
INTRODUCTION
One of the most lethal cancers, glioblastoma multiforme (GBM) is the most common cancer of the glial cells, with an incidence of about 3/100,000 persons per year [1–2]. The basic procedure for GBM patients with clinical symptoms caused by the mass effect is surgical treatment (cytoreduction), which is also used to obtain material for histopathological examination. It should be combined with other methods such as radiotherapy, or in another variant chemotherapy with fotemustine or cyclically administered temozolomide (TMZ) or angiogenesis-inhibiting bevacizumab. Other methods, including immunotherapy, continue to be studied [3].
GBM has been subject to highly intense research due to the very low five-year post-operative survival rate, estimated to be only 9.8% [4]. In particular, researchers focus on intercellular signaling in the GBM tumor, i.e. autocrine influence of factors secreted by the GBM cells on themselves and the remaining cells in the niche. This has resulted in significant progress over the last 4 years in the understanding of the previously little known secretory factors such as neurotensin (NT), growth differentiation factor-15 (GDF-15), sphingosine-1-phosphate S1P, and of infection with cytomegalovirus (CMV). In this paper, we begin the discussion of these factors with the issue of intratumoral heterogeneity.
INTRATUMORAL HETEROGENEITY OF GLIOBLASTOMA
The population of tumor cells is not homogenous. It consists of genetically and epigenetically diverse tumor cells [5–6] with different expressions of mRNA [5, 7] and proteins [8]. This intratumoral heterogeneity in GBM was first reported in the 1980s [9]. Thanks to increasingly precise and sensitive research methods in which proteome, transcriptome and genome analysis can be performed on single cells, recent research shows in detail the differentiation of cancer cells in a GBM tumor [10].
Formation of intratumoral heterogeneity
Due to the uncontrolled division of cancer cells, a tumor has a much larger number of changed cells at the onset of neoplasm. The divisions result in the accumulation of genetic changes, and over time the environment within the tumor becomes increasingly diverse. In particular, selection pressure is exerted by the distribution of necrotic areas, different concentrations of oxygen including hypoxia [11], metabolic compounds, and tissue hormones, and the placement of unaltered tumor-building cells. Selected in a Darwinian-like manner [10, 12], different tumor cell lines are formed with various mechanisms of bypassing cancer resistance mechanisms, exhibiting properties described as the ‘hallmarks of cancer’ [13, 14].
Intratumoral heterogeneity seems to depend primarily on cancer stem cells, forming a small and rarely dividing population in a tumor [15]. During division they form a stem cell and a rapidly dividing cancer cell. The latter cells have a limited number of divisions and by definition do not form tumors in animals inoculated with them. However, according to most recent research, the differentiated GBM cells are able to dedifferentiate into glioblastoma stem cells (GSC) [16]. This partly refutes the theory of intratumoral heterogeneity based solely on cancer stem cells, and indicates that both stem and differentiated cells are responsible for the diversity of tumor cell lines [16].
Intratumoral heterogeneity in the development of glioblastoma
Mutations in the development of individual GBM lines are not haphazard. Sottoriva et al. show that they can be organized into three stages [10]. First, very characteristic changes occur on chromosome 7, with the amplification of the fragment with epidermal growth factor receptor (EGFR), cyclin-dependent kinase (CDK)6, and MET genes. It is also highly likely that deletion occurs on chromosome 10 with the PTEN gene. This stage is also characterized by deletion of the chromosome 9 fragment with the cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A/B) gene.
The next stages of tumor growth include very different mutations on different chromosomes, which results in a very large diversity of tumor cell lines within a single tumor. These include changes on chromosome 17 with P53 and neurofibromin 1 (NF1) genes, or on chromosome 4 with solute carrier family 2 member 9 (SLC2A9/GLUT9) gene, and platelet-derived growth factor (PDGFR)A amplification [10]. Also mutations of this type occur later in GBM recurrences, resulting in considerable genetic differences between the GBM cells in the relapse sites and the parent tumor [8].
The probability of each mutation depends on the tumor microenvironment and the selection of individual clones by anti-cancer mechanisms. Of particular significance is the location of the tumor in the brain; e.g. periventricularly located GBM has a higher expression of factors such as vascular endothelial growth factor (VEGF)-C or hepatocyte growth factor (HGF) than at cortical locations [17].
Intratumoral heterogeneity results in the creation of a tumor with a specific cell distribution pattern. GBM cells with amplified PDGFRA form a compact population surrounded by cells with amplified EGFR [18]. The accumulation of changes results in the formation of specific GBM subtypes: classical, mesenchymal, neural, and proneural [5]. In each GBM tumor there is a proneural cell population [5], while the other subtypes may occur in very low numbers or not at all. However, there have been no studies showing the detailed structures formed by cancer cells.
Functional domains of the tumor
Experiments on neurospheres derived from stabilized GBM cell lines demonstrate that these tumor cells are interdependent and specialized in specific functions [19]. In particular, tumor cells co-operate with each other for specific purposes in cancer development [20]. An example of this are the mesenchymal GBM cells, which contain many more proteins associated with immunosuppression [21]. Thanks to this they can participate in cancer immune evasion. However, intratumoral functional domains require further research which could open new possibilities for effective antitumor therapies.
Impact on therapy
GBM cell differentiation in a single tumor in terms of resistance to anti-cancer drugs has very negative consequences for therapy. It is estimated that 1/4 of tumor clones are resistant to TMZ and only 1/10 are very susceptible to the drug [22]. Such a scope of resistance in a GBM tumor is similar for other anti-cancer drugs [22] This has important implications for therapy, because the use of an anti-cancer drug, including TMZ, destroys only those cells which are susceptible to the drug, but leaves other cells that are resistant to it [22]. Within a few months of chemotherapy, new tumors in relapse sites are formed by GBM cells which survive treatment [4]. This results in a five-year survival rate of 10% in patients after chemotherapy with TMZ.
Some hope lies in studying the cancer microenvironment, in particular interactions between the tumor niche and cancer cells, and the intercellular signaling in the tumor microenvironment. These processes depend on many secretion factors (Figure 1).
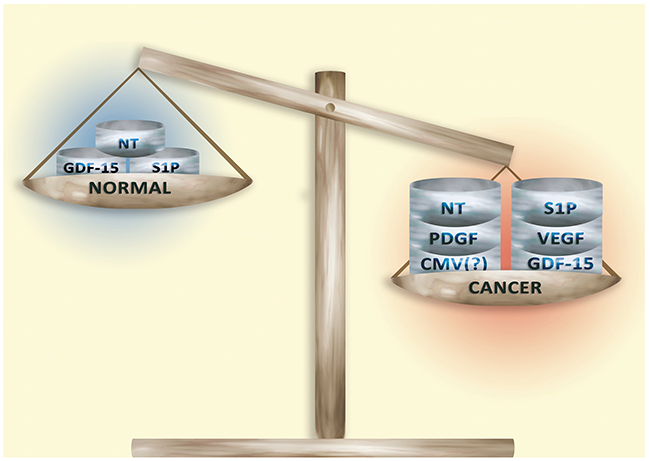
Figure 1: Secretory factors in normal tissue and in the tumor microenvironment. Secretory factors responsible for the ‘hallmarks of cancer’ occur in low concentrations in non-cancerous tissue. However, the development of a tumor increases the concentration of these factors. This process is non-specific and so the combinations and levels of secretory factors vary among tumors and even within a single tumor.
GBM has been studied extensively for NT, GDF-15, S1P, and infection with CMV, which play important roles in tumor processes, in particular the viability, migration and invasion of tumor cells, GSC, angiogenesis, and tumor immune escape (Figure 2).
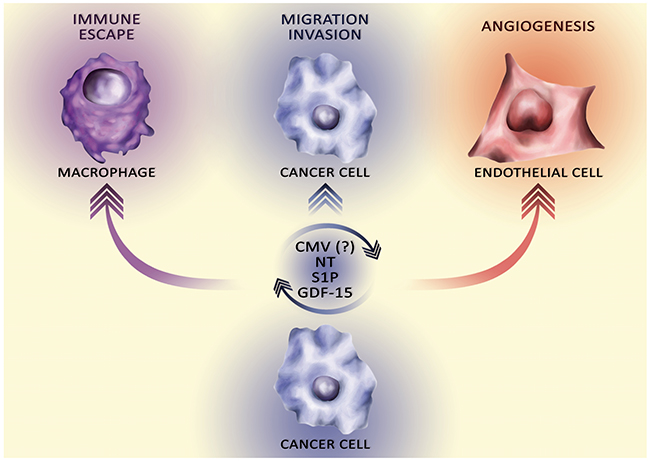
Figure 2: The influence of secretory factors on the ‘hallmarks of cancer’. Cancer cells secrete various secretory factors into the tumor microenvironment. The total pool of these secretory factors affects the hallmarks of cancer, in particular via autocrine stimulation of tumor cell proliferation, angiogenesis, migration and invasion, and tumor immune escape.
CYTOMEGALOVIRUS
Cytomegalovirus as a carcinogenic factor in glioblastoma
CMV is a DNA virus of the β-herpesvirinae subfamily, carried by more than half the global population [23]. Since Cobbs et al. demonstrated the expression of CMV in all GBMs [24], the incidence of CMV infection in the tumor has been widely discussed. Several research groups have confirmed the occurrence of CMV DNA and the expression of antigens of the proteins encoded by its genome in almost 100% GBMs [25–32].
Other research groups have also confirmed the presence of CMV in GBM, although only in 36% (27/75) [33], 51% (25/49) [34] and 75% (12/16) [35] of the tumors studied. In addition, other research groups have shown a lack of CMV infection in samples of brain slices affected by GBM [36–39]. They also postulate a false positive in other research groups due to the cross-reactivity of antibodies with non-viral proteins such as myelin basic protein or human serum albumin [38]. Some also postulate a false positive caused by non-specific immunocytochemistry staining of glial cells with gemistocytic morphology [36].
Epidemiology of the cytomegalovirus
CMV infection occurs in more than a half of the global population, with the likelihood of infection increasing with age [40–44]. Forty percent of people under 10 years of age are carriers of this virus. Higher age is associated with a higher likelihood of contracting and carrying this virus. In older people, the virus is estimated to have infected 70%-90% of the population, depending on the population studied. Although this high number of CMV carriers is not reflected in the incidence of GBM (3/100,000 persons/year [1, 2]), numerous studies show that the virus does increase the aggressiveness of GBM [45–51].
Tumor microenvironment and cytomegalovirus
The appearance of CMV in GBM may be caused by an immunosuppressive microenvironment of the tumor, as CMV infection is completely controlled by a healthy immune system [52–55]. Particularly crucial here is the NK cell response [52, 53, 55]. As a result, the virus exists in the body in a latent form and is reactivated when immunity reduces, e.g. as a result of the action of immunosuppressive drugs after transplantation [56, 57]. The tumor microenvironment, in particular in GBM, also involves intensive immunosuppression processes that cause the immune evasion of cancer cells (Figure 3) [58, 59]. This allows an intensive replication of CMV in GBM [60].
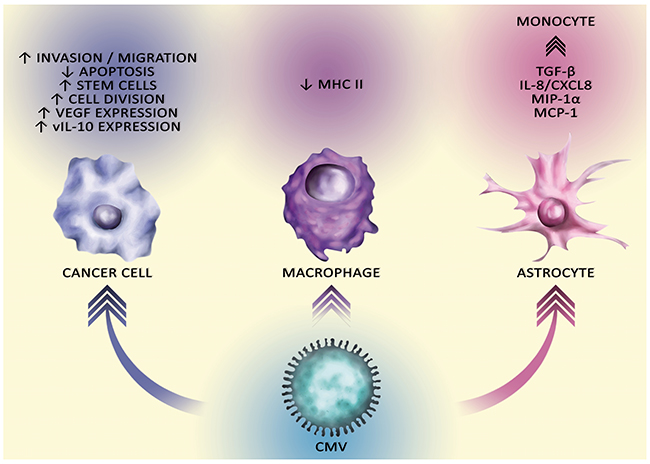
Figure 3: The carcinogenic effect of CMV on different cells. CMV affects cells in the tumor niche, particularly macrophages and astrocytes which affects the tumor immune escape. CMV also affects cancer cells. It stimulates migration and invasion GSC, stimulates divisions and disturbs apoptosis. CMV in the tumor cell is responsible for the tumor immune escape via the production of vIL-10.
Effect of cytomegalovirus on the tumor niche and tumor immune escape
Due to the lack of appropriate research models, little is known about the direct influence of CMV on processes occurring in the GBM tumor niche. Nevertheless, in vitro experiments with CMV infection of various cell types have made it possible to develop a model of the interaction between this factor and processes occurring in the tumor niche.
From monocytes with latent infection to lytic infection in the tumor
CMV is present in CD34+ bone-marrow progenitors, and consequently it also occurs in peripheral blood mononuclear cells during differentiation of the progenitors into monocytes [61, 62]. CMV does not cause lytic infection of monocytes; it is latent in these cells [63]. However, even during latent infections, the CMV reprograms the expression profile of some of the genes in the monocytes. In this state, only a small number of viral genes [64] are expressed.
Studies have shown increased expression of viral chemokine scavenger receptor US28 that stimulates the migration of infected cells in response to a wide spectrum of chemokines [65, 66]. US28 increases migration especially in response to C-X3-C motif chemokine ligand (CX3CL)1/fractalkine [67] and to a lesser extent in response to chemokines such as CC motif chemokine ligand (CCL)2/monocyte chemoattractant protein 1 (MCP-1), CCL3/macrophage inflammatory protein (MIP)-1α, CCL4/MIP-1β and CCL5/regulated on activation, normal T-cell expression and secretion (RANTES) [65, 67].
The effect of individual chemokines is cell-specific and depends on the type of cell where the expression of US28 has taken place [67]. Also, simultaneous activation of CX3CL1/fractalkine together with CCL2/MCP-1 or CCL5/RANTES on US28 results in no migration of monocytes or macrophages that are expressing this receptor [67, 68]. If in the tumor microenvironment there are also other chemokines (i.e. in addition to CX3CL1/fractalkine), then it results in the inhibition of US28-dependent monocyte and macrophage migration.
CX3CL1/fractalkine is mainly secreted by neurons [69]. In physiological conditions it shows a neuroprotective action, suppressing excessive activation of microglial cells by proinflammatory agents, e.g., LPS or CMV [70, 71]. CX3CL1/fractalkine is also produced by GSC [72] and TAM in the GBM niche [73], which may result in the recruitment of monocytes with latent CMV infection expressing US28. The action of this chemokine may also affect the location of infected TAM and other cells throughout the tumor niche [66, 67].
The use of antibodies neutralizing CX3CL1/fractalkine results in a decrease in the intensity of the migration of TAM and microglia isolated from GBM tumors. Nevertheless, the use of CX3CR1-neutralizing antibodies, which are the specific receptor for this chemokine, causes the same decrease in the intensity of migration of these cells [74]. This demonstrates that in the tumor microenvironment in these cells, it is CX3CL1/fractalkine with CX3CR1, but not viral US28, that are responsible for the migration of TAM and microglial cells. The effect of CX3CL1/fractalkine-dependent TAM and microglial cells in the context of CMV infection requires further research, especially with regard to the recruitment of monocytes with latent CMV infection.
After the migration into the tumor niche, monocytes differentiate into macrophages [75]. This differentiation of monocytes with latent infection into macrophages often causes CMV reactivation [61, 62]. CMV reactivation can also be caused by granulocyte-colony stimulating factor (G-CSF) [76], a cytokine produced in the GBM tumor [77]. These facts may explain the presence of active CMV infection in GBM.
Cytomegalovirus as a oncogenic factor: effect on apoptosis and proliferation
Some researchers suggest that CMV occurs in almost all GBMs and the incidence of CMV in GBM tumors is positively correlated with the grade of the tumor. Indeed, almost all GBM samples with the highest grades have been reported to contain the antigens or DNA of this virus [25–32]. This indicates that CMV plays an important role in the development of GBM. As demonstrated by in vitro experiments, the virus enters GBM cells through EGFR [78] or PDGFR-α (Figure 4) [79]. These receptors are important for GBM cells and are often amplified and overexpressed [80, 81]. CMV has a particular tropism to GSC in which it enhances the stem cell phenotype [46, 48–50]. CMV in tumor cells disrupts apoptosis in many ways, especially via the viral proteins, such as activation of the viral inhibitor caspase-8 and the viral mitochondria-localized inhibitor of apoptosis, homolog of anti-apoptotic Bcl-2 [82]. In addition, the immediate early 86 (IE86) viral protein initiates activating transcription factor 5 (ATF5), an anti-apoptotic protein commonly found in GBM [31, 83]. The IE86 protein also causes changes in the level of histone acetylation, which changes the expression of many genes in GBM cells [31]. In addition to its effect on apoptosis, CMV also affects cell division. It enhances the expression of telomerase, an enzyme essential for unlimited cancer cell divisions [84]. Viral proteins also reduce the expression of Rb and p53 proteins, which are important for regulating cell division [45, 85–87]. In addition, CMV proteins alter the expression of cell cycle cyclins, halting the division of normal cells and favoring viral DNA replication [88, 89]. However, as a result of tumor changes, this mechanism is impaired and CMV in some GBM cell lines induces cell division [45]. Another mechanism in which viral proteins promote GBM growth is the activation of the PDGFR-α and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-protein kinase B (PKB) pathways, i.e., pathways crucial for the stimulation of GBM proliferation [45, 90, 91].
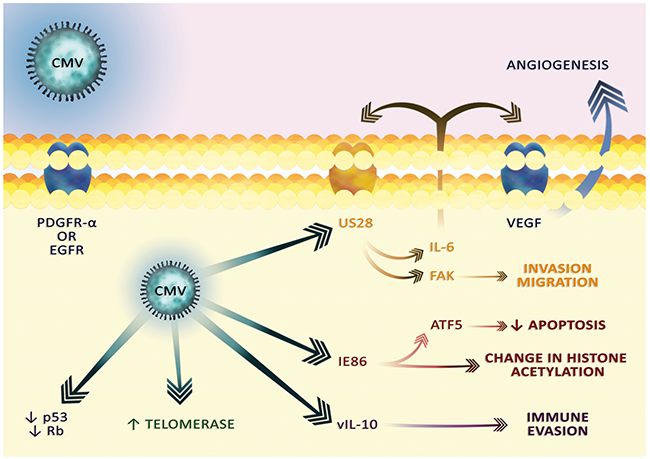
Figure 4: The cytoplasmic effect of CMV infection. CMV virions enter cells via the PDGFR-α or EGFR receptors. The viral proteins transmit the signal that causes changes characteristic for cancer. In particular, the US28 viral receptor is involved in angiogenesis, migration and invasion. vIL-10 is involved in tumor immune escape.
Effect on angiogenesis and tumor dissemination
In addition to the effect on replication, CMV increases angiogenesis and GBM dissemination. The CMV genome encodes the US28 receptor, a homolog of the receptor for the CC chemokine family, responsible for the disruption of the immune response against CMV [23]. US28 increases the expression of VEGF by increasing interleukin (IL)-6 expression and activating the hypoxia inducible factor (HIF)-1α/pyruvate kinase M2 (PKM2) pathway [51, 92–94]. IL-6 activates its receptor and signal transducer and activator of transcription (STAT)3, which then expresses VEGF. In addition to the effects on VEGF, CMV also reduces the expression of thrombospondin-1, an angiogenesis inhibitor [87]. CMV infection of the GBM cells also results in increased expression of endocan, a compound associated with the remodeling of the blood vessels and angiogenesis [95].
CMV also participates in a very characteristic sign of GBM, i.e., early cancer dissemination. US28 activates focal adhesion kinase (FAK) via phospholipase C-β (PLC-β), an enzyme reducing the adhesion of cells, which results in the migration of GBM [93, 94, 96, 97]. Another way of dissemination is in increased expression of matrix metalloproteinase (MMP)2 [35]. However, the mechanisms of CMV’s influence on GBM cells requires further research and more detailed understanding, in particular regarding the activation of the human endogenous retrovirus [98].
Another mechanism of CMV-induced angiogenesis in GBM is intensification of the stem cell phenotype [48]. Finally, the CMV genome contains a UL7 protein, similar to the N-terminal V-like domain of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), inducing vasculogenesis and migration of endothelial cells [99].
Effect on cancer cells in cancer immune evasion
CMV affects the communication between the tumor cell and the cell of the tumor microenvironment. In particular, the virus causes immune evasion of the infected cancer cell, especially GSC [46]. It disrupts major histocompatibility complex (MHC) I: human leukocyte antigen (HLA)-A, HLA-B and HLA-C, expression of which makes it impossible to recognize the altered antigens on the tumor cell by immune cells [100–104].
In infected cells CMV also reduces the expression of surface HLA-G and increases the levels of soluble HLA-G, as evidenced by experiments on U-373 MG astrocytoma cells [105, 106]. HLA-G is a non-classical molecule of MHC I that plays an important role in maternal-fetal tolerance [107] but is also a carcinogenic factor [108]. Lowering surface HLA-G expression and increasing the expression of soluble HLA-G by CMV is a mechanism of viral attack on the host immune response.
There are cytotoxic T cells in the human body that recognize CMV antigens response restricted by HLA-G [109]. However, in the tumor microenvironment, the increased amount of soluble HLA-G has an immunosuppressive effect [108, 110]. Although the degradation of surface HLA-G may stimulate an antitumor immune response, at the same time numerous cancer immune evasion mechanisms occur in the tumor microenvironment.
CMV infection of GBM cells also results in increased expression of arginase-2 [111] and FasL [112], which interfere with cancer-related immunosuppression. In an infected cell, CMV also induces the production of viral interleukin-10 (vIL-10) with an immunosuppressive action [47, 113, 114]. Infected cells also produce various chemokines that cause chemotaxis of various cells to the site of the CMV infection. If a CMV infection focus is present in the GBM tumor, then these infected cells maybe be recruited into the tumor niche.
Effect of cytomegalovirus on astrocytes
CMV has a high tropism for astrocytes and therefore these cells play an important role in CMV infection of the brain [115]. CMV has tropism for neural stem cells and immature glia cells in the subventricular zone [116, 117], and for GSC in the tumor niche [46]. CMV is also replicated in other cells, including nerve cells [118]. Infection in the brain is followed by chronic inflammation in this organ. Viral processes and the fight against CMV crucially depend on the production of chemokines, i.e. cytokines with chemotactic activity.
In the first stage of infection, astrocytes increase the expression of chemokines such as CCL2/MCP-1, CCL3/MIP-1α and IL-8/CXC motif chemokine ligand (CXCL)8, but not CCL5/RANTES. However, the infected astrocytes do not produce an increased amount of proinflammatory cytokines such as IL-1β, IL-6 or tumor necrosis factor α (TNF-α) [119]. Chemokines enable the chemotaxis of immune cells responsible for the fight against CMV infection. In combination with pro-inflammatory cytokines [120, 121] produced by other immune system cells such as NK cells, NKT cells, microglial cells [89, 122], CD4+ T-cells [123] and CD8+ T-cells [124], the fight against CMV infection can proceed. CMV also causes a decrease in the expression of CCR5, which interferes with the chemotaxis of these cells in response to chemokines [125]. This effect is dependent on viral protein UL128, which is included in the envelope of the CMV virion [126]. During cell infection, this protein degrades several chemokine receptors, not just CCR5, to interfere with chemo-dependent migration of infected cells.
Chemokines which play an important role in fighting CMV infection in an immunosuppressive cancer microenvironment can support tumor processes. Secreted by cells infected with CMV, chemokines act as chemoattractants for regulatory T cells (Treg), microglia, neutrophils and monocytes. These cells are recruited into the GBM tumor niche where they participate in tumor processes [59, 127–129]. CMV infection results in increased expression and secretion of cytokines that may contribute to the formation of the immunosuppressive cancer microenvironment. In particular, expression of vIL-10 in astrocytes plays a crucial role in immunosuppressive mechanisms [122, 130]. This cytokine reduces the production of CXCL10/IP-10 chemokine in infected microglial cells and thus reduces recruitment of lymphocytes that fight against CMV and tumor cells [122]. vIL-10 also causes differentiation of monocytes into immunosuppressive macrophages with a M2c phenotype [47, 130]. In the infected astrocytes transforming growth factor β (TGF-β) is expressed, an immunosuppressive cytokine having a key function in the development of GBM [131].
In addition to the effects on immune responses, CMV in astrocytes disrupts the uptake of glutamate [132]. This results in an increase in the concentration of this amino acid and cell toxicity in the CMV infection microenvironment [132]. This feature is shared with GBM [133]. However, there are no studies on CMV dependence on cytotoxic concentrations of glutamate in the GBM microenvironment.
Effect of cytomegalovirus on microglial cells
CMV infection causes major changes in microglial function. CCL2/MCP-1 and CCL5/RANTES chemokines produced in infected astrocytes cause migration of microglial cells to the site of CMV infection [119, 134, 135]. Influenced by CMV, microglia produce pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, which help to control CMV infection. Infected microglial cells also produce chemokines such as CCL2/MCP-1, CCL3/MIP-1α, CCL5/RANTES, IL-8/CXCL8 [119] and CXCL10/IP-10 [122]. CMV infection also results in the degradation of CCR5 and CXCR4 in microglia [125], as well as other chemokine receptors, which interferes with chemokine-dependent chemotaxis of infected cells [126]. This process depends on the aforementioned CMV virion envelope protein: UL128.
CCL2/MCP-1 and CCL5/RANTES cause monocyte migration, which results in the accumulation of these cells in the site of CMV infection [136]. In turn, CXCL10/IP-10 induces migration of T and NK cells [122], which results in the accumulation of CD8+ T-cells, responsible for chronic activation of microglial cells via the production of interferon gamma (IFN)-γ [124] which helps to control CMV infection. However, chronic inflammation is toxic to the cells in the brain, and one of the mechanisms that protects the brain from damage are immunosuppressive reactions involving Treg [124].
Chronic inflammation can assist in the development of many cancers, including GBM. Prolonged inflammation activates immunosuppressive mechanisms. In particular, the effect of IFN-γ changes to oncogenic [137]. Chronic inflammation is also accompanied by the recruitment of Treg [138] and an increase in the expression of immunosuppressive IL-10 [139]. In addition to the inflammatory effect, infected cells produce chemokines: CCL2/MCP-1 and CCL5/RANTES and also chemokines encoded by the CMV genome, that help in recruiting various GBM-associated cells, including neutrophils [139–144], macrophages [135, 136, 145, 146], microglial cells [119, 134, 135] and Treg [129]. After migration, these cells are included in the carcinogenic mechanisms.
In particular, this refers to the presence of anti-inflammatory cytokines in GBM, such as IL-10 and TGF-β, and also vIL-10 expression during CMV infection. The latter cytokine reduces expression of CXCL10/IP-10 in microglial cells and thus reduces recruitment of lymphocytes fighting against CMV infection and responsible for tumor destruction [122]. Also, vIL-10 causes monocyte differentiation into immunosuppressive macrophages with M2c phenotype [47, 130]. The anti-inflammatory cytokines, in particular vIL-10, are responsible for the expression of programmed death ligand-1 (PD-L1)/B7-H1 in microglia [46, 147]. This is an immunosuppressive molecule that reduces the antiviral response of CD8+ T-cells but also the antiviral activity of other immune cells. This mechanism also contributes to GBM tumor immune evasion. However, due to the lack of appropriate research models, this requires confirmation, just as the other CMV activities in GBM.
Influence of infection in tumor niche on monocytes and macrophages
In addition to CX3CL1/fractalkine, other chemokines also affect monocyte chemotaxis into the GBM tumor. In particular, CMV-infected GSC cells [46], astrocytes [119], macrophages [63, 148, 149] and microglia [119] secrete increased amounts of CCL2/MCP-1 and CCL5/RANTES. It seems that expression of these chemokines, at least with regard to CCL2/MCP-1, is highest in the early phase of cell infection and is dependent on the pp71 viral protein [63, 150, 151]. The expression of some chemokines is then reduced by other viral proteins at a later stage of infection [63, 150, 151].
CCL2/MCP-1 and CCL5/RANTES are chemokines that cause chemotaxis of monocytes from the blood and the subsequent accumulation of these cells in the focus of CMV infection. These chemokines also play an important role in the recruitment of monocytes into the cancer niche [135, 136, 145, 146]. Also, the CMV genome encodes viral chemokines that affect monocytes and macrophages. In particular, the murine CMV genome encodes murine cytomegalovirus chemokine (MCK)-1 and MCK-2 [152, 153]. This shows the identical mechanisms of CMV and GBM on blood monocytes. However, further studies are required to understand the effects of the aforementioned chemokines secreted during CMV infection on the recruitment of monocytes into the GBM niche.
After chemotaxis of monocytes into the tumor niche, they differentiate into macrophages. This process is induced by IL-10 and vIL-10, encoded by the CMV genome. These cytokines, in particular vIL-10, induce differentiation of monocytes into macrophages with phenotype M2c [46, 47, 113, 114]. Differentiation of monocytes by vIL-10 results in the activation of the PI3K and STAT3 pathways resulting in the increased expression of heme oxygenase-1 (HO-1) [47, 130]. Expression of this enzyme maintains this state of macrophage polarization.
These cells exhibit increased expression of IL-10 [130] TGF-β [46] and VEGF [46] and increased expression of immunosuppressive protein PD-L1/B7-H1 [46]. This is also accompanied by reduced expression of TNF-α [154] and a reduction in the expression of MHC II components [46, 101, 102, 155, 156]. As a result, the impaired MHC II presentation of antigens interferes with the antiviral response and also disrupts the antitumor response of the immune system [104].
One should not forget that CMV infection is not the only factor causing monocyte differentiation into macrophages. In GBM there are other factors that are produced by GBM cells and cells that accompany this tumor. In particular, these are factors such as M-CSF [157] or IL-10 [158]. However, further research is required to understand this problem.
Influence of direct infection on monocytes and macrophages
Direct infection with CMV affects monocytes [159]. CMV does not replicate in infected monocytes, but these cells are subject to latent infection with this virus [63]. After the infection of monocytes, CMV inhibits the apoptosis of these short-lived cells [160, 161]. This virus disrupts the expression of antigens by the infected monocytes; in particular it lowers the expression of MHC II components, in particular HLA-DR [62, 162].
During direct infection with this virus, monocytes dependent on NF-κB and PI3K differentiate into macrophages that simultaneously secrete cytokines and chemokines of M1 and M2 macrophages [148, 149, 163]. The gene expression profile is more similar to M1 than to M2 polarization [148, 159]. There is an increase in the expression of cytokines associated with M1 polarization, i.e. IL-1β, IL-6, IL-15, TNF-α, and an increase in the expression of M1 marker: CD80. However, infected monocytes also secrete factors associated with M2 polarity, such as IL-10 [148].
Infected monocytes begin to produce larger amounts of chemokines, in particular CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β and CCL5/RANTES but also large amounts of CCL8/MCP-2, CCL19/ELC, CCL20/MIP-3α, CCL23/MPIF-1, with a reduced secretion of CXCL1/GROα [63, 148]. They also secrete CXCL10/IP-10 and CXCL11/I-TAC, causing T-cell and NK cell migration with a possible antiviral effect. Expression of CCL2/MCP-1, CCL4/MIP-1β and CCL8/MCP-2 is highest at the onset of monocyte infection and decreases with time [63, 150, 151]. CMV also causes an increase in cyclooxygenase-2 (COX-2) expression in infected monocytes, but also a decrease in VEGF expression [148].
Infection of monocytes with CMV virions results in reduced expression of many receptors for chemokines such as CCR1, CCR2, CCR5 and CXCR4 [148, 164], which interferes with the action of chemokines directly after CMV infection. CMV does not affect the expression of CCR7 and CX3CR, which is already low in monocytes [164]. This effect is dependent on viral protein UL128, which is included in the CMV envelope complexes [126]. During infection, this protein causes the degradation of many chemokine receptors, which may be very important in the GBM tumor, where CMV is intensely replicated.
CMV infects the already polarized macrophages M1 and M2, with a higher tropism for M2 macrophages [149, 165]. After macrophage infection, CMV inhibits apoptosis in these cells [166]. In infected macrophages CMV further increases the expression of surface and soluble HLA-G, which impairs the immune response in the microenvironment of the infected cells. [105]. This may explain HLA-G expression in TAM and microglial cells in GBM tumor sections [167].
Soluble HLA-G also causes monocyte differentiation to immunosuppressive M2 macrophages, which may be significant in a tumor microenvironment with active CMV infection [108, 110]. The infection of macrophages is pro-inflammatory, which stimulates the immune system and thus may have an antitumor effect. The increased expression of MHC I components (HLA-A, HLA-B and HLA-C) and CD80 and CD86 helps in the presentation of antigens by these cells. However, it appears that the effect of CMV on the amount of MHC I is cell-specific because in the U-373 MG astrocytoma cells [105, 106] or primary murine fibroblasts [168], CMV causes MHC I degradation. In M2 macrophages, CMV reduces the expression of their markers: CD163 and CD206 [149].
Cytokine expression also changes in infected macrophages. In M1 there is an increase in the expression of chemokines and pro-inflammatory cytokines [149]. The same effect is exerted by CMV infection of M2 macrophages. There is an increase in the expression of chemokines such as CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES but not CXCL10/IP-10 [149]. This helps in recruiting monocytes from the blood to the CMV infection focus, but this mechanism is also common in cancer, not just GBM [136].
Infection of M2 macrophages results in increased secretion of pro-inflammatory cytokines such as IL-1β, IL-2, IL-6, IL-12, IL-15, TNF-α and IFN-γ and anti-inflammatory IL-10. Infected M2 macrophages secrete larger amounts of VEGF, which affects angiogenesis. It is also worth noting that the chemokines secreted by CMV-infected cells affect angiogenesis [169–171]. In particular, CCL2/MCP-1 and CCL5/RANTES cause vascular remodeling which may affect angiogenesis in GBM. Factors secreted by infected M2 macrophages are capable of enhancing immune responses in immune cells, which may have antiviral and antitumor effects.
In addition to the effects on the secretion of cytokines and chemokines, CMV interferes with chemotaxis in infected macrophages. In particular, it reduces the expression of CCR1 and CCR5 [172]. This effect is dependent on the expression of CMV genes. CMV replication also results in the expression and secretion of macrophage migration inhibitory factor (MIF) [172]. In this way, macrophages (also uninfected macrophages) are insensitive to many chemokines such as CCL2/MCP-1, CCL5/RANTES, CX3CL1/fractalkine, as well as to CCL19/MIP-3, CXCL1/GROα, CXCL12/SDF-IL-8/CXCL8 and macrophage-colony stimulating factor (M-CSF) [172]. Nevertheless, CX3CL1/fractalkine causes an in vitro increase in the migration of TAM and microglial cells isolated from GBM tumors [74].
The results of studies on the expression profile of different genes in TAM and microglia from a GBM tumor partially coincide with in vitro studies on the infection of macrophages. In particular, TAM and microglia isolated from GBM exhibit the expression of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α at a level similar to M1 macrophages [73]. Similar observations in TAM from in vivo models in mice show a mixed gene expression profile. In these models TAM simultaneously express genes specific for different macrophage phenotypes, with a predominance of M1 phenotype [173, 174]. However, TAM isolated from postoperative human GBM tumors do not express genes associated with immune activation [175].
TAM from GBM postoperative tumors are significantly different from CMV-infected and non-infected M2 macrophages. In particular, these TAM do not express TNF-α, although 20% of microglial cells and myeloid-derived suppressor cells (DMSC) isolated from GBM tumors do express this cytokine [176]. In vitro macrophage infection by CMV increases TNF-α expression [149] which indicates that CMV infection, if present in GBM tumors, does not affect TNF-α expression in TAM. But microglial cells infected in vitro by CMV do exhibit enhanced TNF-α expression, indicating the influence of CMV infection [119]. TAM have reduced expression of CD163 and CD206. In particular, these markers are expressed by a very small percentage of TAM isolated from proneural and neural GBM [176]. This is similar to the in vitro observation of CMV-infected M2 monocytes, in which CD163 and CD206 expression was reduced [149].
The effect of CMV monocyte infection on the differentiation of these cells in the GBM tumor, as well as the effect on infected TAM, still needs to be investigated further. Nevertheless, some of the findings on TAM isolated from postoperative GBM tumors were in contrast to those expected from in vitro studies on CMV-infected macrophages. On the other hand, studies on microglia and DMSC [176] have shown that inflammation caused by some factors match CMV infection. Research on CMV infection in GBM should be continued, with particular regard to the location of the infection in a particular type of cell in the tumor niche.
CMV has different tropisms for different cells. Also, the replication rate of this virus varies between cell types [177]. CMV lytic infection is destructive to the cells to which the virus has a particularly high tropism and a high rate of replication. However, in some cells, the virus immediately goes into a latent state and is activated only by some undiscovered factors. This results in a certain intratumoral heterogeneity in the CMV infection focus.
Effect of cytomegalovirus on regulatory T cells
Acute CMV infection results in inflammatory reactions and, in particular, chronic activation of microglial cells [124]. Immunosuppressive reactions, in particular recruitment of Treg, help to reduce excessive inflammatory response and thus protect against brain damage [138]. During cessation of the inflammatory response, the concentration of Treg in the inflammatory focus returns to physiological levels.
In a GBM tumor there is an increased number of Treg that have a role in cancer immune evasion [178]. Recruitment of these cells is accomplished via CCL2/MCP-1 [129], i.e. a chemokine that is produced by CMV-infected cells [89]. Further research is required to determine whether CMV affects the recruitment of Treg into the tumor niche or the expression of CCL2/MCP-1 is the result of CMV-independent cancer mechanisms. Further studies are also needed with regard to Treg populations in GBM tumors and how they are influenced by CMV. CMV carriers, particularly older adults, have increased numbers of cytomegalovirus-induced regulatory T cells (iTreg) [179–181]. These are Treg which alleviate inflammatory reactions. However, iTreg are specifically activated by CMV antigens, which causes them to only act in the focus of the CMV infection.
CMV has been shown to activate a certain T-cell subpopulation to produce IL-10 and thereby to alleviate the immune response. These cells do not express Foxp3, a Treg marker [182]. This T-cell subpopulation is activated in response to IL-27, which in turn is induced by type I IFN. These chemokines are produced by infected cells. In particular, IFN-α is produced in infected monocytes [148, 183] and IFN-β in infected M2 macrophages [165]. Further studies on the effect of CMV on Treg and on other immune system cells in a GBM tumor are required.
Effect of cytomegalovirus on neutrophils
Neutrophils play an important role in reactions caused by CMV which infects these cells and thus is spread throughout the body [184]. They also play an important role in GBM. Neutrophils are recruited near CD133+ GSC [185], i.e., near the same cells for which CMV has tropism [46]. The elevated number of neutrophils in the GBM tumor increases the aggressiveness of this tumor and, in addition, worsens the prognosis for the patient [186, 187]. Neutrophils in tumors are involved in angiogenesis, migration and invasion of cancer cells, and cancer immune evasion [127]. However, very little research has been devoted to the relation between these cells and cancer.
Neutrophils have been shown to be recruited under the influence of chemokines which are expressed in CMV-infected cells. The chemokines that are important for neutrophils include IL-8/CXCL8 [140], CCL2/MCP-1 [139] and viral CXC motif chemokine ligand 1 (vCXCL1) [141–144]. The CMV genome contains the UL146 gene which encodes protein vCXCL1. This viral chemokine, which works specifically as a chemoattractant for neutrophils, allows CMV to infect neutrophils and spread throughout the body in these cells [184]. If CMV infection is present in the GBM tumor, then neutrophils may be recruited into the tumor niche. However, the exact effect of CMV on neutrophil recruitment, as well as the effect of this carcinogenic factor on already recruited neutrophils in GBM is poorly understood and requires further investigation [186].
Correlation between cytomegalovirus infection and glioblastoma epidemiology
All of the discussed mechanisms may play a crucial role in GBM growth, which may be confirmed by the fact that some research groups estimate that 100% of GBM are infected with CMV [25–30, 32]. This virus also very often causes congenital neuronal disorders. It is striking that CMV has tropism for neural stem cells and immature glia cells in the subventricular zone [116, 117]. This region of the brain is considered to be the source of stem cells from which cancerous tumors such as gliomas (including GBM) are produced via carcinogenesis [188]. However, over 50% of the population has a latent CMV infection [40–43] and the number of GBM cases is only about 3/100,000 persons per year [1, 2], which shows a poor correlation between CMV infection and GBM epidemiology. CMV infection models in GBM in mice should answer further questions about the exact role of CMV in GBM development [189].
NEUROTENSIN
Neurotensin, receptors, functions
Neurotensin (NT) is a peptide hormone consisting of 13 amino acids. There are currently 4 known receptors of this hormone: NT receptor types 1-4 (NTSR1-4) [190]. NTSR1 has a high (0.1-0.3nM) affinity for NT, and NTSR2 has a low (3-10nM) affinity. Both these receptors are G-protein-coupled. Two other receptors, NTSR3/sortilin and NTSR4/SorLA, contain the Vps10p domain [191]. The extracellular domain of the NTSR3/sortilin can be released by its proteinase, and as a result can occur as soluble NT receptor type 3 (sNTSR3), performing biological functions without the involvement of NT [192, 193].
NT regulates the function of the digestive tract [194, 195]. In particular it stimulates the small bowel as well as colonic mucosa growth, and increases the production of digestive enzymes by the pancreas. NT is also produced in the brain where it influences the secretion and action of neurotransmitters [196–198]. In particular, NT reduces the effect of dopamine [197, 198]. NT also causes an increase in extracellular glutamate levels, associated with neurotoxic effects in pathological conditions [199–201]. NT is therefore associated with neurodegenerative diseases, in particular Parkinson’s disease as well as schizophrenia or drug abuse [197]. Finally, NT is also associated with cancer, which has been best researched in pancreatic, colorectal, breast, lung, prostate, and liver cancers [194]. Recent research shows that NT has important functions in gliomas, especially in GBM [202].
Neurotensin and cancer cell
Expression of NT and NTSR1 in gliomas increases with increasing tumor grade [202]. Among the gliomas, the highest expression of NT and NTSR1 occurs in GBM, which positively correlates with increased postoperative mortality [202]. In addition, different cell lines express different NT receptors. The GL261, U-87 MG, U-118 MG and A172 lines express NTSR1 [202–204]. The C6 line does not express NTSR1 but rather NTSR2 [205]. The U-373 MG line expresses three different NT receptors: NTSR1, NTSR2 and NTSR3/sortilin [206].
Effect on signal transduction in tumor cell
Exact NT signal transduction in GBM cells is not well known. Exact mechanisms have been established in other cancers, mainly lung, breast, colon and pancreatic adenocarcinoma cell lines [192, 207–210]. Activation of the NTSR1 receptor leads to activation of the EGFR family: in particular EGFR, ErbB2/HER2, and ErbB3/HER3, which in turn are responsible for signal transduction within the tumor cell [207, 209]. The PI3K-PKB pathway and extracellular signal-regulated kinase 1 and 2 (ERK1/2) mitogen-activated protein kinase (MAPK) are activated, and are responsible for the all properties of NT described in the following sections of this article.
Activation of the EGFR family by NTSR1 is dependent on the PLC-β-protein kinase C (PKC) pathway, which increases expression and activates MMP1 and MMP9 (Figure 5) [207, 209–212]. In particular, the increase in MMP9 expression is responsible for PKC activation of the PI3K-PKB and ERK1/2 MAPK pathways [212]. MMP1 and MMP9 release epidermal growth factor (EGF)-like ligands, in particular heparin-binding EGF-like growth factor (HB-EGF), neuregulin 1 and neuregulin 2, which activate the EGFR family [209, 210]. As a result, these receptors activate ERK1/2 MAPK and the PI3K-PKB pathway [207].
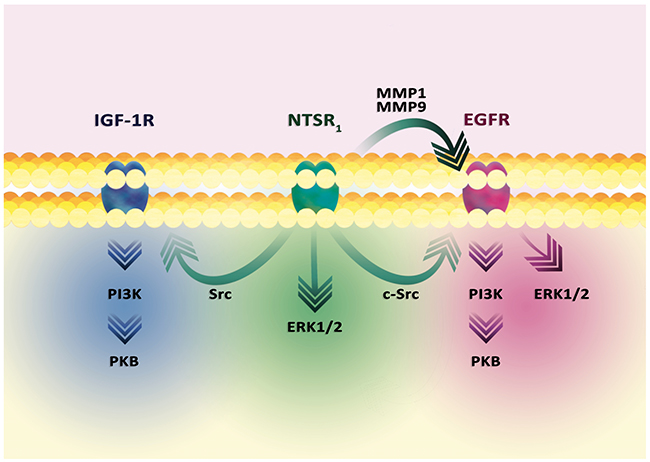
Figure 5: Signal transduction in the tumor cell from the NTSR1 receptor. In general, the activation of NTSR1 leads to activation of ERK1/2 MAPK and PI3K-PKB cascades. The signal transmission involves EGFR, activated via c-Src. EGFR can also be activated by MMP1 and MMP9. These metalloproteins release the EGF-like ligands, thus activating these receptors. As a result, the ERK1/2 MAPK cascade and the PI3K-PKB pathway are activated. Nevertheless, the ERK1/2 MAPK cascade can be directly activated by the NTSR1-PLCβ-PKC pathway, without the involvement of other receptors. Similarly, the PI3K-PKB pathway can be activated by signal transduction to IGF-1R.
EGFR activation may occur in a different manner. In prostate tumor PC3 cells, NT causes EGFR activation via c-Src [213]. This kinase causes phosphorylation of Tyr845 EGFR which results in STAT5b activation. Also, activated NTSR1 causes NF-κB activation which results in the increased expression of miR-21 and miR-155 [214]. miR-21 inhibits PTEN expression, a phosphatase degrading the PKB activator, which allows NT to increase the activity of this kinase. miR-155 reduces expression of the protein phosphatase 2 catalytic subunit α (PPP2CA), the suppressor of PKB activity.
There is also an EGFR-independent mechanism of signal transduction from NTSR1, i.e. via the activation of the PLCβ-PKC-ERK1/2 MAPK pathway. PI3K-PKB is also activated [207], which may involve another receptor with tyrosine kinase activity. An example of this is the insulin-like growth factor 1 receptor (IGF-1R) activated by Src in human colonic epithelial NCM460 cells [215].
GBM cells have been shown to express ErbB/HER2 while ErbB3/HER3 is more abundant on GSC [216–218]. Expression of these receptors, as well as the importance of EGFR amplification [81] in tumor processes in GBM, gives strong evidence that NT also acts through these receptors in this type of tumor.
Neurotensin and glioblastoma stem cells
Expression of NT as well as receptors of this hormone in GBM occur mainly in GSC (Figure 6) [203, 219]. NTSR1 regulates the carcinogenic properties of GSC of various cells lines. The exact mechanism of NT effect on GSC is dependent on IL-8/CXCL8 [203]. NT after activation of NTSR1 and EGFR increases expression of IL-8/CXCL8 in GSC. Following the secretion of IL-8/CXCL8, this chemokine activates the CXCR1 receptor in an autocrine manner, which activates the STAT3 transcriptional factor. As a consequence, the expression of stem cell markers increases, especially nestin and Sox2, and sphere-forming ability is increased [203]. IL-8/CXCL8 also supports proliferation, migration and invasion [220]. Also, this chemokine is involved in angiogenesis and tumor immunosuppression [221, 222].
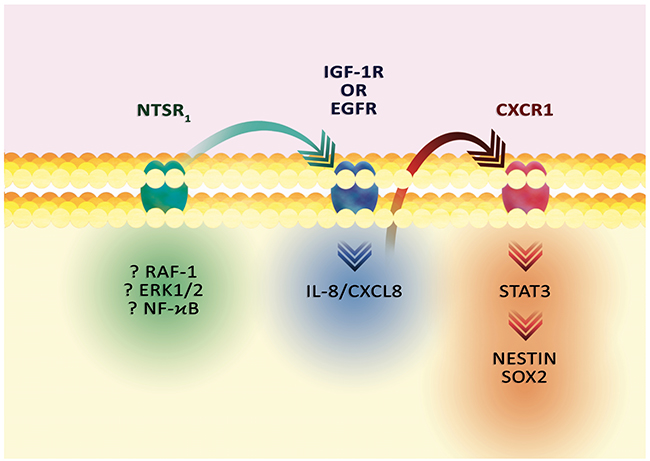
Figure 6: The effect of NT on GSC markers. Activation of NTSR1 results in signal transmission to IGF-1R or EGFR and increased IL-8/CXCL8 expression. Then, the activation of CXCR1, an IL-8/CXCL8 receptor, activates STAT3 and increases the expression of stem cell markers: nestin and Sox2.
The exact mechanism of increased expression of IL-8/CXCL8 by NT in GBM has not been well understood. Experiments on other types of cancer show that ERK1/2 MAPK cascade, in particular ERK1/2 and RAF-1, are important in signaling, as demonstrated by stem cells of hepatocellular carcinoma and HCT116 human colorectal cancer [208, 223]. Also, the effect of NT on IL-8/CXCL8 expression may depend on NF-κB activation as demonstrated by transfected NCM460-line colonocytes and HCT116 human colorectal cancer [223, 224]. Nevertheless, the expression of IL-8/CXCL8 may also be activated by other receptors other than the EGFR family. In particular, the Src activation of the IGF-1R receptor can activate PKB, which increases expression of IL-8/CXCL8 in the colonic epithelial NCM460 cells [215].
Effect on proliferation
NT stimulates the proliferation of GBM cells [202]. Activation of NTSR1 enhances the expression of CDK4 and CDK6 [204]. This effect is associated with a decrease in miR-129-3p expression and a reduction in miR-29b-1 expression via the NTSR1-c-myc pathway. These miRNAs reduce the expression of CDK6. This makes NT proliferate via the increased expression of CDK6. Also, NTSR1 via c-myc increases expression of CDK4 [204]. Thanks to these pathways, NT stimulates GBM cells to cross the G1/S checkpoint. In addition to the effects on proliferation, NT inhibits apoptosis by increasing Bcl-2 expression, as demonstrated on breast cancer MCF-7 cells [225].
Effect on glutamate concentration
One of the features of gliomas, including GBM, is increased glutamate concentrations in the tumor environment [133]. This causes a neurotoxic effect. However, the impact of NT in this process is controversial. In the brain, NT increases glutamate concentration [199–201]. However, the induction of expression and activation of NTSR1 on GBM C6 cells results in an increase in the amount of excitatory amino acid carrier 1 (EAAC1) on the cell membrane, resulting in the uptake of aspartate and glutamate [226]. The involvement of NT in the neurotoxic effects of glutamate within GBM and gliomas requires further research.
Effect on dissemination
NT stimulates GBM cells to invasion [202]. NT causes changes in the cytoskeleton organization. In particular, by activating Rac1 and cell division cycle 42 (Cdc42) increases the motility of U373 cells on laminin substrate [206]. Cells cultured on a plastics-only medium had more fibrillar actin and filopodial protrusions, and so showed lower motility [206].
Information on the effect of NT on GBM dissemination is incomplete, as no specific inhibitor studies have been conducted. Studies on other types of cancer show that NT acts via NTSR1 and NTSR3/sortilin which caused epithelial-mesenchymal transition [193, 212, 227, 228]. NT via NTSR1 in lung cancer cell lines NCI-H1299 activates FAK resulting in cell migration [229]. Also the migration of tumor cells is enhanced by sNTSR3 [192, 193]. Irrespective of NT or signal transduction from EGFR, NT increases FAK phosphorylation and activation [192]. Also, sNTSR3 decreases the expression of integrins, E-cadherin localization disorder, and changes in the desmosome structure, resulting in tumor cell release and migration [193]. Importantly, sNTSR3 does not affect tumor cell proliferation as it does not activate ERK1/2 MAPK [192].
In addition to FAK activation and changes in cytoskeleton organization and expression integration, NT also affects GBM dissemination by other means. In particular, NT induces an increase in IL-8/CXCL8 expression. By the action of this chemokine, the expression of MMP or activity of the uroplasminogen activation system is enhanced, as was the case in pancreatic adenocarcinoma BxPC-3 and PANC-1 lines [230].
Neurotensin and angiogenesis
To date, the effect of NT on angiogenesis has not been unambiguously determined. However, it may be inferred that – similar to immune evasion – it is cell-specific. In experiments on human umbilical vein endothelial cells (HUVEC) NT has not been reported to cause angiogenesis [231]. However, in experimental colitis of the large intestine, NTSR1 is a factor that does enhance angiogenesis [232]. In particular, NTSR1 activity stabilizes and increases HIF-1α expression. This results in increased expression of genes dependent on this protein, especially VEGF-A which is involved in angiogenesis [232].
NT can also indirectly influence tumor angiogenesis through IL-8/CXCL8. NT induces an increase in IL-8/CXCL8 expression in GSC [203]. This chemokine causes the recruitment of tumor-associated neutrophils which secrete various hormones involved in angiogenesis [222, 233].
Neurotensin and tumor immune evasion
Effect on macrophages
The effect of immunological processes is cell-specific. NT does not affect macrophages in tumor immune evasion but instead enhances the already induced immune response by increasing macrophage activation, although this effect is about 10 times smaller than at 100 U/ml IFN-γ [234–236]. In experiments on RAW 264.7, NT does not affect migration, expression of TNF-α, IL-10, nor IL-12 in macrophages [236]. In contrast, NT increases IL-1β and IL-6 expression [236]. NT also strengthens the immune response as demonstrated on lipopolysaccharide (LPS)-activated RAW 264.7 macrophages [236] and rat alveolar macrophages [237]. NT increases the production of TNF-α, IL-1β and IL-12 [236]. However, NT does not modify the expression of IL-1 caused by IFN-γ nor that caused by leukotriene B4 [237]. NT also increases the migration and expression of COX-2 and inducible nitric oxide synthase (iNOS) in LPS and IFN-γ activated macrophages [235]. The NT effect on macrophages is associated with NTSR1 activation, which affects LPS-activated NF-κB factor and the activated JAK2-STAT1 pathway [235]. However, due to immunosuppressive processes in tumors [238], NT does not directly play any direct role in the activation or migration of macrophages in GBM tumors.
Effect on microglial cells
In a similar way NT affects microglia. This is a heterogeneous population of cells; with only 8% of adult mouse brain cells and 13% of neonatal C57Bl/6 mouse brain reacting to NT [239]. Activation of microglial cells of these populations, especially in the neonatal brain of mice, may be impaired by the action of a previously anti-inflammatory cytokine such as IL-4 [239]. Nevertheless, the number of NT-responsive microglial cells may increase. If adult mouse brain microglial cells are activated with a proinflammatory cytokine such as IFN-γ, the number of NT-responsive cells increases 3 times [239]. This effect does not occur under the influence of LPS or IFN-γ on microglia isolated from a neonatal mouse brain.
Microglial cells express NTSR3/sortilin but not NTSR1 [240–242]. Activation of this receptor causes migration of microglial cells. This effect is triggered by the activation of PI3K and MAPK cascades [240]. This results in changes in F-actin polymerization and filopodia formation. Via NTSR3/sortilin activation, NT induces an increase in the expression of IL-1, TNF-α, CCL2/MCP-1, CCL5/RANTES, IL-8/CXCL8, CXCL2/MIP-2 but not altering the expression of IL-6, CCL3/MIP-1α nor CCL4/MIP-1β [241, 242]. The effect on the expression of these hormones is dependent on the activation of ERK1/2 MAPK and PI3K in microglial cells [241]. Chemokines produced by microglial cells participate in the migration and recruitment of other microglial cells (CCL2/MCP-1 and CCL5/RANTES) [134] as well as macrophages (CCL2/MCP-1 and CCL5/RANTES) [134, 136, 146], neutrophils (IL-8/CXCL8, CCL2/MCP-1 and CXCL2/MIP-2) [139, 140] and Treg (CCL2/MCP-1) [129]. In contrast, pro-inflammatory cytokines enhance the immune response. Nevertheless, the immunosuppressive tumor microenvironment decreases the number of NT-responsive microglial cells [238].
Effect on neutrophils
NT also has an effect on neutrophils. However, this action is poorly understood and requires further investigation in GBM tumors. It has been shown that in vitro NT at a concentration as low as 0.1nM acts as a chemoattractant for neutrophils, increasing their targeted migration [243]. This effect is direct via NT receptors on these cells and also indirect via IL-8/CXCL8 [203, 222]. NT expression, occurring predominantly in GSC [203, 219], may explain the accumulation of neutrophils in the GBM tumor near the GSC [185]. Further studies on the effect of NT on the migration of neutrophils to the tumor niche and the location of these cells in the tumor are required.
After the migration of neutrophils near GBM, NT increases adherence and diapedesis of neutrophils, thereby increasing the infiltration of these cells within the tumor. This has been demonstrated by in vitro experiments in which NT caused adherence to bronchial epithelial cells [244]. It seems that NT induces neutrophil activation, in particular phagocytosis of these cells [243].
Effect on dendritic cells
NT has immunosuppressive properties on fetal-skin dendritic cells [245]. NT reduces the production of TNF-α, IL-10 and VEGF in these cells, which is anti-angiogenic [245]. On the other hand, NT enhances the synthesis of EGF in these cells, which may have a significant effect on GBM with amplified EGFR near dendritic cells. NT also interferes with LPS activity in dendritic cells. Incubation of NT together with LPS or pre-incubation of NT completely abolishes dendritic cell response to LPS, in particular the expression of TNF-α, IL-6 and IL-10 [245]. This shows that NT can interfere with the antitumor immune response.
GROWTH DIFFERENTIATION FACTOR-15 / MACROPHAGE INHIBITORY CYTOKINE-1
Growth differentiation factor-15 as an antineoplastic agent
GDF-15/macrophage inhibitory cytokine-1 (MIC-1) is a member of a TGF-β superfamily hormone [246]. To date, no receptor for this extracellular protein has been identified [247, 248]. GDF-15 seems to act via the TGF-β receptor type II (TGFβRII) [249, 250]. Heterodimers of TGFβRI/activin receptor-like kinase (ALK-5) and TGFβRII are also important in the signal transduction. According to a recent study, the GDNF family receptor α-like (GFRAL) is a specific GDF-15 receptor [251–253]. Thanks to this, overexpression of GDF-15 results in decreased appetite and weight loss [254, 255]. GDF-15 is also associated with the cachexia associated with cancer. This property of GDF-15 may explain the decreased appetite and drastic weight loss in some glioma and GBM patients [256].
GDF-15 plays a very important and diverse function in cancer processes. At the beginning, it has antitumor properties, as it inhibits tumor cell division (Table 1). It induces phosphorylation of Smad3 (a protein that participates in tumor suppression) and apoptosis via the intrinsic mitochondrial pathway, as demonstrated on U-87 MG, U-118 MG, U-251 MG, U-373 MG and T98G cell lines [248, 257]. GDF-15 also disrupts connective tissue growth factor (CTGF)-induced angiogenesis in HUVEC [258]. In particular, GDF-15 decreases FAK activation and decreases clustering of the αVβ3 integrin. Expression of GDF-15 is enhanced by the action of the p53 protein in an antitumor mechanism [259]. However, the resulting mutations in the P53 gene and hypermethylation of the GDF15 gene promoter result in reduced expression of this protein in cancer cells [257]. In GBM cells the epigenetic silencing of Egr-1 and Sp-1 transcription factors results in a decrease in expression of GDF-15, as demonstrated with the use of histone deacetylase inhibitor Trichostatin A [260].
Table 1: Anti- and pro-cancer properties of GDF-15
Anti-cancer properties |
Pro-cancer properties |
|
|---|---|---|
Cell viability |
Inducted intrinsic mitochondrial apoptosis pathway |
Increased viability via increased activation of the PI3K-PKB pathway |
Neutralization of the cytotoxic action of TGF-β1 |
||
Angiogenesis |
Reduction in CTGF-dependent angiogenesis |
In hypoxia, an increased expression of HIF-1α and VEGF |
Migration and invasion |
Decreased FAK activity |
Increased expression of MMP2 and MMP9 |
Decreased intensity of integrin αVβ3clustering |
Inducted epithelial-mesenchymal transition |
|
Tumor immune escape |
Reduced IL-2 synthesis |
|
Increased IL-10 synthesis |
||
Reduced IL-12 synthesis |
||
Increased TGF-β1 synthesis |
||
NK dysfunction |
||
Reduced infiltration of macrophages and T cells. |
Growth differentiation factor-15 as a progression of cancer
As cancer progresses, tumor cell resistance to GDF-15 and its elevated synthesis increases [261, 262]. This is an indication of the progression of cancer; hence a high correlation of this hormone level and a reduction in survival of patients after GBM removal [262, 263]. The expression of GDF-15 in secondary glioblastomas is much higher than in primary glioblastomas [261]. Nevertheless, expression of GDF-15 is not the same for all GBM cells in the tumor. The highest expression occurs in the mesenchymal subtype, with the lowest in the proneural subtype [262]. Another significant source of GDF-15 are TAM, as shown in esophageal squamous cell carcinomas [264].
Related to the progression of tumor formation, resistance to GDF-15 is associated with changes in the pathways activated by this hormone. In particular, GDF-15 no longer causes Smad3 phosphorylation [263]. As a result, the hormone no longer causes apoptosis of the GBM lines, particularly lines A172 and LN-229 [261–263, 265]. These lines, unlike the U-87 MG and T98G lines (not-resistant to GDF-15), have a high expression of this protein [261, 262, 265].
It has also been shown that GDF-15 stimulates the intensity of proliferation not only of LN-229 and A172, but also of LN-319, U-87 MG, D-247 MG, LN-308, LN-428, LN-18 and U-373 MG [261]. On the other hand, studies by Kadowaki et al. and Zhang et al. show an opposite effect. GDF-15 reduces the proliferation of U-87 MG and U-373 MG cells lines [248, 257]. In addition to Smad3 pathways, GDF-15 also increases the activation of the PI3K-PKB pathway which increases the viability of the cells [248]. Another pathway of GDF-15 proliferative activity is TGF-β1 dysfunction [266]. Accumulated in the cancer cell nucleus, GDF-15 causes a disorder in the expression of genes associated with Smad factors [266]. Smad factors are associated with the transduction of a signal from the TGF-β1 receptor. In this way, GDF-15 abolishes the inhibitory effect on cell division TGF-β1.
Effect on angiogenesis
In addition to proliferation, GDF-15 also induces angiogenesis in advanced tumor processes. GDF-15 stimulates the proliferation of HUVEC via increased expression of cyclins D1 and E [267, 268]. This effect depends on the activation of PI3K-PKB and ERK1/2 and JNK MAPK pathways. In anoxia, expression of GDF-15 increases in GBM cells independently of p53 and HIF-1, as demonstrated in the LN-Z308 cell line [269]. Then, this hormone causes angiogenesis in hypoxic conditions which corresponds to increasing peritumoral angiogenesis in region of raised regional tissue tension followed by regional cerebral blood flow failure causing hypoxia [270]. Through the stabilization of the p53-MDM2 complex, it disrupts the p53 function in vascular cells [271]. This is followed by an increase in HIF-1α expression and an increase in VEGF expression, as demonstrated in HUVEC [271]. In hypoxia, this causes angiogenesis in the tumor.
Effect on migration and invasion
GDF-15 also promotes migration and invasion of GBM cells [262]. Anti-GDF-15 antibodies induce a decrease in the invasive capabilities of lines such as U-373 MG and LN-308 while increasing the invasion capability of the LN-428 line [261]. This indicates that depending on tumor cell changes, GDF-15 inhibits or enhances the migration and invasion of GBM cells. Nevertheless, GBM is a tumor with high intratumoral heterogeneity. As a result, this cytokine can cause the migration of certain tumor lines sensitive to GDF-15 in the GBM tumor. Linking such changes in the cancer cell to the effect of GDM-15 requires further investigation. It is known that GDF-15 affects the activity of the uroplasminogen activation system in LNT-229 and LN-308 glioma cells. GDF-15 induces an increase in miRNA expression, silencing plasminogen activator inhibitor-1 (PAI-1), and a less pronounced silencing ofthe expression of urokinase-type plasminogen activator (uPAR) receptor [262]. More precise studies have shown that GDF-15 affects GBM cell migration independently of uroplasminogen activation system expression [262].
GDF-15 can have an affect on invasion by using other pathways as demonstrated in experiments using other cancers. GDF-15 causes epithelial-mesenchymal transition (EMT) of colorectal cancer in HT29 and SW480 cell lines [272] and carcinoma cell line HepG2 [273]. GDF-15 results in decreased expression of E-cadherin and increased expression of N-cadherin and vimentin.
The adhesion of GBM cells is performed by integrins [274]. GDF-15 may interfere with integrin activation [250, 275]. However, the impact on GBM migration via this pathway still needs to be investigated further. This cytokine may also increase the expression of MMP2 and MMP9 by activation of the PI3K-mTOR pathway, as demonstrated in ovarian cancer cells [276]. However, the impact on GBM via this pathway is yet to be confirmed.
Effect on tumor immune escape
GDF-15 also causes tumor immune escape. In experiments on splenocytes, it reduced IL-2 synthesis and increased the synthesis of immunosuppressive IL-10 [265]. GDF-15 has been shown to impair NK function and reduce malignant infiltration of macrophages and T cells in tumors [265]. In addition, GDF-15 causes dendritic cell function abnormalities. It reduces the synthesis of IL-12 and increases the synthesis of TGF-β1, a cytokine that also strongly disrupts the immune function [277]. GDF-15 also decreases the expression on dendritic cell membrane proteins, particularly CD25, CD83, CD86 and HLA-DR [277, 278]. These changes cause disorders in the stimulation of the antitumor immune response, in particular a reduction in the stimulation of cytotoxic T lymphocytes and other immune cells [277]. This causes immunosuppression in the tumor microenvironment.
In addition to silencing the antitumor immune response, GDF-15 affects cell migration to the tumor niche. In particular, it enhances expression of CCL2/MCP-1 chemokine via TGFβRII-SMAD-3, as demonstrated on RAW 264.7 macrophages [249]. GDF-15 action via TGFβRII differs from the activation of this receptor by TGF-β (which does not increase the expression of CCL2/MCP-1). In addition to its effects on chemokine expression, GDF-15 exhibits increased expression of CCL2/MCP-1 receptor (CCR2) in macrophages [249]. This cytokine also changes CCR2 phosphorylation, which increases the intensity of activation of this receptor [249].
GDF-15 may interfere with the recruitment of monocytes and neutrophils into the tumor niche. In particular, GDF-15 disrupts integrin activation on THP-1 monocytes and murine neutrophils [250, 275]. This results in abnormal adherence and diapedesis of these cells and hence a decrease in infiltration of monocytes and neutrophils to other tissues. This effect is dependent on TGFβRI/ALK-5 and TGFβRII receptors [250].
SPHINGOSINE-1-PHOSPHATE
Sphingosine-1-phosphate synthesis, degradation, and receptors
Sphingosine-1-phosphate (S1P) is a hormone; a sphingolipid synthesized from sphingosine by two sphingosine kinase isoforms: sphingosine kinase 1 (SphK1) and sphingosine kinase 2 (SphK2) [279, 280]. These enzymes catalyze the same reaction but have different cellular locations and functions [280]. Activated SphK1 attaches to the cell membrane and catalyzes the formation of S1P; it is responsible for the concentration of S1P outside the cell. The activity and product of the reaction catalyzed by this enzyme have antiapoptotic and promitogenic properties. In contrast, SphK2 is primarily a nuclear enzyme. Its inactive form is attached to biological membranes (in particular to the cell membrane) and to the endoplasmic reticulum via the BH3 domain. SphK2 activity has proapoptotic properties and inhibits cell division.
S1P is inactivated in two ways. First, it can be dephosphorylated by S1P-catalyzed phosphohydrolase (SPP)1 or SPP2. Another way to inactivate this hormone is through breakdown by S1P lyase (SPL).
The synthesized S1P can act as a second messenger, as well in an autocrine or paracrine manner via S1P receptors on the surface of cells. As the second messenger, S1P activates peroxisome proliferator-activated receptor γ (PPARγ) and thus performs important functions in HUVEC physiology [281]. However, S1P is also secreted outside the cell. Then, in an autocrine or paracrine manner it activates five of its receptors (S1PR1-5), coupled with different small G proteins and thus differing in signal transduction and function [282].
Sphingosine-1-phosphate-related enzymes in the glioblastoma multiforme tumor
S1P plays a very important role in apoptosis [283–285], homeostasis of the immune system [284, 286] and blood vessel physiology [287]. An increasing number of papers show that S1P plays a very important role in the pathogenesis of cancers, including the development of brain tumors. GBM is associated with the overexpression of S1PR1, S1PR2, S1PR3 and S1PR5 and higher S1P concentrations than in the rest of the brain [288, 289]. At the same time, S1PR4 is not expressed in this tumor or in normal brain tissue [288, 289].
In contrast, SphK1 expression is higher in recurrent and secondary GBM, whereas SphK2 is higher in primary GBM [288]. Also, the expression of S1PRs differs in GBM. The expression of S1PR1 and S1PR5 is elevated in all types of GBM, mostly in secondary GBM [288]. In contrast, increased expression of S1PR2 and S1PR3 occurs only in secondary GBM [288].
A reduction in S1PR1 expression is associated with a shorter postoperative survival time of patients [289–291]. Also the overexpression S1PR2 [289], S1PR5 [288] SphK1 [292–294] and SPP1 [289] is associated with short postoperative survival time. Nevertheless, different studies indicate different proteins related to the survival of patients. Research by Bien-Möller et al. shows that the expression of S1PR3, S1PR5 and the enzymes SphK1, SphK2, SPP2 and SPL1 is not related to survival [289]. In contrast, Quint et al. show that S1PR1, S1PR2, S1PR3, SphK1 and SphK2 have no such effect [288].
Effect on glioblastoma cell viability
In vitro experiments show that S1P and enzyme expression involved in the biochemistry of this hormone influence the viability and behavior of GBM cell lines. The induction of expression and activity of SphK1 are influenced by various factors, in particular activation of the receptors of PDGFR [295] EGFR [296], and the expression of variant III of EGFR mutation (EGFRvIII) [294]. These receptors are closely involved in the development of GBM [80, 81, 297].
In addition to growth factors, inflammatory reactions also increase the expression of SphK1. IL-1 enhances the expression of SphK1 in GBM cells via c-Jun terminal kinase (JNK) MAPK and AP-1, independently of NF-κB [298]. Hypoxic stress also increases the expression of SphK1 [299, 300]. It increases the expression and activity of SphK1 and thus the extracellular concentration of S1P. SphK1 increases the rate of cell proliferation, increases migration and invasion, and inhibits multiple glioma cell lines, in particular LN-229, LN-382, U-87 MG, U-373 MG, U-1242 MG, and primary human non-established GBM GBM6 cells [292, 301, 302]. In particular, the activation of PKB by S1PRs results in inactivation of FOXO3a and consequently a decrease in the expression of proapoptotic Bcl-2-like protein 11 (Bim) [302]. The expression of S1PRs, mostly S1PR1, increases the rate of proliferation of U-118 MG and U-373 MG cells [303]. This effect is related to the activation of ERK1/2 MAPK. S1PR5 has been shown to inhibit proliferation by inhibiting ERK1/2 MAPK activation [303]. Nevertheless, Yoshida et al. showed the opposite results, with S1PR1 decreasing tumor cell proliferation rates in U-87 MG and U-251 MG cancer cell lines [291].
S1P has different effects on different cell lines. LN18 cell proliferation is not affected by signal transduction from any of the S1PRs [289]. This is due to mutations in the P53 gene, which results in the independence of proliferation and apoptosis of cells with mutations in that gene from the level of S1P [304].
Effect on glioblastoma stem cells
The action of S1P differs according to the cell type. The greatest synthesis and secretion of S1P occurs in GSC [305]. The expression of S1PR1, S1PR2 and S1PR4 also occurs predominantly in these cells, which results in the fact that it is GSC that mainly react to S1P [306]. For example, S1P in an autocrine manner increases the life span of the GSC. It stimulates the expression of GSC markers [307]. S1P also stimulates GSC proliferation and has antiapoptotic and antinecrotic effects [307]. As a result of this action, S1P causes resistance of GSC to TMZ, which is independent of methylguanine-methyltransferase (MGMT) expression [305, 308]. These S1P properties are due to the Notch pathway in GSC [309], crucial because of its role in promoting proliferation and self-renewal of these cells in the GBM tumor (Figure 7) [310]. Signal transduction from S1PR3 induces the p38 MAPK-dependent ADAM17 activation in the signal transmission from Notch1. This ADAM17 activation mechanism is independent of Notch1 receptor activation.
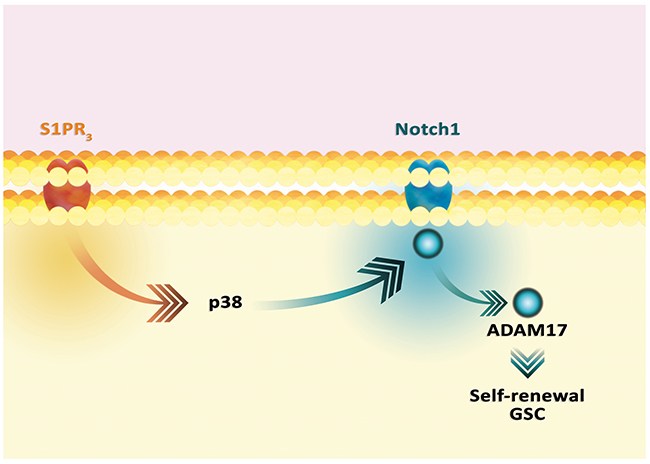
Figure 7: Activation of Notch1 pathway by S1P. The activation of S1PR3 activates the Notch1 pathway. This activation is independent of the Notch1 ligand and is dependent on the ADAM17 protein, which results in GSC self-renewal.
Hypoxia and angiogenesis
S1P also participates in angiogenesis. In the model of hypoxia using CoCl2 in GBM cells, the expression of SphK2 decreased and expression of SphK1 increased [299, 300]. This results in an increase in the synthesis and concentration of S1P in the tumor microenvironment [299]. The increase in SphK1 expression was due to an increase in HIF-2α activity that binds to the promoter of the SphK1 gene [299]. HIF-1α likewise has this effect. Increasing S1P concentrations stimulates GBM cells to proliferate and inhibit apoptosis by ERK1/2 MAPK and PKB activation [300, 302, 303].
S1P not only affects tumor cells in an autocrine manner but also blood vessel cells. In particular, it initiates endothelial cell sprouting and migration, and formation of ‘tubes’ as shown on HUVEC, human dermal microvascular endothelial cells, and mouse embryonic fibroblasts [299, 311, 312]. This process is mainly triggered by S1PR1 activation under hypoxic conditions [299].
However, in HUVEC, S1P may participate in angiogenesis independently of its specific receptors. S1P in these cells directly activates PPARγ, resulting in an increase in PAI-1 expression [281]. Importantly, S1P does not affect the expression of proteins involved in lipid metabolism. A consequence of PAI-1 expression is the stimulation of angiogenesis [313].
The endothelial structures formed by cells are not stable, and so S1P is not a sufficient factor for the entire course of angiogenesis. It is other proangiogenic factors that support the formation of new vessels in the tumor [311]. That is why in hypoxia VEGF expression occurs later than SphK1 expression [299]. Nevertheless, it seems that VEGF and S1P work together in angiogenesis, mutually enhancing each other’s action [299, 314].
Effect on migration and invasion
GBM is a tumor that always recurs after surgery. The migration and invasion of GBM cells depends on several factors, including S1P and the proteins involved in the biochemistry of this hormone. The expression of S1PR1 and S1PR3 is responsible for the migration and invasion of U-118 MG and U-373 MG cells [303]. This process depends on the plasminogen activation system. In particular, in A172, U-118 MG, and U-373 MG cells, the overexpression of S1PR1, and to a lesser extent that of S1PR2 and S1PR3, results in an increase in urokinase-type plasminogen activator (uPA) and uPAR activity, independently of S1P [293, 315]. In contrast, in U-373 MG cells the overexpression of S1PR1 and S1PR2, as well as activation of S1PR2 by S1P, result in increased expression and activity of uPAR and PAI-1 [293, 315, 316]. This is due to the activation of MEK1/2 and Rho by S1PRs [316]. This signal transduction also involves protein kinase D2 (PKD2) [317]. The activation of S1PRs activates PKD2. As a consequence, GBM cells express the proteins involved in migration and invasion, in particular proteins associated with plasminogen, integrin α-2, integrin α-4, and MMP1 [317].
S1P-dependent GBM cell migration and invasion is influenced by other signaling pathways; in particular EGFR-Src-PKCδ activates SphK1 [315]. As a consequence, PAI-1 is expressed in U-373 MG and A172 cells [315]. S1P-dependent expression of PAI-1 and uPAR may also be enhanced by IL-1 [316].
In some models, S1PR1 and S1PR2 activation inhibits migration but increases invasiveness by increasing the adhesion of U-87 MG, U-118 MG, U-251 MG and U-373 MG cells [293, 303, 318]. This process depends on the induction of cysteine-rich angiogenic inducer 61/CCN family member 1 (Cyr61/CCN1) expression [293, 303].
Another very important route in the migration and transfiguration of GBM cells by S1P is Ca2+ mobilization [295, 296, 306]. Activation of S1PRs results in signal transductions involving MAPK, RhoA/ROK, and phospholipase C [296]. This signaling also involves membrane-type-1 MMP and the glucose-6-phosphate transporter; the silencing of their expression impairs the effect of S1P on Ca2+ mobilization [296].
The migration of GBM cells via Ca2+ mobilization may partly depend on cytoplasmic S1P, i.e. independently of S1PRs [295]. In particular, S1P activates transient receptor potential C1 (TRPC1), which causes Ca2+ mobilization in the cytoplasm [295]. In U-252 MG cells, this mechanism is induced by PDGFR activation, in particular the synthesis of S1P.
Tumor immune evasion: effect on macrophages
In GBM, TAM play an important role in immune modulation and tumor development [75]. The amount of TAM increases with the grade of the glioma [319]. Thus, they constitute a significant percentage of cells in the GBM tumor [176, 320, 321].
There are currently no studies showing the effects of S1P from glioma or GBM on TAM. Nevertheless, on the basis of work on melanoma [322], breast cancer [323] and S1P biochemistry research in GBM, it can be deduced that this hormone significantly influences macrophage behavior in brain tumors particularly angiogenesis [292–294].
No sudden angiogenesis occurs during a growth in tumor volume. Angiogenesis is only induced by hypoxia and signaling pathways activated by tumor microenvironment [324]. Cell apoptosis occurs very often in tumor microenvironment. Apoptotic bodies contain S1P produced by SphK2, which affects macrophages [325–329]. However, SphK1 may also be activated during apoptosis, especially during the action of antitumor drugs [283]. S1P in apoptotic bodies causes large changes in macrophages; it activates S1PR1, which results in an increase in HIF-1α expression even in normoxia [328]. As S1P alone does not increase the expression of HIF-1α, in order to induce a given effect the apoptotic bodies also activate other non-S1P signaling pathways. With regard to the effect of S1P on HIF-1α expression, the second factor is TGF-β. An increase in HIF-1α expression activates mechanisms leading to angiogenesis in the tumor.
SphK activity is not important in the differentiation of progenitor cells into monocytes [330]. In contrast, S1P is important in the egress of monocytes from the spleen and bone marrow, as demonstrated by the use of FTY720. This process is dependent on S1PRs, with the exception of S1PR3 [331]. In monocytes already circulating in the blood, particularly in immunosuppressive mouse monocytes (CD45+CD11b+Gr1−), activation of receptors S1PR2 and S1PR3 by S1P activates PI3K and induces migration of these cells [332–334]. Activation of S1PR1 and S1PR5 does not result in the migration of monocytes [334, 335].
Targeted monocyte migration induced by S1P is partly dependent on thrombin. S1P induces increased expression of protease-activated receptor-4 (PAR-4), a thrombin receptor [336], which directs the migration of monocytes to the site of elevated thrombin activity. This receptor also increases expression of COX-2. Monocyte migration mediated by S1P and thrombin can exist within the GBM tumor because this cancer has elevated thrombin activity and increased activity of SphK1 and S1P levels [337].
Activating S1PR1 and S1PR3 on macrophages, S1P acts as a chemoattractant for these cells [283, 332, 338–340]. Activation of S1PRs in macrophages results in ADP secretion and the synthesis of extracellular ATP via adenylate kinase activity. In consequence, the P2X7 receptor is activated on macrophages [341]. This results in changes in actin polymerization, which facilitates migration. In contrast, S1PR2 activation reduces macrophage migration [338, 342], which is associated with increased cAMP levels and decreased PKB phosphorylation.
In addition to the effect on chemotaxis, S1P results in increased expression of intercellular adhesion molecule I (ICAM-1), which increases monocyte adhesion to these cells. This effect has been demonstrated on HUVEC, where ICAM-1 expression was dependent on S1PR1 [343]. S1P has also been shown to increase expression of E-selectin in HUVEC [344]. This effect depended on S1PR1 which activated PI3K-PKB and ERK1/2 MAPK pathways. It induced an increase in SphK activity with intracellular S1P playing the role of a second messenger [344]. Intracellular S1P activates NF-κB, which increases the expression of genes dependent on this transcription factor, including an increased expression of E-selectin [345–347].
S1P has also been shown to increase ICAM-1 expression on human pulmonary alveolar epithelial cells in a process mediated by S1PR1 and S1PR3 [348]. In these cells, ICAM-1 expression depended on the activation of ERK1/2 MAPK, p38 MAPK and JNK MAPK, and on c-Src kinase, EGFR, PDGFR and PKB [348]. However, S1P seems to disrupt the adhesion of monocytes to the walls of blood vessels [349]. It causes rearrangement on HUVEC integrins α5β1 and αVβ3 cells, which impairs monocyte adhesion to these cells [350]. In addition to the effect on adhesion proteins, S1P increases the expression of chemokines that attract monocytes and macrophages. Via S1PR1 and S1PR3, S1P causes an increase in the expression of CCL2/MCP-1 in HUVEC [343]. An increase in the expression of other chemokines may also be involved in the S1P-induced migration of monocytes. CYM-5442, an S1PR1 agonist, reduces the expression of CCL2/MCP-1 and CCL7/MCP-3 in HUVEC [351].
Macrophages phagocytose the apoptotic bodies, which enables the removal of cells that are subject to apoptosis. S1P from apoptotic bodies activates S1PR1 on macrophages and inhibits their apoptosis [326]. This effect is dependent on the level of intracellular Ca2+ and the activation of ERK1/2 and PI3K. It increases the expression of Bcl-2 and Bcl-xL and causes the phosphorylation of Bcl-2-associated death promoter (BAD) in these cells. High density lipoproteins (HDL), which contain S1P, also inhibit macrophage apoptosis [352]. In particular, S1PR1, S1PR2 and S1PR3 are activated, which results in the activation of the STAT3-JAK2 pathway and therefore an increase in survivin expression.
Nevertheless, the phagocytosis itself also partly depends on S1P. S1PR2 reduces the intensity of Escherichia coli phagocytosis by increasing the RhoA-GTP level. This causes a contraction of the macrophages [353]. S1PR2 reduces the amount of Rac1-GTP, which inhibits actin polymerization and therefore disrupts phagocytosis in macrophages [353]. However, S1PR2 stimulates antibody-dependent phagocytosis [354].
S1P reduces the pro-inflammatory immune response in macrophages. Via S1PR1, but not S1PR2, S1P reduces the expression of inflammatory cytokines from LPS-activated macrophages. In particular, it reduces the expression of IL-12, TNF-α and CCL2/MCP-1 and increases the expression of arginase I. Which enzyme reduces nitric oxide (NO) production in macrophages. S1P also reduces the activation of NF-κB in LPS-treated macrophages, which reduces the expression of iNOS [355].
Activation of macrophages by proinflammatory agents, such as LPS or TNF-α, results in increased production and secretion of proinflammatory cytokines. Nevertheless, SphKs do not participate in the signal transduction induced by LPS or TNF-α as demonstrated in vivo and in vitro using gene knockouts of these enzymes in murine monocytes and macrophages [330]. Similar results have been obtained on RAW 264.7 macrophages [356]. SphK1 does not affect the LPS-induced prostaglandin E2 (PGE2) production. On the other hand, increased SphK1 activity is required in RAW 264.7 to increase PGE2 production in response to TNF-α [356], which is related to S1P involvement as a second messenger in TNF-α–induced activation of NF-κB [345–347]. Then, after chemotaxis, this hormone silences excessive immune response in macrophages [285]. However, this property of S1P also plays an important role in cancer immune evasion [322, 323, 357]. S1P polarizes to M2 macrophages [322, 323, 326, 329], with a significant role of increased HO-1 expression in this process. HO-1 expression is due to the activation of two pathways by S1PR1: a p38 MAPK-dependent pathway and another which activates STAT1 and increases VEGF expression [329]. Then, an increase in HO-1 activity causes an increase in expression of antiapoptotic proteins in macrophages, in particular Bcl-xL and Bcl-2 [329]. HO-1 also increases the expression of adenosine A2A receptor in macrophages, one of the immunosuppressive mechanisms in immune cells [329].
S1P significantly changes the secretory profile of macrophages. In apoptotic bodies S1P increases the expression of COX-2 protein, in part by stabilizing COX-2 mRNA [356, 327, 358]. In addition to the effect on this enzyme, S1P also increases the activity of microsomal prostaglandin E synthase-1 (mPGES1) and decreases the activity of prostaglandin D synthase (PGDS) and 15-hydroxyprostaglandin dehydrogenase (15-PGDH). In the tumor microenvironment, this increases the production of PGE2, a compound which participates in many mechanisms of GBM development [359, 360]. S1P increases the expression of lipocalin 2 (LCN2) in macrophages, resulting in lymphangiogenesis in the breast tumor model [361].
S1P also influences NO production in macrophages. The activation of S1PR2 on macrophages by the apoptotic bodies [362] induces the activation of extracellular signal-regulated kinase 5 (ERK5) and subsequent activation of the cAMP responsive element binding protein (CREB). This, in turn, increases the expression of arginase II, an enzyme that metabolizes L-arginine, an amino acid that serves as a substrate for nitric oxide synthase. This results in a reduction in iNOS activity and NO production. ERK5 also increases expression of CD206 and VEGF. Independently of ERK5, activation of S1PR2 results in an increase in IL-10 expression [362].
As a result of exposure to S1P, indoleamine 2,3-dioxygenase (IDO), IL-8/CXCL8, IL-10 and CD206 expression increases, NO levels decrease, TNF-α and IL-12 expression decreases, i.e. polarization to M2 macrophages occurs [284, 322, 323, 326, 329].
In resident peritoneal macrophages, S1P reduces the production of poinflammatory cytokines via S1PR2, as has been demonstrated on a model with an abnormal activity of this receptor. Knockout of Gα12/13, a significant protein in the transduction of signals from S1PR2 increased the expression of iNOS and COX-2, and the expression of pro-inflammatory cytokines such as IL-6, TNF-α and IFN-γ, and chemokines CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES and CXCL10/IP-10 [363]. This shows an immunosuppressive effect of S1P on these cells. However, in mouse bone marrow-derived macrophages, knockout of Gα12/13 caused an increase in the expression of iNOS and IL-6 but not COX-2 nor the cytokines IL-1β, IL-10 and TNF-α [363]. At physiological concentrations, S1P alone did not result in an increased production of TNF-α, IL-6, IL-10, IL-12, or CCL2/MCP-1 in bone marrow-derived macrophages or in these cells differentiated to M1 or M2 macrophages [364]. S1P also did not affect phagocytic capacity or iNOS expression in that model.
FTY720 as an immunosuppressive drug
Knowledge of the effect of S1P on individual immune cells, including cells in the tumor niche, is incomplete. So far, some studies have been based on the influence of FTY720, a compound with complex effects on S1PR. Therefore, here we will discuss the mechanisms of FTY720 action to better understand the effect of S1P on various immune system cells.
2-amino-2-[2-(4-octylphenyl)]-1,3-propanediol hydrochloride (FTY720/fingolimod) is an S1P analogue which exhibits immunosuppressive activity. FTY720 is phosphorylated by both SphK isoforms [365, 366], but phosphorylation by SphK2 has better reaction parameters than SphK1. Therefore, in the human body FTY720 is phosphorylated mainly by SphK2 [367–371]. A reverse reaction, i.e. the dephosphorylation of FTY720-P, is carried out by lipid phosphatase 3 (LPP3) and to a lesser extent by SPP1 in cells [372]. Due to the uneven distribution of both SphK isoforms across human organs, FTY720 is mostly phosphorylated in the spleen, brain and lung [365, 373]. To a lesser degree, phosphorylation occurs in blood and lymph nodes, and is very low in other organs.
FTY720-P is an agonist of S1PR1, S1PR3, S1PR4 and S1PR5 [373]. However, studies on S1PR2 show that FTY720-P at a concentration of 40nM can activate some signaling pathways through this receptor [374, 375]. Binding affinities of FTY720-P for receptors S1PR1, S1PR3 and S1PR5 are about 10nM, while100nM for S1PR4 [373]. At higher concentrations also non-phosphorylated form of FTY720 can activate S1PR1 (binding affinities of 300±51nM), and S1PR5 (binding affinities 2623 ± 317nM) [376]. In this way, it activates these receptors and acts similarly to S1P. Nanomolar concentrations of FTY720 cause permanent internalization, downregulation and finally degradation of S1PR1 and S1PR5, and to a lesser extent S1PR2 [377]. FTY720-P also shows similar properties against S1PR1 [378–380]. As a result, FTY720 and FTY720-P disrupt the signal transmission from these receptors. Eventually, FTY720 is inactivated in the liver via ω-hydroxylation catalyzed by CYP4F2 and to a lesser extent by CYP4F3B [381].
Due to its properties, FTY720 has been investigated as an immunosuppressive agent in organ and tissue transplants [382–386]. In particular, FTY720 accumulates in lymph nodes and inhibits the egress of lymphocytes [387]. This reduces the number of these cells in the blood and thereby reduces the immune response [388]. FTY720 is a potential anti-inflammatory drug in ischemia-reperfusion injury [389, 390]. FTY720 can also penetrate the blood-brain barrier [391], reducing inflammation in the brain [392]. Therefore, it can be used as a drug against relapsing-remitting multiple sclerosis, and has already been approved by the FDA for universal use [393–395].
Sphingosine-1-phosphate and microglial cells
S1P is a hormone involved in the activation of the microglia by pro-inflammatory factors. An activation of these cells by a pro-inflammatory factor such as LPS results in an increase in the expression and activity of SphK1 and thus an increase in the production of S1P [396]. The effect of LPS on IL-1β and TNF-α production in BV2 microglial cells is cancelled by SphK1 gene knockout, or the use of an inhibitor of this enzyme [397, 398]. Without pro-inflammatory factor LPS, S1P alone only slightly increases TNF-α and IL-1β production [396].
SphK1 gene knockout, or the use of an inhibitor of this enzyme, only partially suppress the effect of LPS on iNOS expression [396]. This shows that SphK1 activity only partially participates in the LPS-induced expression of this enzyme. In contrast, SphK1 gene knockout significantly lowers iNOS expression. Blocking of S1PR1 activity does not completely suppress the effect of LPS on the expression of proinflammatory cytokines in microglial cells. Probably, other S1PRs are involved in this mechanism, or, in part, this effect depends on the S1P intracellular pool. However, further research is necessary in this area.
Based on current knowledge, it can be concluded that S1P plays the role of a second messenger within the microglial cell. TNF-α and cerebral ischemia reperfusion and oxygen-glucose deprivation reperfusion result in increased SphK1 activity. As a result, intracellular S1P levels increase, which in turn increases the activity of TNF receptor-associated factor 2 (TRAF2) [345, 346, 347]. The subsequent activation of E3 ubiquitin ligase eventually results in the activation of NF-κB, followed by the expression of genes dependent on this transcription factor. An example of the effect of this signal transmission pathway is the production of IL-17 which acts neurotoxically [347, 399].
The importance of SphK1, S1P and S1PR1 for inflammatory reactions in the brain allows for the development of a therapeutic approach that could protect this organ from damage. An example of such a therapeutic approach is the use of FTY720, which has been shown to inhibit LPS-induced microglia activation in vitro [397, 400, 401] and in vivo in mice with ischemic lesion [402]. FTY720 acts partly via the disruption of S1PR1, as demonstrated by the use of W146, an antagonist of this receptor [400]. Due to the effect on S1PRs, FTY720 interferes with signal transmission from p38 MAPK without affecting JNK1/2 MAPK [401].
In addition to the effects on inflammatory cytokines in inflammations, FTY720 increases expression of brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF), which both have a neuroprotective effect [400]. In contrast, FTY720 does not alter the production of IL-6, IL-10, IL-12p40 and TNF-α in microglial cells activated by CD40L or toll-like receptor 3 (TLR3) ligand [403]. This shows that the effect of S1PRs and so the action of FTY720 can occur only in some immune responses.
Sphingosine-1-phosphate and myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSC) are a very important element in GBM mechanisms. These cells are present in significant numbers in the GBM tumor. It is estimated that they represent 40%±20% of all CD11b+ cells in this tumor [176]. GBM patients also have an elevated number of these cells in the blood. MDSC are mainly involved in cancer immune evasion but also in angiogenesis and cancer cell migration [404, 405].
In the functioning of MDSC, an important role is played by S1P, as evidenced by experiments involving FTY720. This drug caused an in vivo increase in MDSC activity in the spleen of murine sclerodermatous chronic graft-versus-host disease [406] and in the spleen and liver of the immune-mediated hepatic injury model [407] and in tumors [408]. Due to these properties, FTY720 silences the immune response, protecting the organs from damage, but also participating in tumor processes.
MDSC accumulation is dependent on increased expression of CXCL1/GROα and CXCL2/GROβ as well as increased expression on the MDSC receptor for these chemokines: CXCR2 [407]. In the tumor, activation of the S1PR3-ERK1/2 MAPK pathway on MDSC by FTY720 results in increased expression of granulocyte-macrophage colony-stimulating factor (GM-CSF), resulting in MDSC accumulation in the tumor niche and autocrine stimulation of immunosuppressive functions of these cells [408–410]. These results show that S1PR3 activation by carcinogenic S1P can stimulate MDSC immunosuppressive activity in the tumor niche.
Prolonged exposure to FTY720 also causes a disturbance in the transmission of signals from S1PR1 that independently of PI3K-PKB reduces the activity of mTOR, thus increasing the expression of iNOS in the MDSC [407]. An increase in the concentration of NO causes the differentiation and stimulation of Treg function, which is as an important mechanism of the immunosuppressive effect of FTY720 on these cells. However, such an action can help facilitate the development of cancer [411, 412].
The balance of the effect of S1P on S1PR1 and S1PR3 in the activity of MDSC in tumor niche requires further studies.
Sphingosine-1-phosphate and regulatory T cells
S1P also influences the function of Treg cells. In naïve CD4+ T cells S1PR1 causes activation of mTOR [413]. The activation of this pathway causes Smad3 malfunction. Thus, S1P and FTY720 disrupt in vitro and in vivo cell differentiation to Treg but stimulate differentiation into cytotoxic TH1. This mechanism is an element of a negative feedback mechanism that inhibits overly extensive TGF-β action, as the latter causes an increase in SphK1 expression. S1P then disrupts the action of TGF-β [413]. However, FTY720 in other experimental conditions causes permanent down-regulation and degradation of S1PR1 which interferes with signal transduction through the receptor [377–380]. This enhances the immunosuppressive effect.
In vivo experiments show that FTY720 causes differentiation and an increase in Treg numbers in the spleen [414], but FTY720 does not affect the proliferation of these cells [414, 415]. Wolf et al. have shown that FTY720 does disrupt Treg proliferation by inhibiting IL-2-dependent STAT5 phosphorylation [416]. In inflammatory reactions, FTY720 causes in vivo retention of Treg in lymph nodes near the inflammatory sites, but not from the spleen [417]. An increase in Treg numbers results in an immune response near the lymph nodes [418].
S1P participates in the silencing of the immune response in the tumor microenvironment. S1P causes the S1PR1-mediated activation of STAT3, and thereby accumulation of Treg in the tumor niche, as evidenced in an in vivo model of the B16 melanoma cell line, MB49 bladder carcinoma line, and in patients with breast cancer [419, 420]. So far no study has analyzed the effect of S1P on Treg in the tumor niche. Research on FTY720 show that this drug supports the functions of these cells. The active form of FTY720-P induces an increased expression of TGF-β1 and FoxP3 marker in Treg cells [415, 421]. On the other hand it does not cause a significant increase in IL-10 production in these cells. In addition, FTY720 expresses the Foxp3 marker on Foxp3-CD4+T cells in vivo [417]. This indicates that S1P has an opposite, anti-cancer effect by disturbing the functions of Treg.
Sphingosine-1-phosphate and neutrophils
The effect of S1P on neutrophils in the tumor microenvironment is poorly understood. Also, the effect of S1P on neutrophil functions is still debatable. In vivo and in vitro murine models showed that the knockout of SphK1 or SphK2 had no effect on the migration and respiratory burst of neutrophils [422]. However, other experiments show the importance of the SphK1-S1P pathway in the physiology of these cells. The activation of S1PR1 results in the in vivo infiltration of neutrophils during inflammatory reactions [423]. Different inflammatory reactions depend on different mechanisms. During an allergic response, neutrophil infiltration is dependent on SphK1 activity but not on S1PR1, S1PR2 and S1PR3 activity [424].
The direct effect of S1P on neutrophils consists of a moderate inhibition of neutrophil migration via HUVEC and inhibits chemotaxis stimulated by IL-8/CXCL8 or formyl-methionyl-leucyl-phenylalanine (fMLP) [425]. Indirectly, S1P acts on neutrophil migration by causing increased expression of IL-8/CXCL8, a chemokine acting on neutrophils. The effect of S1P on IL-8/CXCL8 production has been demonstrated in normal epithelial virus-transformed BEAS-2B cell line [426–428], A549 lung carcinoma line [429] and human airway smooth muscle [430]. This mechanism is involved in airway inflammation. In BEAS-2B cells the effect of S1P on the expression of IL-8/CXCL8 depends on the activation S1PR2 [428]. This enables the activation of NF-κB and an increase in IL-8/CXCL8 expression. Importantly, this effect is independent of EGFR. In BEAS-2B cells S1P can also activate ERK1/2 MAPK, depending on phospholipase D (PLD) in these cells [426, 427]. ERK1/2 MAPK and PLD activation may also involve an increase in intracellular Ca2+ concentration, as demonstrated in experiments on A549 cells [429].
Activation of ERK1/2 MAPK results in an increase in IL-8/CXCL8 expression. The mechanism of S1P effect on IL-8/CXCL8 expression is cell dependent. In HUVEC S1P increases expression of IL-8/CXCL8 by activating S1PR1 and S1PR3 [343]. In human airway smooth muscle isolated from patients, the effect of S1P on the expression of IL-8/CXCL8 was dependent on p38 and ERK1/2 MAPK, but independent of NF-κB [430]. p38 and ERK1/2 MAPK activate mitogen and stress activated kinase 1 (MSK1) which results in an increase in IL-8/CXCL8 expression.
In addition to the effects on chemokines, S1P increases the expression of ICAM-1 on cells such as A549 [429] and HUVECs [343]. This helps in the diapedesis of neutrophils. S1P also increases IL-8/CXCL8 expression in ovarian cancer cells such as HEY, OCC1 and SKOV3 [431]. The effect of S1P on cells in the GBM niche requires further studies. S1P is mainly synthesized by GSC [305]. If S1P exerts a chemotactic effect on neutrophils via IL-8/CXCL8 then this may explain the presence of these cells near the GSC [185]. However, the association of S1P with the recruitment and distribution of neutrophils in GBM has yet to be investigated.
Neutrophils are short-lived cells that undergo rapid apoptosis [432]. Activation of these cells by pro-inflammatory factors blocks the apoptosis, with an important role played by SphK1: an LPS-induced increase in the expression and activity of SphK1 inhibits the intensity of neutrophil apoptosis via activation of PI3K [433] and p38 MAPK [434, 435]. A similar mechanism occurs in the activity of GM-CSF [433].
Extracellular S1P and SphK1 activity in cells increases the respiratory burst in activated neutrophils. In particular, studies on neutrophil activation by fMLP [433, 436, 437] and activation of the receptor for immunoglobulin Fcγ [438] show an increase in the production of S1P in immune responses, which augments the respiratory burst in activated neutrophils. S1P affects the activity of NADPH oxidase in two ways. It activates the PI3K-PKB pathway [433, 437] and independently of PI3K it increases intracellular Ca2+ concentration [433, 434]. The increase in intracellular Ca2+ concentration results in activation of p38 MAPK and consequently S100A8/A9 translocation and thereby an increase in NADPH oxidase activity [434]. However, this impact still requires further research because Zemann et al. had earlier shown that a knockout of the SphK1 gene did not affect the intensity of the respiratory burst induced by fMLP [422].
Enzymes involved in S1P production may also inhibit the respiratory burst. In particular, LPS causes increased expression of SphK1 in neutrophils [439]. This protein, regardless of its enzymatic activity, stabilizes JNK MAPK and thus distorts the signal transmission through this kinase. Consequently, it reduces NADPH oxidase activation.
Neutrophils accumulate in GBM tumors, which results in a deterioration in prognosis for patients [186, 187]. In the tumor niche, neutrophils secrete many substances involved in angiogenesis, migration and invasion of tumor cells and in tumor immune evasion [127, 440]. Nevertheless, the significance of these cells in the context of cancer processes is poorly understood. The impact of S1P on tumor neutrophils is even less understood. However, in vitro studies show that S1P activity on neutrophil is similar to the behavior (migration and apoptosis inhibition) of these cells in the tumor niche. Significantly, the respiratory burst in neutrophils associated with cancers is at a low level [127]. S1P does increase the respiratory burst, but in the tumor microenvironment there are no substances that stimulate it.
MULTI-DRUG THERAPY AGAINST SECRETORY FACTORS
Therapeutic strategies for the treatment of glioblastoma multiforme
Treatment limitations such as high average age onset, tumor localization, and still inadequate knowledge of GBM pathophysiology, are cited as factors contributing to the short median survival [441]. Currently, standard therapeutic procedures in GBM include surgical resection of tumors followed by radiotherapy and chemotherapy. Surveys so far confirm that tumor resection should be performed to the maximum extent possible [442]. The next step in GBM treatment is radiotherapy, i.e. external beam radiation therapy [443] or stereotactic radiosurgery (gamma knife) [444]. Radiotherapy is combined with chemotherapy, in particular fotemustine or cyclically administered TMZ [445, 446]. Both these compounds are alkylating agents and thus, by damaging DNA, they inhibit cell proliferation. Nevertheless, the currently used therapeutic approach to GBM treatment is very ineffective, with very low 5-year survival [4]. Therefore, new therapeutic methods are being sought.
Novel therapies
Novel therapies are being developed to support the classic GBM treatment. Many of these therapies are still at clinical level [447]. The novel therapies include, among others, calorie restricted ketogenic diet [448–451], immunotherapy [452–455] and the use of oncolytic viruses [456–458]. New chemotherapeutics are also being developed to generate personalized therapy [459–460].
Calorie restricted ketogenic diet
Changes in the metabolism of carbohydrates and fats are one of the ‘hallmarks of cancer’ [13, 14]. First demonstrated by Otto Warburg, after whom it was named the Warburg effect [461, 462], the phenomenon is based on the intense anaerobic glycolysis that produces lactic acid and acidification in the tumor microenvironment. Lactic acid and low pH in the tumor are one of the most important elements of the tumor microenvironment which cause cancer immune evasion [463]. To some extent, the Warburg effect also makes tumor cells dependent on carbohydrates as a major source of energy, as cancer cells are not able to use ketone bodies as a source of energy. Therefore the implementation of the ketogenic diet, i.e. carbohydrate-restricted diet, causes the ‘starvation’ of cancer cells, including GBM [449, 464]. Normal cells, including nerve cells, are able to metabolize ketone bodies. Due to the metabolic difference between GBM and non-cancer cells, a combination of a calorie restricted ketogenic diet with a standard therapeutic approach is proposed [448–451].
Immunotherapy
Certain hopes are also linked to two therapeutic approaches, which may act on non-cancer cells or directly on tumor cells. The first approach targets cells associated with the tumor, particularly Treg, macrophages and microglia, which have a significant effect on tumor immune evasion [59, 465]. The second approach aims at stimulating the cells of the immune system to destroy cancer cells [452–455]. The combination of these two strategies is also advocated because of tumor immune evasion processes that compromise the effects of immunotherapy [238, 454, 466]. Therefore, the use of antitumor immunostimulant drugs, especially the use of pro-inflammatory cytokines, should increase the therapeutic effects of immunotherapy. This therapeutic approach, as well as immunotherapy itself, specifically destroy tumor cells. As a result, it has fewer side effects compared to non-specific drugs destroying dividing cells [467–470].
Multi-drug therapy as a strategy against glioblastoma multiforme: personalized therapy
The ongoing research on GBM continues to reveal specific mechanisms in the development of GBM, which helps develop therapies targeted at a specific enzyme, tissue hormone, or other specific tumorigenic agent in a particular patient. This is known as personalized therapy [459, 460].
Nevertheless, GBM is a tumor with a very high intratumoral heterogeneity. GBM cells in each patient exhibit a different sensitivity to a given drug. It is estimated that 1/4 of all GBM cells in a given patient are resistant to TMZ and 1/10 are very susceptible to this drug [22]. Therefore, the use of a single drug in GBM results in unsatisfactory therapeutic outcomes. An example of this is TMZ, which, when given to patients undergoing radiotherapy and neurosurgical intervention, results in an increase in the 5-year survival from 1.9% to 9.8% [4]. One also should not forget about the serious side effect of antineoplastic drugs. The use of many drugs and therapeutic approaches at the same time will result in compounding side effects [471].
Multi-drug therapy as a strategy against intratumoral heterogeneity
The extension of the personalized therapy may be a multi-drug therapy, with particular emphasis on the secretory factors in a tumor. Using only one drug often causes GBM recurrence, because a significant percentage of tumor cells are resistant to the drug [22]. It is much less likely to find a tumor cell resistant to two drugs at the same time, and even less so to five drugs. If TMZ is used in addition to radiotherapy, it can increase the 5-year survival rate 5 times. The use of an additional drug can further increase this rate [308, 472–476]. It is best to include a drug that attacks a GBM specific target that does not have a significant function in healthy cells. As a result, the side effects of this drug will be smaller. One example of this is the use of drugs against CMV infection [475, 477, 478].
When choosing drugs for a multidrug therapy, how they interwork should be considered. One should be chosen from the ‘hallmarks of cancer’ and then match all drugs to the selected target. At the same time, GBM contains many mechanisms that trigger the stimulation of proliferation, apoptosis inhibition or tumor immune evasion. The use of four drugs inhibiting proliferation and one specifically impairing tumor immune evasion results in the response of tumor cells similar to when only anti-proliferative drugs are used. Tumor immune evasion mechanisms vary in the tumor microenvironment. Blocking of one signaling particle leads to the drug’s action only in a small part of the tumor (i.e. due to intratumoral heterogeneity), or a lack of therapeutic effects associated with the complementary action of other immune evasion mechanisms.
NT, S1P, GDF-15 and CMV infection have almost identical properties and functions. Within GBM, their concentrations are increased, and the expression of their receptors and enzyme activity responsible for their production also increase. All these factors have implications for all significant ‘hallmarks of cancer’ such as stimulated proliferation, inhibited apoptosis, tumorigenic effect on GSC, angiogenesis, migration, invasion, and tumor immune evasion. In addition, the increase in the concentrations of these factors is not local, but gradually occurs throughout the tumor. This offsets certain problems associated with intratumoral heterogeneity. One may even assume that in the tumor microenvironment there is a pool of all the secretory factors that complement and cooperate with one another. Therefore, multi-drug therapy may be used to interfere with various secretory factors. As a result, tumorigenic and antitumoral imbalance in the tumor microenvironment may be impaired, consequently leading to the destruction of all tumor cells [479].
Antineoplastic agents fighting cytomegalovirus infection
Based on knowledge used to develop the currently used therapies, cytostatics are used to treat cancer [480]. These drugs or X-rays destroy only dividing cells. Due to the fact that GSC are rarely-dividing cells with drug resistance enzymes, this therapeutic approach has only the short-term effect of decreasing tumor mass [481]. During such therapy, GSC are not destroyed, which results in the recurrence of cancer. Evaluated on the basis of available literature, the role of CMV in tumoral mechanisms in GBM brings some therapeutic hopes. In particular, tropism of CMV for CD133+ GSC and the significance of this virus in GSC functions make these cells significant in CMV/GBM therapy [46, 48–50].
The growth of a tumor associated with chronic CMV infection takes years. During this process Darwinian-like selection of cells occurs, in terms of tumor processes, resulting in the formation of advanced cancer [12]. In these, tumor processes are fully dependent on the pro-tumor properties of CMV. This leads to the susceptibility of such tumors to antiviral drugs used against CMV [473, 475]. Currently, the proposed approach is to combine radiotherapy and TMZ with the use of antiviral drugs or immunotherapy against CMV.
Cidofovir and valganciclovir are being tested as antiviral drugs in CMV infection, while other new drugs are also being developed. Cidofovir is an analog of cytosine. It inhibits DNA polymerase activity not only in CMV but also in other viruses [482]. This counteracts CMV replication. However, the activity of cidofovir is very non-specific [473]. This drug is also a substrate for non-viral DNA polymerases in dividing cells. As a result, cidofovir causes in vitro DNA double-stranded breaks and apoptosis of U-87 MG and SF7796 cells, independently of CMV infection [473]. Also, this drug in vivo enhances the survival of athymic mice intracranially inoculated with U-87 MG and SF7796 cells [473].
Another anti-CMV drug tested against GBM is valganciclovir. This drug is specifically phosphorylated by the UL97 kinase viral protein [482]. This reaction is necessary to convert this prodrug into active ganciclovir. Because valganciclovir penetrates the blood-brain barrier, it can be used in GBM therapy [483, 484]. Combined with standard therapy in clinical trials, valganciclovir brings a significant increase in mean survival rate. The effects of valganciclovir can occur after only 6 months of therapy with this prodrug. At this point the 4 year postoperative survival and median overall survival increase from 5.9% and 13.1 months to 27.3% and 24.1 months, respectively [475]. Continuation of valganciclovir therapy can significantly increase median overall survival to 56.4 months [474]. Also the combination of valganciclovir with bevacizumab, radiotherapy and TMZ increases the 6-month progression-free survival and average survival [485].
In addition to the use of antiviral drugs, researchers also recommend the use of immunotherapy against CMV in GBM treatment, especially the use of autologous dendritic cells [466] or autologous cytotoxic T cells [478, 486–488] vaccinated with specific CMV antigens. Autologous dendritic cells are sensitized to the pp65 viral protein and then are introduced into the body of the patient. The combination of this therapeutic approach with neurosurgery, radiotherapy and TMZ increases overall survival from 19.2 months to 41.1 months and long-term progression-free survival from 8.0 months to 25.3 months [466]. In addition to dendritic cells, CMV/GBM immunotherapy uses autologous cytotoxic T cells that are sensitized to CMV antigens by autologous dendritic cells [488–490] or autologous peripheral blood mononuclear cells [478, 486, 487]. The use of autologous cytotoxic T cells in GBM therapy increases the mean overall survival from 4.3 months to 79.8 months [478].
Antitumor agents directed against neurotensin and neurotensin receptor
Many anti-cancer drugs directed against NT and NTSR1 (Table 2) are currently being tested. In the research of new GBM therapies, the most significant in this group of compounds is a NTSR1 antagonist: SR48692 [202, 491–493]. This compound exhibits antitumor properties in vitro by inhibiting proliferation and cell migration of U-87 MG GBM cells and GL261 gliomas [202]. Also SR48692 has therapeutic properties in vivo in C57BL/6 mice intracranially inoculated with GL261 cells [202].
Table 2: Experimental anti-cancer drugs and pharmaceutical agents against NT and NTSR1
Drug |
Mechanism of action |
Research model |
Bibliography |
|---|---|---|---|
SR48692 |
NTSR1 antagonist |
A375 melanoma cell line in vitro |
202, 491-493 |
Neurotensin analogs |
NTR1-targeted drug |
HT-29 colorectal adenocarcinoma cell line in vitro and in vivo |
494-499 |
DOTA- and DTPA- chelated neurotensin analogs |
NTR1-targeted drug |
HT-29 colorectal adenocarcinoma cell line in vitro and in vivo |
500-502 |
Neurotensin Branched Peptides |
NTR1-targeted drug |
HT-29 human adenocarcinoma cell line in vitro and in vivo |
503, 504 |
Neurotensin polyplex |
Gene transfection |
N1E-115 neuroblastoma cell line in vitro and in vivo |
505-507 |
Compounds that destroy tumor cells which overexpress NTSRs are also being tested on models of other tumors. These are NT derivatives labelled with radioactive isotopes or cytostatic drugs such as methotrexate or gemcitabine. An example of NT derivatives is a modified fragment of this hormone that does not undergo rapid proteolytic degradation [494–499]. Such an NT analog may be further chelated by diethylenetriamine pentaacetic acid (DTPA) or 1,4,7,10-tetraazacyldodecane-1,4,7,10-tetraacetic acid (DOTA) to enhance stability [500–502]. Another possibility are oligobranched peptides containing a NT fragment in their sequence which is recognized by NTSR1 [503, 504]. By labeling such NT derivatives with radioactive isotopes or cytostatic drugs, such drugs specifically destroy tumor cells that overexpress NTSR1. In addition to this therapeutic approach, a gene therapy is being tested in which a NT polyplex is used, i.e. a vector composed of NT, poly-L-lysine, and a plasmid encoding an antitumor protein such as thymidine kinase [505–507]. Nevertheless, these NT derivatives, used in the treatment of other cancers, do not cross the blood-brain barrier and so cannot be used in GBM therapy. Hence the search for the new methods of weakening the blood-brain barrier or carrying drugs through this barrier [508–511].
Drugs directed against growth differentiation factor-15
The GDF-15 receptor is currently unknown. Therefore, the most important route in anticancer therapy directed against this secretory factor are antibodies neutralizing GDF-15 [512]. Nevertheless, the blood-brain barrier prevents the use of these antibodies in GBM therapy [509–511].
Drugs targeted at the sphingosine-1-phosphate pathway
In anti-cancer therapy directed against S1P, much attention is given to the inhibitors of SphKs [513–516]. In particular, the best known is the specific inhibitor SphK1: 2R,3S,4E)-N-methyl-5-(4’-pentylphenyl)-2-aminopent-4-ene-1,3-diol (SK1-I) both SphK: 2-(p-hydroxyanilino)-4-p-chlorophenyl)thiazole (SKI-II) [517, 518]. Their efficacy against GBM has also been confirmed in vitro on various cell lines such as A-172, LN-18, LN-229, U-87 MG, U-251 MG and T98G [301, 302, 308, 472]. Also on the in vivo model, SphKs inhibitors have shown antitumor properties against GBM. SK1-I reduces tumor mass, inhibits angiogenesis and causes apoptosis of tumor cells, and increases the survival of nude mice intracranially inoculated with LN-229 cells [301]. In addition, SKI-II destroys tumor cells in vivo in nude mice inoculated subcutaneously with U-87 MG cells [302]. In GBM therapy with SphK inhibitors, it is also proposed to combine these drugs with the currently applied therapy, in particular with TMZ, to increase the therapeutic effect [308, 472].
In vitro and in vivo studies indicate that FTY720/fingolimod micromolar concentration has antitumor properties [519, 520]. FTY720 in vitro inhibits proliferation and migration, and causes apoptosis of GBM cell lines such as U-87 MG, U-251 MG, T98G and GSC isolated from GBM tumors [521–525]. This in vivo compound reduces tumor mass, causes apoptosis and necrosis of tumor cells, and increases survival of nude mice intracranially inoculated with GSC from GBM tumors [522]. Also, FTY720 produces the same effects in nude mice subcutaneously inoculated with U-87 MG and U-251 MG cells [524]. FTY720 penetrates the blood-brain barrier and can therefore be used in GBM therapy [391, 522, 524]. Activation of S1PRs causes GBM cell proliferation. Nevertheless, tumor cells are characterized by frequent mutations in the p53 protein [526]. This results in a lack of stimulation of cell proliferation by S1PRs activation [304] and thereby enhances the antitumor activity of FTY720 that is dependent and independent of these receptors [520]. At nanomolar concentrations, FTY720 causes down-regulation and degradation of S1PRs [377]. Its antineoplastic properties can only be observed at micromolar concentrations, which indicates the the mechanisms of the antineoplastic action of FTY720 is independent of S1RPs
FTY720 also inhibits angiogenesis and cancels the action of VEGF by reducing vascular permeability and reduced sprouting of HUVEC at concentrations below 1 nM, by acting on CXCR4 and S1PR [367, 527]. CXCR4 receptors are receptors whose activation may be involved in angiogenesis. This receptor may be regulated by S1PRs [528–530]. Nevertheless, the effect of FTY720 on CXCR4 in angiogenesis inhibition should be further explored. Also in an in vivo model, FTY720 inhibited tumor growth and angiogenesis in mice inoculated with PLC/PRF/5 and Huh7 human hepatocellular carcinoma lines at a dose of 10mg/kg per day [531], B16/BL6 murine melanoma at 3mg/kg daily [378], and Lewis lung carcinoma LLC1 line at a dose of 10 mg/kg daily [527].
However, the GBM tumor does not consist only of tumor cells but also of tumor-associated cells, in particular immune cells. Immune reactions also play a very important role in tackling cancer. The use of immunosuppressive drugs such as FTY720 results in the impairment of the immune system and consequently may facilitate the development of GBM as well as other tumors [411, 412].
In addition to SphK inhibitors, researchers postulate the use of S1PR antagonists in the treatment of tumors [532, 533]. It is also advocated to use S1P-neutralizing antibodies acting on many types of cancer [534–537]. However, this therapeutic approach has not been studied in terms of glioma and GBM, because the blood-brain barrier significantly impedes the transmission of antibodies to the microenvironment of these tumors. On the other hand, some hope may lie in the search for new methods of transmitting various substances through the barrier [509–511].
Anti-cancer drugs directed against other secretory factors
Nonsteroidal anti-inflammatory drugs (NSAIDs), in particular selective COX-2 inhibitors and nonselective cyclooxygenase inhibitors, have been reported to reduce the production of PGE2 [359, 360, 538–540], or CD39 and CD73 inhibitors reducing adenosine production [541–544]. All of the therapeutic agents that have been mentioned so far can be used in combination with drugs that interfere with the action of tissue hormones with a greater importance for GBM, for example, tyrosine kinase inhibitors or anti-EGFR or anti-EGFRvIII antibodies [545–547].
Problem I: blood-brain barrier
The blood-brain barrier protects the central nervous system against various toxic and biological chemicals. It is impervious to antibodies and a significant number of drugs [509–511]. This greatly hampers the treatment of diseases in this organ. Although within GBM the barrier is suppressed, some GBM parts are still protected by it [509–511]. Therefore, NT labelled with radioactive isotopes or cytostatic drugs, as well as specific anti-S1P or anti-GDF-15 antibodies or some of the aforementioned anti-cancer drugs, are ineffective in GBM therapy [507]. This is why researchers are looking for new therapeutic substances that are able to penetrate this barrier. Another field is the search for drugs that would weaken the action of the blood-brain barrier, or for compounds that would carry conjugated substances through this barrier. One example is angiopep-2, which carries NT through this barrier [508]. Advancement of knowledge in solving this problem is necessary in the development of new therapeutic methods in GBM.
Problem II: compounding side effects
The use of multidrug therapies that target physiological factors presents a high risk of side effects compounding [471]. Therefore, lower concentrations of all drugs should be used so that only fewer of the enzymes are inactivated. Lower antibody concentrations should also neutralize some of the hormones. As a result of the development of tumoral processes, there are many more aforementioned enzymes, tissue hormones or receptors in the tumor niche or a cancer cell then in non-cancer tissue [202, 262, 263, 288, 289, 292–294]. If these molecules are chosen as the target of therapy, it is more likely that the drug acts at lower concentrations in the tumor cell than in a healthy cell [493]. In this case, the effect of such a therapy on cancer cells would be more toxic than for healthy cells. By reducing the concentration of secretory factors such as NT, S1P, GDF-15, from very high to physiological or even lower levels, it may have a destructive effect on the viability of the tumor cell. In contrast, in the healthy cell a slight decrease in the aforementioned hormones is going to have a much smaller adverse effect. The tumor microenvironment selects tumor cells in a certain direction [479]. As a result, tumor cells are dependent on this environment, in particular on secretory factors, which are very often elevated during cancer development and act on all ‘hallmarks of cancer’.
Problem III: therapy duration
GBM is a cancer that recurs despite surgical intervention, radiotherapy and chemotherapy. This is associated with the dissemination of cancer cells across many areas of the brain. After the excision of the main tumor, tumor cells are distributed throughout the entire brain. Over time, they become activated and a new relapse site emerges. Therefore, the effects of some experimental therapies are only visible after more than 6 months of taking the drug [474, 475].
A therapeutic approach based on the interference with the tumor microenvironment disturbs the development of cancer and the formation of a relapse site. Nevertheless, to achieve some therapeutic success, it is required to destroy all cancer cells which create relapse sites. Therefore, therapy must last until this goal is completed.
CONCLUSION - INTRATUMORAL HETEROGENEITY AS A TARGET OF RESEARCH
To better understand the effects of multidrug therapies, it is important to focus on the changes that occur in tumor processes, and in particular those that lead to intratumoral heterogeneity. Many changes in cancer cells are interrelated, and so GBM tumor cell subtypes exhibit specialization and play different functions [20, 21]. The discovery of patterns of changes in tumor cells will help divide them according to their susceptibility to particular drugs [548]. This will also allow an understanding of the interrelationships between the individual cells in the tumor and, consequently an ability to interfere with the communication between the cells [20, 21]. In this way, it will be possible to develop adequate multidrug therapies with 100% effectiveness and minimal side effects.
Abbreviations
ATF5 - Activating transcription factor 5; CCL - CC motif chemokine ligand; CDK - Cyclin-dependent kinase; CMV - Cytomegalovirus; CTGF - Connective tissue growth factor; CX3CL - C-X3-C motif chemokine ligand; CXCL - C-X-C motif chemokine ligand; EGF - Epidermal growth factor; EGFR - Epidermal growth factor receptor; ERK1/2 - Extracellular signal-regulated kinase 1 and 2; FAK - Focal adhesion kinase; GBM - Glioblastoma multiforme; GDF-15 - Growth differentiation factor-15; GSC - Glioblastoma stem cells; HIF - Hypoxia inducible factor; HLA - Human leukocyte antigen; HO-1 - Heme oxygenase 1; HUVEC - Human umbilical vein endothelial cells; IDO - Indoleamine 2,3-dioxygenase; IE86 - Immediate early 86; IGF-1R - Insulin-like growth factor 1 receptor; IL - Interleukin; LPS - Lipopolysaccharide; MAPK - Mitogen-activated protein kinase; MCP-1 - Monocyte chemoattractant protein 1; MHC - Major histocompatibility complex; MIP - Macrophage inflammatory protein; MMP - Matrix metalloproteinase; NT - Neurotensin; NTSR1-4 - NT receptor subtype 1-4; PAI-1 - Plasminogen activator inhibitor-1; PDGF - Platelet-derived growth factor; PI3K - Phosphatidylinositol-4,5-bisphosphate 3-kinase; PKB - Protein kinase B; PKC - Protein kinase C; PLC-β - Phospholipase C-β; PPARγ - Peroxisome proliferator-activated receptor γ; RANTES - Regulated on activation, normal T-cell expressed and secreted; S1P - Sphingosine-1-phosphate; S1PR1-5 - S1P receptor 1-5; sNTSR3 - Soluble NT receptor subtype 3; SphK - Sphingosine kinase; SPL - S1P lyase; SPP - S1P-catalyzed phosphohydrolase; STAT - Signal transducer and activator of transcription; TAM - Tumor-associated macrophages; TGF-β - Transforming growth factor β; TMZ - Temozolomide; TNF-α - Tumor necrosis factor α; UPA - Urokinase-type plasminogen activator; uPAR - Receptor for urokinase-type plasminogen activator; VEGF - Vascular endothelial growth factor; vIL-10 - Viral interleukin-10.
Author contributions
Jan Korbecki - literature search and review, writing the manuscript, final acceptance of the manuscript.
Izabela Gutowska - participated in writing the manuscript.
Ireneusz Kojder - participated in writing the manuscript.
Dariusz Jeżewski - final acceptance of the manuscript.
Marta Goschorska - participated in writing the manuscript.
Agnieszka Łukomska - participated in writing the manuscript.
Anna Lubkowska - final acceptance of the manuscript.
Dariusz Chlubek - final acceptance of the manuscript.
Irena Baranowska-Bosiacka - manuscript concept, literature search and review, writing the manuscript, final acceptance of the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was supported by the statutory budget of the Department of Biochemistry and Medical Chemistry, Pomeranian Medical University.
REFERENCES
1. Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006; 2:494–503.
2. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014; 16:iv1–iv63.
3. Chen J, Xu T. Recent therapeutic advances and insights of recurrent glioblastoma multiforme. Front Biosci (Landmark Ed). 2013; 18:676–684.
4. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459–466.
5. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014; 344:1396–1401.
6. Parker NR, Hudson AL, Khong P, Parkinson JF, Dwight T, Ikin RJ, Zhu Y, Cheng ZJ, Vafaee F, Chen J, Wheeler HR, Howell VM. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci Rep. 2016; 6:22477.
7. Hu LS, Ning S, Eschbacher JM, Baxter LC, Gaw N, Ranjbar S, Plasencia J, Dueck AC, Peng S, Smith KA, Nakaji P, Karis JP, Quarles CC, et al. Radiogenomics to characterize regional genetic heterogeneity in glioblastoma. Neuro Oncol. 2017; 19:128–137.
8. de Aquino PF, Carvalho PC, Nogueira FC, da Fonseca CO, de Souza Silva JC, Carvalho Mda G, Domont GB, Zanchin NI, Fischer Jde S. A time-based and intratumoral proteomic assessment of a recurrent glioblastoma multiforme. Front Oncol. 2016; 6:183.
9. Paulus W, Peiffer J. Intratumoral histologic heterogeneity of gliomas. A quantitative study. Cancer. 1989; 64:442–447.
10. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavaré S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013; 110:4009–4014.
11. Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR, Park DM. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017; 19:887–896.
12. Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999; 9:M57–M60.
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
15. Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma stem cells: signaling, microenvironment, and therapy. Stem Cells Int. 2016; 2016:7849890.
16. Schneider M, Ströbele S, Nonnenmacher L, Siegelin MD, Tepper M, Stroh S, Hasslacher S, Enzenmüller S, Strauss G, Baumann B, Karpel-Massler G, Westhoff MA, Debatin KM, et al. A paired comparison between glioblastoma “stem cells” and differentiated cells. Int J Cancer. 2016; 138:1709–1718.
17. Denicolaï E, Tabouret E, Colin C, Metellus P, Nanni I, Boucard C, Tchoghandjian A, Meyronet D, Baeza-Kallee N, Chinot O, Figarella-Branger D. Molecular heterogeneity of glioblastomas: does location matter? Oncotarget. 2016; 7:902–913. https://doi.org/10.18632/oncotarget.6433.
18. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, Leversha MA, Mikkelsen T, Brennan CW. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012; 109:3041–3046.
19. Sugimori M, Hayakawa Y, Boman BM, Fields JZ, Awaji M, Kozano H, Tamura R, Yamamoto S, Ogata T, Yamada M, Endo S, Kurimoto M, Kuroda S. Discovery of power-law growth in the self-renewal of heterogeneous glioma stem cell populations. PLoS One. 2015; 10:e0135760.
20. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011; 71:4055–4060.
21. Doucette T, Rao G, Rao A, Shen L, Aldape K, Wei J, Dziurzynski K, Gilbert M, Heimberger AB. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res. 2013; 1:112–122.
22. Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, Cusimano M, Squire JA, Scherer SW, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015; 112:851–856.
23. Landolfo S, Gariglio M, Gribaudo G, Lembo D. The human cytomegalovirus. Pharmacol Ther. 2003; 98:269–297.
24. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002; 62:3347–3350.
25. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, Sampson JH. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008; 10:10–18.
26. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008; 116:79–86.
27. Bhattacharjee B, Renzette N, Kowalik TF. Genetic analysis of cytomegalovirus in malignant gliomas. J Virol. 2012; 86:6815–6824.
28. Rahbar A, Stragliotto G, Orrego A, Peredo I, Taher C, Willems J, Söderberg-Naucler C. Low levels of human cytomegalovirus infection in glioblastoma multiforme associates with patient survival; -a case-control study. Herpesviridae. 2012; 3:3.
29. Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer-Solberg N, Strååt K, Stragliotto G, Söderberg-Nauclér C. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J Clin Virol. 2013; 57:36–42.
30. dos Santos CJ, Stangherlin LM, Figueiredo EG, Corrêa C, Teixeira MJ, da Silva MC. High prevalence of HCMV and viral load in tumor tissues and peripheral blood of glioblastoma multiforme patients. J Med Virol. 2014; 86:1953–1961.
31. Huang R, Qian D, Hu M, Zhang X, Song J, Li L, Chen H, Wang B. Association between human cytomegalovirus infection and histone acetylation level in various histological types of glioma. Oncol Lett. 2015; 10:2812–2820.
32. Shamran HA, Kadhim HS, Hussain AR, Kareem A, Taub DD, Price RL, Nagarkatti M, Nagarkatti PS, Singh UP. Detection of human cytomegalovirus in different histopathological types of glioma in Iraqi patients. Biomed Res Int. 2015; 2015:642652.
33. Fonseca RF, Kawamura MT, Oliveira JA, Teixeira A, Alves G, Carvalho Mda G. The prevalence of human cytomegalovirus DNA in gliomas of Brazilian patients. Mem Inst Oswaldo Cruz. 2012; 107:953–954.
34. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol. 2011; 103:231–238.
35. Ahani N, Karimi Arzenani M, Shirkoohi R, Rokouei M, Alipour Eskandani M, Nikravesh A. Expression of insulin-like growth factor binding protein-2 (IGFBP-2) gene in negative and positive human cytomegalovirus glioblastoma multiforme tissues. Med Oncol. 2014; 31:812.
36. Baumgarten P, Michaelis M, Rothweiler F, Starzetz T, Rabenau HF, Berger A, Jennewein L, Braczynski AK, Franz K, Seifert V, Steinbach JP, Allwinn R, Mittelbronn M, et al. Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics. Neuro Oncol. 2014; 16:1469–1477.
37. Cimino PJ, Zhao G, Wang D, Sehn JK, Lewis JS Jr, Duncavage EJ. Detection of viral pathogens in high grade gliomas from unmapped next-generation sequencing data. Exp Mol Pathol. 2014; 96:310–315.
38. Yamashita Y, Ito Y, Isomura H, Takemura N, Okamoto A, Motomura K, Tsujiuchi T, Natsume A, Wakabayashi T, Toyokuni S, Tsurumi T. Lack of presence of the human cytomegalovirus in human glioblastoma. Mod Pathol. 2014; 27:922–929.
39. Strong MJ, Blanchard E 4th, Lin Z, Morris CA, Baddoo M, Taylor CM, Ware ML, Flemington EK. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus - tumor association. Acta Neuropathol Commun. 2016; 4:71.
40. de Ory F, Ramírez R, García Comas L, León P, Sagües MJ, Sanz JC. Is there a change in cytomegalovirus seroepidemiology in Spain? Eur J Epidemiol. 2004; 19:85–99.
41. Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis. 2006; 43:1143–1151.
42. Zhao P, Ma D, Xue F, Ji C, Wang S, Zhang X, Zhou Y, Yu X. Seroprevalence and risk factors of human cytomegalovirus infection in the eastern Chinese population. Arch Virol. 2009; 154:561–564.
43. Lübeck PR, Doerr HW, Rabenau HF. Epidemiology of human cytomegalovirus (HCMV) in an urban region of Germany: what has changed? Med Microbiol Immunol. 2010; 199:53–60.
44. Bates M, Brantsaeter AB. Human cytomegalovirus (CMV) in Africa: a neglected but important pathogen. J Virus Erad. 2016; 2:136–142.
45. Cobbs CS, Soroceanu L, Denham S, Zhang W, Kraus MH. Modulation of oncogenic phenotype in human glioma cells by cytomegalovirus IE1-mediated mitogenicity. Cancer Res. 2008; 68:724–730.
46. Dziurzynski K, Wei J, Qiao W, Hatiboglu MA, Kong LY, Wu A, Wang Y, Cahill D, Levine N, Prabhu S, Rao G, Sawaya R, Heimberger AB. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin Cancer Res. 2011; 17:4642–4649.
47. Avdic S, Cao JZ, McSharry BP, Clancy LE, Brown R, Steain M, Gottlieb DJ, Abendroth A, Slobedman B. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J Virol. 2013; 87:10273–10282.
48. Matlaf LA, Harkins LE, Bezrookove V, Cobbs CS, Soroceanu L. Cytomegalovirus pp71 protein is expressed in human glioblastoma and promotes pro-angiogenic signaling by activation of stem cell factor. PLoS One. 2013; 8:e68176.
49. Fiallos E, Judkins J, Matlaf L, Prichard M, Dittmer D, Cobbs C, Soroceanu L. Human cytomegalovirus gene expression in long-term infected glioma stem cells. PLoS One. 2014; 9:e116178.
50. Soroceanu L, Matlaf L, Khan S, Akhavan A, Singer E, Bezrookove V, Decker S, Ghanny S, Hadaczek P, Bengtsson H, Ohlfest J, Luciani-Torres MG, Harkins L, et al. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res. 2015; 75:3065–3076.
51. de Wit RH, Mujić-Delić A, van Senten JR, Fraile-Ramos A, Siderius M, Smit MJ. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1α/PKM2 axis in glioblastoma cells. Oncotarget. 2016; 7:67966–67985. https://doi.org/10.18632/oncotarget.11817.
52. Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. Human NK cells can control CMV infection in the absence of T cells. Blood. 2008; 112:914–915.
53. Wu Z, Sinzger C, Frascaroli G, Reichel J, Bayer C, Wang L, Schirmbeck R, Mertens T. Human cytomegalovirus-induced NKG2C(hi) CD57(hi) natural killer cells are effectors dependent on humoral antiviral immunity. J Virol. 2013; 87:7717–7725.
54. Vieira Braga FA, Hertoghs KM, van Lier RA, van Gisbergen KP. Molecular characterization of HCMV-specific immune responses: parallels between CD8(+) T cells, CD4(+) T cells, and NK cells. Eur J Immunol. 2015; 45:2433–2445.
55. Wu Z, Sinzger C, Reichel JJ, Just M, Mertens T. Natural killer cells can inhibit the transmission of human cytomegalovirus in cell culture by using mechanisms from innate and adaptive immune responses. J Virol. 2015; 89:2906–2917.
56. Lischka P, Zimmermann H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr Opin Pharmacol. 2008; 8:541–548.
57. Fisher RA. Cytomegalovirus infection and disease in the new era of immunosuppression following solid organ transplantation. Transpl Infect Dis. 2009; 11:195–202.
58. Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front Oncol. 2015; 5:153.
59. See AP, Parker JJ, Waziri A. The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. J Neurooncol. 2015; 123:405–412.
60. Romo N, Magri G, Muntasell A, Heredia G, Baía D, Angulo A, Guma M, López-Botet M. Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J Leukoc Biol. 2011; 90:717–726.
61. Sinclair J, Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol. 2006; 87:1763–1779.
62. Hargett D, Shenk TE. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A. 2010; 107:20039–20044.
63. Noriega VM, Haye KK, Kraus TA, Kowalsky SR, Ge Y, Moran TM, Tortorella D. Human cytomegalovirus modulates monocyte-mediated innate immune responses during short-term experimental latency in vitro. J Virol. 2014; 88:9391–9405.
64. Goodrum F. Human cytomegalovirus latency: approaching the gordian knot. Annu Rev Virol. 2016; 3:333–357.
65. Kuhn DE, Beall CJ, Kolattukudy PE. The cytomegalovirus US28 protein binds multiple CC chemokines with high affinity. Biochem Biophys Res Commun. 1995; 211:325–330.
66. Beisser PS, Laurent L, Virelizier JL, Michelson S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J Virol. 2001; 75:5949–5957.
67. Vomaske J, Melnychuk RM, Smith PP, Powell J, Hall L, DeFilippis V, Früh K, Smit M, Schlaepfer DD, Nelson JA, Streblow DN. Differential ligand binding to a human cytomegalovirus chemokine receptor determines cell type-specific motility. PLoS Pathog. 2009; 5:e1000304.
68. Hjortø GM, Kiilerich-Pedersen K, Selmeczi D, Kledal TN, Larsen NB. Human cytomegalovirus chemokine receptor US28 induces migration of cells on a CX3CL1-presenting surface. J Gen Virol. 2013; 94:1111–1120.
69. Marchesi F, Locatelli M, Solinas G, Erreni M, Allavena P, Mantovani A. Role of CX3CR1/CX3CL1 axis in primary and secondary involvement of the nervous system by cancer. J Neuroimmunol. 2010; 224:39–44.
70. Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000; 29:305–315.
71. Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003; 979:65–70.
72. Erreni M, Solinas G, Brescia P, Osti D, Zunino F, Colombo P, Destro A, Roncalli M, Mantovani A, Draghi R, Levi D, Rodriguez Y, Baena R, et al. Human glioblastoma tumours and neural cancer stem cells express the chemokine CX3CL1 and its receptor CX3CR1. Eur J Cancer. 2010; 46:3383–3392.
73. Hattermann K, Sebens S, Helm O, Schmitt AD, Mentlein R, Mehdorn HM, Held-Feindt J. Chemokine expression profile of freshly isolated human glioblastoma-associated macrophages/microglia. Oncol Rep. 2014; 32:270–276.
74. Held-Feindt J, Hattermann K, Müerköster SS, Wedderkopp H, Knerlich-Lukoschus F, Ungefroren H, Mehdorn HM, Mentlein R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res. 2010; 316:1553–1566.
75. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008; 216:15–24.
76. Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe. 2010; 8:284–291.
77. Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol. 1999; 155:1557–1567.
78. Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003; 424:456–461.
79. Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008; 455:391–395.
80. Nazarenko I, Hede SM, He X, Hedrén A, Thompson J, Lindström MS, Nistér M. PDGF and PDGF receptors in glioma. Ups J Med Sci. 2012; 117:99–112.
81. Liffers K, Lamszus K, Schulte A. EGFR amplification and glioblastoma stem-like cells. Stem Cells Int. 2015; 2015:427518.
82. McCormick AL. Control of apoptosis by human cytomegalovirus. Curr Top Microbiol Immunol. 2008; 325:281–295.
83. Wang T, Qian D, Hu M, Li L, Zhang L, Chen H, Yang R, Wang B. Human cytomegalovirus inhibits apoptosis by regulating the activating transcription factor 5 signaling pathway in human malignant glioma cells. Oncol Lett. 2014; 8:1051–1057.
84. Strååt K, Liu C, Rahbar A, Zhu Q, Liu L, Wolmer-Solberg N, Lou F, Liu Z, Shen J, Jia J, Kyo S, Björkholm M, Sjöberg J, et al. Activation of telomerase by human cytomegalovirus. J Natl Cancer Inst. 2009; 101:488–497.
85. Kalejta RF, Shenk T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A. 2003; 100:3263–3268.
86. Kalejta RF, Bechtel JT, Shenk T. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol. 2003; 23:1885–1895.
87. Lee K, Jeon K, Kim JM, Kim VN, Choi DH, Kim SU, Kim S. Downregulation of GFAP, TSP-1, and p53 in human glioblastoma cell line, U373MG, by IE1 protein from human cytomegalovirus. Glia. 2005; 51:1–12.
88. Bain M, Sinclair J. The S phase of the cell cycle and its perturbation by human cytomegalovirus. Rev Med Virol. 2007; 17:423–434.
89. Spector DH. Human cytomegalovirus riding the cell cycle. Med Microbiol Immunol. 2015; 204:409–419.
90. Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol. 2001; 75:6022–6032.
91. Cobbs C, Khan S, Matlaf L, McAllister S, Zider A, Yount G, Rahlin K, Harkins L, Bezrookove V, Singer E, Soroceanu L. HCMV glycoprotein B is expressed in primary glioblastomas and enhances growth and invasiveness via PDGFR-alpha activation. Oncotarget. 2014; 5:1091–1100. https://doi.org/10.18632/oncotarget.1787.
92. Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, Lira SA, Söderberg-Nauclér C, Smit MJ. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci Signal. 2010; 3:ra58.
93. Maussang D, Langemeijer E, Fitzsimons CP, Stigter-van Walsum M, Dijkman R, Borg MK, Slinger E, Schreiber A, Michel D, Tensen CP, van Dongen GA, Leurs R, Smit MJ. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009; 69:2861–2869.
94. Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, Soteropoulos P, Cobbs CS. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res. 2011; 71:6643–6653.
95. Xing Y, Wang Y, Wang S, Wang X, Fan D, Zhou D, An J. Human cytomegalovirus infection contributes to glioma disease progression via upregulating endocan expression. Transl Res. 2016; 177:113–126.
96. Cobbs CS, Soroceanu L, Denham S, Zhang W, Britt WJ, Pieper R, Kraus MH. Human cytomegalovirus induces cellular tyrosine kinase signaling and promotes glioma cell invasiveness. J Neurooncol. 2007; 85:271–280.
97. Miller WE, Zagorski WA, Brenneman JD, Avery D, Miller JL, O’Connor CM. US28 is a potent activator of phospholipase C during HCMV infection of clinically relevant target cells. PLoS One. 2012; 7:e50524.
98. Assinger A, Yaiw KC, Göttesdorfer I, Leib-Mösch C, Söderberg-Nauclér C. Human cytomegalovirus (HCMV) induces human endogenous retrovirus (HERV) transcription. Retrovirology. 2013; 10:132.
99. MacManiman JD, Meuser A, Botto S, Smith PP, Liu F, Jarvis MA, Nelson JA, Caposio P. Human cytomegalovirus-encoded pUL7 is a novel CEACAM1-like molecule responsible for promotion of angiogenesis. MBio. 2014; 5:e02035.
100. Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J Virol. 2006; 80:951–963.
101. Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011; 157:151–160.
102. Noriega V, Redmann V, Gardner T, Tortorella D. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res. 2012; 54:140–151.
103. Barel MT, Ressing M, Pizzato N, van Leeuwen D, Le Bouteiller P, Lenfant F, Wiertz EJ. Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J Immunol. 2003; 171:6757–6765.
104. Rölle A, Olweus J. Dendritic cells in cytomegalovirus infection: viral evasion and host countermeasures. APMIS. 2009; 117:413–426.
105. Onno M, Pangault C, Le Friec G, Guilloux V, André P, Fauchet R. Modulation of HLA-G antigens expression by human cytomegalovirus: specific induction in activated macrophages harboring human cytomegalovirus infection. J Immunol. 2000; 164:6426–6434.
106. Pizzato N, Garmy-Susini B, Le Bouteiller P, Lenfant F. Down-regulation of HLA-G1 cell surface expression in human cytomegalovirus infected cells. Am J Reprod Immunol. 2003; 50:328–333.
107. Zhang YH, He M, Wang Y, Liao AH. Modulators of the balance between M1 and M2 macrophages during pregnancy. Front Immunol. 2017; 8:120.
108. Marchesi M, Andersson E, Villabona L, Seliger B, Lundqvist A, Kiessling R, Masucci GV. HLA-dependent tumour development: a role for tumour associate macrophages? J Transl Med. 2013; 11:247.
109. Lenfant F, Pizzato N, Liang S, Davrinche C, Le Bouteiller P, Horuzsko A. Induction of HLA-G-restricted human cytomegalovirus pp65 (UL83)-specific cytotoxic T lymphocytes in HLA-G transgenic mice. J Gen Virol. 2003; 84:307–317.
110. Lee CL, Guo Y, So KH, Vijayan M, Guo Y, Wong VH, Yao Y, Lee KF, Chiu PC, Yeung WS. Soluble human leukocyte antigen G5 polarizes differentiation of macrophages toward a decidual macrophage-like phenotype. Hum Reprod. 2015; 30:2263–2274.
111. Costa H, Xu X, Overbeek G, Vasaikar S, Patro CP, Kostopoulou ON, Jung M, Shafi G, Ananthaseshan S, Tsipras G, Davoudi B, Mohammad AA, Lam H, et al. Human cytomegalovirus may promote tumour progression by upregulating arginase-2. Oncotarget. 2016; 30:47221–47231. https://doi.org/10.18632/oncotarget.9722.
112. Cinatl J Jr, Blaheta R, Bittoova M, Scholz M, Margraf S, Vogel JU, Cinatl J, Doerr HW. Decreased neutrophil adhesion to human cytomegalovirus-infected retinal pigment epithelial cells is mediated by virus-induced up-regulation of Fas ligand independent of neutrophil apoptosis. J Immunol. 2000; 165:4405–4413.
113. Lin YL, Chang PC, Wang Y, Li M. Identification of novel viral interleukin-10 isoforms of human cytomegalovirus AD169. Virus Res. 2008; 131:213–223.
114. Slobedman B, Barry PA, Spencer JV, Avdic S, Abendroth A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J Virol. 2009; 83:9618–9629.
115. Aiba-Masago S, Baba S, Li RY, Shinmura Y, Kosugi I, Arai Y, Nishimura M, Tsutsui Y. Murine cytomegalovirus immediate-early promoter directs astrocyte-specific expression in transgenic mice. Am J Pathol. 1999; 154:735–743.
116. Kawasaki H, Kosugi I, Arai Y, Tsutsui Y. The amount of immature glial cells in organotypic brain slices determines the susceptibility to murine cytomegalovirus infection. Lab Invest. 2002; 82:1347–1358.
117. Tsutsui Y, Kosugi I, Kawasaki H, Arai Y, Han GP, Li L, Kaneta M. Roles of neural stem progenitor cells in cytomegalovirus infection of the brain in mouse models. Pathol Int. 2008; 58:257–267.
118. van Den Pol AN, Mocarski E, Saederup N, Vieira J, Meier TJ. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J Neurosci. 1999; 19:10948–10965.
119. Cheeran MC, Hu S, Yager SL, Gekker G, Peterson PK, Lokensgard JR. Cytomegalovirus induces cytokine and chemokine production differentially in microglia and astrocytes: antiviral implications. J Neurovirol. 2001; 7:135–147.
120. Cheeran MC, Hu S, Gekker G, Lokensgard JR. Decreased cytomegalovirus expression following proinflammatory cytokine treatment of primary human astrocytes. J Immunol. 2000; 164:926–933.
121. Stacey MA, Marsden M, Pham N TA, Clare S, Dolton G, Stack G, Jones E, Klenerman P, Gallimore AM, Taylor PR, Snelgrove RJ, Lawley TD, Dougan G, et al. Neutrophils recruited by IL-22 in peripheral tissues function as TRAIL-dependent antiviral effectors against MCMV. Cell Host Microbe. 2014; 15:471–483.
122. Cheeran MC, Hu S, Sheng WS, Peterson PK, Lokensgard JR. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J Virol. 2003; 77:4502–4515.
123. Cheeran MC, Gekker G, Hu S, Yager SL, Peterson PK, Lokensgard JR. CD4(+) lymphocyte-mediated suppression of cytomegalovirus expression in human astrocytes. Clin Diagn Lab Immunol. 2000; 7:710–713.
124. Mutnal MB, Hu S, Little MR, Lokensgard JR. Memory T cells persisting in the brain following MCMV infection induce long-term microglial activation via interferon-γ. J Neurovirol. 2011; 17:424–437.
125. Lecointe D, Dugas N, Leclerc P, Hery C, Delfraissy JF, Tardieu M. Human cytomegalovirus infection reduces surface CCR5 expression in human microglial cells, astrocytes and monocyte-derived macrophages. Microbes Infect. 2002; 4:1401–1408.
126. Straschewski S, Patrone M, Walther P, Gallina A, Mertens T, Frascaroli G. Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J Virol. 2011; 85:5150–5158.
127. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012; 33:949–955.
128. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014; 6:1670–1690.
129. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, Han Y, Wu M, Zhang L, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016; 76:5671–5682.
130. Avdic S, McSharry BP, Steain M, Poole E, Sinclair J, Abendroth A, Slobedman B. Human cytomegalovirus-encoded human interleukin-10 (IL-10) homolog amplifies its immunomodulatory potential by upregulating human IL-10 in monocytes. J Virol. 2016; 90:3819–3827.
131. Kossmann T, Morganti-Kossmann MC, Orenstein JM, Britt WJ, Wahl SM, Smith PD. Cytomegalovirus production by infected astrocytes correlates with transforming growth factor-beta release. J Infect Dis. 2003; 187:534–541.
132. Zhang L, Li L, Wang B, Qian DM, Song XX, Hu M. HCMV induces dysregulation of glutamate uptake and transporter expression in human fetal astrocytes. Neurochem Res. 2014; 39:2407–2418.
133. Maus A, Peters GJ. Glutamate and α-ketoglutarate: key players in glioma metabolism. Amino Acids. 2017; 49:21–32.
134. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, Weller M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol. 2003; 54:388–392.
135. Pham K, Luo D, Liu C, Harrison JK. CCL5, CCR1 and CCR5 in murine glioblastoma: immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J Neuroimmunol. 2012; 246:10–17.
136. Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006; 25:357–371.
137. Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. 2011; 17:6118–6124.
138. Lokensgard JR, Schachtele SJ, Mutnal MB, Sheng WS, Prasad S, Hu S. Chronic reactive gliosis following regulatory T cell depletion during acute MCMV encephalitis. Glia. 2015.
139. Mutnal MB, Cheeran MC, Hu S, Little MR, Lokensgard JR. Excess neutrophil infiltration during cytomegalovirus brain infection of interleukin-10-deficient mice. J Neuroimmunol. 2010; 227:101–110.
140. Craigen JL, Yong KL, Jordan NJ, MacCormac LP, Westwick J, Akbar AN, Grundy JE. Human cytomegalovirus infection up-regulates interleukin-8 gene expression and stimulates neutrophil transendothelial migration. Immunology. 1997; 92:138–145.
141. Sparer TE, Gosling J, Schall TJ, Mocarski ES. Expression of human CXCR2 in murine neutrophils as a model for assessing cytomegalovirus chemokine vCXCL-1 function in vivo. J Interferon Cytokine Res. 2004; 24:611–620.
142. Miller-Kittrell M, Sai J, Penfold M, Richmond A, Sparer TE. Functional characterization of chimpanzee cytomegalovirus chemokine, vCXCL-1(CCMV). Virology. 2007; 364:454–465.
143. Lüttichau HR. The cytomegalovirus UL146 gene product vCXCL1 targets both CXCR1 and CXCR2 as an agonist. J Biol Chem. 2010; 285:9137–9146.
144. Heo J, Dogra P, Masi TJ, Pitt EA, de Kruijf P, Smit MJ, Sparer TE. Novel human cytomegalovirus viral chemokines, vCXCL-1s, display functional selectivity for neutrophil signaling and function. J Immunol. 2015; 195:227–236.
145. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol. 2011; 104:83–92.
146. Krasnikova TL, Arefieva TI, Pylaeva EA, Sidorova MV. Ingramon, a peptide inhibitor of MCP-1 chemokine, reduces migration of blood monocytes stimulated by glioma-conditioned medium. Bull Exp Biol Med. 2016; 160:480–482.
147. Schachtele SJ, Hu S, Sheng WS, Mutnal MB, Lokensgard JR. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia. 2014; 62:1582–1594.
148. Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J Immunol. 2008; 181:698–711.
149. Bayer C, Varani S, Wang L, Walther P, Zhou S, Straschewski S, Bachem M, Söderberg-Naucler C, Mertens T, Frascaroli G. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J Virol. 2013; 87:67–79.
150. Hamilton ST, Scott GM, Naing Z, Rawlinson WD. Human cytomegalovirus directly modulates expression of chemokine CCL2 (MCP-1) during viral replication. J Gen Virol. 2013; 94:24495–24503.
151. Naing Z, Webel R, Hamilton S, Schmeiser C, Scott G, Marschall M, Rawlinson W. Stimulatory effects of human cytomegalovirus tegument protein pp71 lead to increased expression of CCL2 (monocyte chemotactic protein-1) during infection. J Gen Virol. 2015; 96:1855–1862.
152. Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc Natl Acad Sci U S A. 1999; 96:10881–10886.
153. Wagner FM, Brizic I, Prager A, Trsan T, Arapovic M, Lemmermann NA, Podlech J, Reddehase MJ, Lemnitzer F, Bosse JB, Gimpfl M, Marcinowski L, MacDonald M, et al. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 2013; 9:e1003493.
154. Spencer JV. The cytomegalovirus homolog of interleukin-10 requires phosphatidylinositol 3-kinase activity for inhibition of cytokine synthesis in monocytes. J Virol. 2007; 81:2083–2086.
155. Zuo J, Rowe M. Herpesviruses placating the unwilling host: manipulation of the MHC class II antigen presentation pathway. Viruses. 2012; 4:1335–1353.
156. Odeberg J, Söderberg-Nauclér C. Reduced expression of HLA class II molecules and Iinterleukin-10- and transforming growth factor beta1-independent suppression of T-cell proliferation in human cytomegalovirus-infected macrophage cultures. J Virol. 2001; 75:5174–5181.
157. Nijaguna MB, Patil V, Urbach S, Shwetha SD, Sravani K, Hegde AS, Chandramouli BA, Arivazhagan A, Marin P, Santosh V, Somasundaram K. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J Biol Chem. 2015; 290:23401–23415.
158. Qiu B, Zhang D, Wang C, Tao J, Tie X, Qiao Y, Xu K, Wang Y, Wu A. IL-10 and TGF-β2 are overexpressed in tumor spheres cultured from human gliomas. Mol Biol Rep. 2011; 38:3585–3591.
159. Stevenson EV, Collins-McMillen D, Kim JH, Cieply SJ, Bentz GL, Yurochko AD. HCMV reprogramming of infected monocyte survival and differentiation: a Goldilocks phenomenon. Viruses. 2014; 6:782–807.
160. Collins-McMillen D, Kim JH, Nogalski MT, Stevenson EV, Chan GC, Caskey JR, Cieply SJ, Yurochko AD. Human cytomegalovirus promotes survival of infected monocytes via a distinct temporal regulation of cellular Bcl-2 family proteins. J Virol. 2015; 90:2356–2371.
161. Cojohari O, Peppenelli MA, Chan GC. Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J Virol. 2016; 90:6443–6452.
162. Buchmeier NA, Cooper NR. Suppression of monocyte functions by human cytomegalovirus. Immunology. 1989; 66:278–283.
163. Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res. 2009; 144:329–333.
164. Frascaroli G, Varani S, Moepps B, Sinzger C, Landini MP, Mertens T. Human cytomegalovirus subverts the functions of monocytes, impairing chemokine-mediated migration and leukocyte recruitment. J Virol. 2006; 80:7578–7589.
165. Poglitsch M, Weichhart T, Hecking M, Werzowa J, Katholnig K, Antlanger M, Krmpotic A, Jonjic S, Hörl WH, Zlabinger GJ, Puchhammer E, Säemann MD. CMV late phase-induced mTOR activation is essential for efficient virus replication in polarized human macrophages. Am J Transplant. 2012; 12:1458–1468.
166. McCormick AL, Roback L, Livingston-Rosanoff D, St Clair C. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J Virol. 2010; 84:5108–5123.
167. Kren L, Muckova K, Lzicarova E, Sova M, Vybihal V, Svoboda T, Fadrus P, Smrcka M, Slaby O, Lakomy R, Vanhara P, Krenova Z, Michalek J. Production of immune-modulatory nonclassical molecules HLA-G and HLA-E by tumor infiltrating ameboid microglia/macrophages in glioblastomas: a role in innate immunity? J Neuroimmunol. 2010; 220:131–135.
168. Hengel H, Reusch U, Geginat G, Holtappels R, Ruppert T, Hellebrand E, Koszinowski UH. Macrophages escape inhibition of major histocompatibility complex class I-dependent antigen presentation by cytomegalovirus. J Virol. 2000; 74:7861–7868.
169. Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006; 42:768–778.
170. Schober A, Zernecke A. Chemokines in vascular remodeling. Thromb Haemost. 2007; 97:730–737.
171. Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol. 2008; 28:1928–1936.
172. Frascaroli G, Varani S, Blankenhorn N, Pretsch R, Bacher M, Leng L, Bucala R, Landini MP, Mertens T. Human cytomegalovirus paralyzes macrophage motility through down-regulation of chemokine receptors, reorganization of the cytoskeleton, and release of macrophage migration inhibitory factor. J Immunol. 2009; 182:477–488.
173. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008; 83:1136–1144.
174. Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, Holtman IR, Wang X, Eggen BJ, Boddeke HW, Hambardzumyan D, Wolf SA, Kettenmann H. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015; 10:e0116644.
175. Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, Holland EC, Synowitz M, Wolf SA, Kettenmann H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia. 2016; 64:1416–1436.
176. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, Liebelt BD, Yaghi NK, Ezhilarasan R, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016.
177. Towler JC, Ebrahimi B, Lane B, Davison AJ, Dargan DJ. Human cytomegalovirus transcriptome activity differs during replication in human fibroblast, epithelial and astrocyte cell lines. J Gen Virol. 2012; 93:1046–1058.
178. Li Z, Liu X, Guo R, Wang P. CD4+Foxp3- type 1 regulatory T cells in glioblastoma multiforme suppress T cell responses through multiple pathways and are regulated by tumor-associated macrophages. Int J Biochem Cell Biol. 2016; 81:1–9.
179. Tovar-Salazar A, Patterson-Bartlett J, Jesser R, Weinberg A. Regulatory function of cytomegalovirus-specific CD4+CD27-CD28- T cells. Virology. 2010; 398:158–167.
180. Schwele S, Fischer AM, Brestrich G, Wlodarski MW, Wagner L, Schmueck M, Roemhild A, Thomas S, Hammer MH, Babel N, Kurtz A, Maciejewski JP, Reinke P, et al. Cytomegalovirus-specific regulatory and effector T cells share TCR clonality--possible relation to repetitive CMV infections. Am J Transplant. 2012; 12:669–681.
181. Terrazzini N, Bajwa M, Vita S, Cheek E, Thomas D, Seddiki N, Smith H, Kern F. A novel cytomegalovirus-induced regulatory-type T-cell subset increases in size during older life and links virus-specific immunity to vascular pathology. J Infect Dis. 2014; 209:1382–1392.
182. Clement M, Marsden M, Stacey MA, Abdul-Karim J, Gimeno Brias S, Costa Bento D, Scurr MJ, Ghazal P, Weaver CT, Carlesso G, Clare S, Jones SA, Godkin A, et al. Cytomegalovirus-specific IL-10-producing CD4+ T cells are governed by type-I IFN-induced IL-27 and promote virus persistence. PLoS Pathog. 2016; 12:e1006050.
183. Gredmark S, Tilburgs T, Söderberg-Nauclér C. Human cytomegalovirus inhibits cytokine-induced macrophage differentiation. J Virol. 2004; 78:10378–10389.
184. van der Strate BW, Hillebrands JL, Lycklama à Nijeholt SS, Beljaars L, Bruggeman CA, Van Luyn MJ, Rozing J, The TH, Meijer DK, Molema G, Harmsen MC. Dissemination of rat cytomegalovirus through infected granulocytes and monocytes in vitro and in vivo. J Virol. 2003; 77:11274–11278.
185. Lee SY, Kim JK, Jeon HY, Ham SW, Kim H. CD133 regulates IL-1β signaling and neutrophil recruitment in glioblastoma. Mol Cells. 2017; 40:515–522.
186. Rahbar A, Cederarv M, Wolmer-Solberg N, Tammik C, Stragliotto G, Peredo I, Fornara O, Xu X, Dzabic M, Taher C, Skarman P, Söderberg-Nauclér C. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. Oncoimmunology. 2015; 5:e1075693.
187. Zhang J, Zhang S, Song Y, He M, Ren Q, Chen C, Liu Z, Zeng Y, Xu J. Prognostic role of neutrophil lymphocyte ratio in patients with glioma. Oncotarget. 2017; 8:59217–59224. https://doi.org/10.18632/oncotarget.19484.
188. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, Alvarez-Buylla A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006; 51:187–199.
189. Price RL, Chiocca EA. Modeling cytomegalovirus infection in mouse tumor models. Front Oncol. 2015; 5:61.
190. Kleczkowska P, Lipkowski AW. Neurotensin and neurotensin receptors: characteristic, structure-activity relationship and pain modulation--a review. Eur J Pharmacol. 2013; 716:54–60.
191. Hermey G. The Vps10p-domain receptor family. Cell Mol Life Sci. 2009; 66:2677–2689.
192. Massa F, Devader C, Béraud-Dufour S, Brau F, Coppola T, Mazella J. Focal adhesion kinase dependent activation of the PI3 kinase pathway by the functional soluble form of neurotensin receptor-3 in HT29 cells. Int J Biochem Cell Biol. 2013; 45:952–959.
193. Massa F, Devader C, Lacas-Gervais S, Béraud-Dufour S, Coppola T, Mazella J. Impairement of HT29 cancer cells cohesion by the soluble form of neurotensin receptor-3. Genes Cancer. 2014; 5:240–249. https://doi.org/10.18632/genesandcancer.22.
194. Evers BM. Neurotensin and growth of normal and neoplastic tissues. Peptides. 2006; 27:2424–2433.
195. Kalafatakis K, Triantafyllou K. Contribution of neurotensin in the immune and neuroendocrine modulation of normal and abnormal enteric function. Regul Pept. 2011; 170:7–17.
196. Uhl GR. Distribution of neurotensin and its receptor in the central nervous system. Ann N Y Acad Sci. 1982; 400:132–149.
197. St-Gelais F, Jomphe C, Trudeau LE. The role of neurotensin in central nervous system pathophysiology: what is the evidence? J Psychiatry Neurosci. 2006; 31:229–245.
198. Boules M, Li Z, Smith K, Fredrickson P, Richelson E. Diverse roles of neurotensin agonists in the central nervous system. Front Endocrinol (Lausanne). 2013; 4:36.
199. Ferraro L, Tomasini MC, Siniscalchi A, Fuxe K, Tanganelli S, Antonelli T. Neurotensin increases endogenous glutamate release in rat cortical slices. Life Sci. 2000; 66:927–936.
200. Antonelli T, Fuxe K, Tomasini MC, Mazzoni E, Agnati LF, Tanganelli S, Ferraro L. Neurotensin receptor mechanisms and its modulation of glutamate transmission in the brain: relevance for neurodegenerative diseases and their treatment. Prog Neurobiol. 2007; 83:92–109.
201. Ferraro L, Beggiato S, Tomasini MC, Fuxe K, Tanganelli S, Antonelli T. Neurotensin regulates cortical glutamate transmission by modulating N-methyl-D-aspartate receptor functional activity: an in vivo microdialysis study. J Neurosci Res. 2011; 89:1618–1626.
202. Ouyang Q, Gong X, Xiao H, Zhou J, Xu M, Dai Y, Xu L, Feng H, Cui H, Yi L. Neurotensin promotes the progression of malignant glioma through NTSR1 and impacts the prognosis of glioma patients. Mol Cancer. 2015; 14:21.
203. Zhou J, Yi L, Ouyang Q, Xu L, Cui H, Xu M. Neurotensin signaling regulates stem-like traits of glioblastoma stem cells through activation of IL-8/CXCR1/STAT3 pathway. Cell Signal. 2014; 26:2896–2902.
204. Ouyang Q, Chen G, Zhou J, Li L, Dong Z, Yang R, Xu L, Cui H, Xu M, Yi L. Neurotensin signaling stimulates glioblastoma cell proliferation by upregulating c-Myc and inhibiting miR-29b-1 and miR-129-3p. Neuro Oncol. 2016; 18:216–226.
205. Ayala-Sarmiento AE, Martinez-Fong D, Segovia J. The internalization of neurotensin by the low-affinity neurotensin receptors (NTSR2 and vNTSR2) activates ERK 1/2 in glioma cells and allows neurotensin-polyplex transfection of tGAS1. Cell Mol Neurobiol. 2015; 35:785–795.
206. Servotte S, Camby I, Debeir O, Deroanne C, Lambert CA, Lapière CM, Kiss R, Nusgens B, Decaestecker C. The in vitro influences of neurotensin on the motility characteristics of human U373 glioblastoma cells. Neuropathol Appl Neurobiol. 2006; 32:575–584.
207. Müller KM, Tveteraas IH, Aasrum M, Ødegård J, Dawood M, Dajani O, Christoffersen T, Sandnes DL. Role of protein kinase C and epidermal growth factor receptor signalling in growth stimulation by neurotensin in colon carcinoma cells. BMC Cancer. 2011; 11:421.
208. Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, Lo CM, Guan XY, Chan KW. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012; 55:807–820.
209. Dupouy S, Doan VK, Wu Z, Mourra N, Liu J, De Wever O, Llorca FP, Cayre A, Kouchkar A, Gompel A, Forgez P. Activation of EGFR, HER2 and HER3 by neurotensin/neurotensin receptor 1 renders breast tumors aggressive yet highly responsive to lapatinib and metformin in mice. Oncotarget. 2014; 5:8235–8251. https://doi.org/10.18632/oncotarget.1632.
210. Younes M, Wu Z, Dupouy S, Lupo AM, Mourra N, Takahashi T, Fléjou JF, Trédaniel J, Régnard JF, Damotte D, Alifano M, Forgez P. Neurotensin (NTS) and its receptor (NTSR1) causes EGFR, HER2 and HER3 over-expression and their autocrine/paracrine activation in lung tumors, confirming responsiveness to erlotinib. Oncotarget. 2014; 5:8252–8269. https://doi.org/10.18632/oncotarget.1633.
211. Moody TW, Chan DC, Mantey SA, Moreno P, Jensen RT. SR48692 inhibits non-small cell lung cancer proliferation in an EGF receptor-dependent manner. Life Sci. 2014; 100:25–34.
212. Akter H, Park M, Kwon OS, Song EJ, Park WS, Kang MJ. Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumour Biol. 2015; 36:6053–6062.
213. Amorino GP, Deeble PD, Parsons SJ. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene. 2007; 26:745–756.
214. Bakirtzi K, Hatziapostolou M, Karagiannides I, Polytarchou C, Jaeger S, Iliopoulos D, Pothoulakis C. Neurotensin signaling activates microRNAs-21 and -155 and Akt, promotes tumor growth in mice, and is increased in human colon tumors. Gastroenterology. 2011; 141:1749–1761.
215. Zhao D, Bakirtzi K, Zhan Y, Zeng H, Koon HW, Pothoulakis C. Insulin-like growth factor-1 receptor transactivation modulates the inflammatory and proliferative responses of neurotensin in human colonic epithelial cells. J Biol Chem. 2011; 286:6092–6099.
216. Andersson U, Guo D, Malmer B, Bergenheim AT, Brännström T, Hedman H, Henriksson R. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol. 2004; 108:135–142.
217. Mineo JF, Bordron A, Quintin-Roué I, Maurage CA, Buhé V, Loisel S, Dubois F, Blond S, Berthou C. Increasing of HER2 membranar density in human glioblastoma U251MG cell line established in a new nude mice model. J Neurooncol. 2006; 76:249–255.
218. Duhem-Tonnelle V, Bièche I, Vacher S, Loyens A, Maurage CA, Collier F, Baroncini M, Blond S, Prevot V, Sharif A. Differential distribution of erbB receptors in human glioblastoma multiforme: expression of erbB3 in CD133-positive putative cancer stem cells. J Neuropathol Exp Neurol. 2010; 69:606–622.
219. Yi L, Xiao H, Xu M, Ye X, Hu J, Li F, Li M, Luo C, Yu S, Bian X, Feng H. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J Neuroimmunol. 2011; 232:75–82.
220. Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, Gursel D, Boockvar JA, Fischbach C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013; 73:7079–7089.
221. Zhang M, Ye G, Li J, Wang Y. Recent advance in molecular angiogenesis in glioblastoma: the challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015; 32:229–236.
222. Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, Wu K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016; 31:61–71.
223. Wang X, Wang Q, Ives KL, Evers BM. Curcumin inhibits neurotensin-mediated interleukin-8 production and migration of HCT116 human colon cancer cells. Clin Cancer Res. 2006; 12:5346–5355.
224. Zhao D, Keates AC, Kuhnt-Moore S, Moyer MP, Kelly CP, Pothoulakis C. Signal transduction pathways mediating neurotensin-stimulated interleukin-8 expression in human colonocytes. J Biol Chem. 2001; 276:44464–44471.
225. Somaï S, Gompel A, Rostène W, Forgez P. Neurotensin counteracts apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2002; 295:482–488.
226. Najimi M, Maloteaux JM, Hermans E. Cytoskeleton-related trafficking of the EAAC1 glutamate transporter after activation of the G(q/11)-coupled neurotensin receptor NTS1. FEBS Lett. 2002; 523:224–228.
227. Kamimae S, Yamamoto E, Kai M, Niinuma T, Yamano HO, Nojima M, Yoshikawa K, Kimura T, Takagi R, Harada E, Harada T, Maruyama R, Sasaki Y, et al. Epigenetic silencing of NTSR1 is associated with lateral and noninvasive growth of colorectal tumors. Oncotarget. 2015; 6:29975–29990. https://doi.org/10.18632/oncotarget.5034.
228. Ye Y, Long X, Zhang L, Chen J, Liu P, Li H, Wei F, Yu W, Ren X, Yu J. NTS/NTR1 co-expression enhances epithelial-to-mesenchymal transition and promotes tumor metastasis by activating the Wnt/β-catenin signaling pathway in hepatocellular carcinoma. Oncotarget. 2016; 7:70303–70322. https://doi.org/10.18632/oncotarget.11854.
229. Leyton J, Garcia-Marin L, Jensen RT, Moody TW. Neurotensin causes tyrosine phosphorylation of focal adhesion kinase in lung cancer cells. Eur J Pharmacol. 2002; 442:179–186.
230. Olszewski U, Hamilton G. Neurotensin signaling induces intracellular alkalinization and interleukin-8 expression in human pancreatic cancer cells. Mol Oncol. 2009; 3:204–213.
231. Busby JE, Shih SJ, Yang JC, Kung HJ, Evans CP. Angiogenesis is not mediated by prostate cancer neuropeptides. Angiogenesis. 2003; 6:289–293.
232. Bakirtzi K, West G, Fiocchi C, Law IK, Iliopoulos D, Pothoulakis C. The neurotensin-HIF-1α-VEGFα axis orchestrates hypoxia, colonic inflammation, and intestinal angiogenesis. Am J Pathol. 2014; 184:3405–3414.
233. Tazzyman S, Niaz H, Murdoch C. Neutrophil-mediated tumour angiogenesis: subversion of immune responses to promote tumour growth. Semin Cancer Biol. 2013; 23:149–158.
234. Peck R. Neuropeptides modulating macrophage function. Ann N Y Acad Sci. 1987; 496:264–270.
235. Kim HS, Yumkham S, Choi JH, Lee SH, Kim TH, Ryu SH, Suh PG. Neurotensin enhances nitric oxide generation via the JAK2-STAT1 pathway in murine macrophage Raw264.7 cells during costimulation with LPS and IFNgamma. Neuropeptides. 2006; 40:221–229.
236. Moura LI, Silva L, Leal EC, Tellechea A, Cruz MT, Carvalho E. Neurotensin modulates the migratory and inflammatory response of macrophages under hyperglycemic conditions. Biomed Res Int. 2013; 2013:941764.
237. Lemaire I. Neurotensin enhances IL-1 production by activated alveolar macrophages. J Immunol. 1988; 140:2983–2988.
238. Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune evasion strategies of glioblastoma. Front Surg. 2016; 3:11.
239. Pannell M, Szulzewsky F, Matyash V, Wolf SA, Kettenmann H. The subpopulation of microglia sensitive to neurotransmitters/neurohormones is modulated by stimulation with LPS, interferon-γ, and IL-4. Glia. 2014; 62:667–679.
240. Martin S, Vincent JP, Mazella J. Involvement of the neurotensin receptor-3 in the neurotensin-induced migration of human microglia. J Neurosci. 2003; 23:1198–1205.
241. Dicou E, Vincent JP, Mazella J. Neurotensin receptor-3/sortilin mediates neurotensin-induced cytokine/chemokine expression in a murine microglial cell line. J Neurosci Res. 2004; 78:92–99.
242. Patel AB, Tsilioni I, Leeman SE, Theoharides TC. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc Natl Acad Sci U S A. 2016.
243. Goldman R, Bar-Shavit Z, Romeo D. Neurotensin modulates human neutrophil locomotion and phagocytic capability. FEBS Lett. 1983; 159:63–67.
244. Robbins RA, Nelson KJ, Gossman GL, Rubinstein I. Neurotensin stimulates neutrophil adherence to bronchial epithelial cells in vitro. Life Sci. 1995; 56:1353–1359.
245. da Silva L, Neves BM, Moura L, Cruz MT, Carvalho E. Neurotensin downregulates the pro-inflammatory properties of skin dendritic cells and increases epidermal growth factor expression. Biochim Biophys Acta. 2011; 1813:1863–1871.
246. Strelau J, Böttner M, Lingor P, Suter-Crazzolara C, Galter D, Jaszai J, Sullivan A, Schober A, Krieglstein K, Unsicker K. GDF-15/MIC-1 a novel member of the TGF-beta superfamily. J Neural Transm Suppl. 2000.
247. Wang X, Baek SJ, Eling TE. The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem Pharmacol. 2013; 85:597–606.
248. Zhang Z, Wu L, Wang J, Li G, Feng D, Zhang B, Li L, Yang J, Ma L, Qin H. Opposing effects of PI3K/Akt and Smad-dependent signaling pathways in NAG-1-induced glioblastoma cell apoptosis. PLoS One. 2014; 9:e96283.
249. de Jager SC, Bermúdez B, Bot I, Koenen RR, Bot M, Kavelaars A, de Waard V, Heijnen CJ, Muriana FJ, Weber C, van Berkel TJ, Kuiper J, Lee SJ, et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med. 2011; 208:217–225.
250. Artz A, Butz S, Vestweber D. GDF-15 inhibits integrin activation and mouse neutrophil recruitment through the ALK-5/TGF-βRII heterodimer. Blood. 2016; 128:529–541.
251. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, Coskun T, Hamang MJ, Sindelar DK, Ballman KK, Foltz LA, Muppidi A, Alsina-Fernandez J, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med. 2017; 23:1215–1219.
252. Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, Beck SC, South VJ, Dinh TQ, Cash-Mason TD, Cavanaugh CR, Nelson S, Huang C, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med. 2017; 23:1150–1157.
253. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjær SB, Wu X, Huang T, Hultman K, Paulsen SJ, Wang J, Bugge A, Frantzen JB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. 2017; 23:1158–1166.
254. Tsai VW, Husaini Y, Manandhar R, Lee-Ng KK, Zhang HP, Harriott K, Jiang L, Lin S, Sainsbury A, Brown DA, Breit SN. Anorexia/cachexia of chronic diseases: a role for the TGF-β family cytokine MIC-1/GDF15. J Cachexia Sarcopenia Muscle. 2012; 3:239–243.
255. Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, Nicoletti R, Chiu MI, Gyuris J, Garcia JM. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J Cachexia Sarcopenia Muscle. 2015; 6:317–324.
256. Griffith JL, Hochberg FH. Anorexia and weight loss in glioma patients. Psychosomatics. 1988; 29:335–337.
257. Kadowaki M, Yoshioka H, Kamitani H, Watanabe T, Wade PA, Eling TE. DNA methylation-mediated silencing of nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) in glioma cell lines. Int J Cancer. 2012; 130:267–277.
258. Whitson RJ, Lucia MS, Lambert JR. Growth differentiation factor-15 (GDF-15) suppresses in vitro angiogenesis through a novel interaction with connective tissue growth factor (CCN2). J Cell Biochem. 2013; 114:1424–1433.
259. Cheng JC, Chang HM, Leung PC. Wild-type p53 attenuates cancer cell motility by inducing growth differentiation factor-15 expression. Endocrinology. 2011; 152:2987–2995.
260. Yoshioka H, Kamitani H, Watanabe T, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) expression is increased by the histone deacetylase inhibitor trichostatin A. J Biol Chem. 2008; 283:33129–33137.
261. Strelau J, Schmeer C, Peterziel H, Sackmann T, Herold-Mende C, Steiner H, Weller M, Unsicker K. Expression and putative functions of GDF-15, a member of the TGF-beta superfamily, in human glioma and glioblastoma cell lines. Cancer Lett. 2008; 270:30–39.
262. Codó P, Weller M, Kaulich K, Schraivogel D, Silginer M, Reifenberger G, Meister G, Roth P. Control of glioma cell migration and invasiveness by GDF-15. Oncotarget. 2016; 7:7732–7746. https://doi.org/10.18632/oncotarget.6816.
263. Shnaper S, Desbaillets I, Brown DA, Murat A, Migliavacca E, Schluep M, Ostermann S, Hamou MF, Stupp R, Breit SN, de Tribolet N, Hegi ME. Elevated levels of MIC-1/GDF15 in the cerebrospinal fluid of patients are associated with glioblastoma and worse outcome. Int J Cancer. 2009; 125:2624–2630.
264. Urakawa N, Utsunomiya S, Nishio M, Shigeoka M, Takase N, Arai N, Kakeji Y, Koma Y, Yokozaki H. GDF15 derived from both tumor-associated macrophages and esophageal squamous cell carcinomas contributes to tumor progression via Akt and Erk pathways. Lab Invest. 2015; 95:491–503.
265. Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, Tabatabai G, Wick W, Weller M, Wischhusen J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res. 2010; 16:3851–3859.
266. Min KW, Liggett JL, Silva G, Wu WW, Wang R, Shen RF, Eling TE, Baek SJ. NAG-1/GDF15 accumulates in the nucleus and modulates transcriptional regulation of the Smad pathway. Oncogene. 2016; 35:377–388.
267. Jin YJ, Lee JH, Kim YM, Oh GT, Lee H. Macrophage inhibitory cytokine-1 stimulates proliferation of human umbilical vein endothelial cells by up-regulating cyclins D1 and E through the PI3K/Akt-, ERK-, and JNK-dependent AP-1 and E2F activation signaling pathways. Cell Signal. 2012; 24:1485–1495.
268. Wang S, Li M, Zhang W, Hua H, Wang N, Zhao J, Ge J, Jiang X, Zhang Z, Ye D, Yang C. Growth differentiation factor 15 promotes blood vessel growth by stimulating cell cycle progression in repair of critical-sized calvarial defect. Sci Rep. 2017; 7:9027.
269. Albertoni M, Shaw PH, Nozaki M, Godard S, Tenan M, Hamou MF, Fairlie DW, Breit SN, Paralkar VM, de Tribolet N, Van Meir EG, Hegi ME. Anoxia induces macrophage inhibitory cytokine-1 (MIC-1) in glioblastoma cells independently of p53 and HIF-1. Oncogene. 2002; 21:4212–4219.
270. Nowacki P, Kojder I. Peritumoral angiogenesis around primary and metastatic brain neoplasms. Morphometric analysis. Folia Neuropathol. 2001; 39:95–102.
271. Song H, Yin D, Liu Z. GDF-15 promotes angiogenesis through modulating p53/HIF-1? signaling pathway in hypoxic human umbilical vein endothelial cells. Mol Biol Rep. 2012; 39:4017–4022.
272. Li C, Wang J, Kong J, Tang J, Wu Y, Xu E, Zhang H, Lai M. GDF15 promotes EMT and metastasis in colorectal cancer. Oncotarget. 2016; 7:860–872. https://doi.org/10.18632/oncotarget.6205.
273. Wang L, Liu Y, Li W, Song Z. Growth differentiation factor 15 promotes cell viability, invasion, migration, and angiogenesis in human liver carcinoma cell line HepG2. Clin Res Hepatol Gastroenterol. 2017; 41:408–414.
274. Tate MC, Aghi MK. Biology of angiogenesis and invasion in glioma. Neurotherapeutics. 2009; 6:447–457.
275. Subramanian P, Mitroulis I, Hajishengallis G, Chavakis T. Regulation of tissue infiltration by neutrophils: role of integrin α3β1 and other factors. Curr Opin Hematol. 2016; 23:36–43.
276. Griner SE, Joshi JP, Nahta R. Growth differentiation factor 15 stimulates rapamycin-sensitive ovarian cancer cell growth and invasion. Biochem Pharmacol. 2013; 85:46–58.
277. Zhou Z, Li W, Song Y, Wang L, Zhang K, Yang J, Zhang W, Su H, Zhang Y. Growth differentiation factor-15 suppresses maturation and function of dendritic cells and inhibits tumor-specific immune response. PLoS One. 2013; 8:e78618.
278. Segerer SE, Rieger L, Kapp M, Dombrowski Y, Müller N, Dietl J, Kämmerer U. MIC-1 (a multifunctional modulator of dendritic cell phenotype and function) is produced by decidual stromal cells and trophoblasts. Hum Reprod. 2012; 27:200–209.
279. Le Stunff H, Milstien S, Spiegel S. Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem. 2004; 92:882–899.
280. Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005; 280:37118–37129.
281. Parham KA, Zebol JR, Tooley KL, Sun WY, Moldenhauer LM, Cockshell MP, Gliddon BL, Moretti PA, Tigyi G, Pitson SM, Bonder CS. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015; 29:3638–3653.
282. Young N, Van Brocklyn JR. Signal transduction of sphingosine-1-phosphate G protein-coupled receptors. ScientificWorldJournal. 2006; 6:946–966.
283. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008; 22:2629–2638.
284. Weigert A, Weis N, Brüne B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology. 2009; 214:748–760.
285. Ley S, Weigert A, Weichand B, Henke N, Mille-Baker B, Janssen RA, Brüne B. The role of TRKA signaling in IL-10 production by apoptotic tumor cell-activated macrophages. Oncogene. 2013; 32:631–640.
286. Arlt O, Schwiebs A, Japtok L, Rüger K, Katzy E, Kleuser B, Radeke HH. Sphingosine-1-phosphate modulates dendritic cell function: focus on non-migratory effects in vitro and in vivo. Cell Physiol Biochem. 2014; 34:27–44.
287. Thuy AV, Reimann CM, Hemdan NY, Gräler MH. Sphingosine 1-phosphate in blood: function, metabolism, and fate. Cell Physiol Biochem. 2014; 34:158–171.
288. Quint K, Stiel N, Neureiter D, Schlicker HU, Nimsky C, Ocker M, Strik H, Kolodziej MA. The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumour Biol. 2014; 35:8979–8989.
289. Bien-Möller S, Lange S, Holm T, Böhm A, Paland H, Küpper J, Herzog S, Weitmann K, Havemann C, Vogelgesang S, Marx S, Hoffmann W, Schroeder HW, Rauch BH. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget. 2016; 7:13031–13046. https://doi.org/10.18632/oncotarget.7366.
290. Yoshida Y, Nakada M, Harada T, Tanaka S, Furuta T, Hayashi Y, Kita D, Uchiyama N, Hayashi Y, Hamada J. The expression level of sphingosine-1-phosphate receptor type 1 is related to MIB-1 labeling index and predicts survival of glioblastoma patients. J Neurooncol. 2010; 98:41–47.
291. Yoshida Y, Nakada M, Sugimoto N, Harada T, Hayashi Y, Kita D, Uchiyama N, Hayashi Y, Yachie A, Takuwa Y, Hamada J. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int J Cancer. 2010; 126:2341–2352.
292. Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005; 64:695–705.
293. Young N, Pearl DK, Van Brocklyn JR. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res. 2009; 7:23–32.
294. Estrada-Bernal A, Lawler SE, Nowicki MO, Ray Chaudhury A, Van Brocklyn JR. The role of sphingosine kinase-1 in EGFRvIII-regulated growth and survival of glioblastoma cells. J Neurooncol. 2011; 102:353–366.
295. Lepannetier S, Zanou N, Yerna X, Emeriau N, Dufour I, Masquelier J, Muccioli G, Tajeddine N, Gailly P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium. 2016; 60:373–383.
296. Fortier S, Labelle D, Sina A, Moreau R, Annabi B. Silencing of the MT1-MMP/G6PT axis suppresses calcium mobilization by sphingosine-1-phosphate in glioblastoma cells. FEBS Lett. 2008; 582:799–804.
297. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009; 16:748–754.
298. Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM, Singh SK, Kapitonov D, Rokita H, Wright S, Griswold-Prenner I, Milstien S, Spiegel S, Kordula T. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem. 2009; 284:3408–3417.
299. Anelli V, Gault CR, Cheng AB, Obeid LM. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J Biol Chem. 2008; 283:3365–3375.
300. Zhang H, Li W, Sun S, Yu S, Zhang M, Zou F. Inhibition of sphingosine kinase 1 suppresses proliferation of glioma cells under hypoxia by attenuating activity of extracellular signal-regulated kinase. Cell Prolif. 2012; 45:167–175.
301. Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, Zipkin RE, Dent P, Kordula T, Milstien S, Spiegel S. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009; 69:6915–6923.
302. Guan H, Song L, Cai J, Huang Y, Wu J, Yuan J, Li J, Li M. Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim pathway and contributes to apoptosis resistance in glioma cells. PLoS One. 2011; 6:e19946.
303. Young N, Van Brocklyn JR. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res. 2007; 313:1615–1627.
304. Bernhart E, Damm S, Wintersperger A, Nusshold C, Brunner AM, Plastira I, Rechberger G, Reicher H, Wadsack C, Zimmer A, Malle E, Sattler W. Interference with distinct steps of sphingolipid synthesis and signaling attenuates proliferation of U87MG glioma cells. Biochem Pharmacol. 2015; 96:119–130.
305. Riccitelli E, Giussani P, Di Vito C, Condomitti G, Tringali C, Caroli M, Galli R, Viani P, Riboni L. Extracellular sphingosine-1-phosphate: a novel actor in human glioblastoma stem cell survival. PLoS One. 2013; 8:e68229.
306. Annabi B, Lachambre MP, Plouffe K, Sartelet H, Béliveau R. Modulation of invasive properties of CD133+ glioblastoma stem cells: a role for MT1-MMP in bioactive lysophospholipid signaling. Mol Carcinog. 2009; 48:910–919.
307. Marfia G, Campanella R, Navone SE, Di Vito C, Riccitelli E, Hadi LA, Bornati A, de Rezende G, Giussani P, Tringali C, Viani P, Rampini P, Alessandri G, et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia. 2014; 62:1968–1981.
308. Bektas M, Johnson SP, Poe WE, Bigner DD, Friedman HS. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemother Pharmacol. 2009; 64:1053–1058.
309. Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014; 5:4806.
310. Dimov I, Tasić-Dimov D, Conić I, Stefanovic V. Glioblastoma multiforme stem cells. ScientificWorldJournal. 2011; 11:930–958.
311. Anelli V, Gault CR, Snider AJ, Obeid LM. Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 2010; 24:2727–2738.
312. Abuhusain HJ, Matin A, Qiao Q, Shen H, Kain N, Day BW, Stringer BW, Daniels B, Laaksonen MA, Teo C, McDonald KL, Don AS. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J Biol Chem. 2013; 288:37355–37364.
313. Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost. 2005; 3:35–45.
314. Igarashi J, Erwin PA, Dantas AP, Chen H, Michel T. VEGF induces S1P1 receptors in endothelial cells: implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci U S A. 2003; 100:10664–10669.
315. Paugh BS, Paugh SW, Bryan L, Kapitonov D, Wilczynska KM, Gopalan SM, Rokita H, Milstien S, Spiegel S, Kordula T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008; 22:455–465.
316. Bryan L, Paugh BS, Kapitonov D, Wilczynska KM, Alvarez SM, Singh SK, Milstien S, Spiegel S, Kordula T. Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: implications for invasiveness. Mol Cancer Res. 2008; 6:1469–1477.
317. Bernhart E, Damm S, Wintersperger A, DeVaney T, Zimmer A, Raynham T, Ireson C, Sattler W. Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp Cell Res. 2013; 319:2037–2048.
318. Malchinkhuu E, Sato K, Maehama T, Mogi C, Tomura H, Ishiuchi S, Yoshimoto Y, Kurose H, Okajima F. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem Biophys Res Commun. 2008; 366:963–968.
319. Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, Heber-Katz E, Curtis MT, Cotzia P, Hooper DC. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013; 19:3776–3786.
320. Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000; 46:957–961.
321. Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg. 2009; 110:572–582.
322. Mrad M, Imbert C, Garcia V, Rambow F, Therville N, Carpentier S, Ségui B, Levade T, Azar R, Marine JC, Diab-Assaf M, Colacios C, Andrieu-Abadie N. Downregulation of sphingosine kinase-1 induces protective tumor immunity by promoting M1 macrophage response in melanoma. Oncotarget. 2016; 7:71873–71886. https://doi.org/10.18632/oncotarget.12380.
323. Weigert A, Schiffmann S, Sekar D, Ley S, Menrad H, Werno C, Grosch S, Geisslinger G, Brüne B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int J Cancer. 2009; 125:2114–2121.
324. Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem. 2009; 107:1053–1062.
325. Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brüne B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood. 2006; 108:1635–1642.
326. Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G, Brüne B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell. 2007; 18:3810–3819.
327. Johann AM, Weigert A, Eberhardt W, Kuhn AM, Barra V, von Knethen A, Pfeilschifter JM, Brüne B. Apoptotic cell-derived sphingosine-1-phosphate promotes HuR-dependent cyclooxygenase-2 mRNA stabilization and protein expression. J Immunol. 2008; 180:1239–1248.
328. Herr B, Zhou J, Werno C, Menrad H, Namgaladze D, Weigert A, Dehne N, Brüne B. The supernatant of apoptotic cells causes transcriptional activation of hypoxia-inducible factor-1alpha in macrophages via sphingosine-1-phosphate and transforming growth factor-beta. Blood. 2009; 114:2140–2148.
329. Weis N, Weigert A, von Knethen A, Brüne B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell. 2009; 20:1280–1288.
330. Xiong Y, Lee HJ, Mariko B, Lu YC, Dannenberg AJ, Haka AS, Maxfield FR, Camerer E, Proia RL, Hla T. Sphingosine kinases are not required for inflammatory responses in macrophages. J Biol Chem. 2013; 288:32563–32573.
331. Lewis ND, Haxhinasto SA, Anderson SM, Stefanopoulos DE, Fogal SE, Adusumalli P, Desai SN, Patnaude LA, Lukas SM, Ryan KR, Slavin AJ, Brown ML, Modis LK. Circulating monocytes are reduced by sphingosine-1-phosphate receptor modulators independently of S1P3. J Immunol. 2013; 190:3533–3540.
332. Keul P, Lucke S, von Wnuck Lipinski K, Bode C, Gräler M, Heusch G, Levkau B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res. 2011; 108:314–323.
333. Awojoodu AO, Ogle ME, Sefcik LS, Bowers DT, Martin K, Brayman KL, Lynch KR, Peirce-Cottler SM, Botchwey E. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proc Natl Acad Sci U S A. 2013; 110:13785–13790.
334. Yang L, Han Z, Tian L, Mai P, Zhang Y, Wang L, Li L. Sphingosine 1-phosphate receptor 2 and 3 mediate bone marrow-derived monocyte/macrophage motility in cholestatic liver injury in mice. Sci Rep. 2015; 5:13423.
335. Debien E, Mayol K, Biajoux V, Daussy C, De Aguero MG, Taillardet M, Dagany N, Brinza L, Henry T, Dubois B, Kaiserlian D, Marvel J, Balabanian K, et al. S1PR5 is pivotal for the homeostasis of patrolling monocytes. Eur J Immunol. 2013; 43:1667–1675.
336. Mahajan-Thakur S, Sostmann BD, Fender AC, Behrendt D, Felix SB, Schrör K, Rauch BH. Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression to enhance cell migration and COX-2 formation in human monocytes. J Leukoc Biol. 2014; 96:611–618.
337. Itsekson-Hayosh Z, Shavit-Stein E, Last D, Goez D, Daniels D, Bushi D, Gera O, Zibly Z, Mardor Y, Chapman J, Harnof S. Thrombin activity and thrombin receptor in rat glioblastoma model: possible markers and targets for intervention? J Mol Neurosci. 2015; 56:644–651.
338. Weichand B, Weis N, Weigert A, Grossmann N, Levkau B, Brüne B. Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur J Immunol. 2013; 43:3306–3313.
339. Xie B, Shen J, Dong A, Rashid A, Stoller G, Campochiaro PA. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J Cell Physiol. 2009; 218:192–198.
340. Murakami M, Saito T, Tabata Y. Controlled release of sphingosine-1-phosphate agonist with gelatin hydrogels for macrophage recruitment. Acta Biomater. 2014; 10:4723–4729.
341. Kuehnel MP, Reiss M, Anand PK, Treede I, Holzer D, Hoffmann E, Klapperstueck M, Steinberg TH, Markwardt F, Griffiths G. Sphingosine-1-phosphate receptors stimulate macrophage plasma-membrane actin assembly via ADP release, ATP synthesis and P2X7R activation. J Cell Sci. 2009; 122:505–512.
342. Michaud J, Im DS, Hla T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J Immunol. 2010; 184:1475–1483.
343. Lin CI, Chen CN, Lin PW, Lee H. Sphingosine 1-phosphate regulates inflammation-related genes in human endothelial cells through S1P1 and S1P3. Biochem Biophys Res Commun. 2007; 355:895–901.
344. Weis T, Völker W, Holtwick R, Al Chahaf M, Schmidt A. Sphingosine 1-phosphate (S1P) induces expression of E-selectin and adhesion of monocytes via intracellular signalling pathways in vascular endothelial cells. Eur J Cell Biol. 2010; 89:733–741.
345. Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010; 465:1084–1088.
346. Park ES, Choi S, Shin B, Yu J, Yu J, Hwang JM, Yun H, Chung YH, Choi JS, Choi Y, Rho J. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J Biol Chem. 2015; 290:9660–9673.
347. Su D, Cheng Y, Li S, Dai D, Zhang W, Lv M. Sphk1 mediates neuroinflammation and neuronal injury via TRAF2/NF-κB pathways in activated microglia in cerebral ischemia reperfusion. J Neuroimmunol. 2017; 305:35–41.
348. Lin CC, Lee IT, Hsu CH, Hsu CK, Chi PL, Hsiao LD, Yang CM. Sphingosine-1-phosphate mediates ICAM-1-dependent monocyte adhesion through p38 MAPK and p42/p44 MAPK-dependent Akt activation. PLoS One. 2015; 10:e0118473.
349. Bolick DT, Srinivasan S, Kim KW, Hatley ME, Clemens JJ, Whetzel A, Ferger N, Macdonald TL, Davis MD, Tsao PS, Lynch KR, Hedrick CC. Sphingosine-1-phosphate prevents tumor necrosis factor-{alpha}-mediated monocyte adhesion to aortic endothelium in mice. Arterioscler Thromb Vasc Biol. 2005; 25:976–981.
350. Aoki S, Yatomi Y, Shimosawa T, Yamashita H, Kitayama J, Tsuno NH, Takahashi K, Ozaki Y. The suppressive effect of sphingosine 1-phosphate on monocyte-endothelium adhesion may be mediated by the rearrangement of the endothelial integrins alpha(5)beta(1) and alpha(v)beta(3). J Thromb Haemost. 2007; 5:1292–1301.
351. Cheng Q, Ma S, Lin D, Mei Y, Gong H, Lei L, Chen Y, Zhao Y, Hu B, Wu Y, Yu X, Zhao L, Liu H. The S1P1 receptor-selective agonist CYM-5442 reduces the severity of acute GVHD by inhibiting macrophage recruitment. Cell Mol Immunol. 2015; 12:681–691.
352. Feuerborn R, Becker S, Potì F, Nagel P, Brodde M, Schmidt H, Christoffersen C, Ceglarek U, Burkhardt R, Nofer JR. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis. 2017; 257:29–37.
353. Hou J, Chen Q, Zhang K, Cheng B, Xie G, Wu X, Luo C, Chen L, Liu H, Zhao B, Dai K, Fang X. Sphingosine 1-phosphate receptor 2 signaling suppresses macrophage phagocytosis and impairs host defense against sepsis. Anesthesiology. 2015; 123:409–422.
354. McQuiston T, Luberto C, Del Poeta M. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology. 2011; 157:1416–1427.
355. Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. 2008; 102:950–958.
356. Hammad SM, Crellin HG, Wu BX, Melton J, Anelli V, Obeid LM. Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat. 2008; 85:107–114.
357. Rodriguez YI, Campos LE, Castro MG, Aladhami A, Oskeritzian CA, Alvarez SE. Sphingosine-1 phosphate: a new modulator of immune plasticity in the tumor microenvironment. Front Oncol. 2016; 6:218.
358. Brecht K, Weigert A, Hu J, Popp R, Fisslthaler B, Korff T, Fleming I, Geisslinger G, Brüne B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011; 25:2408–2417.
359. Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009; 30:377–386.
360. Qiu J, Shi Z, Jiang J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov Today. 2017; 22:148–156.
361. Jung M, Ören B, Mora J, Mertens C, Dziumbla S, Popp R, Weigert A, Grossmann N, Fleming I, Brüne B. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal. 2016; 9:ra64.
362. Barra V, Kuhn AM, von Knethen A, Weigert A, Brüne B. Apoptotic cell-derived factors induce arginase II expression in murine macrophages by activating ERK5/CREB. Cell Mol Life Sci. 2011; 68:1815–1827.
363. Grimm M, Tischner D, Troidl K, Albarrán Juárez J, Sivaraj KK, Ferreirós Bouzas N, Geisslinger G, Binder CJ, Wettschureck N. S1P2/G12/13 signaling negatively regulates macrophage activation and indirectly shapes the atheroprotective B1-cell population. Arterioscler Thromb Vasc Biol. 2016; 36:37–48.
364. Müller J, von Bernstorff W, Heidecke CD, Schulze T. Differential S1P receptor profiles on M1- and M2-polarized macrophages affect macrophage cytokine production and migration. Biomed Res Int. 2017; 2017:7584621.
365. Billich A, Bornancin F, Dévay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003; 278:47408–47415.
366. Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003; 554:189–193.
367. Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003; 278:47281–47290.
368. Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004; 279:52487–52492.
369. Kharel Y, Lee S, Snyder AH, Sheasley-O’neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, Tung KS, Engelhard VH, Macdonald TL, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005; 280:36865–36872.
370. Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006; 107:1454–1458.
371. Högenauer K, Billich A, Pally C, Streiff M, Wagner T, Welzenbach K, Nussbaumer P. Phosphorylation by sphingosine kinase 2 is essential for in vivo potency of FTY720 analogues. ChemMedChem. 2008; 3:1027–1029.
372. Mechtcheriakova D, Wlachos A, Sobanov J, Bornancin F, Zlabinger G, Baumruker T, Billich A. FTY720-phosphate is dephosphorylated by lipid phosphate phosphatase 3. FEBS Lett. 2007; 581:3063–3068.
373. Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002; 277:21453–21457.
374. Sobel K, Menyhart K, Killer N, Renault B, Bauer Y, Studer R, Steiner B, Bolli MH, Nayler O, Gatfield J. Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J Biol Chem. 2013; 288:14839–14851.
375. Sobel K, Monnier L, Menyhart K, Bolinger M, Studer R, Nayler O, Gatfield J. FTY720 phosphate activates sphingosine-1-phosphate receptor 2 and selectively couples to Gα12/13/Rho/ROCK to induce myofibroblast contraction. Mol Pharmacol. 2015; 87:916–927.
376. Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002; 296:346–349.
377. Gräler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004; 18:551–553.
378. LaMontagne K, Littlewood-Evans A, Schnell C, O’Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G, Billy E, Theuer A, Hla T, et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006; 66:221–231.
379. Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009; 5:428–434.
380. Sykes DA, Riddy DM, Stamp C, Bradley ME, McGuiness N, Sattikar A, Guerini D, Rodrigues I, Glaenzel A, Dowling MR, Mullershausen F, Charlton SJ. Investigating the molecular mechanisms through which FTY720-P causes persistent S1P1 receptor internalization. Br J Pharmacol. 2014; 171:4797–4807.
381. Jin Y, Zollinger M, Borell H, Zimmerlin A, Patten CJ. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug Metab Dispos. 2011; 39:191–198.
382. Kunikata S, Nagano T, Nishioka T, Akiyama T, Kurita T. Immunosuppressive action of FTY720 for renal allograft a rat model. Transplant Proc. 1999; 31:1157–1159.
383. Suzuki T, Shimamura T, Jin MB, Yokota R, Fukai M, Iida J, Taniguchi M, Magata S, Horiuchi H, Yamashita K, Nomura M, Omura T, Kishida A, et al. Dose-dependent study of a novel immunosuppressant, FTY720, with the canine renal allograft transplantation model. Transplant Proc. 1999; 31:1208–1209.
384. Tamura A, Li XK, Funeshima N, Enosawa S, Amemiya H, Kitajima M, Suzuki S. Immunosuppressive therapy using FTY720 combined with tacrolimus in rat liver transplantation. Surgery. 2000; 127:47–54.
385. Liu L, Wang C, He X, Shang W, Bi Y, Wang D. Long-term effect of FTY720 on lymphocyte count and islet allograft survival in mice. Microsurgery. 2007; 27:300–304.
386. Lopes CT, Gallo AP, Palma PV, Cury PM, Bueno V. Skin allograft survival and analysis of renal parameters after FTY720 + tacrolimus treatment in mice. Transplant Proc. 2008; 40:856–860.
387. Sensken SC, Bode C, Gräler MH. Accumulation of fingolimod (FTY720) in lymphoid tissues contributes to prolonged efficacy. J Pharmacol Exp Ther. 2009; 328:963–969.
388. Claes N, Dhaeze T, Fraussen J, Broux B, Van Wijmeersch B, Stinissen P, Hupperts R, Hellings N, Somers V. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One. 2014; 9:e111115.
389. Kaudel CP, Frink M, van Griensven M, Schmiddem U, Probst C, Bergmann S, Krettek C, Klempnauer J, Winkler M. FTY720 application following isolated warm liver ischemia improves long-term survival and organ protection in a mouse model. Transplant Proc. 2007; 39:493–498.
390. Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, Guo S, Qin T, Alsharif N, Brinkmann V, Liao JK, Lo EH, Waeber C. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011; 69:119–129.
391. Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, Reuschel R, Beerli C, Schwartz M, Billich A. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007; 323:469–475.
392. Das A, Arifuzzaman S, Kim SH, Lee YS, Jung KH, Chai YG. FTY720 (fingolimod) regulates key target genes essential for inflammation in microglial cells as defined by high-resolution mRNA sequencing. Neuropharmacology. 2017; 119:1–14.
393. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010; 33:91–101.
394. Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, Vollmer T, Agius MA, Kappos L, Stites T, Li B, Cappiello L, von Rosenstiel P, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014; 13:545–556.
395. Khatri BO. Fingolimod in the treatment of relapsing-remitting multiple sclerosis: long-term experience and an update on the clinical evidence. Ther Adv Neurol Disord. 2016; 9:130–147.
396. Nayak D, Huo Y, Kwang WX, Pushparaj PN, Kumar SD, Ling EA, Dheen ST. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience. 2010; 166:132–144.
397. Marfia G, Navone SE, Hadi LA, Paroni M, Berno V, Beretta M, Gualtierotti R, Ingegnoli F, Levi V, Miozzo M, Geginat J, Fassina L, Rampini P, et al. The adipose mesenchymal stem cell secretome inhibits inflammatory responses of microglia: evidence for an involvement of sphingosine-1-phosphate signalling. Stem Cells Dev. 2016; 25:1095–1107.
398. Gaire BP, Lee CH, Sapkota A, Lee SY, Chun J, Cho HJ, Nam TG, Choi JW. Identification of sphingosine 1-phosphate receptor subtype 1 (S1P1) as a pathogenic factor in transient focal cerebral ischemia. Mol Neurobiol. 2017.
399. Lv M, Zhang D, Dai D, Zhang W, Zhang L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm Res. 2016; 65:551–562.
400. Noda H, Takeuchi H, Mizuno T, Suzumura A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol. 2013; 256:13–18.
401. Cipriani R, Chara JC, Rodríguez-Antigüedad A, Matute C. FTY720 attenuates excitotoxicity and neuroinflammation. J Neuroinflammation. 2015; 12:86.
402. Czech B, Pfeilschifter W, Mazaheri-Omrani N, Strobel MA, Kahles T, Neumann-Haefelin T, Rami A, Huwiler A, Pfeilschifter J. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Commun. 2009; 389:251–256.
403. Durafourt BA, Lambert C, Johnson TA, Blain M, Bar-Or A, Antel JP. Differential responses of human microglia and blood-derived myeloid cells to FTY720. J Neuroimmunol. 2011; 230:10–16.
404. Jiang J, Guo W, Liang X. Phenotypes, accumulation, and functions of myeloid-derived suppressor cells and associated treatment strategies in cancer patients. Hum Immunol. 2014; 75:1128–1137.
405. Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, de Gruijl TD. Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim Biophys Acta. 2014; 1846:560–575.
406. Huu DL, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, Fujimoto M. FTY720 ameliorates murine sclerodermatous chronic graft-versus-host disease by promoting expansion of splenic regulatory cells and inhibiting immune cell infiltration into skin. Arthritis Rheum. 2013; 65:1624–1635.
407. Liu G, Bi Y, Wang R, Yang H, Zhang Y, Wang X, Liu H, Lu Y, Zhang Z, Chen W, Chu Y, Yang R. Targeting S1P1 receptor protects against murine immunological hepatic injury through myeloid-derived suppressor cells. J Immunol. 2014; 192:3068–3079.
408. Li Y, Zhou T, Wang Y, Ning C, Lv Z, Han G, Morris JC, Taylor EN, Wang R, Xiao H, Hou C, Ma Y, Shen B, et al. The protumorigenic potential of FTY720 by promoting extramedullary hematopoiesis and MDSC accumulation. Oncogene. 2017; 36:3760–3771.
409. Gargett T, Christo SN, Hercus TR, Abbas N, Singhal N, Lopez AF, Brown MP. GM-CSF signalling blockade and chemotherapeutic agents act in concert to inhibit the function of myeloid-derived suppressor cells in vitro. Clin Transl Immunology. 2016; 5:e119.
410. Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, Katz SC. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016; 23:188–198.
411. Haebich G, Mughal A, Tofazzal N. Superficial spreading malignant melanoma in a patient on fingolimod therapy for multiple sclerosis. Clin Exp Dermatol. 2016; 41:433–434.
412. Sharim J, Tashjian R, Golzy N, Pouratian N. Glioblastoma following treatment with fingolimod for relapsing-remitting multiple sclerosis. J Clin Neurosci. 2016; 30:166–168.
413. Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010; 11:1047–1056.
414. Kim MG, Lee SY, Ko YS, Lee HY, Jo SK, Cho WY, Kim HK. CD4+ CD25+ regulatory T cells partially mediate the beneficial effects of FTY720, a sphingosine-1-phosphate analogue, during ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2011; 26:111–124.
415. Liu Y, Jiang J, Xiao H, Wang X, Li Y, Gong Y, Huang Y. The sphingosine-1-phosphate receptor agonist FTY720 and its phosphorylated form affect the function of CD4+CD25+ T cells in vitro. Int J Mol Med. 2012; 30:211–219.
416. Wolf AM, Eller K, Zeiser R, Dürr C, Gerlach UV, Sixt M, Markut L, Gastl G, Rosenkranz AR, Wolf D. The sphingosine 1-phosphate receptor agonist FTY720 potently inhibits regulatory T cell proliferation in vitro and in vivo. J Immunol. 2009; 183:3751–3760.
417. Sun Y, Wang W, Shan B, Di J, Chen L, Ren L, Li W, Li DJ, Lin Y. FTY720-induced conversion of conventional Foxp3- CD4+ T cells to Foxp3+ regulatory T cells in NOD mice. Am J Reprod Immunol. 2011; 66:349–362.
418. Geng S, Zhong Y, Zhou X, Zhao G, Xie X, Pei Y, Liu H, Zhang H, Shi Y, Wang B. Induced regulatory T cells superimpose their suppressive capacity with effector T cells in lymph nodes via antigen-specific S1p1-dependent egress blockage. Front Immunol. 2017; 8:663.
419. Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M, Yu H. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep. 2014; 6:992–999.
420. Rathinasamy A, Domschke C, Ge Y, Böhm HH, Dettling S, Jansen D, Lasitschka F, Umansky L, Gräler MH, Hartmann J, Herold-Mende C, Schuetz F, Beckhove P. Tumor specific regulatory T cells in the bone marrow of breast cancer patients selectively upregulate the emigration receptor S1P1. Cancer Immunol Immunother. 2017; 66:593–603.
421. Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007; 178:2458–2468.
422. Zemann B, Urtz N, Reuschel R, Mechtcheriakova D, Bornancin F, Badegruber R, Baumruker T, Billich A. Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol Lett. 2007; 109:56–63.
423. Finley A, Chen Z, Esposito E, Cuzzocrea S, Sabbadini R, Salvemini D. Sphingosine 1-phosphate mediates hyperalgesia via a neutrophil-dependent mechanism. PLoS One. 2013; 8:e55255.
424. Sun WY, Abeynaike LD, Escarbe S, Smith CD, Pitson SM, Hickey MJ, Bonder CS. Rapid histamine-induced neutrophil recruitment is sphingosine kinase-1 dependent. Am J Pathol. 2012; 180:1740–1750.
425. Kawa S, Kimura S, Hakomori S, Igarashi Y. Inhibition of chemotactic motility and trans-endothelial migration of human neutrophils by sphingosine 1-phosphate. FEBS Lett. 1997; 420:196–200.
426. Cummings RJ, Parinandi NL, Zaiman A, Wang L, Usatyuk PV, Garcia JG, Natarajan V. Phospholipase D activation by sphingosine 1-phosphate regulates interleukin-8 secretion in human bronchial epithelial cells. J Biol Chem. 2002; 277:30227–30235.
427. Wang L, Cummings R, Usatyuk P, Morris A, Irani K, Natarajan V. Involvement of phospholipases D1 and D2 in sphingosine 1-phosphate-induced ERK (extracellular-signal-regulated kinase) activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem J. 2002; 367:751–760.
428. O’Sullivan MJ, Hirota N, Martin JG. Sphingosine 1-phosphate (S1P) induced interleukin-8 (IL-8) release is mediated by S1P receptor 2 and nuclear factor κB in BEAS-2B cells. PLoS One. 2014; 9:e95566.
429. Milara J, Mata M, Mauricio MD, Donet E, Morcillo EJ, Cortijo J. Sphingosine-1-phosphate increases human alveolar epithelial IL-8 secretion, proliferation and neutrophil chemotaxis. Eur J Pharmacol. 2009; 609:132–139.
430. Rahman MM, Alkhouri H, Tang F, Che W, Ge Q, Ammit AJ. Sphingosine 1-phosphate induces neutrophil chemoattractant IL-8: repression by steroids. PLoS One. 2014; 9:e92466.
431. Schwartz BM, Hong G, Morrison BH, Wu W, Baudhuin LM, Xiao YJ, Mok SC, Xu Y. Lysophospholipids increase interleukin-8 expression in ovarian cancer cells. Gynecol Oncol. 2001; 81:291–300.
432. Bugl S, Wirths S, Müller MR, Radsak MP, Kopp HG. Current insights into neutrophil homeostasis. Ann N Y Acad Sci. 2012; 1266:171–178.
433. MacKinnon AC, Buckley A, Chilvers ER, Rossi AG, Haslett C, Sethi T. Sphingosine kinase: a point of convergence in the action of diverse neutrophil priming agents. J Immunol. 2002; 169:6394–6400.
434. Schenten V, Melchior C, Steinckwich N, Tschirhart EJ, Bréchard S. Sphingosine kinases regulate NOX2 activity via p38 MAPK-dependent translocation of S100A8/A9. J Leukoc Biol. 2011; 89:587–596.
435. Lin WC, Lin CF, Chen CL, Chen CW, Lin YS. Inhibition of neutrophil apoptosis via sphingolipid signaling in acute lung injury. J Pharmacol Exp Ther. 2011; 339:45–53.
436. Niwa M, Kozawa O, Matsuno H, Kanamori Y, Hara A, Uematsu T. Tumor necrosis factor-alpha-mediated signal transduction in human neutrophils: involvement of sphingomyelin metabolites in the priming effect of TNF-alpha on the fMLP-stimulated superoxide production. Life Sci. 2000; 66:245–256.
437. Wang Z, Fan H, Xie R, Yang J, Ren Y, Yang Y, Li W. The effect of sphingosine 1-phosphate/sphingosine 1-phosphate receptor on neutrophil function and the relevant signaling pathway. Acta Haematol. 2015; 134:49–56.
438. Florey O, Haskard DO. Sphingosine 1-phosphate enhances Fc gamma receptor-mediated neutrophil activation and recruitment under flow conditions. J Immunol. 2009; 183:2330–2336.
439. Di A, Kawamura T, Gao XP, Tang H, Berdyshev E, Vogel SM, Zhao YY, Sharma T, Bachmaier K, Xu J, Malik AB. A novel function of sphingosine kinase 1 suppression of JNK activity in preventing inflammation and injury. J Biol Chem. 2010; 285:15848–15857.
440. Hurt B, Schulick R, Edil B, El Kasmi KC, Barnett C Jr. Cancer-promoting mechanisms of tumor-associated neutrophils. 2017; 214:938–944.
441. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016; 131:803–820.
442. Orringer D, Lau D, Khatri S, Zamora-Berridi GJ, Zhang K, Wu C, Chaudhary N, Sagher O. Extent of resection in patients with glioblastoma: limiting factors, perception of resectability, and effect on survival. J Neurosurg. 2012; 117:851–859.
443. Verburg FA, Sweeney R, Hänscheid H, Dießl S, Israel I, Löhr M, Vince GH, Flentje M, Reiners C, Samnick S. Patients with recurrent glioblastoma multiforme. Initial experience with p-[(131)I]iodo-L-phenylalanine and external beam radiation therapy. Nuklearmedizin. 2013; 52:36–42.
444. Imber BS, Kanungo I, Braunstein S, Barani IJ, Fogh SE, Nakamura JL, Berger MS, Chang EF, Molinaro AM, Cabrera JR, McDermott MW, Sneed PK, Aghi MK. Indications and efficacy of gamma knife stereotactic radiosurgery for recurrent glioblastoma: 2 decades of institutional experience. Neurosurgery. 2017; 80:129–139.
445. Addeo R, Caraglia M, De Santi MS, Montella L, Abbruzzese A, Parlato C, Vincenzi B, Carraturo M, Faiola V, Genovese M, Cennamo G, Del Prete S. A new schedule of fotemustine in temozolomide-pretreated patients with relapsing glioblastoma. J Neurooncol. 2011; 102:417–424.
446. Lombardi G, Bellu L, Pambuku A, Della Puppa A, Fiduccia P, Farina M, D’Avella D, Zagonel V. Clinical outcome of an alternative fotemustine schedule in elderly patients with recurrent glioblastoma: a mono-institutional retrospective study. J Neurooncol. 2016; 128:481–486.
447. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014; 6:1359–1370.
448. Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ, Glass J, Kim L, Shi W. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J Neurooncol. 2014; 117:125–131.
449. Maroon JC, Seyfried TN, Donohue JP, Bost J. The role of metabolic therapy in treating glioblastoma multiforme. Surg Neurol Int. 2015; 6:61.
450. Martuscello RT, Vedam-Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov O, Deleyrolle LP, Reynolds BA. A supplemented high-fat low-carbohydrate diet for the treatment of glioblastoma. Clin Cancer Res. 2016; 22:2482–2495.
451. Winter SF, Loebel F, Dietrich J. Role of ketogenic metabolic therapy in malignant glioma: a systematic review. Crit Rev Oncol Hematol. 2017; 112:41–58.
452. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuño MA, Richardson JE, Fan X, Ji J, Chu RM, Bender JG, Hawkins ES, Patil CG, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013; 62:125–135.
453. Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, Hayashi K, Komita H, Watanabe N, Suzuki Y, Yamamoto Y, Mori R, Arai T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother. 2016; 65:1499–1509.
454. Kamran N, Calinescu A, Candolfi M, Chandran M, Mineharu Y, Asad AS, Koschmann C, Nunez FJ, Lowenstein PR, Castro MG. Recent advances and future of immunotherapy for glioblastoma. Expert Opin Biol Ther. 2016; 16:1245–1264.
455. Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV. Engineering challenges for brain tumor immunotherapy. Adv Drug Deliv Rev. 2017; 114:19–32.
456. Ning J, Wakimoto H. Oncolytic herpes simplex virus-based strategies: toward a breakthrough in glioblastoma therapy. Front Microbiol. 2014; 5:303.
457. Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic virotherapy for the treatment of malignant glioma. Neurotherapeutics. 2017; 14:333–344.
458. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, Neumann JO, Schöning T, Hüsing J, Beelte B, Kiprianova I, Roscher M, Bhat R, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther. 2017.
459. Jue TR, McDonald KL. The challenges associated with molecular targeted therapies for glioblastoma. J Neurooncol. 2016; 127:427–434.
460. Weathers SS, Gilbert MR. Toward personalized targeted therapeutics: an overview. Neurotherapeutics. 2017; 14:256–264.
461. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927; 8:519–530.
462. Warburg O. On respiratory impairment in cancer cells. Science. 1956; 124:269–270.
463. Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol. 2013; 230:350–355.
464. Seyfried TN, Mukherjee P. Targeting energy metabolism in brain cancer: review and hypothesis. Nutr Metab (Lond). 2005; 2:30.
465. Poon CC, Sarkar S, Yong VW, Kelly JJ. Glioblastoma-associated microglia and macrophages: targets for therapies to improve prognosis. Brain. 2017; 140:1548–1560.
466. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, Norberg P, Xie W, Herndon JE 2nd, Healy P, McLendon RE, Friedman AH, Friedman HS, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017; 23:1898–1909.
467. Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, Meuli R, Janzer R, Pizzolato G, Miralbell R, Porchet F, Regli L, de Tribolet N, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002; 20:1375–1382.
468. Lanzetta G, Campanella C, Rozzi A, Nappa M, Costa A, Fedele F, Innocenzi G, Gagliardi FM, Salvati M, Minniti G, Frati A, Frati L, Vecchione A. Temozolomide in radio-chemotherapy combined treatment for newly-diagnosed glioblastoma multiforme: phase II clinical trial. Anticancer Res. 2003; 23:5159–5164.
469. Athanassiou H, Synodinou M, Maragoudakis E, Paraskevaidis M, Verigos C, Misailidou D, Antonadou D, Saris G, Beroukas K, Karageorgis P. Randomized phase II study of temozolomide and radiotherapy compared with radiotherapy alone in newly diagnosed glioblastoma multiforme. J Clin Oncol. 2005; 23:2372–2377.
470. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
471. dos Santos MA, Pignon JP, Blanchard P, Lefeuvre D, Levy A, Touat M, Louvel G, Dhermain F, Soria JC, Deutsch E, Le Teuff G. Systematic review and meta-analysis of phase I/II targeted therapy combined with radiotherapy in patients with glioblastoma multiforme: quality of report, toxicity, and survival. J Neurooncol. 2015; 123:307–314.
472. Noack J, Choi J, Richter K, Kopp-Schneider A, Régnier-Vigouroux A. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis. 2014; 5:e1425.
473. Hadaczek P, Ozawa T, Soroceanu L, Yoshida Y, Matlaf L, Singer E, Fiallos E, James CD, Cobbs CS. Cidofovir: a novel antitumor agent for glioblastoma. Clin Cancer Res. 2013; 19:6473–6483.
474. Söderberg-Nauclér C, Rahbar A, Stragliotto G. Survival in patients with glioblastoma receiving valganciclovir. N Engl J Med. 2013; 369:985–986.
475. Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, Bjurman B, Tammik C, Skarman P, Peredo I, Söderberg-Nauclér C. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: a randomized, double-blind, hypothesis-generating study. Int J Cancer. 2013; 133:1204–1213.
476. Yu Z, Zhao G, Xie G, Zhao L, Chen Y, Yu H, Zhang Z, Li C, Li Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget. 2015; 6:32930–32943. https://doi.org/10.18632/oncotarget.5405.
477. Schuessler A, Walker DG, Khanna R. Cytomegalovirus as a novel target for immunotherapy of glioblastoma multiforme. Front Oncol. 2014; 4:275.
478. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, Jones L, Crough T, Dasari V, Klein K, Smalley A, Alexander H, Walker DG, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014; 74:3466–3476.
479. Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res. 2016; 76:3136–3144.
480. Fine HA. New strategies in glioblastoma: exploiting the new biology. Clin Cancer Res. 2015; 21:1984–1988.
481. Colak S, Medema JP. Cancer stem cells--important players in tumor therapy resistance. FEBS J. 2014; 281:4779–4791.
482. Prichard MN, Kern ER. The search for new therapies for human cytomegalovirus infections. Virus Res. 2011; 157:212–221.
483. Gynther M, Kääriäinen TM, Hakkarainen JJ, Jalkanen AJ, Petsalo A, Lehtonen M, Peura L, Kurkipuro J, Samaranayake H, Ylä-Herttuala S, Rautio J, Forsberg MM. Brain pharmacokinetics of ganciclovir in rats with orthotopic BT4C glioma. Drug Metab Dispos. 2015; 43:140–146.
484. Peredo I, Helldén A, Wolmer-Solberg N, Pohanka A, Stragliotto G, Rahbar A, Ståhle L, Bellander BM, Söderberg-Nauclér C. Ganciclovir concentrations in the cerebral extracellular space after valganciclovir treatment; a case study. BMJ Case Rep. 2015.
485. Peng C, Wang J, Tanksley JP, Mobley BC, Ayers GD, Moots PL, Clark SW. Valganciclovir and bevacizumab for recurrent glioblastoma: a single-institution experience. Mol Clin Oncol. 2016; 4:154–158.
486. Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol Cell Biol. 2012; 90:872–880.
487. Ghazi A, Ashoori A, Hanley PJ, Brawley VS, Shaffer DR, Kew Y, Powell SZ, Grossman R, Grada Z, Scheurer ME, Hegde M, Leen AM, Bollard CM, et al. Generation of polyclonal CMV-specific T cells for the adoptive immunotherapy of glioblastoma. J Immunother. 2012; 35:159–168.
488. Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, Liu R, Johnson LA, Weinhold K, Archer GE, Sampson JH, Mitchell DA. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res. 2014; 20:2684–2694.
489. Carlsson B, Hou M, Giandomenico V, Nilsson B, Totterman TH, Essand M. Simultaneous generation of cytomegalovirus-specific CD8+ and CD4+ T lymphocytes by use of dendritic cells comodified with pp65 mRNA and pp65 protein. J Infect Dis. 2005; 192:1912–1920.
490. Heine A, Grünebach F, Holderried T, Appel S, Weck MM, Dörfel D, Sinzger C, Brossart P. Transfection of dendritic cells with in vitro-transcribed CMV RNA induces polyclonal CD8+- and CD4+-mediated CMV-specific T cell responses. Mol Ther. 2006; 13:280–288.
491. Moody TW, Chiles J, Casibang M, Moody E, Chan D, Davis TP. SR48692 is a neurotensin receptor antagonist which inhibits the growth of small cell lung cancer cells. Peptides. 2001; 22:109–115.
492. Wang JG, Li NN, Li HN, Cui L, Wang P. Pancreatic cancer bears overexpression of neurotensin and neurotensin receptor subtype-1 and SR 48692 counteracts neurotensin induced cell proliferation in human pancreatic ductal carcinoma cell line PANC-1. Neuropeptides. 2011; 45:151–156.
493. Zhang Y, Zhu S, Yi L, Liu Y, Cui H. Neurotensin receptor1 antagonist SR48692 reduces proliferation by inducing apoptosis and cell cycle arrest in melanoma cells. Mol Cell Biochem. 2014; 389:1–8.
494. García-Garayoa E, Allemann-Tannahill L, Bläuenstein P, Willmann M, Carrel-Rémy N, Tourwé D, Iterbeke K, Conrath P, Schubiger PA. In vitro and in vivo evaluation of new radiolabeled neurotensin(8-13) analogues with high affinity for NT1 receptors. Nucl Med Biol. 2001; 28:75–84.
495. Bergmann R, Scheunemann M, Heichert C, Mäding P, Wittrisch H, Kretzschmar M, Rodig H, Tourwé D, Iterbeke K, Chavatte K, Zips D, Reubi JC, Johannsen B. Biodistribution and catabolism of (18)F-labeled neurotensin(8-13) analogs. Nucl Med Biol. 2002; 29:61–72.
496. García-Garayoa E, Bläuenstein P, Bruehlmeier M, Blanc A, Iterbeke K, Conrath P, Tourwé D, Schubiger PA. Preclinical evaluation of a new, stabilized neurotensin(8--13) pseudopeptide radiolabeled with (99m)tc. J Nucl Med. 2002; 43:374–383.
497. Buchegger F, Bonvin F, Kosinski M, Schaffland AO, Prior J, Reubi JC, Bläuenstein P, Tourwé D, García Garayoa E, Bischof Delaloye A. Radiolabeled neurotensin analog, 99mTc-NT-XI, evaluated in ductal pancreatic adenocarcinoma patients. J Nucl Med. 2003; 44:1649–1654.
498. García-Garayoa E, Maes V, Bläuenstein P, Blanc A, Hohn A, Tourwé D, Schubiger PA. Double-stabilized neurotensin analogues as potential radiopharmaceuticals for NTR-positive tumors. Nucl Med Biol. 2006; 33:495–503.
499. Zhang K, An R, Gao Z, Zhang Y, Aruva MR. Radionuclide imaging of small-cell lung cancer (SCLC) using 99mTc-labeled neurotensin peptide 8-13. Nucl Med Biol. 2006; 33:505–512.
500. de Visser M, Janssen PJ, Srinivasan A, Reubi JC, Waser B, Erion JL, Schmidt MA, Krenning EP, de Jong M. Stabilised 111In-labelled DTPA- and DOTA-conjugated neurotensin analogues for imaging and therapy of exocrine pancreatic cancer. Eur J Nucl Med Mol Imaging. 2003; 30:1134–1139.
501. Deng H, Wang H, Wang M, Li Z, Wu Z. Synthesis and evaluation of 64Cu-DOTA-NT-Cy5.5 as a dual-modality PET/fluorescence probe to image neurotensin receptor-positive tumor. Mol Pharm. 2015; 12:3054–3061.
502. Jia Y, Shi W, Zhou Z, Wagh NK, Fan W, Brusnahan SK, Garrison JC. Evaluation of DOTA-chelated neurotensin analogs with spacer-enhanced biological performance for neurotensin-receptor-1-positive tumor targeting. Nucl Med Biol. 2015; 42:816–823.
503. Brunetti J, Falciani C, Lelli B, Minervini A, Ravenni N, Depau L, Siena G, Tenori E, Menichetti S, Pini A, Carini M, Bracci L. Neurotensin branched peptide as a tumor-targeting agent for human bladder cancer. Biomed Res Int. 2015; 2015:173507.
504. Falciani C, Fabbrini M, Pini A, Lozzi L, Lelli B, Pileri S, Brunetti J, Bindi S, Scali S, Bracci L. Synthesis and biological activity of stable branched neurotensin peptides for tumor targeting. Mol Cancer Ther. 2007; 6:2441–2448.
505. Navarro-Quiroga I, Antonio González-Barrios J, Barron-Moreno F, González-Bernal V, Martinez-Arguelles DB, Martinez-Fong D. Improved neurotensin-vector-mediated gene transfer by the coupling of hemagglutinin HA2 fusogenic peptide and Vp1 SV40 nuclear localization signal. Brain Res Mol Brain Res. 2002; 105:86–97.
506. Arango-Rodriguez ML, Navarro-Quiroga I, Gonzalez-Barrios JA, Martinez-Arguelles DB, Bannon MJ, Kouri J, Forgez P, Rostene W, Garcia-Villegas R, Jimenez I, Martinez-Fong D. Biophysical characteristics of neurotensin polyplex for in vitro and in vivo gene transfection. Biochim Biophys Acta. 2006; 1760:1009–1020.
507. Rubio-Zapata HA, Rembao-Bojorquez JD, Arango-Rodriguez ML, Dupouy S, Forgez P, Martinez-Fong D. NT-polyplex: a new tool for therapeutic gene delivery to neuroblastoma tumors. Cancer Gene Ther. 2009; 16:573–584.
508. Demeule M, Beaudet N, Régina A, Besserer-Offroy É, Murza A, Tétreault P, Belleville K, Ché C, Larocque A, Thiot C, Béliveau R, Longpré JM, Marsault É, et al. Conjugation of a brain-penetrant peptide with neurotensin provides antinociceptive properties. J Clin Invest. 2014; 124:1199–1213.
509. Leten C, Struys T, Dresselaers T, Himmelreich U. In vivo and ex vivo assessment of the blood brain barrier integrity in different glioblastoma animal models. J Neurooncol. 2014; 119:297–306.
510. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015; 19:1–12.
511. Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. 2016; 18:27–36.
512. Wang XB, Jiang XR, Yu XY, Wang L, He S, Feng FY, Guo LP, Jiang W, Lu SH. Macrophage inhibitory factor 1 acts as a potential biomarker in patients with esophageal squamous cell carcinoma and is a target for antibody-based therapy. Cancer Sci. 2014; 105:176–185.
513. Li PH, Wu JX, Zheng JN, Pei DS. A sphingosine kinase-1 inhibitor, SKI-II, induces growth inhibition and apoptosis in human gastric cancer cells. Asian Pac J Cancer Prev. 2014; 15:10381–10385.
514. Yang L, Weng W, Sun ZX, Fu XJ, Ma J, Zhuang WF. SphK1 inhibitor II (SKI-II) inhibits acute myelogenous leukemia cell growth in vitro and in vivo. Biochem Biophys Res Commun. 2015; 460:903–908.
515. Ju T, Gao D, Fang ZY. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem Biophys Res Commun. 2016; 470:728–734.
516. Liu H, Zhang CX, Ma Y, He HW, Wang JP, Shao RG. SphK1 inhibitor SKI II inhibits the proliferation of human hepatoma HepG2 cells via the Wnt5A/β-catenin signaling pathway. Life Sci. 2016; 151:23–29.
517. Hengst JA, Wang X, Sk UH, Sharma AK, Amin S, Yun JK. Development of a sphingosine kinase 1 specific small-molecule inhibitor. Bioorg Med Chem Lett. 2010; 20:7498–7502.
518. Pyne S, Bittman R, Pyne NJ. Sphingosine kinase inhibitors and cancer: seeking the golden sword of Hercules. Cancer Res. 2011; 71:6576–6582.
519. Dickson MA, Carvajal RD, Merrill AH Jr, Gonen M, Cane LM, Schwartz GK. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin Cancer Res. 2011; 17:2484–2492.
520. Zhang L, Wang HD, Ji XJ, Cong ZX, Zhu JH, Zhou Y. FTY720 for cancer therapy (Review). Oncol Rep. 2013; 30:2571–2578.
521. Sonoda Y, Yamamoto D, Sakurai S, Hasegawa M, Aizu-Yokota E, Momoi T, Kasahara T. FTY720, a novel immunosuppressive agent, induces apoptosis in human glioma cells. Biochem Biophys Res Commun. 2001; 281:282–288.
522. Estrada-Bernal A, Palanichamy K, Ray Chaudhury A, Van Brocklyn JR. Induction of brain tumor stem cell apoptosis by FTY720: a potential therapeutic agent for glioblastoma. Neuro Oncol. 2012; 14:405–415.
523. Zhang L, Wang H, Zhu J, Ding K, Xu J. FTY720 reduces migration and invasion of human glioblastoma cell lines via inhibiting the PI3K/AKT/mTOR/p70S6K signaling pathway. Tumour Biol. 2014; 35:10707–10714.
524. Zhang L, Wang H, Ding K, Xu J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol Lett. 2015; 236:43–59.
525. Sordillo LA, Sordillo PP, Helson L. Sphingosine kinase inhibitors as maintenance therapy of glioblastoma after ceramide-induced response. Anticancer Res. 2016; 36:2085–2095.
526. Montgomery RM, Queiroz Lde S, Rogerio F. EGFR, p53, IDH-1 and MDM2 immunohistochemical analysis in glioblastoma: therapeutic and prognostic correlation. Arq Neuropsiquiatr. 2015; 73:561–568.
527. Schmid G, Guba M, Ischenko I, Papyan A, Joka M, Schrepfer S, Bruns CJ, Jauch KW, Heeschen C, Graeb C. The immunosuppressant FTY720 inhibits tumor angiogenesis via the sphingosine 1-phosphate receptor 1. J Cell Biochem. 2007; 101:259–270.
528. Walter DH, Rochwalsky U, Reinhold J, Seeger F, Aicher A, Urbich C, Spyridopoulos I, Chun J, Brinkmann V, Keul P, Levkau B, Zeiher AM, Dimmeler S, et al. Sphingosine-1-phosphate stimulates the functional capacity of progenitor cells by activation of the CXCR4-dependent signaling pathway via the S1P3 receptor. Arterioscler Thromb Vasc Biol. 2007; 27:275–282.
529. Ryser MF, Ugarte F, Lehmann R, Bornhäuser M, Brenner S. S1P(1) overexpression stimulates S1P-dependent chemotaxis of human CD34+ hematopoietic progenitor cells but strongly inhibits SDF-1/CXCR4-dependent migration and in vivo homing. Mol Immunol. 2008; 46:166–171.
530. Mierzejewska K, Klyachkin YM, Ratajczak J, Abdel-Latif A, Kucia M, Ratajczak MZ. Sphingosine-1-phosphate-mediated mobilization of hematopoietic stem/progenitor cells during intravascular hemolysis requires attenuation of SDF-1-CXCR4 retention signaling in bone marrow. Biomed Res Int. 2013; 2013:814549.
531. Ho JW, Man K, Sun CK, Lee TK, Poon RT, Fan ST. Effects of a novel immunomodulating agent, FTY720, on tumor growth and angiogenesis in hepatocellular carcinoma. Mol Cancer Ther. 2005; 4:1430–1438.
532. Kennedy PC, Zhu R, Huang T, Tomsig JL, Mathews TP, David M, Peyruchaud O, Macdonald TL, Lynch KR. Characterization of a sphingosine 1-phosphate receptor antagonist prodrug. J Pharmacol Exp Ther. 2011; 338:879–889.
533. Watters RJ, Wang HG, Sung SS, Loughran TP, Liu X. Targeting sphingosine-1-phosphate receptors in cancer. Anticancer Agents Med Chem. 2011; 11:810–817.
534. O’Brien N, Jones ST, Williams DG, Cunningham HB, Moreno K, Visentin B, Gentile A, Vekich J, Shestowsky W, Hiraiwa M, Matteo R, Cavalli A, Grotjahn D, et al. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res. 2009; 50:2245–2257.
535. Sabbadini RA. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol. 2011; 162:1225–1238.
536. Zhang L, Wang X, Bullock AJ, Callea M, Shah H, Song J, Moreno K, Visentin B, Deutschman D, Alsop DC, Atkins MB, Mier JW, Signoretti S, et al. Anti-S1P antibody as a novel therapeutic strategy for VEGFR TKI-resistant renal cancer. Clin Cancer Res. 2015; 21:1925–1934.
537. Pal SK, Drabkin HA, Reeves JA, Hainsworth JD, Hazel SE, Paggiarino DA, Wojciak J, Woodnutt G, Bhatt RS. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer. 2017; 123:576–582.
538. Joki T, Heese O, Nikas DC, Bello L, Zhang J, Kraeft SK, Seyfried NT, Abe T, Chen LB, Carroll RS, Black PM. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000; 60:4926–4931.
539. Zhou R, Zhang LZ, Wang RZ. Effect of celecoxib on proliferation, apoptosis, and survivin expression in human glioma cell line U251. Chin J Cancer. 2010; 29:294–299.
540. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR, Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011; 71:2664–2674.
541. Braganhol E, Tamajusuku AS, Bernardi A, Wink MR, Battastini AM. Ecto-5’-nucleotidase/CD73 inhibition by quercetin in the human U138MG glioma cell line. Biochim Biophys Acta. 2007; 1770:1352–1359.
542. Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF, Wink MR, Delgado-Cañedo A, Battastini AM. The role of ecto-5’-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem. 2008; 319:61–68.
543. Xu S, Shao QQ, Sun JT, Yang N, Xie Q, Wang DH, Huang QB, Huang B, Wang XY, Li XG, Qu X. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol. 2013; 15:1160–1172.
544. Blacher E, Ben Baruch B, Levy A, Geva N, Green KD, Garneau-Tsodikova S, Fridman M, Stein R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int J Cancer. 2015; 136:1422–1433.
545. Kalman B, Szep E, Garzuly F, Post DE. Epidermal growth factor receptor as a therapeutic target in glioblastoma. Neuromolecular Med. 2013; 15:420–434.
546. Padfield E, Ellis HP, Kurian KM. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol. 2015; 5:5.
547. Sepúlveda-Sánchez JM, Vaz MÁ, Balañá C, Gil-Gil M, Reynés G, Gallego Ó, Martínez-García M, Vicente E, Quindós M, Luque R, Ramos A, Ruano Y, Pérez-Segura P, et al. Phase II trial of dacomitinib, a pan-HER (human epidermal growth factor receptor) tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017; 19:1522–1531.
548. Oh YT, Cho HJ, Kim J, Lee JH, Rho K, Seo YJ, Choi YS, Jung HJ, Song HS, Kong DS, Seol HJ, Lee JI, Yoon Y, et al. Translational validation of personalized treatment strategy based on genetic characteristics of glioblastoma. PLoS One. 2014; 9:e103327.