumed at day 5 (Figure 5F, 5G, 5H, 5I). While long-term treatment with daid002 caused proliferation defects in cells pretreated with HA and moesin KD, (Figure 5F, 5G, 5J). These data suggest that daid002 caused defects in cell proliferation via down-regulation of moesin.
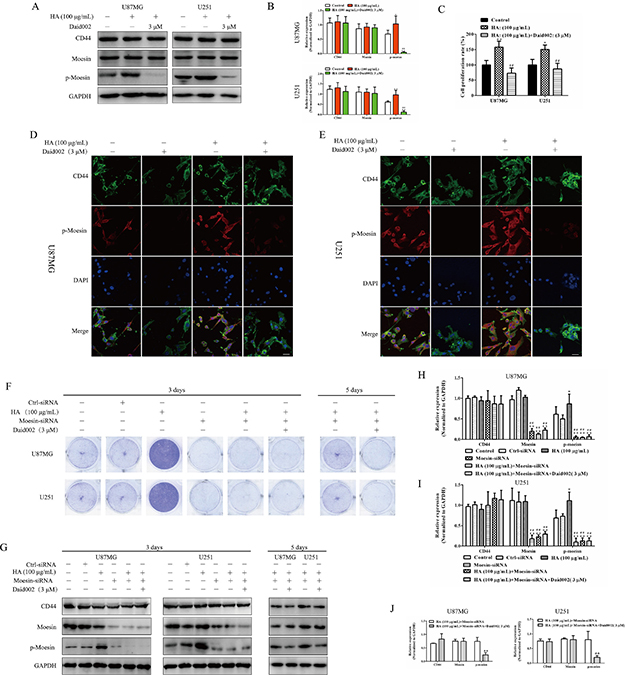
Figure 5: Daid002 reversed the over proliferation induced by HA via inhibiting p-moesin. U87MG and U251 cells were treated with HA and daid002 (3 μM), CD44, moesin, p-moesin were detected by western blot (A) and quantitated (B). (C) U87MG and U251 cells were treated with HA, or with daid002 (3 μM), cell viability was determined by the MTT assay for 24 h. Meanwhile, CD44 and p-moesin expression were determined by immunofluorescence assay (D and E), scale bar: 20 μm). U87MG and U251 cells were pretreat with HA (100 μg/mL), then cells were stimulated with daid002 (3 μM) and knocked-down with moesin-siRNA for 3 days or 5 days, cells were stained with crystal violet (F), and CD44, moesin, p-moesin were detected by western blot. GAPDH as a loading control. (G). (H, I and J) Quantitation of western bolt results in Figure 5G (H and I: 3 days, J: 5 days). Results were presented as means ± SD (n = 3). *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group, #P < 0.05, ##P < 0.01 indicate statistically significant differences versus HA group, &P < 0.05, &&P < 0.01 indicate statistically significant differences versus HA + moesin-siRNA group.
Daid002 disrupted the connection between moesin and CD44
The oncogenic effects of moesin is mediated through its interaction with CD44 [12]. To examine whether the anti-proliferative effect of daid002 was mediated by disrupting the connection between moesin and CD44 in GBM cells, we used an immunoprecipitation (IP) assay. Moesin interacted with CD44 in U87MG and U251 cells (Figure 6A, 6B), consistent with the findings of previous studies.
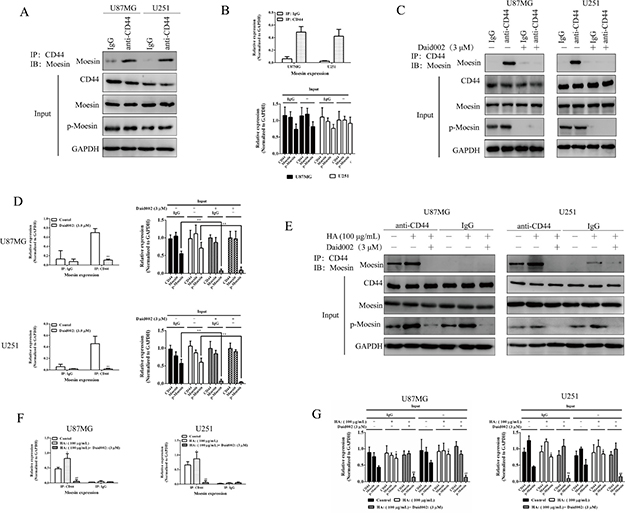
Figure 6: Daid002 disrupted the interaction between CD44 and moesin. The interaction between CD44 and moesin were detected with IP, and western blot was used to determined expression of moesin, CD44, p-moesin (A and B). Cells were simulated with daid002 (3 μM), IP assay was used to determine the interaction of CD44/moesin (C and D). HA (100 μg/mL) and daid002 (3 μM) were treated with cells separately or in combination, IP assay was used to detect the interaction of CD44/moesin (E, F and G). GAPDH as a loading control. Results were presented as means ± SD (n = 3). *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group, #P < 0.05, ##P < 0.01 indicate statistically significant differences versus HA group.
To investigate the mechanism underlying the specific effect of daid002 on moesin/CD44, U87MG and U251 cells were stimulated with daid002. At a concentration of 3 μM, daid002 suppressed the interaction of moesin and CD44 (Figure 6C, 6D). HA enhanced the interaction between CD44 with moesin and this interaction was specifically inhibited by daid002 (Figure 6E, 6F, 6G). Collectively, these data suggest that daid002 specifically disrupts the connection between CD44 and moesin.
Daid002 disturbed the combination of CD44 with the FERM domain
To determine the possibility of combination of daid002 with CD44 or moesin, we incubated daid002 with cell lysates containing various moesin proteins or CD44 protein and protein A+G agarose (beads), after which CD44 or moesin protein was incubated with all beads (Figure 7A). We determined levels of moesin or CD44 by western blot. In U87MG and U251 cells, beads with CD44 antibody were used to bind CD44 proteins, then cell lysates with knocked down CD44 (Supplementary Figure 3A and 3B) were incubated with bead-antibody-CD44 complexes. No differences were found on beads-bond moesin and moesin in supernatants with or without daid002 treatment in U87MG and U251 cells (Figure 7B and 7C). Afterwards, beads with moesin antibody were used to bind moesin proteins, then cell lysates in which knocked-down moesin was incubated with bead-antibody-moesin complexes. It was found that beads-bond CD44 was reduced, while CD44 in supernatants was increased with daid002 treatment (Figure 7D and 7E). These results demonstrated that daid002 bond to moesin instead of CD44 to regulate interaction of CD44/moesin.
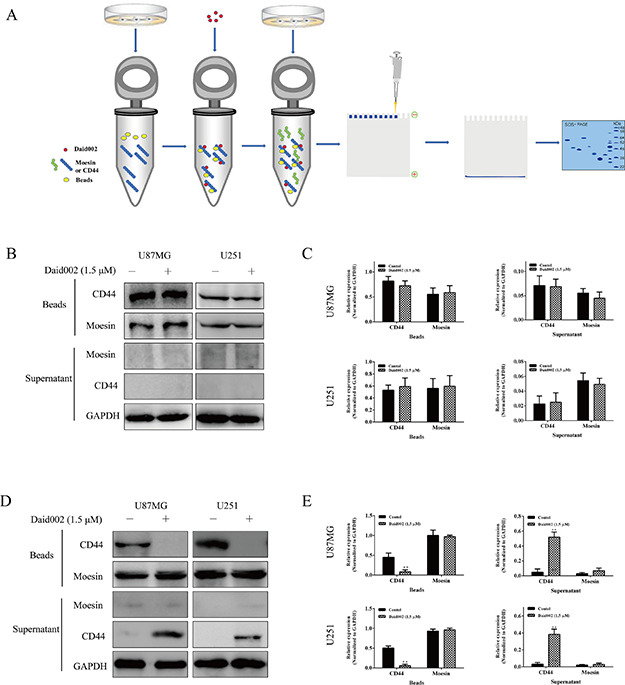
Figure 7: Daid002 combined with moesin but not CD44. Protein of CD44 was incubate with daid002 (1.5 μM) and beads (A), and then moesin protein was incubated, CD44, moesin expression on beads and in supernatant were detected by western blot (B and C). Protein of moesin was incubate with daid002 (1.5 μM) and beads (A), and then CD44 protein was incubated, CD44 or moesin expression on beads and in supernatant were detected by western blot (D and E). GAPDH as a loading control. Results were presented as means ± SD (n = 3). *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group.
Moesin contains three important domains: The N-terminal membrane binding domain (FERM, F for 4.1 protein, E for ezrin, R for radixin, and M for moesin) consisting of three subdomains, designated F1, F2, F3 or A, B, C, which bind membrane and transmembrane proteins via phosphatidylinositol (4,5)-bisphosphate (PIP2); a α-helical linker region; and a C-terminal actin-binding domain containing a phosphorylation site [20–22]. CD44 was reported to bind with the FERM domain and regulated downstream signaling [23]. Hence, we speculated that daid002 targeted the FERM domain to disrupt the interaction of CD44 with moesin. Indeed, computer-simulated co-crystal structures revealed that daid002 occupied the FERM domain (Figure 8A, 8B).
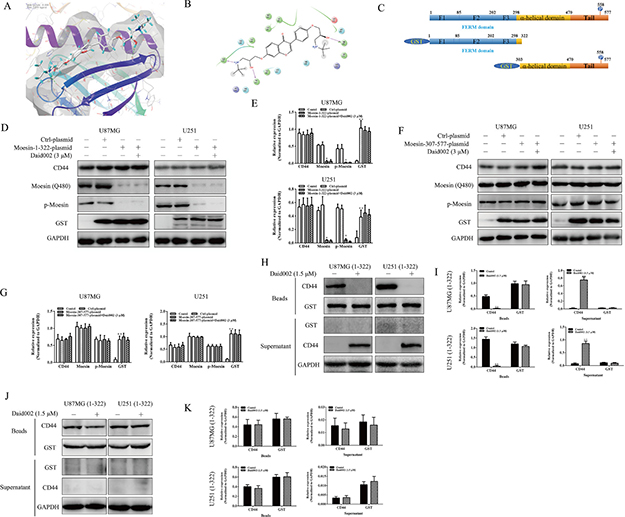
Figure 8: Daid002 disturbed the combination of CD44 with moesin binding sites. Co-crystal structures of daid002 bounded to FERM domain (A and B). The structures of moesin and two truncated moesin proteins (C). U87MG and U251 cells were transfected with moesin-1-322-plasmid and moesin-307-577-plasmid, and then cells were stimulated with daid002 (3 μM), protein levels of CD44, moesin, p-moesin and GST were determined by western blot (D, E, F and G). Protein of GST was incubate with daid002 (1.5 μM) and beads, and then CD44 protein was incubated, CD44, GST expression on beads and in supernatant were detected by western blot (H and I). Protein of CD44 was incubate with daid002 (1.5 μM) and beads, and then GST protein was incubated, CD44 and GST expression on beads and in supernatant were detected by western blot (J and K). GAPDH as a loading control. Results were presented as means ± SD (n = 3). *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group, #P < 0.05, ##P < 0.01 indicate statistically significant differences versus 1-322 cell group, &P < 0.05, &&P < 0.01 indicate statistically significant differences versus 307-577 cell group.
To confirm whether disruption of the interaction between CD44 and moesin was due to the colonization of daid002 with the FERM domain, we used two truncated plasmids (pGEX4T-moesin-1-322 and pGEX4T-moesin-307-577, Figure 8C) to establish stable transfected cell lines named U87MG (1-322), U87MG (307-577), U251 (1-322) and U251 (307-577). The pGEX4T-moesin-1-322 plasmid induced cell to express truncated moesin protein from 1-322 without the C-terminal actin-binding domain with glutathione s-transferase (GST) expression (Figure 8C). pGEX4T-moesin-307-577 plasmid transfected cells expressed truncated moesin protein from 307 to 577 with GST expression, without the FERM domain (Figure 8C). An antibody against moesin (Q480) bound to glutamine (gln, Q) at the 480th site in the tail region of moesin. GST expression was detected in 1-322 cells, but protein levels of p-moesin and moesin (Q480) were downregulated (Figure 8D and 8E). In 307-577 cells, GST, p-moesin and moesin (Q480) were expressed (Figure 8F and 8G). All these cells were simulated with daid002 (3 μM), compared with plasmid transfection only. No differences in p-moesin expression were observed in 1-322s and 307-577s (Figure 8D–8G). Meanwhile, protein level of GST in transfected GBM cells were not altered by daid002 treatment (Figure 8D–8G). These results suggest that daid002 did not directly affect the phosphorylation site of moesin, while it might bind to the FERM domain and affect the interaction of CD44 with moesin.
To further verify whether daid002 targets the FERM domain, we incubated daid002 with cell lysates containing various GST-moesin proteins or CD44 protein and protein A+G agarose (beads), after which CD44 or GST protein was incubated with all beads. We determined levels of GST or CD44 by western blot. In stably transfected cell lines (1-322) cells, we found that CD44 expression with beads was reduced, but was increased in supernatants with daid002 (Figure 8H and 8I). To exclude the possibility of combination with CD44 and daid002 in stably transfected cell lines, CD44 antibody was used instead of GST antibody to repeat the experiments. With daid002, no differences were found on beads and in supernatants (Figure 8J and 8K).
Taken together, these data suggest that daid002 binds to 1-322 protein residues of moesin and blocks CD44 from combining with the FERM domain.
Daid002 induced cell cycle arrest via CD44/moesin/β-catenin
CD44/moesin pathway modulates β-catenin to regulate cell cycle [12]. To determine this potential mechanism, U87MG and U251 cells were stimulated with daid002. Cyclin D1 protein and p-β-catenin expression were significantly decreased as well as nuclear translocation of β-catenin (Figure 9A, 9B). When cells were pretreated with HA, expression of p-β-catenin (S552) and nuclear translocation of β-catenin were increased (Figure 9C, 9D). Daid002 decreased protein levels of p-β-catenin as well as of cyclin D1. A β-catenin TOP/FOP luciferase reporter assay suggested that HA-activated β-catenin signaling and daid002 blocked activation of β-catenin in both HA-pretreated and untreated cells (Figure 9E). These results suggest that daid002 regulates expression of p-β-catenin and cyclin D1 via CD44/moesin.
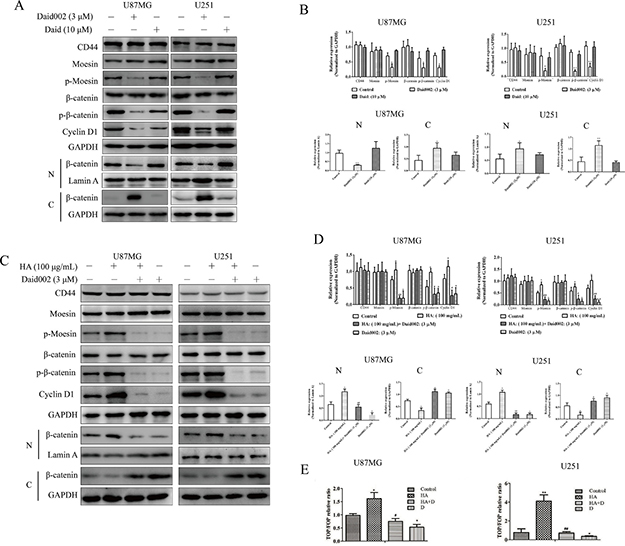
Figure 9: Daid002 induced cell cycle arrest via CD44/moesin/β-catenin. Daid002 and daid were used to simulate cells for 24 h, CD44, moesin, p-moesin, β-catenin, p-β-catenin and expression of β-catenin in nucleus together with cytoplasm were detected by western blot (A and B). HA and daid002 were simulated with cells, CD44, moesin, p-moesin, β-catenin, p-β-catenin and expression of β-catenin in nucleus together with cytoplasm were determined (C and D). β-catenin TOP/FOP luciferase reporter assay with or without HA (100 μg/mL, HA) pretreatment and daid002 (3 μM, D) treatment was conducted. The luciferase activity was normalized to renilla activity, and the luciferase activity of control cells was set to 1 (E). N: nucleus, C: cytoplasm. Results were presented as means ± SD (n = 3). *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group, #P < 0.05, ##P < 0.01 indicate statistically significant differences versus HA group.
Daid002 inhibited the GBM growth in vivo
To access the acute toxicity of daid002 in vivo, daid002 was given intravenously to ICR mice, and the median lethal dose (LD50) was calculated as 5.573 mg/kg (Table 2). Next, U251 tumor-bearing mice were randomly divided into five groups. Each group was treated with various concentrations of daid002, temozolomide, and solvent through caudal veins, once every two days for 20 days. The 0.5 mg/kg and 1 mg/kg daid002 groups exhibited significant inhibition on GBM growth. The inhibitory effect of 1 mg/kg daid002 was greater than that of 20 mg/kg temozolomide (TMZ) (Figure 10A, 10B, 10C). At day 14, the tumor growth was markedly inhibited at 0.5 mg/kg and 1 mg/kg daid002 (Figure 10C). By comparison, body weight increased with 1 mg/kg daid002 treatment. We measured serum levels of aspartate transaminase (AST), alanine transaminase (ALT), and γ-glutamyltransferase (γ-GT) to represent liver function, while glucose, blood urea nitrogen (BUN) and creatinine (Cr) were measured to represent kidney function. No significant differences were found among the five groups (Figure 10D, 10E), suggesting that daid002 and TMZ did not have substantial toxicity on tumor-bearing nude mice. In addition, expression of ki67 and proliferating cell nuclear antigen (PCNA) were decreased with daid002 treatment (Figure 10F). Protein levels of p-moesin, p-β-catenin and nuclear translocation of β-catenin were reduced in daid002-treated groups, and no significant differences were found among the five groups for CD44, moesin and β-catenin expression (Figure 10H, 10G and 10I). The results of immunohistochemical (IHC) analysis showed that nuclear translocation of β-catenin was decreased with daid002 treatment (Figure 10J and 10K). However, TMZ treatment had no effect on β-catenin nuclear translocation (Figure 10J and 10K). Taken together, these data suggest that daid002 inhibited GBM growth via inhibiting p-moesin in vivo.
Table 2: The acute animal experiment of daid002
Dose of daid002 (mg/kg) |
Total animals |
Death of animals |
|---|---|---|
16.00 |
10 |
10 |
12.00 |
10 |
10 |
8.00 |
10 |
8 |
4.00 |
10 |
2 |
2.00 |
10 |
0 |
1.00 |
10 |
0 |
0.50 |
10 |
0 |
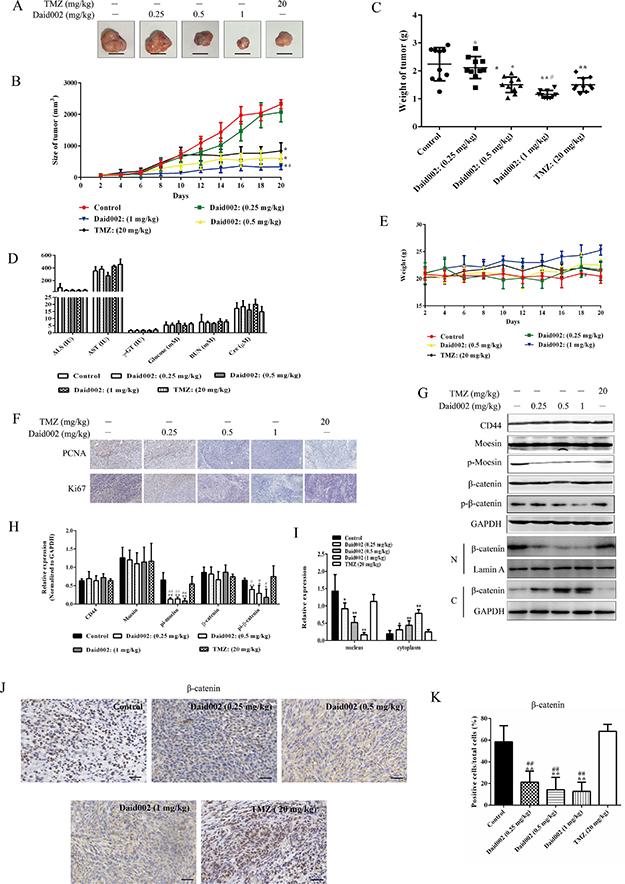
Figure 10: Daid002 inhibited the GBM growth in vivo. Tumor size of mice was measure by vernier caliper for 20 days during the treatment time and tumor were collected at day 21 (A and B, scale bar: 1 cm). Tumor mass was weighed (C). Six serum biochemical levels were measured respectively in different groups (D). Weight changes of mice in five separate groups were detected for 20 days (E). The expression of PCNA and ki67 were measure by immunohistochemistry (F, scale bar: 50 μm). The expression of CD44, moesin, p-moesin, β-catenin, p-β-catenin and nuclear translocation of β-catenin detected by western blot and quantitated (G, H and I). (J) Subcellular localization of beta-catenin was determined by immunohistochemistry on sections of xenografted tumors (Scale bar: 30 μm). (K) The total and positive cells (DAB stained in nucleus) which were stained with DAB in nucleus were count, and the percentage of positive cells in total cells were calculated in all section in five group. Results were presented as means ± SD (n = 10), *P < 0.05, **P < 0.01 indicate statistically significant differences versus control group, #P < 0.05, ##P < 0.01 indicate statistically significant differences versus TMZ group.
DISCUSSION
Glioblastoma has a very poor prognosis. Though some patients could benefit from temozolomide, those with O6-methylguanine-DNA methyltransferase (MGMT) overexpression are poor-responders [24–26]. Nowadays, efforts are focused on new targets for therapy. Here, we showed that daid002 suppressed GBM growth in vivo and in vitro via inhibiting CD44/moesin/β-catenin signaling pathway (Figure 11).
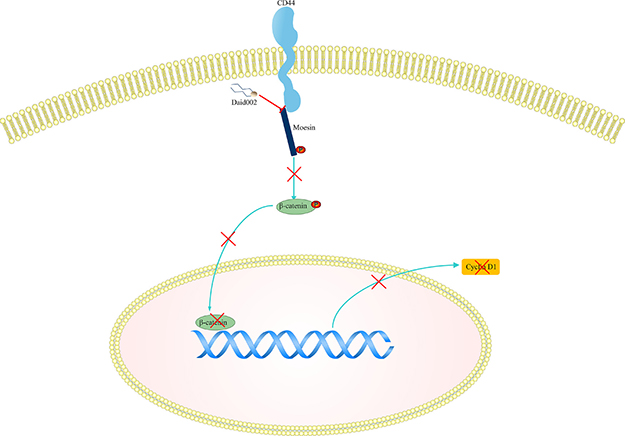
Figure 11: The pathway involved in daid002 treatment. Daid002 disturbed interaction of CD44 with moesin to decrease expression of p-β-catenin and cyclin D1.
It is known that CD44 phosphorylates and actives ERM protein [27, 28]. Previous studies added moesin and CD44 on the map of relevant molecular changes in GBM [12]. Moreover, disturbing the moesin/CD44 pathway was identified as an effective strategy to treat glioblastoma [13, 29]. RNA interference, blocking antibodies and peptides were applied to the treatments [30, 31]. Few studies reported new compounds to treat GBM through targeting the CD44–moesin interaction. Here, we identified a new anti-GBM agent, daid002, as a potential agent for disrupting CD44-moesin. Daid002 significantly inhibited the phosphorylation of moesin in GBM. Both in vivo and vitro study pinpointed that daid002 suppressed the proliferation of GBM cells via disrupting the combination of FERM domain of moesin between CD44.
We demonstrated daid002 induced G0/G1 arrest of GBM cells. This effect mostly dependent on nuclear translocation of β-catenin-mediated cycle proteins cyclin D1. In our study, elevated phosphorylation of moesin increased β-catenin phosphorylation (ser552), and in turn induced β-catenin nuclear translocation, which were consistence with previous studies [12, 32]. Based on the target genes screening in moesin overexpressing cells, besides β-catenin signaling, there are many signaling pathways, including mitogen-activated protein kinase (MAPK) and AKT regulated by moesin, [12]. Those signaling pathways induced by moesin phosphorylation needs to be further studied. Even though ERM proteins play important roles in cell division [34–37]. the specific regulation of moesin on β-catenin-induced cell cycle arrest is worth investigating.
Daid inhibited the proliferation of breast cancer and murine neuroblastoma cells [33]. The structure of daid is similar to 17β-estradiol, which can bind to estrogen receptors (ER) and modulate the cell cycle, survival and apoptosis [34, 35]. GBM expresses both ERα and ERβ [36–38]. ERβ agonists was reported to inhibit the growth of gliomas cells in vivo [36]. Estrogen also increased the survival rate in glioblastoma orthotopic animal model [37]. Recent studies demonstrated that high expression of ERβ was an independent favorable prognostic factor in brain tumors [39, 40]. Thus, whether the effects of daid002 on GBM were dependent on ER should be further studied.
The first inhibitor of radixin/CD44, DX-52-1, a quinocarmycin analog, was shown to disrupt the interaction between radixin and CD44 [41]. In moesin-overexpressing cells, it had a similar effect on cell growth [12]. However, a clinical trial of DX-52-1 failed due to high toxicity [42–44]. In our study, we reported that daid002, sharing similar mechanism with DX-52-1, could be used for the treatment of GBM. Treatment with 0.5 mg/kg daid002 did not exhibit significant toxicity on tumor-bearing nude mice in terms of weight change, liver function, or renal function, suggesting that daid002 has a better safety profile than does DX-52-1.
One of the biggest challenges in the development of therapeutics for GBM is achieving sufficient blood–brain barrier (BBB) penetration. while in our study, we examined the effects of daid002 in transplanted GBM, but did not in situ. Hence, in situ GBM animal models should be adopted to assess the effects of daid002 in the future.
In summary, we demonstrated that daid002 inhibited CD44/moesin pathway and suggested that daid002 could be a potential candidate for the treatment of GBM. Our data also pointed toward the design of new and less-toxic compounds targeting CD44/moesin for cancer therapeutics.
MATERIALS AND METHODS
Chemicals and antibodies
Daid002 and daid was obtained from Organic Chemistry Laboratory of China Pharmaceutical University. Samples containing 98% or higher daid002 were used in all experiments unless otherwise indicated. Daid002 and daid were dissolved in 100% dimethyl sulfoxide (DMSO) at a concentration of 200 mM, and stored at – 20ºC. The working solution was freshly prepared in the basal medium with a final DMSO concentration less than 0.1%. TMZ was obtained from Changzhou new area Jili Chemical, China. For in vivo study, TMZ (98 %) and daid002 were dissolved in PBS (pH = 4.0) for use immediately upon preparation.
DMEM (219-010-LL) was from Wisent Inc. (Nanjing, China). Fetal bovine serum was obtained from Gibco Life Technologies (NY, USA). ATRA, MTT and HA were obtained from Sigma life science (MO, USA). Sources of the antibodies are as follows: anti-β-catenin (ab32572, 1:1000 for western blot, 1:200 for IHC) was from Abcam Ltd (MA, USA). anti-cyclin D1 (BZ00998, 1:1000 for western blot), anti-PCNA (LM1013, 1:100 for IHC), anti-Ki-67 (LM0690, 1:200 for IHC), anti-GST (AP0006, 1:1500 for western blot) was from Bioworld Technology Inc (MN, USA). Anti-CD44 (BBA10, 1:1000 for western blot, 1:300 for IHC, 1:250 for IF) was from R & D systems (MN, USA). GAPDH (KC-5G5, 1:3000 for western blot) and anti-mouse IgG (H+L) (KC-MM-035) were from Kangchen (Shanghai, China). p-β-catenin (Ser552, 9566S, 1:2000 for western blot, 1:300 for IHC) and moesin (Q480, 3150S, 1:2000 for western blot, 1:200 for IHC) were obtained from Cell Signaling Technology (MA, USA). Control siRNA (sc-37007, 50 nM), moesin siRNA (sc-35955, 40 nM), and p-moesin (Ser 558, SC-12895-R, 1:400 for western blot, 1:100 for IHC, 1:150 for IF) were from Santa Cruz Biotechnology (CA, USA). Alexa fluor 546 donkey anti-rabbit lgG (A-11056, 1:300 for IF) and Alexa fluor 488 donkey anti-rabbit lgG (A-11055, 1:200 for IF) were from Life Technologies (NY, USA). Cell cycle PI detection kit (KGA512) was from KeyGen Biotech (Nanjing, China). Millicell EZ slide was obtained from Millipore Corporation (Darmstadt, Germany). Protein A+G Agarose, mouse/rabbit IgG, crystal violet staining solution, radio immunoprecipitation assay lysis buffer (RIPA lysis buffer) and trypan blue solution were obtained from Beyotime (Hanzhou, China).
Cell culture
Lung carcinoma cells (A596), colon cancer cells (HCT116), hepatoma carcinoma cells (HepG2) and gastric carcinoma cells (MKN-28) were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Human GBM U87MG, U251, U-118MG and A-172 cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China), routinely cultured in DMEM containing 10% fetal bovine serum, 1% penicillin and streptomycin, and maintained in a humidified atmosphere of 95% air, 5% CO2 at 37ºC. Cells in passages 2–4 from their arrivals were used for experiments. Cells were plated in 6-well plates, then pretreated with HA (100 μg/ml) and treated with the various concentrations of daid002 and ATRA.
Proliferation assay
GBM cells were seeded into 6-well plates and treated as indicated. For cell viability assay, cells were plated into 96-well plates at approximately 4000/well in 100 μL medium. Daid002 was added to the cells at indicated concentrations and durations. Cell viability was measured using an MTT assay following the manufacturer’s protocol. Absorbance was measured at 570 nm with the Universal Microplate Reader (BioTek Instruments Inc., VT, USA). Cell survival ratio was calculated as treated/control × 100%. The IC50 was taken as the concentration that caused 50% inhibition of cell proliferation and was calculated using the Graphpad Prism 7.01. All assays were performed in triplicate. For crystal violet staining assays, cells were seeded in 12-well plates and stimulated with various treatments. Plates were further cultured in an incubator for an appropriate time and stained with crystal violet following the manufacturer’s protocol. For trypan blue staining assays, cells were seeded in 6-well plates and stimulated with various treatments, cells were collected and stained with trypan blue. Then cells were counted in hematocytometer and analyzed. All assays were performed in triplicate.
Flow cytometry analysis
Cells were starved in serum-free medium for 6 h to be synchronized. After treatment with daid002 and ATRA, cells were harvested and resuspended with 70% ethanol overnight at 4ºC. Cell apoptosis PI detection kit (KeyGen Biotech) was used to dye the DNA for 30 min at room temperature. Finally, cells were analyzed by Accuri C6 Software flow cytometry (Becton Dickinson, CA, USA).
Western blot assay
Proteins were separated by SDS–polyacrylamide gel electrophoresis and electroblotted onto PVDF membranes by standard procedures. After blocking with 5% BSA in PBS for 1.5 h at 37ºC, the membranes were incubated for another 1.5 h with the primary antibody at 4ºC overnight, followed by peroxidase-conjugated secondary antibody for 1 h at 37ºC. Bands were detected with Tanon 5200 Chemiluminescence imaging system (Shanghai, China) and quantified by imageJ.
Knockdown and overexpression assay
U87MG and U251 cells were transfected with siRNA using a transfection reagent (Vazyme Biotech, Nanjing, China). Cells were also transfected with experiment and control plasmid (obtained from Dr. Haiwei Zhang, Chongqing Cancer Hospital) with transfection reagent (Vazyme Biotech, Nanjing, China) in 6-well plates. The pGEX4T moesin 307-577 plasmid was a gift from Vijaya Ramesh (Addgene plasmid # 11639, 5 μg for each well). The pGEX4T moesin 1-322 plasmid was a gift from Vijaya Ramesh (Addgene plasmid # 11638, 3 μg for each well). The sequence of CD44 siRNA was 5′-UGCCUUUGAUGGACCAAUU-3′, synthesized by Shanghai Sangon (25 nM for transfection, Shanghai, China). Moesin siRNA (h) is a pool of three different siRNA duplexes: sc-35955A: Sense: GGAACAUCUCUUUCAAUGAtt, antisense: UCAUU GAAAGAGAUGUUCCtt. sc-35955B, Sense: GCUAGAAUAUCCUACUUCUtt, Antisense: AGAAGUAGGAUAUUCUAGCtt. sc-35955C: Sense: CCCAUCCCGUU UAGAGUUAtt, Antisense: UAACUCUAAACGGGAUGGGtt.
Dual-luciferase reporter assay
TOP-FLASH (5 μg for each well, with three repeats of the Tcf-binding site) or FOP-FLASH (6 μg for each well, with three repeats of a mutated Tcf-binding site) plasmids (obtained from Dr. Haiwei Zhang, Chongqing Cancer Hospital) were transfected into cells in 6-well plates. PRL-TK vector was used as an internal control reporter (Promega, WI, USA). Luciferase activity was measured 48 h after transfection with a dual Luciferase Reporter Gene Assay Kit (Beyotime, Hangzhou, China). Renilla luciferase activity was used as an internal control. TCF activity was calculated as fold of activity (TOP/FOP Luc).
In vivo study
ICR and nude mice (BALB/cJNju-Foxn1nu/Nju) were obtained from Model Animal Research Center of Nanjing University (MARC). For acute toxicity experiments, ICR mice were divided into eight groups (10 mouse per group, half male and female) and daid002 was given intravenously (16, 12, 8, 4, 2, 1, 0.5 mg/kg). LD50 was calculated by SPSS 22.0 via regression analysis. For transplanted tumor study, nude mice weighing from 18 to 22 g were group-housed in a temperature-controlled room with a 12 h: 12 h light–dark cycle. U251 cells were subcutaneously injected into 50 female and male nude mice. Each group was treated with various concentrations of daid002 (0.25, 0.5, 1 mg/kg), temozolomide (20 mg/kg) and solvent through caudal veins once every two days for 20 days. The treatment was performed once every two days for 20 days. The tumor volume was measured with a caliper every two day, using the following formula: volume = length × width2/2. All tumor-bearing mice were sacrificed at day 21. Tumors, serum and organs were collected. Animal treatment and maintenance were carried out with ethics approval from the Experimentation Ethics Review Committee of China Pharmaceutical University.
Immunofluorescent staining
Cells were washed with PBS and fixed with methyl alcohol for 20 min at – 20ºC. TritonX-100 for 20 min at 4°C was used for permeabilization. Cells were then blocked with PBS containing 3% BSA for 1 h at 37ºC. The primary antibodies were diluted to a certain concentration according to the instructions. Cells were treated with the secondary antibody at appropriate dilution in 1% BSA for 1 h at 37ºC. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, Santa Cruz Biotechnology, CA, USA). Immunofluorescence photomicrographs and were captured using a fluorescent microscope (Carl Zeiss, Germany).
Immunohistochemistry staining
Tissues were sent to make paraffin section by the Nanjing Drug Toxicology Research Institute. Blocking was performed by incubating 2 h at room temperature with PBS containing 10% goat serum. Then, sections were incubated with primary antibodies for 2 h at 4 ×degrees celsius. Then the immunohistochemistry kit of Maixin Biotech was used. The immunoreactivity was finally examined by NanoZoomer 2.0 RS (Hamamatsu Photonics Co., Ltd, Shizuoka, Japan).
Immunoprecipitation assay
Cell lysates were prepared for immunoprecipitation. Aliquots of cell lysates were incubated with 2 μg of anti-CD44 antibody at 4ºC for 2 h and then precleared with protein-A/D-Sepharose at 4ºC overnight. Immunoprecipitated complex was washed five times with lysis buffer and then boiled in SDS sample buffer for 8 min. Then the immunoprecipitated complex was subjected to western blot with anti-moesin antibody, followed by peroxidase-conjugated appropriate secondary antibody and visualized by Tanon 5200 Chemiluminescence imaging system (Shanghai, China).
Co-crystal structures analyze of daid002 with moesin
The compound was prepared by using OPLS 2005 FF. 3D structures of the small molecules were prepared by using LigPrep prior to docking simulation to obtain the accessible least-energy ionized conformer and tautomeric states of the ligands. The complex structure obtained was first prepared with the Protein Preparation Wizard workflow. The subsequent step involved removal of water molecules without any contact and addition of hydrogen atoms to the structure. The optimized model structure was minimized until the average root-mean-square deviation (RMSD) of the non-hydrogen atoms reached 0.30 Å using the OPLS – all atom (AA) force field. The binding region was defined by a box centered on the centroid of the crystal ligand and in similar size with the polypeptide. The standard precision (SP) mode was used for the docking and scoring. All other parameters were kept default. The best pose was output based on Glide score and the protein−ligand interactions.
Study of daid002 bind with FERM domain
U251 and U87MG cells were transfected with two truncated plasmids pGEX4T-moesin-1-322 and pGEX4T-moesin-307-577. The cell lysates were extracted. For extracting cell lysis contained CD44 protein, cells were knocked down moesin and incubated with agarose-moesin-antibody overnight. For extracting cell lysis contained various moesin protein, cells were knocked down CD44 and incubated with agarose-CD44-antibody overnight. Daid002 was used to incubate with different moesin proteins which were bound to beads, and then CD44 protein were also incubated with beads. Western blot analysis was used to detected moesin or CD44 expression in supernatant or beads.
Statistical analysis
All experiments reported here were performed with at least triplicate independent replications. Results are presented as the mean ± S.D. Significance between two groups was assessed by the Student’s two-tailed t-test. The non-parametric Mann-Whitney U-test was used to assess significance between two means of data sets lacking a normal distribution and having a small sample size. Data sets consisting of more than 2 groups were analyzed by analysis of variance (ANOVA) with Tukey-Kramer HSD post-test for multiple comparisons if significance was determined. P value that was less than 0.05 was considered statistically significant for all data sets. All statistical analysis was performed using GraphPad Prism software.
Abbreviations
GBM, Glioblastoma multiforme; ERM, ezrin-radixin-moesin; NF2, neurofibromatosis type 2; TCGA, the cancer genome atlas; HA, hyaluronic acid; MTT, methyl thiazolyl tetrazolium; IC50, half maximal inhibitory concentration; daid, daidzein; ATRA, all-trans retinoic acid; IF, immunofluorescence; IP, immunoprecipitation; FERM, 4.1 protein/ezrin/radixin/moesin; PIP2, phosphatidylinositol (4,5)-bisphosphate; Q, glutamine; GST, glutathione s-transferase; LD50, median lethal dose; TMZ, temozolomide; AST, aspartate transaminase; ALT, alanine transaminase; γ-GT, γ-glutamyltransferase; BUN, blood urea nitrogen; Cr, creatinine; IHC, immunohistochemical; MGMT, O6-methylguanine-DNA methyltransferase; MAPK, mitogen-activated protein kinase; ER, estrogen receptors; BBB, blood–brain barrier; DMSO, dimethyl sulfoxide; PCNA, proliferating cell nuclear antigen; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PI, propidium iodide; RIPA, radio immunoprecipitation assay lysis buffer; ICR, institute of cancer research; DAPI, 4’,6-diamidino-2-phenylindole; ANOVA, analysis of variance
Author contributions
RH and QW conceived the project. XZ and XTL performed the study. JHZ, WZ, analyzed the data. JRL provided daid002 and daid. XZ wrote the paper. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We thank the Cellular and Molecular Biology Center of China Pharmaceutical University for assistance with IHC and IF works and we are grateful to Xiao-Nan Ma for her technical help. We would like to thank Dr. Haiwei Zhang for providing plasmids.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the National Natural Science Foundation of China (No. 81372268 and No. 81672816), the Program for Jiangsu Province Innovative Research (KYLX16_1120), the Natural Science Foundation for Distinguished Young Scholars of Jiangsu Province (No. BK20130026), the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. ZJ11173).
REFERENCES
1. Altieri R, Agnoletti A, Quattrucci F, Garbossa D, Calamo Specchia FM, Bozzaro M, Fornaro R, Mencarani C, Lanotte M, Spaziante R, Ducati A. Molecular biology of gliomas: present and future challenges. Transl Med UniSa. 2014; 10:29–37.
2. Salacz ME, Watson KR, Schomas DA. Glioblastoma. Part II: Future directions. Mo Med. 2011; 108:289–91.
3. Binello E, Green S, Germano IM. Radiosurgery for high-grade glioma. Surg Neurol Int. 2012; 3:S118–26.
4. Baumert BG, Hegi ME, van den Bent MJ, von Deimling A, Gorlia T, Hoang-Xuan K, Brandes AA, Kantor G, Taphoorn MJ, Hassel MB, Hartmann C, Ryan G, Capper D, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033–26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016; 17:1521–32.
5. Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000; 101:259–70.
6. Roubinet C, Decelle B, Chicanne G, Dorn JF, Payrastre B, Payre F, Carreno S. Molecular networks linked by Moesin drive remodeling of the cell cortex during mitosis. J Cell Biol. 2011; 195:99–112.
7. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010; 11:276–87.
8. Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010; 463:318–25.
9. Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010; 18:655–68.
10. Jijiwa M, Demir H, Gupta S, Leung C, Joshi K, Orozco N, Huang T, Yildiz VO, Shibahara I, de Jesus JA, Yong WH, Mischel PS, Fernandez S, et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS One. 2011; 6:e24217.
11. Jung WY, Kang Y, Lee H, Mok YJ, Kim HK, Kim A, Kim BH. Expression of moesin and CD44 is associated with poor prognosis in gastric adenocarcinoma. Histopathology. 2013; 63:474–81.
12. Zhu X, Morales FC, Agarwal NK, Dogruluk T, Gagea M, Georgescu MM. Moesin is a glioma progression marker that induces proliferation and Wnt/beta-catenin pathway activation via interaction with CD44. Cancer Res. 2013; 73:1142–55.
13. DeSouza LV, Matta A, Karim Z, Mukherjee J, Wang XS, Krakovska O, Zadeh G, Guha A, Siu KM. Role of moesin in hyaluronan induced cell migration in glioblastoma multiforme. Mol Cancer. 2013; 12:74.
14. Mahmoud AM, Yang W, Bosland MC. Soy isoflavones and prostate cancer: a review of molecular mechanisms. J Steroid Biochem Mol Biol. 2014; 140:116–32.
15. Dixon-Shanies D, Shaikh N. Growth inhibition of human breast cancer cells by herbs and phytoestrogens. Oncol Rep. 1999; 6:1383–7.
16. Uifalean A, Schneider S, Gierok P, Ionescu C, Iuga CA, Lalk M. The Impact of Soy Isoflavones on MCF-7 and MDA-MB-231 Breast Cancer Cells Using a Global Metabolomic Approach. Int J Mol Sci. 2016; 17.
17. Siegelin MD, Gaiser T, Habel A, Siegelin Y. Daidzein overcomes TRAIL-resistance in malignant glioma cells by modulating the expression of the intrinsic apoptotic inhibitor, bcl-2. Neurosci Lett. 2009; 454:223–8.
18. Tedeschi-Blok N, Lee M, Sison JD, Miike R, Wrensch M. Inverse association of antioxidant and phytoestrogen nutrient intake with adult glioma in the San Francisco Bay Area: a case-control study. BMC Cancer. 2006; 6:148.
19. Zohar R, Suzuki N, Suzuki K, Arora P, Glogauer M, McCulloch CA, Sodek J. Intracellular osteopontin is an integral component of the CD44-ERM complex involved in cell migration. J Cell Physiol. 2000; 184:118–30.
20. Ponuwei GA. A glimpse of the ERM proteins. J Biomed Sci. 2016; 23:35.
21. McClatchey AI. ERM proteins at a glance. J Cell Sci. 2014; 127:3199–204.
22. Clucas J, Valderrama F. ERM proteins in cancer progression. J Cell Sci. 2014; 127:267–75.
23. Mori T, Kitano K, Terawaki S, Maesaki R, Hakoshima T. Crystallographic characterization of the radixin FERM domain bound to the cytoplasmic tail of adhesion molecule CD44. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007; 63:844–7.
24. Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol. 2016; 134:381–97.
25. Ludwig K, Kornblum HI. Molecular markers in glioma. J Neurooncol. 2017; 134:505–512.
26. Shen D, Liu T, Lin Q, Lu X, Wang Q, Lin F, Mao W. MGMT promoter methylation correlates with an overall survival benefit in Chinese high-grade glioblastoma patients treated with radiotherapy and alkylating agent-based chemotherapy: a single-institution study. PLoS One. 2014; 9:e107558.
27. Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, Tung K, Tsokos GC. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007; 178:1938–47.
28. Jensen PV, Larsson LI. Actin microdomains on endothelial cells: association with CD44, ERM proteins, and signaling molecules during quiescence and wound healing. Histochem Cell Biol. 2004; 121:361–9.
29. Ranuncolo SM, Ladeda V, Specterman S, Varela M, Lastiri J, Morandi A, Matos E, Bal de Kier Joffe E, Puricelli L, Pallotta MG. CD44 expression in human gliomas. J Surg Oncol. 2002; 79:30–5; discussion 5–6.
30. Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, Ghatak S. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011; 278:1429–43.
31. Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. Eur J Cancer. 2010; 46:1271–7.
32. Wang Q, Lu X, Zhao S, Pang M, Wu X, Wu H, Hoffman RM, Yang Z, Zhang Y. Moesin Expression Is Associated with Glioblastoma Cell Proliferation and Invasion. Anticancer Res. 2017; 37:2211–8.
33. Brownson DM, Azios NG, Fuqua BK, Dharmawardhane SF, Mabry TJ. Flavonoid effects relevant to cancer. J Nutr. 2002; 132:3482S-9S.
34. Rajan RK, M SS, Balaji B. Soy isoflavones exert beneficial effects on letrozole-induced rat polycystic ovary syndrome (PCOS) model through anti-androgenic mechanism. Pharm Biol. 2017; 55:242–51.
35. Handayani R, Rice L, Cui Y, Medrano TA, Samedi VG, Baker HV, Szabo NJ, Shiverick KT. Soy isoflavones alter expression of genes associated with cancer progression, including interleukin-8, in androgen-independent PC-3 human prostate cancer cells. J Nutr. 2006; 136:75–82.
36. Sareddy GR, Li X, Liu J, Viswanadhapalli S, Garcia L, Gruslova A, Cavazos D, Garcia M, Strom AM, Gustafsson JA, Tekmal RR, Brenner A, Vadlamudi RK. Selective Estrogen Receptor beta Agonist LY500307 as a Novel Therapeutic Agent for Glioblastoma. Sci Rep. 2016; 6:24185.
37. Barone TA, Gorski JW, Greenberg SJ, Plunkett RJ. Estrogen increases survival in an orthotopic model of glioblastoma. J Neurooncol. 2009; 95:37–48.
38. Li Q, Jedlicka A, Ahuja N, Gibbons MC, Baylin SB, Burger PC, Issa JP. Concordant methylation of the ER and N33 genes in glioblastoma multiforme. Oncogene. 1998; 16:3197–202.
39. Batistatou A, Kyzas PA, Goussia A, Arkoumani E, Voulgaris S, Polyzoidis K, Agnantis NJ, Stefanou D. Estrogen receptor beta (ERbeta) protein expression correlates with BAG-1 and prognosis in brain glial tumours. J Neurooncol. 2006; 77:17–23.
40. Assimakopoulou M, Sotiropoulou-Bonikou G, Maraziotis T, Varakis J. Does sex steroid receptor status have any prognostic or predictive significance in brain astrocytic tumors? Clin Neuropathol. 1998; 17:27–34.
41. Kahsai AW, Zhu S, Wardrop DJ, Lane WS, Fenteany G. Quinocarmycin analog DX-52–1 inhibits cell migration and targets radixin, disrupting interactions of radixin with actin and CD44. Chem Biol. 2006; 13:973–83.
42. Fuse E, Nishiie H, Kobayashi H, Ikeda S, Saito H, Covey J, Kobayashi S. Disposition of DX-52–1, a novel anticancer agent, after intravenous administration to mice and dogs. Eur J Drug Metab Pharmacokinet. 1997; 22:53–63.
43. Bunnell CA, Supko JG, Eder JP Jr, Clark JW, Lynch TJ, Kufe DW, Shulman LN. Phase I clinical trial of 7-cyanoquinocarcinol (DX-52–1) in adult patients with refractory solid malignancies. Cancer Chemother Pharmacol. 2001; 48:347–55.
44. Mirsalis JC, Schindler-Horvat J, Hill JR, Green CE, Mitoma C, Tomaszewski JE, Tyson CA, Donohue SJ. Toxicity of a quinocarmycin analog, DX-52–1, in rats and dogs in relation to clinical outcome. Cancer Chemother Pharmacol. 2003; 51:193–201.