
INTRODUCTION
Dendritic cells (DCs) are versatile cells that initiate adaptive immune responses, maintain steady state immunity, and mediate inflammatory responses as innate immune cells [1]. DC function is generally determined by localized environmental factors. DCs are mainly tolerant under steady-state conditions, and promote the generation of immunosuppressive T-regulatory cells (Tregs) that inhibit effector CD4+ and CD8+ T cell proliferation and activation [2]. Under inflammatory conditions, DCs are stimulated to activate and mature, and can induce either innate or adaptive responses. Several self-regulatory mechanisms limit inflammatory response expansion, and are critical for immunohomeostasis maintenance.
Hypoxia is an important microenvironment factor commonly observed in many pathological contexts, including solid tumors and inflammation, as well as some normal physiological processes [3, 4]. Tissue-resident DCs and blood DC precursors, including monocytes, can be recruited to sites of inflammation or solid tumor tissues that may be hypoxic. The effects of hypoxia on DCs are unclear, possibly due to differences in DC lineages, O2 tensions, and time courses across experiments. DCs also exhibit distinct behaviors in the hypoxic sites of solid tumors versus inflamed joints in rheumatoid arthritis (RA) cases. DCs in solid tumors are tolerant and inhibit adaptive Th1 cells [5], whereas they stimulate inflammatory Th17 cells in the joints of RA patients [6]. Thus, understanding how hypoxia finely regulates DC function is important for the development of effective anti-hypoxia-associated disease therapies.
Tolerogenic DCs execute their immunosuppressive functions via inhibitory molecules, such as programmed death ligand receptors (PD-L1 and PD-L2), CD103, arginase, or indoelamine 2,3-dioxygenase (IDO) [7–9]. IDO, a rate-limiting enzyme in tryptophan degradation, impacts peripheral tolerance and immune regulation [10]. IDO activation may deplete local concentrations of the essential amino acid tryptophan, and tryptophan metabolites, such as kynurenine, 3-hydroxyanthranilic acid, and 3-hydroxykynurenine, can act as immunosuppressants [11]. Under normal physiological conditions, IDO production is also critical for maintenance of immunohomeostasis, fetal and oral tolerance, and an immunosuppressive environment in the gut. However, IDO overproduction is detrimental to mounting an effective immune response against tumors and some invading pathogens [12]. Some DC subsets produce more IDO than others, such as CD103+ DCs in the gut that express higher IDO levels than CD103- DCs. Still, most DC subsets can be induced to produce IDO by immunosuppressive cytokines, such as TGF-β, immunosuppressive signals, such as CTLA-4, STAT3, and Foxo3, and some inflammatory stimuli, such as IFN-γ, IFN-α, and TLR agonists [13–17]. The role of hypoxia in regulating IDO production by DCs is as yet unclear.
Several mechanisms have been suggested to explain attenuation of hypoxia-mediated tissue inflammation and injury. Increased production of extracellular adenosine, a well-known anti-inflammatory factor, is one such self-protective response to hypoxia. Extracellular adenosine is produced from successive phosphohydrolysis of ATP by ectonucleotidases such as CD39 and CD73, which are upregulated by hypoxia [18]. The effects of extracellular adenosine are mediated by the four P1 purinergic receptors, A1, A2A, A2B, and A3. Adenosine A2a and A2b receptors mediate adenosine inhibition of inflammatory cytokine release (IL-12, IL-6, and TNF) and Th1 cell activation by DCs [19, 20]. Adenosine receptors are likely involved in hypoxia-regulated IDO production in DCs.
In some inflammatory microenvironments, such as inflamed joints in RA, hypoxia, GM-CSF and/or IL-4 may coexist, stimulating monocyte differentiation into DCs. This study explored the effects of hypoxia on IDO production in monocyte-derived DCs, and showed that hypoxia increased IDO production via adenosine A3R.
RESULTS
Hypoxia (1% O2) enhances IDO production
IDO depletes tryptophan, an amino acid essential for cell growth. IDO expression reportedly mediates T cell tolerance by DCs. Many immune microenvironment factors induce IDO production in DCs, such as IFN-γ, TNF-α, and PGE2 [21]. Given the roles of hypoxia in immune tolerance and inflammation, we evaluated IDO production in DCs during DC differentiation from monocytes under hypoxic conditions.
DC differentiation from monocytes via GM-CSF and IL-4 takes approximately seven days under normoxic conditions (21% O2). We therefore cultured monocytes in hypoxic conditions for seven days. We also cultured monocytes in sequential hypoxia conditions over seven days to mimic hypoxia in circulation (9–5% O2) and tissues (2–1% O2), conditions to which monocytes might be exposed during their migration from blood into tissues. Hypoxia dose-dependently upregulated IDO expression in DCs during differentiation from monocytes. The lowest O2 concentration tested (1% O2) had the strongest stimulatory effect (Figure 1A). To rule out the possibility that increased IDO production was associated with the differentiation process and not hypoxia, monocytes were cultured with 2% O2 for five days, followed by 1% or 2% O2 for the last two days. IDO expression was still stimulated by 1% O2, but not by 2% O2 (Figure 1B).
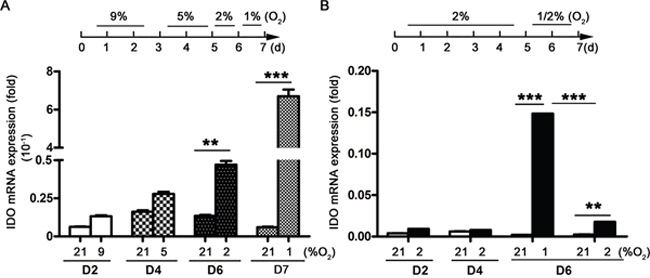
Figure 1: IDO production during DC differentiation from monocytes under hypoxic conditions. Monocytes were cultured under conditions of normoxia (21% O2; controls), 9-5-2-1% O2 hypoxia (A), or 2-1% O2 hypoxia (B) IDO expression relative to that of GAPDH was detected using real-time PCR. Data are shown as means ± SD of three replicates. **P<0.01, ***P<0.001.
Effects of IDO on mature DCs under hypoxic conditions
The costimulatory and MHC phenotypes are critical for DC-mediated T cell responses. Although IDO contributes to T cell tolerance by DCs, the effects of IDO on DC maturation are still unclear. We blocked IDO production in DCs before exposure to hypoxia (1% O2) using an IDO inhibitor (1-MT) on day five. We detected mature DC phenotypes via flow cytometry two days later. Hypoxia (1% O2) upregulated MHC-II, CD86, and CD54 in DCs and inhibited CD40 and CD83 compared to normoxia (Figure 2). Hypoxia (1% O2) also upregulated CD274 (PD-L1), which reduces T cell proliferation and induces T cell apoptosis. Blocking IDO production in hypoxia-conditioned DCs reversed MHC-II, CD86, CD54, and CD274 upregulation, but further downregulated CD40 and CD83 (Figure 2).
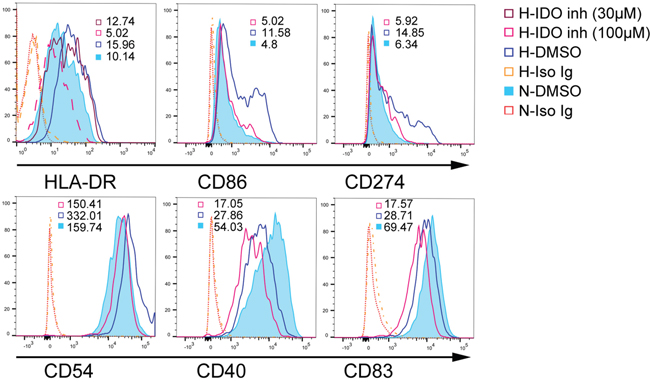
Figure 2: IDO effects on mature DCs under hypoxic conditions. Monocytes were cultured under conditions of normoxia (21% O2; controls) or 2% O2 hypoxia for 5 d. The IDO inhibitor, 1-MT (100 or 30 μM), was added to hypoxia-conditioned DCs for another 2 d at 1% O2, and CD86, CD40, CD54, HLA-DR, CD83, and CD274 expression was detected using FACS. Hypoxia-conditioned DCs with isotype Ig staining were pooled from those cells treated with DMSO or 1-MT. Results are shown as relative median fluorescence intensities (normoxia- or hypoxia-conditioned DCs with specific Ig staining vs. normoxia- or hypoxia-conditioned DCs with isotype Ig staining) and represent three experiments.
HIF-1 is non-essential for IDO production in DCs under hypoxic conditions
To explore the underlying mechanisms of hypoxia-induced IDO production in DCs, we evaluated the effects of HIF-1 signaling, a well-described hypoxia-induced pathway that controls expression of >100 hypoxia-associated genes. We found that HIF-1α knockdown via siRNA (Figure 3A, 3B) inhibited IDO production in DCs under normoxic conditions, but had no effect on IDO under hypoxic conditions as compared to control siRNA-treated DCs (Figure 3C). This indicated that HIF-1 signaling regulates IDO production in DCs under normoxic conditions, but is not essential for increased IDO expression during hypoxia (1% O2).
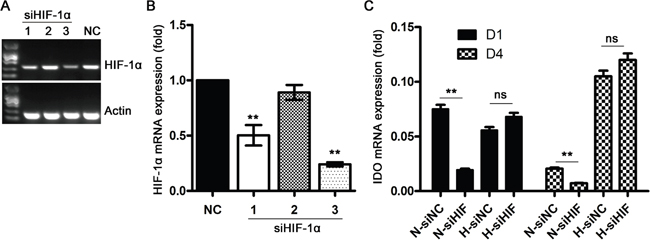
Figure 3: Effects of HIF-1 signaling on IDO production in DCs during hypoxia. Monocytes were transfected with control (NC) or HIF-1α-specific siRNAs under normoxic conditions for 48 h, and HIF-1α expression was detected using real-time PCR (A & B) Knockdown efficiency was calculated by normalizing HIF-1α expression to that of Actin; (C) DCs were transfected with NC or HIF-1α siRNA (pooled HIF-1α siRNA1+2 as indicated in (B) on d 1 or d 4 for 48 h under normoxic (21% O2) or hypoxic conditions (5 d of 2% O2 then 1 d of 1% O2), and then IDO expression was detected relative to that of GAPDH using real-time PCR Data are shown as means ± SD of three replicates. **P<0.01.
Adenosine A3 receptor controls IDO production during hypoxia
Extracellular adenosine is increased by tissue hypoxia. To investigate the roles of adenosine in IDO production, we dynamically detected adenosine receptor (AR) expression in DCs during differentiation. We found that hypoxia upregulated four adenosine receptors, A1R, A2aR, A2bR, and A3R. A2aR and A2bR expression increased the most at 1% O2 late in the differentiation process, and A3R was the most abundant at this late stage (Figure 4A). We then evaluated the roles of A2aR, A2bR, and A3R in IDO production during hypoxia (1% O2) using specific inhibitors of A2bR and A3R, and an A2aR agonist. A3R blockade during hypoxia inhibited IDO production in DCs, while A2bR blockade further enhanced IDO production. Activating A2aR had no effect on IDO production (Figure 4B). These results indicated that adenosine released during hypoxia (1% O2) promotes IDO production via A3R.
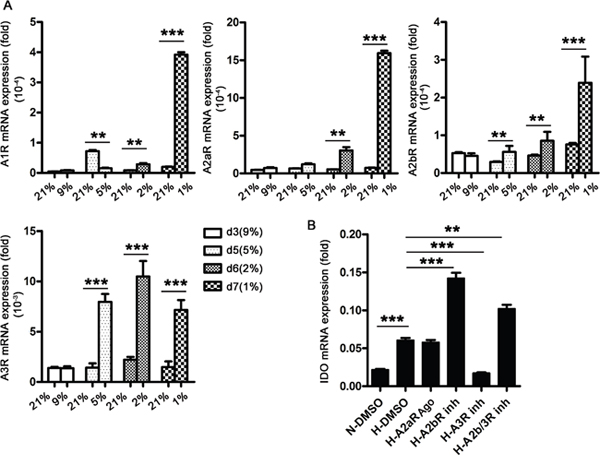
Figure 4: Adenosine A3R is involved in IDO production in DCs during hypoxia. Monocytes were cultured under conditions of normoxia (21% O2; control) or 9-5-2-1% O2 sequential hypoxia for 7 d, and A1R, A2aR, A2bR, and A3R expression relative to that of GAPDH was detected using real-time PCR. (A) DCs on d 5 were treated with specific AR inhibitors or an agonist, and IDO expression relative to that of GAPDH was detected using real-time PCR. (B) Data are shown as means ± SD of three replicates and represent three experiments. **P<0.01, ***P<0.001.
A3R signaling modulates DC phenotype
Adenosine-treated DCs are reportedly impaired in stimulating T cell responses. As costimulatory and MHC-II molecules are critical for antigen presentation by DCs, we investigated whether and which adenosine receptors were involved in IDO-upregulated CD86, CD274, MHC-II and CD54 expression. We treated DCs on day five (before 1% O2 hypoxia) with A2bR or A3R inhibitors, or an A2aR agonist. Similar to the effects of IDO inhibition in DCs, CD86, MHC-II, CD54, and CD274 upregulation was reversed by A3R inhibition, but was enhanced or unaffected by A2bR blockade or A2aR activation (Figure 5). Moreover, blocking A3R had no effect on CD40 and CD83 downregulation. These findings indirectly confirmed that hypoxia induced IDO production in DCs via A3R.
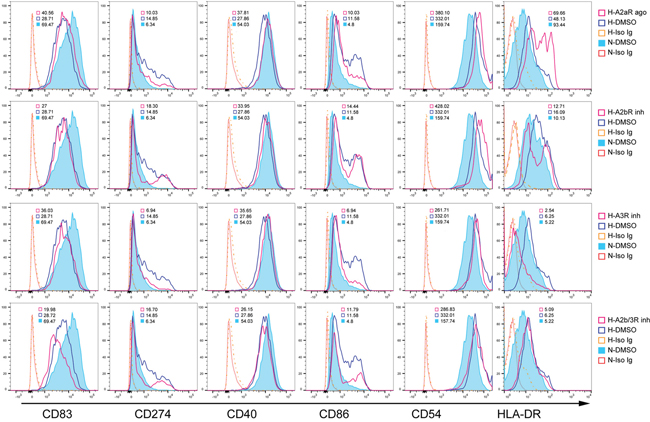
Figure 5: Adenosine A3R modulates DC phenotypes during hypoxia. Monocytes were cultured under conditions of normoxia (21% O2; control) or 2% hypoxia for 5 d. A2bR or A3R inhibitors, or an A2aR agonist was added to hypoxia-conditioned DCs for another 2 d at 1% O2, and CD86, CD40, CD54, HLA-DR, CD83, and CD274 expression was detected using FACS. Data are shown as relative median fluorescence intensities to isotype Ig staining, and represent three experiments.
DISCUSSION
Hypoxia-associated metabolic reprogramming helps determine bioenergetic needs during the immune response and other physiological and pathological processes. Hypoxia and increased extracellular adenosine are both features of inflammation and solid tumors [22–24]. We found that hypoxia divergently regulates mature DCs via extracellular adenosine-induced IDO production in an autocrine manner. The auto-regulatory effects of IDO in DCs might be associated with tryptophan metabolites, such as kynurenine, 3-hydroxyanthranilic acid, and 3-hydroxykynurenine. Our observations of crosstalk between hypoxia, adenosine receptors, and DC-produced IDO suggest a novel mechanism through which hypoxia-related metabolites in the microenvironment modulate immune responses in cooperation with other immune cells, particularly T cells. In addition to directly inhibiting effector T cell responses, IDO upregulates PD-L1 in DCs. The PD-L1/PD-1 interaction in T cells transmits an inhibitory signal that suppresses T cell proliferation and may induce apoptosis [25, 26].
Our findings also indicated that IDO upregulates CD86 and HLA-DR, the costimulatory and MHC-II molecules critical for DC-triggered effector CD4+ and CD8+ T cell responses. However, such upregulation may have no effect during extreme hypoxia, because our previous work showed that DCs did not efficiently stimulate T cell responses under <5% O2 conditions. Impaired T cell responses during hypoxia may be due in part to direct inhibition of IDO released from DCs or other cell types, and/or regulatory T cell activity. In contrast to effector T cells, CD4+CD25+ regulatory T cell potency is reportedly enhanced under hypoxic conditions [27, 28]. Additionally, IDO-producing DCs are essential for induction of regulatory T-cell responses [29]. Thus, in hypoxic microenvironments, such as in some solid tumors, DCs are tolerant not only due to inhibitory signals from adenosine/A2aR and A2bR, but also from autocrine IDO. This promotes regulatory T cell survival and function, but inhibits the generation of effector CD4+ and CD8+ T helper cells, which may promote tumor cell growth and expansion.
Hypoxia may also induce IDO production in non-immune cells via adenosine/A3R signaling, and this metabolic reprogramming mechanism may participate in many other pathophysiological processes. Obesity, dyslipidemia, hypertension, and type 2 diabetes are described as metabolic syndromes, which may involve inducible IDO production in adipose tissues. During chronic inflammation, a common factor in metabolic syndromes, the local hypoxic microenvironment may upregulate extracellular adenosine production. Adenosine controls insulin signals in adipose tissue, muscle, and liver [30, 31], and all four adenosine receptors are reportedly involved in glucose homeostasis, adipogenesis, and insulin resistance [32]. Increased levels of IDO-generated tryptophan metabolites, the kynurenines, may promote metabolic syndrome development via pro-oxidative, neurotoxic, and apoptotic activities, along with upregulation of inducible nitric oxide synthase, phospholipase A2, arachidonic acid, prostaglandin, 5-lipoxygenase, and leukotriene cascade [33]. Whether or not hypoxic factors increase IDO production via adenosine in metabolic syndromes is as yet unknown.
IDO can be produced by multiple immune cell types, such as macrophages, DCs, and eosinophils, as well as epithelial, endothelial, and tumor cells [34]. Regulation of IDO expression is complex. Antigen-presenting cells, such as DCs and macrophages, are preferentially stimulated to express IDO when challenged with proinflammatory stimuli, including IFN-γ, TNFα, IL-32, and PGE2 [35]. IFN-γ is an important product of activated NK and effector CD4+ and CD8+ T cells, which are induced during viral infections. TNF-α and IL-32 are also critical mediators in some inflammatory diseases, including RA, chronic obstructive pulmonary disease, and graft-vs.-host disease [36]. Along with TNFα and IL-32, hypoxic factors also promote IDO production. Thus, hypoxia-induced IDO in combination with other inflammatory stimuli may limit inflammation expansion and injury in RA as part of key immunoregulatory feedback loops.
Several signaling pathways can induce IDO production. Janus kinase 1 (JAK1) and Stat1 signaling mediate IFN-γ-induced IDO transcription [37]. Stat1 directly binds IFN-γ-activated sites within the IDO promoter or indirectly affects IDO transcription by inducing IFN regulatory factor-1 (IRF-1), which binds the IDO promoter at two IFN-stimulated response element sites [38]. Additionally, the JAK1/2/STAT3, protein kinase Cδ, and NF-κB pathways can also induce IDO expression [39, 40]. Still, the mechanisms by which hypoxia/adenosine/A3R triggers IDO expression in DCs must be further clarified. Our study suggests that decreased intracellular cAMP levels may be involved A3R-mediated IDO upregulation. Prostanoid PGD2 inhibits IDO expression via the PGD2 receptor, DP1, which increases intracellular cAMP levels that is found to be inhibitory in IDO production. This PGD2 inhibitory effect acts through DP1/cAMP/PKA/CREB signaling [41]. Extracellular adenosine relays signals by activating any of its four receptors (A1R, A2aR, A2bR, and A3R). A1R or A3R activation attenuates intracellular cAMP levels, whereas A2aR or A2bR activation elevates cAMP levels [42]. We observed that blocking A2bR further enhanced IDO expression, implying that A3R-induced IDO production is associated with cAMP/PKA pathway inhibition.
In summary, we demonstrated that hypoxia induces IDO production in human DCs via adenosine A3R, overcoming the inhibitory effects of A2bR. The relationship between hypoxia, adenosine, and IDO should be further explored in other cell types. Our findings offer guidance for pharmacological administration of adenosine receptor agonists or antagonists with the goal of achieving immune tolerance or controlling insulin resistance and other metabolic disorders via IDO modulation.
MATERIALS AND METHODS
Monocyte-derived DC induction and culture
CD14+ human monocytes from buffy coats of healthy volunteers (Department of Transfusion, Shanghai Changhai Hospital) were purified using anti-CD14 microbeads (Miltenyi Biotec, Bergisch-Gladbach, Germany) and cultured in RPMI 1640 medium (Gibco) containing 10% FBS (Gibco), 50 ng/mL GM-CSF (R&D Systems), and 10 ng/mL IL-4 (R&D Systems) under normoxic (21% O2, controls) or hypoxic (range from 9% to 5% to 2% to 1%, or from 2% to 1%, or 2% O2) conditions for seven days. In some experiments, cells were transfected with 20 nM siRNA (GenePharm) using INTERFERin (Polyplus-transfection) or treated with a specific inhibitor of IDO (1-methyl-[D]-tryptophan, 1-MT, 100 μM), an adenosine receptor inhibitor (A2bR, MRS1754 from Calbiochem; A3R MRS1191 from Sigma-Aldrich; both 10 μM) or an A2aR agonist (CGS21680, 10 μM, Calbiochem). After 48 h, cells were collected for analysis. Sequences of siRNAs of HIF-1α are: 5’-CCAGUUAUGAUUGUGAAGUUATT-3’ (sense), 5’-UAACUUCACAAUCAUAACUGGTT-3’ (antisense); 5’-GGUGUUGCGUAAAUCAUAUTT-3’ (sense), and 5’-AUAUGAUUUACGCAACACCTT-3’ (antisense); and 5’-GGAGCUAACAGGACAUAGUTT-3 (sense), and 5’-ACUAUGUCCUGUUAGCUCCTT-3’ (antisense). Procedures were approved by the ethics committee of Guizhou Medical University and informed consent was obtained from all donors in this study.
IDO and adenosine receptor quantitative PCR analyses
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was carried out as described previously [43]. Total RNA was extracted using TRIzol (Invitrogen). Quantitative PCR was performed on a LightCycler (Roche, Basel) using the SYBR RT-PCR kit (Takaro). Expression data from each sample were normalized to β-actin or GAPDH. Primer pairs were as follows: β-actin-FW, 5’-GGCGGCACCACCATGTACCCT-3’; β-actin-RV, 5’-AGGGGCCGGACTCGTCATACT-3’; GAPDH-FW, 5’-TGTGGGCATCAATGGATTTGG-3’; GAPDH-RV, 5’-ACACCATGTATTCCGGGTCAAT-3’; IDO-FW, 5’-AGTCCGTGAGTTTGTCCTTTCAA-3’; IDO-RV, 5’-TTTCACACAGGCGTCATAAGCT-3’; A1R-FW, 5’-TCTGGGCGGTGAAGGTGAAC-3’; A1R-RV, 5’-AGTTGCCGTGCGTGAGGAAG-3’; A2aR-FW, 5’-AGGCAGCAAGAACCTTTCAA-3’; A2aR-RV, 5’-CTAAGGAGCTCCACGTCTGG-3’; A2bR-FW, 5’-CCCTTTGCCATCACCATCAG-3’; A2bR-RV, 5’-CCTGACCATTCCCACTCTTGA-3’; A3R-FW, 5’-AGACCACCACCTTCTATTT-3’; A3R-RV, 5’-GACCCTCTTGTATCTGACG-3’.
FACS analysis of DC phenotypes
Human DC phenotypes were assessed via flow cytometry (Becton Dickinson, LSRFortessa). Cells were incubated with fluorochrome-conjugated monoclonal antibodies (CD40-PE-Cy7, CD86-BV510, HLA-DR-APC, CD83-APC-Cy7, CD274-APC, and CD54-PE; BD PharMingen) for 30 min on ice, washed, and analyzed using FlowJo software as previously described [44].
Statistical analyses
Statistical analyses were performed using Graphpad Prism5 software. Differences were assessed using unpaired two-tailed Student t tests for two groups or ANOVAs for more than two groups. Statistical significance was set at P<0.05.
ACKNOWLEDGMENTS
We gratefully acknowledge Wu J for helpful suggestions and discussions.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
GRANT SUPPORT
This work was supported by grants from the National Natural Science Foundation of China (31470858, 81771711) and the Science and Technology Cooperation Project of Guizhou province [2015]7426.
REFERENCES
1. Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017; 17:30-48.
2. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. 2014; 40:642-656.
3. Konisti S, Kiriakidis S, Paleolog EM. Hypoxia--a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol. 2012; 8:153-162.
4. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. 2012; 1826:272-296.
5. Johnson DJ, Ohashi PS. Molecular programming of steady-state dendritic cells: impact on autoimmunity and tumor immune surveillance. Ann N Y Acad Sci. 2013; 1284:46-51.
6. Fan ZD, Zhang YY, Guo YH, Huang N, Ma HH, Huang H, Yu HG. Involvement of P2X7 receptor signaling on regulating the differentiation of Th17 cells and type II collagen-induced arthritis in mice. Sci Rep. 2016; 6:35804.
7. Hobo W, Maas F, Adisty N, de Witte T, Schaap N, van der Voort R, Dolstra H. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010; 116:4501-4511.
8. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, Hagan T, Benitez L, Todor A, Machiah D, Oriss T, Ray A, Bosinger S, et al. mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science. 2017; 357:1014-1021.
9. Lukas D, Yogev N, Kel JM, Regen T, Mufazalov IA, Tang Y, Wanke F, Reizis B, Muller W, Kurschus FC, Prinz M, Kleiter I, Clausen BE, et al. TGF-beta inhibitor Smad7 regulates dendritic cell-induced autoimmunity. Proc Natl Acad Sci U S A. 2017; 114:E1480-E1489.
10. Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol. 2007; 7:817-823.
11. Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci. 2013; 34:136-143.
12. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, Mellor AL. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016; 76:2076-2081.
13. Tu L, Chen J, Zhang H, Duan L. Interleukin-4 inhibits regulatory T cell differentiation through regulating CD103+ dendritic cells. Front Immunol. 2017; 8:214.
14. Nouel A, Pochard P, Simon Q, Segalen I, Le Meur Y, Pers JO, Hillion S. B-cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J Autoimmun. 2015; 59:53-60.
15. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, Yan C, Yan F, Ren X. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014; 193:2574-2586.
16. Ankrum JA, Dastidar RG, Ong JF, Levy O, Karp JM. Performance-enhanced mesenchymal stem cells via intracellular delivery of steroids. Sci Rep. 2014; 4:4645.
17. Wu Y, Chen X, Ge X, Xia H, Wang Y, Su S, Li W, Yang T, Wei M, Zhang H, Gou L, Li J, Jiang X, et al. Isoliquiritigenin prevents the progression of psoriasis-like symptoms by inhibiting NF-kappaB and proinflammatory cytokines. J Mol Med (Berl). 2016; 94:195-206.
18. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002; 110:993-1002.
19. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014; 74:7250-7259.
20. Figueiredo AB, Souza-Testasicca MC, Mineo TW, Afonso LC. Leishmania amazonensis-induced cAMP triggered by adenosine A2B receptor is important to inhibit dendritic cell activation and evade immune response in infected mice. Front Immunol. 2017; 8:849.
21. Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005; 106:2375-2381.
22. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012; 367:2322-2333.
23. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011; 11:201-212.
24. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, Kuchroo VK, Robson SC, Quintana FJ. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013; 14:1054-1063.
25. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999; 5:1365-1369.
26. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002; 8:793-800.
27. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol. 2008; 38:2412-2418.
28. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012; 109:E2784-E2793.
29. Hayashi T, Beck L, Rossetto C, Gong X, Takikawa O, Takabayashi K, Broide DH, Carson DA, Raz E. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest. 2004; 114:270-279.
30. Csoka B, Toro G, Vindeirinho J, Varga ZV, Koscso B, Nemeth ZH, Kokai E, Antonioli L, Suleiman M, Marchetti P, Cseri K, Deak A, Virag L, et al. A2A adenosine receptors control pancreatic dysfunction in high-fat-diet induced obesity. FASEB J. 2017; 31:4985-4997.
31. Csoka B, Koscso B, Toro G, Kokai E, Virag L, Nemeth ZH, Pacher P, Bai P, Hasko G. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes. 2014; 63:850-866.
32. Antonioli L, Blandizzi C, Csoka B, Pacher P, Hasko G. Adenosine signalling in diabetes mellitus--pathophysiology and therapeutic considerations. Nat Rev Endocrinol. 2015; 11:228-241.
33. Oxenkrug GF. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann N Y Acad Sci. 2010; 1199:1-14.
34. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007; 117:1147-1154.
35. Ohmatsu H, Humme D, Gonzalez J, Gulati N, Mobs M, Sterry W, Krueger JG. IL-32 induces indoleamine 2,3-dioxygenase+CD1c+ dendritic cells and indoleamine 2,3-dioxygenase+CD163+ macrophages: relevance to mycosis fungoides progression. Oncoimmunology. 2017; 6:e1181237.
36. Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006; 103:3298-3303.
37. Chon SY, Hassanain HH, Gupta SL. Cooperative role of interferon regulatory factor 1 and p91 (STAT1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J Biol Chem. 1996; 271:17247-17252.
38. Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, Decker T. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci U S A. 2007; 104:2849-2854.
39. Jeong YI, Kim SW, Jung ID, Lee JS, Chang JH, Lee CM, Chun SH, Yoon MS, Kim GT, Ryu SW, Kim JS, Shin YK, Lee WS, et al. Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling pathway in interferon-gamma-stimulated murine dendritic cells. J Biol Chem. 2009; 284:3700-3708.
40. Lee SM, Park HY, Suh YS, Yoon EH, Kim J, Jang WH, Lee WS, Park SG, Choi IW, Choi I, Kang SW, Yun H, Teshima T, et al. Inhibition of acute lethal pulmonary inflammation by the IDO-AhR pathway. Proc Natl Acad Sci U S A. 2017; 114:E5881-E5890.
41. Bassal NK, Hughes BP, Costabile M. Prostaglandin D2 is a novel repressor of IFNgamma induced indoleamine-2,3-dioxygenase via the DP1 receptor and cAMP pathway. Prostaglandins Leukot Essent Fatty Acids. 2016; 110:48-54.
42. Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009; 111:904-915.
43. Zhai Y, Wu B, Li J, Yao XY, Zhu P, Chen ZN. CD147 promotes IKK/IkappaB/NF-kappaB pathway to resist TNF-induced apoptosis in rheumatoid arthritis synovial fibroblasts. J Mol Med (Berl). 2016; 94:71-82.
44. Liu S, Yi L, Ling M, Jiang J, Song L, Liu J, Cao X. HSP70L1-mediated intracellular priming of dendritic cell vaccination induces more potent CTL response against cancer. Cell Mol Immunol. 2016.