
Introduction
One year ago, we published the original CUSP9 treatment protocol for recurrent glioblastoma [1]. CUSP9, Comprehensive Undermining of Survival Paths, was an attempt to block growth facilitating or growth driving signaling systems that have been identified as active in glioblastoma. To develop CUSP9 we found nine re-positioned (re-purposed), already-marketed drugs that had evidence supporting their ability to inhibit one or more of the identified glioblastoma growth and cell survival pathways [1]. CUSP9 has been well-tolerated in the patients that have come to our attention who have been given CUSP9 on a compassionate-use basis. There is no word yet on effectiveness. We present here an update on the rationale and some minor changes to this protocol, designated CUSP9*.
The basic idea behind CUSP9 treatment was detailed in the original document [1] and has not changed for the current CUSP9*. Briefly, given the impasse we have been in since temozolomide introduction as standard initial treatment in 2005 [2] we developed a conceptually new approach. Instead of focusing on finding new cytotoxic drugs or variations combining traditional cancer chemotherapeutic drugs we looked at both native growth-promoting systems and the many escape paths that are mobilized by glioblastoma cells during exposure to current cytotoxic drugs like temozolomide. We then surveyed the research literature looking for already-marketed non-cancer treatment related drugs for which we have data or evidence that that they might block or inhibit one of these identified cytotoxicity circumvention pathways. Using low likelihood of adding to patient side effect burden, good quality of life (QOL) maintenance, and good clinical experience with the drug in question as additional selection criteria, we arrived at nine drugs to augment temozolomide’s anticancer activity: aprepitant, artesunate, auranofin, captopril, Cu gluconate, disulfiram, ketoconazole, nelfinavir, and sertraline [1].
In Fig. 1. the profound effect of the original CUSP9 drugs without temozolomide can be seen. Deep cytotoxicity occurs in glioblastoma cell lines at drug levels expected in vivo. The new, slightly modified CUSP9* shows even greater cytotoxicity to glioblastoma cell lines at even lower concentrations. The deep in vitro cytotoxicity of both CUSP9 and CUSP9* drugs to glioblastoma cell lines is the subject of a separate publication.
Since CUSP9 publication, the European Medicines Agency has delisted ketoconazole, and the manufacturer of nelfinavir has stopped production. Related drugs were therefore substituted in CUSP9*, itraconazole for ketoconazole, ritonavir for nelfinavir. As disulfiram achieves much of its anticancer effectiveness only after one of its metabolites chelates copper, we added copper gluconate to CUSP9. However, disulfiram derivatives chelate copper already in the stomach [references given in Table 4.], negating need for exogenous copper. In CUSP9* therefore, copper gluconate was deleted. In its stead, celecoxib adds further dimensions to our targeting of the many “complementary or redundant pathways” [3] glioblastoma uses to grow and evade our cytotoxicity attempts. CUSP9* therefore consists of simultaneous administration of aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, sertraline, and ritonavir. They are designed to be administered with low-dose, uninterrupted, daily temozolomide. Repositioning already-marketed drugs to block survival and growth paths in glioblastoma remains the watchword.
None of the 22 studies of new cytotoxic drugs, or cytotoxic drug combinations for recurrent glioblastoma reporting in 2012 gave meaningful clinical benefit [1]. We now report similarly sad results for the 27 studies reporting in 2013. Twelve of these 2013 studies are listed in Table 1. Since entry conditions were different for these twelve studies no comparative conclusion can or should be drawn from the differing OS, other than, clearly, no breakthroughs have been made. Not shown in Table 1 are 15 studies that were stopped early for futility, disastrous QOL deterioration, or studies where design vagarities didn’t permit OS determination.
Table 1: Overall survival, OS, in 12 of the 27 clinical studies on new treatments for recurrent glioblastoma reporting in 2013. The remaining 15 studies could not be evaluated for OS but none seemed to show dramatic benefit or evidence that their numbers would be much different from the 12 listed here. It is important not to conclude from the differing OS in this table that one treatment might be different or better than another. Study entry criteria, previous treatments, and other variables make close comparisons between these studies impossible. What we can conclude is that OS is short, glioblastoma is an aggressive disease and better treatments are needed.
Drugs or treatment |
median OS [months] |
reference |
Dose-dense temozolomide + tamoxifen |
17 |
290 19 |
Re-irradiation |
13 |
291 3 |
bevacizumab and temsirolimus |
4 |
292 4 |
open laser ablation |
11 |
293 5 |
temozolomide 100 mg/m2/day x 21 days, 7 days off, repeated |
12 |
294 6 |
erlotinib and sorafenib |
6 |
295 7 |
temozolomide and lapatinib |
6 |
296 8 |
continuous low dose temozolomide 50 mg/m2/day |
7 |
7 9 |
nintedanib |
6 |
297 10 |
bevacizumab and erlotinib |
10 |
298 11 |
sunitinib |
9 |
299 12 |
autologous vaccine+ HSP96 |
12 |
300 13 |
Four additional guiding principles for CUSP9* formulation remain as for CUSP9:
A. Careful attention to using drugs that have a low likelihood of increasing patient side effect burden, a low likelihood of interfering with each other, and a research database allowing reasonable expectation for anti-glioblastoma effects.
B. Importance of having a broad, comprehensive approach, blocking potential as well as actual cytotoxicity escape paths. We aimed to dismantle every glioblastoma cell defense mechanism we could identify for which we had already-marketed inhibitors that also have low inherent risk by themselves and don’t have predictable areas of interference with each other. Although designed to be used with low dose continuous temozolomide 50 mg/m2/day by mouth, recent research is showing anti-glioma activity of the CUSP9* drugs even without temozolomide. This data will be discussed below in the individual drugs’ sections.
C. When one or several growth paths are blocked, a cell, and particularly a cancer cell, shifts reliance to other parallel or compensating paths that have not been blocked. We term this the “Nile Distributary Problem” in that if one distributary at the delta is blocked, total flow into the sea remains unaltered. The unblocked distributaries take up the water that would have flowed through the blocked distributary as can be imagined from satellite image in Fig. 2. Representative quotes in Table 2. are from various authors referring to this phenomenon in cancer growth using different words to discribe this idea. Hence, need for a CUSP-type broad-net protocol.
Table 2: Intratumoral spatial and temporal heterogeneity. Selected recent quotes on the strand of thinking in oncology from the last two years, the principles upon which CUSP9* was developed. Words outside of quotation marks are our addition but we believe in the spirit in which the quoted authors meant their comments. CSC = cancer stem cell; GBM = glioblastoma
Effective “treatment of recurrent GBM includes combination strategies with agents that target complementary or redundant pathways” [3]. |
Writing of a study of renal cell carcinoma...“Gene-expression signatures of good and poor prognosis were detected in different regions of the same tumor.....[there was] extensive intratumor heterogeneity” [301] |
There is “bewildering...heterogeneity” over time following the same tumor and “within individual tumor biopsies [that are] spatially separated” [302]. |
Writing of head/neck squamous cell carcinoma “intratumor heterogeneity showed that a single biopsy may not represent the entire mutational landscape” [303.]. |
Reviewing plasticity and CSC “non-CSCs can reacquire a CSC phenotype” [304.]. |
Writing of cancers generally...there are multiple” dynamic interrelationship[s] between intratumoral cell subpopulations...[that have] clear clinical significance” [305.]. |
Writing of aggressive breast cancer...“coexistence of tumor cells with different phenotypic traits within a primary tumor” with “branched ...[bidirectional] ...evolutionary tumor growth” concluding “primary tumors are ecosystems of evolving clones with different spatial and temporal distributions” that are mutually reinforcing [5]. |
In offering their data and interpretation on why inhibiting growth factor receptor inhibition has not been effective in prolonging glioblastoma survival...”growth factor receptor/PI3K/AKT pathway is complex and nonlinear having many inputs from other pathways (cross covering paths compensating for a particular block), multiple sites of feedback regulation (both positive and negative), and a large number of downstream effectors “[306] (Our comments in parentheses.) |
D. Addressing tumor microregional heterogeneity in time and space, the “multifaceted heterogeneity” of glioblastoma both between different tumors of similar H&E histology and within an individual tumor both in space and over time [3, 4]. A recent study of aggressive human breast cancers by Costa et al is exemplary of this phenomenon [5] that applies to glioblastoma. They demonstrated marked micro-regional heterogeneity within a single tumor mass with respect to a particular proteinase, ADAM-23, showing further that ADAM-23 positive and ADAM-23 negative subpopulations had different but mutually supporting functions [5]. Heterotypic environments create growth vigor in that ADAM-23 negative cells alone are invasion-competent but proliferation-poor while ADAM-23 positive subpopulations are proliferation-competent, invasion-weak [5]. We look to this phenomenon of mutually reinforcing subpopulations with spatial and temporal intratumoral heterogeneity that is also unstable over time as a primary cause of previous chemotherapy failures.
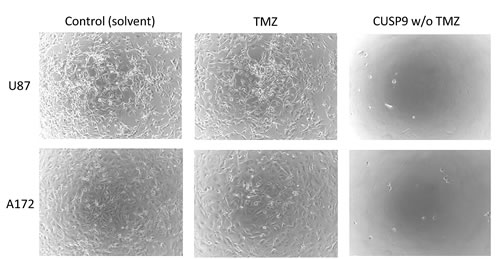
Fig. 1: In vitro activity of CUSP9 drugs compared to that of temozolomide alone. The cell-killing effects of CUSP9 without temozolomide versus temozolomide alone on glioblastoma cell lines in vitro. Two different glioblastoma cell lines were incubated with CUSP9 compounds without temozolomide (TMZ) or with TMZ alone at the respective concentrations commonly achievable in human plasma or cerebrospinal fluid (if the latter data were available). Control cells were treated with the corresponding amount of solvent only. Microphotographs were taken after 7 days of continuous exposure to CUSP9, TMZ, or solvent respectively. While glioblastoma cells grew rapidly under control conditions, all CUSP9 without TMZ-treated cells had died. In comparison, TMZ conferred a markedly weaker inhibition of cell growth.
Although the particular proteinase Costa et al studied might not be a universal or even an important feature of malignancies, we believe that the principle is paradigmatic of cancers. Heterotypic environments within a tumor, shifting over time and space is of extraordinary importance to our understanding of malignant growth. Many researchers have recently come to related conclusions on this matter, selected examples given in Table 2. Hence, we must cast a wide net with CUSP9*, multi targeting to match glioblastoma heterogeneity in time and space.
Also related to this aspect of CUSP9*, concordant in principle and thinking, was the recent COMBAT trial [6] where 74 children with various advanced, treatment-refractory cancers were treated with six drugs- low-dose daily temozolomide, etoposide, celecoxib, vitamin D, fenofibrate and retinoic acid (COMBAT regimen). Temozolomide and etoposide are traditional cytotoxic chemotherapeutic agents. The four ancillary drugs were designed to, as in CUSP9*, block survival paths. It was adequately tolerated enough and authors considered that their study had evidence enough of benefit in this heavily pretreated population to warrant further study [6].
The CUSP9* drugs
Table 1. lists several studies on recurrent glioblastoma that reported in 2013. A summary of the CUSP9* drugs showing half-life, and several core rationale(s) for the individual drugs’ inclusion in, and contribution to, CUSP9* is given in Table 3. Table 4. lists the CUSP9* drugs, P-450 engagements and their common side effects.
The choice of low dose continuous temozolomide [50 mg/m2/day by mouth] was made in view of the low side effect burden imposed by this regimen along with the absence of any other cytotoxic regimen demonstrating significantly longer overall survival (OS) [7, 8]. Note that one of these studies combined temozolomide with another of the CUSP9* drugs, celecoxib [8]. The authors noted that the combination “seems to have activity in recurrent glioblastoma without relevant toxicity” but their progression-free survival at 6 months was only 43% [8]. Clearly better is needed.
Table 3: Overview of CUSP9* drugs with circulating half-life and basic growth paths inhibited. Breast cancer resistance protein, BCRP; Neurokinin-1, synonymous with 11 amino acid, 1.3 kDa peptide substance P; angiotensin converting enzyme, ACE reactive oxygen species, ROS, a term used to refer to any atom with an unpaired valence electron; Translationally controlled tumor protein, TCTP; matrix metaloproteinase -2, -9, MMPs; References are given in the drug’s section in text.
DRUG |
T1/2 |
core survival pathway[s] inhibited... |
ARTESUNATE ------------------ active metabolite dihydroartemisinin |
< 1 hr ------ 1 hr |
phosphoinositide 3-kinase, Akt, increases ROS, NF-κB activation, TNF-alpha, IL-6, TLR2, ------- Same |
APREPITANT |
10 hrs |
NK-1 receptors |
AURANOFIN |
10 days |
thioredoxin, increases ROS, STAT3 |
CAPTOPRIL |
2 hrs |
ACE, AT1 receptors, MMPs |
CELECOXIB |
9 hrs |
COX-1 and -2, carbonic anhydrase -2 and -9 |
DISULFIRAM ----------------- active metabolite diethyldithiocarbamate |
<2 hrs ---- 6 hrs |
ALDH, increases ROS -------- same |
ITRACONAZOLE |
19 hrs |
P-gp efflux transporters, BCRP, Hedgehog, 5-lipoxygenase |
RITONAVIR |
4 hrs |
P-gp efflux transporters [weak], Akt, mTOR, cyclin D3, proteasome, |
SERTRALINE |
1 day |
Akt, mTOR, TCTP |
Discussed below are the nine drugs of CUSP9* with an updated brief review of data on the 6 retained drugs with a fuller exposition of data on two new substitute drugs itraconazole and ritonavir and one new drug, celecoxib. See the original CUSP9 paper [1] for more detailed drug reviews and discussion of thinking behind CUSP.
Table 4: CUSP9* drugs with hepatic P-450 engagements and expected side effect profile based on data and clinical experience with each drug when used individually. * = Alcohol intolerance is not listed as side effect of disulfiram even though this is severe and universal because ALDH inhibition and consequent alcohol intolerance is the main and expected effect of disulfiram. References to the pharmacology and side effect profile of the individual drugs are given in the text. GFR, glomerular filtration rate; LFT, liver function tests;
DRUG |
p-450 inhibited |
metabolism by |
common SE |
rare SE |
ref. |
aprepitant |
3A4 |
3A4 |
hiccups, asthenia, diarrhea |
none |
309, 311, 316 |
artesunate |
2D6 slight |
2A6 slight |
altered taste |
neutropenia ? |
307 |
auranofin |
none known |
unknown |
diarrhea, rash, |
stomatitis, neutropenia thrombocytopenia |
308, 310 312 |
captopril |
none known |
unknown |
orthostatic hypotension, cough |
none |
322 |
celecoxib |
2D6 |
2C9 [3A4 slight] |
headache, edema, reduced GFR |
GI ulceration, thrombosis, rash |
314, 319, 320 |
disulfiram |
2E1 > 1A2 |
2E1 |
metallic taste, fatigue, * |
hepatitis |
139, 312, 323 |
itraconazole |
3A4 |
3A4 |
diarrhea, dizziness, rash, hypokalemia, LFT elevation |
hepatitis |
311, 317 |
ritonavir |
3A4 >2D6 |
3A4, 2D6 |
nausea, paresthesia, elevated cholesterol, LFT elevation |
lipodystrophy |
209, 311, 313, 314, 315 |
sertraline |
2D6 [weak] |
demethylation |
diarrhea, fatigue, nausea, decreased libido |
mania induction |
318, 321 |
Aprepitant
Aprepitant is a 534 Da oral inhibitor of signaling at neurokinin-1 receptors (NK-1) for which 11 amino acid, 1.4 kDa Substance P is the natural ligand [9, 10]. Aprepitant is approved by the FDA, EMA, and widely used as treatment for cancer chemotherapy induced nausea and vomiting for which it is quite effective [9, 10]. It is remarkably free of side effects of its own. Since Substance P signaling at NK-1 is a growth stimulating element in many cancers [11, 12] aprepitant has been suggested as a treatment adjunct for these cancers [13, 14], including glioblastoma [15, 16].
In vitro apoptosis induction by aprepitant and growth stimulation by exogenous Substance P can be demonstrated in other cancers as well [17, 18, 19] but heroic doses well beyond those used in humans seem to be required to demonstrate in vivo tumor suppression. We optimistically interpret this as an example of “Nile Distributary Problem” where other growth stimulating pathways takeover for one that is blocked.
Artesunate
Artesunate is a 384 Da phytoderived drug commonly used worldwide in treatment of malaria [20]. It is one form of the many related drugs derived from the same plant, collectively termed “artemisinins”. Artesunate converts rapidly after ingestion to its active metabolite dihydroartemisinin. Artesunate has inhibitory effects against, and is used in the treatment of various viruses, protozoa, helminths and fungi [21, 22]. Artesunate is particularly active against cytomegalovirus that are resistant to DNA polymerase inhibitors like valganciclovir [23, 24]. Artesunate has demonstrated empirical cytotoxicity against a variety of cancer cells [general review- 22, specific examples: lymphoma and myeloma, 25; pancreatic, 26; hepatocellular, 27; osteosarcoma, 28; gastric, 29; leukemia, 30, 31; glioma, 32].
In malaria studies artesunate is not given alone so its side effect profile is difficult to judge but it seem to be favorable. In short term studies of single agent artesunate 8 mg/kg/day in normals, altered taste and slight decrease in reticulocyte count were the only side effects noted [33].
Although clearly embryotoxic, and genotoxic [34] artesunate behaves clinically differently from traditional cancer chemotoxic agents. Mucositis, nausea, vomiting, bone marrow suppression, hair follicle synchronization typical of genotoxic chemotherapeutic drugs are not features of artesunate.
“If a little force doesn’t work, maybe more force will”. Another of the principles in designing CUSP9 and its first revision CUSP9*, has been “ganging up”, hitting the same system at different points to block, or in the case of ROS increase, the given target mode of glioma cell death. Exposure of epithelial ovarian cancer cells to two of the CUSP drugs, disulfiram and auranofin, increased intracellular reactive oxygen species (ROS) mediated cell death to a greater degree than with either alone, as discussed below in their respective sections [35]. We add artesunate to increase ROS and cell death even further [36-38].
Also built on our ganging up directive was the combination of artesunate with a fourth CUSP9* drug, captopril, in order to inhibit neo-angiogenesis to a greater degree than by treatment with either drug alone [39].
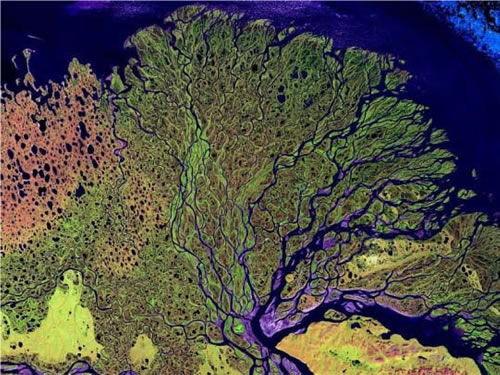
Fig. 2: A satellite image of a river flowing into the Arctic Ocean, illustrating what we call the ¨Nile Distributary Problem¨ where multiple distributaries develop that can cross-cover, maintaining total flow should the flow at one distributary become blocked.
Another area where we use the principle of ganging up is autophagy. Artesunate kills plasmodia and breast cancer cell lines via an autophagic pathway [40]. Artesunate decreased irradiation dose LD50 in LN229 and U87 glioma cell lines [41]. Of central importance to artesunate’s use in CUSP9*, empirically artesunate augments temozolomide cytotoxicity in both U87 and A172 glioma cell lines [32].
Artesunate had good cytotoxicity against a panel of 91 cancer cell lines at in vitro levels well below the artesunate levels in patients treated for malaria [42]. In artesunate mediated plasmodia cytotoxicity, mitochondrial depolarization mediated by an increase in intracellular ROS was identified as mode of action [37, 38].
Artesunate lowered expression of thioredoxin and cytochrome c oxidase mRNA by 95% in schistosomes of infected mice [43]. A fifth CUSP9* drug auranofin is also a strong inhibitor of thioredoxin reductase, increasing intracellular ROS as discussed below.
Auranofin
Auranofin is a 679 Da gold-containing lipophilic drug first marketed in the 1980’s for treatment of rheumatoid arthritis [44]. It remains in use worldwide as one of the so-called DMARDs, disease modifying anti-rheumatic drugs.
Auranofin and thioredoxin reductase
Thioredoxin reductase inhibition results in increased intracellular ROS. The effects of thioredoxin reductase inhibition by auranofin, the primary mode of action in both anti-rheumatoid arthritis and anti-cancer roles [45-48], can be significantly augmented by combination with disulfiram [35], another CUSP9* drug. Auranofin inhibited interleukin-6 induced STAT3 phosphorylation and nuclear translocation via an increased ROS step [49]. Chronic lymphocytic leukemia cells are similarly preferentially killed by auranofin generated ROS [50]. Like auranofin, other structurally unrelated inhibitors of thioredoxin reductase are cytotoxic to cancer cell lines by ROS generation [51-54].
An interesting feedback system [55] reminiscent in principle to that of Schruefer et al [vide infra, 56] and with potentially quite important clinical implications has been delineated since CUSP9 was published. Cell-free medium from glioma cell cultures stimulated synthesis of inducible nitric oxide synthase (iNOS) and consequent increased NO production in non-malignant microglia in vitro but without changing these normal microglia’s proliferation or death rate [55]. This normal microglia produced NO in turn stimulated co-cultured glioma cells’ migration and their synthesis of tumor necrosis factor-alpha (TNF-alpha) and macrophage chemotactic protein-1 (synonymous with CCL2, MCP-1) [55]. MCP-1 has an established growth promoting and negative prognostic effect in glioblastoma [57-59].
Auranofin and 5-lipoxygenase
CUSP9* has been modified from the original CUSP9 to account for a specific reciprocal back-and-forth shunting between cyclooxygenase (COX) and 5-lipoxygenase (5-LO). As one is inhibited, the other tends to become augmented in reciprocal fashion. This is discussed later in Section 2.6. on celecoxib and in Section 4. on reciprocal 5-LO and COX shunting.
Auranofin inhibited 5-LO in stimulated neutrophils [60-62] and in stimulated alveolar macrophages [63]. LTB4 is a major pro-inflammatory leukotriene product of arachidonate metabolism by 5-LO. Activated neutrophils’ migration along an LTB4 gradient is also inhibited by auranofin [61, 64]. Neutrophil aggregation, degranulation, chemotaxis toward LBT4 as well as LBT4 synthesis itself was blocked by auranofin [65-67].
An early study gave evidence of auranofin’s ability to cause arachidonic acid release from macrophages [63]. This was accompanied by some increase in thromboxane and prostaglandin E2, PGE2 [63]. Hence, for this reason also, to limit potential compensatory increase in PGE2, we add celecoxib to CUSP9* as discussed below.
Auranofin and cathepsin B
Cathepsin B is rather prominently upregulated in glioblastoma [16, 68-70 ] and promotes glioblastoma growth by contributing to matrix dissolution, Bcl-2 maintenance, and AKT activation [16, 68-70 ]. Cathepsin B becomes upregulated in a CXCL12 mediated feedback system with vessel endothelium [71] forming yet another positive mutually reinforcing feedback system active in glioblastoma with negative consequences in that glioblastomas are prodigious synthesizers of CXCL12, a target of treatment in its own right [72, 73]. A drawback is that auranofin is only a weak inhibitor of cathepsin B activity [16, 74]. But it is the only one we’ve got.
Captopril
Captopril is 217 Da angiotensin converting enzyme, ACE, inhibitor [75]. It was the first marketed pharmaceutical ACE inhibitor and has remained in wide use since its clinical introduction in the early 1980’s. Captopril and a dozen other marketed ACE inhibitors are supported by a robust database on clinical effectiveness in treating hypertension, chronic heart failure, and as renoprotection during glomerulonephropathies [76]. Although more convenient once-a-day ACE inhibitors have since come on the market, we chose to use captopril because so much of the empirical anti-cancer data was collected specifically on captopril. Although we probably have a class effect, we cannot safely assume this.
Captopril as an anti-cancer agent
We include captopril in CUSP9* based on three not necessarily discrete lines of evidence: a] data showing that captopril inhibits activity of soluble matrix metalloproteinase (MMP) -2 and MMP-9, with a parallel data set showing these MMPs to be a growth facilitating factor in glioblastoma growth, b] ACE inhibitors, including specifically captopril have been shown to inhibit angiogenesis, both normal and cancer-related, c] empirical studies showing growth inhibition in cancer models. These will be reviewed with references below.
As an anti-glioma agent, captopril was first mentioned in 1995 when exposure in vitro resulted in diminished invasion, growth and MMP-2/MMP-9 activity [77]. These results were later replicated [78, 79]. MMP-2 and -9 inhibitory properties of captopril were reviewed in 2012 [80]. Many cancers have been shown to over express MMP-2, -9. The principle of our use of captopril in glioblastoma was active in a study of renal cell carcinoma (RCC), where immunohistochemical evidence for ACE expression on RCC, was first demonstrated in 1983 and this ACE activity was inhibited by captopril [81].
Throughout mammalian physiology, we see the pattern where different organs or organ systems use the same or similar function-mediating system for different ends, ends specific and relevant to the differing goals served by the specific organ or organ system. The renin-angiotensin system (RAS) is one such system. While most commonly thought of in terms of mediating the organ cross-talk between kidney-liver-lung in regulating sodium chloride balance and blood pressure, all components of RAS are endogenously expressed and used within brain and bone for examples, as semi-autonomous systems. Semi-autonomous means that both fully autonomous within bone or brain and elements interacting with systemic RAS are recognized.
Of special importance to CUSP9*, Carpentier et al demonstrated a steroid-sparing effect of ACE inhibitors or the related class of drugs, angiotensin receptor blockers, ARBs in glioblastoma patients [82]. We discuss their findings further below, offering the interpretation that glioblastoma overexpressed ACE functions to enhance peritumoral edema.
In a clinical study of low dose captopril, 12.5 mg twice daily, post-resection PSA rises occurred in 3 of 32 prostate cancer patients in the captopril group versus 10 of 30 in the control group [83]. Crucially, follow up data on OS have not been published. Others are looking at slowing or stopping cancer growth by blocking growth factors with repurposed already-approved drugs. In 2004, Jones et al studied three non-cytotoxic drugs- captopril 50 mg twice daily in combination with marimastat and subcutaneous fragmin- in patients with various terminal metastatic cancers [84]. Whether this treatment regimen slowed disease progress as the authors claimed or not, the drugs were well-tolerated even in this very sick patient population [84].
Marginal reduction in prostate cancer (relative risk was o.7) in those using captopril in hypertension treatment was seen in an epidemiological study of 23,000 men that showed no association- increased or decreased- relative risk in users of any other antihypertensive drug suggesting a captopril-specific rather than class-specific effect [85]. In rat prostate cancer, Wilson et al demonstrated captopril inhibitable ACE expression, conjecturing that ACE matures multiple growth factors by proteolysis of the parent peptide [86], a conjecture with which we agree.
Captopril inhibited tumor growth in a murine colon cancer model [87]. Growth of a human gastric cancer cell line [88] and a myelogenous leukemia cell line [89] were inhibited by low dose captopril. ACE and angiotensin II receptor 1, ATR1 were overexpressed in 19 of 25 resected pancreatic ductal cancers examined by immunohistochemistry and captopril suppressed their proliferation in vitro [90]. Captopril inhibited a renal cell carcinoma cell line’s growth in severe combined immunodeficient mice without showing in vitro proliferation inhibition to that cell line at an equivalent concentration [91].
Estrogen receptor expression and in vitro proliferation of mammary ductal cell carcinoma cell lines were inhibited by captopril [92, 93]. After corneal injury neovascularization at injury site interferes with vision. Captopril inhibited this destructive post-injury corneal vascularization that was tractable to diminished endothelial cell migration at clinical captopril levels <10 micromoles [94]. Building on this observation, Volpert et al examined effects of captopril on a rat fibrosarcoma cell line that was resistant to captopril in vitro. Interestingly, they found in vivo captopril did inhibit this tumor’s growth, suggesting an effect of stroma/host vasculature response rather than the malignant clone itself [94].
Captopril lengthened overall survival in mice with Lewis lung carcinoma and potentiated cyclophosphamide in this model [95]. Very high doses of captopril [~50 mg/kg/day] in mice inhibited fibrosarcoma and squamous cell carcinoma development in irradiated skin [96]. A xenografted human melanoma cell line’s growth was inhibited by captopril [97]. However, and instructively, captopril had no antiproliferative effect on a variety of other cancer cell lines in vitro [98, 99]. Instructive in that clinical cancer growth inhibition via ACE inhibition occurs largely, although not exclusively, through angiogenesis inhibition, not inherent cytotoxicity.
Captopril inhibited the activity of soluble MMP-2 and MMP-9 secreted by a gastric adenocarcinoma cell line [100]. Growth in nude mice of this cell line was inhibited by captopril and inhibited yet further in combination with cisplatin [100.]. Captopril inhibited a mammary ductal cell carcinoma cell line in a Cu++ dependent manner, with indicators this was mediated by increased intracellular ROS [93]. Captopril inhibited soluble MMP-2 and MMP-9 activity from Lewis lung cancer cells in vitro and slowed growth in a murine model [101]. Moreover, in three triple-negative breast cancer cell lines tested, captopril inhibited proliferation only in those cells that expressed outer cell membrane ACE [102]. Importantly for CUSP9*, glioma cells express ACE-like activity [103].
Growth of murine colon cancer metastases to liver [104] and a xenografted non-small cell lung cancer cell line [105] were inhibited by captopril. Of particular importance and interest to the CUSP9* protocol, another CUSP9* drug, artesunate, when added to captopril yielded a synergistic inhibition of angiogenesis in an ovine allantoic membrane angiogenesis model [39].
Captopril and tissue factor
Tissue factor (TF, factor III, also termed thromboplastin), is a 47 kDa outer cell surface receptor for soluble, activated clotting factor VII [106. ]. The TF-factor VII complex then mediates factor X conversion to activated factor Xa. TF is commonly elevated in human glioblastoma [107-110]. TF is yet another of the many growth facilitating factors we aim to inhibit in CUSP9*. In a breast cancer cell line, TF activity and TF mRNA was reduced by about a third by captopril exposure in vitro [111]. TF subserves other growth enhancing aspects of glioblastoma growth in addition to contributing to the excess thrombosis related morbidity associated with glioblastoma [106, 108, 110, 112-115]. This is so particularly in the stem cell subpopulation [114, 115].
The dangers of systemic anticoagulation in glioblastoma are clear [111, 116]. Captopril mediated down-regulation of glioblastoma cell overexpression of TF might be an ideal way to deprive glioblastomas of this growth and migration enhancing system.
Celecoxib
Introduction
New to CUSP9* is celecoxib. Celecoxib is a 381 Da cyclooxygenase (COX) inhibitor commonly and effectively used to treat pain of diverse origins [117, 118]. It lacks any platelet aggregation inhibitory activity as seen with some other COX inhibitors [119]. Clinical and research literature routinely call celecoxib “COX-2 selective” and this is relatively so but to our reading of the data celecoxib inhibits COX-1 to some degree [120-123 vide infra]. Shortly after clinical introduction to clinical practice in symptomatic treatment of pain, anti-cancer effects were noted both empirically and by theoretical reasoning [124 ]. Celecoxib is now being widely used on and off cancer treatment protocols in a variety of different cancers. It has come to our attention that people with glioblastoma are commonly starting celecoxib on their own, often without their oncologist’s knowledge.
Celecoxib and COX-1, COX-2
Celecoxib is currently in 42 open clinical trials as adjuvant to traditional cancer cytotoxic or drugs or treatments [clinicaltrials.gov]. Celecoxib adjuvant trials reporting recently have not been entirely negative but neither have they been strongly encouraging. Many were uncontrolled and had other variables that didn’t permit benefit quantitation. Adjuvant celecoxib was generally well tolerated in these studies.
In an uncontrolled study, children with various advanced cancers were given cyclophosphamide and etoposide as cytotoxic chemotherapy augmented with non-cytotoxic growth factor blocking drugs [thinking along same lines as CUSP9*] celecoxib, thalidomide, and fenofibrate [125]. The authors, experienced pediatric oncologists, considered “Clinical activity was demonstrated in some but not all tumor strata.” An uncontrolled study in advanced metastatic breast cancer of daily cyclophosphamide plus celecoxib 200 mg twice daily “is safe and shows a therapeutic effect in advanced breast cancer patients” [126 ]. In another study, a treatment regimen consisting of celecoxib 200 mg twice daily, 5-fluorouracil, epirubicin and cyclophosphamide followed by docetaxel was reported to be “active and safe for treatment of operable invasive breast cancer” [127]. In addition, in a study of carboplatin, gemcitabine or carboplatin with vinorelbine in advanced non-small cell lung cancer given celecoxib at 400 mg twice daily “The effect on survival by celecoxib in the whole subset of patients was positive” [128].
However, there are also negative studies. For example, a study of advanced epithelial ovarian cancer treated with docetaxel, carboplatin using adjuvant celecoxib at 400 mg twice daily showed no survival benefit from added celecoxib [129]. We refer to the reciprocal relationship between COX and 5-LO, discussed in detail with references in section 4. below as explanation for this failure and how we address this in CUSP9*.
Celecoxib and carbonic anhydrase
Carbonic anhydrase catalyses the reaction CO2 and H2O to H+ and bicarbonate. It has 12 isoforms of which some are soluble in the cytoplasm [isoform -2], some transmembraneous [isoforms -9 and -12]. In either case, the proton is excreted and bicarbonate kept within the cell. The net effect is further acidification of the extracellular milieu. Glioblastoma cell carbonic anhydrase thereby contributes to glioblastoma’s acidic milieu [vide infra]. In 2006, a blind selection of 960 molecules tested for carbonic anhydrase-2 inhibition revealed that celecoxib was among the most potent molecules [130].
Glioblastomas, like many other cancers, express abnormally large amounts of carbonic anhydrase [131-134]. Stronger carbonic anhydrase-9 immunohistochemical staining of human glioblastoma biopsies was correlated with shorter overall survival, a robust finding confirmed by four independent groups [135-138]. In biopsy specimens of endothelium derived from glioblatomas, overexpression of carbonic anhydrase isoform-2 was found and likewise correlated with shorter survival [135]. Overexpression of carbonic anhydrase is thought to be an adaptation allowing better survival in the hypoxic, acidic environment of tumors [134, 135], particularly glioblastomas [138]. Although areas of most intense immunohistochemical staining for carbonic anhydrase tend to be the most hypoxic, and or necrotic, it seems that in fact it is extracellular acidosis that triggers increased expression of carbonic anhydrase [131, 137].
The pan-carbonic anhydrase inhibitor acetazolamide was shown to enhance temozolomide cytotoxicity in a glioma cell line while dexamethasone reduced temozolomide cytotoxicity in this same model [138]. Writing in 2008 these authors suggested clinical use of acetazolamide along with temozolomide to reduce peritumoral edema, enhance temozolomide’s cytotoxicity, and reduce need for [potentially counter-productive] use of dexamethasone. We agree and choose celecoxib partly for its nanomolar inhibition of carbonic anhydrase.
Disulfiram
Disulfiram is a 297 Da drug, used since the 1950’s to treat alcoholism by making ethanol ingestion highly unpleasant [1, 139-142]. As a potent inhibitor of all isoforms of aldehyde dehydrogenase, ALDH, disulfiram stops ethanol metabolism at the acetaldehyde stage. Multi-system dysfunction ensues if ethanol is consumed, as manifest by flushing, hypotension, malaise, nausea, vomiting. Acetaldehyde is poorly tolerated. ALDH normally would transform the toxic ethanol metabolite acetaldehyde into non-toxic acetic acid that is smoothly handled without problem. Since disulfiram chelates Cu++ in the stomach even without adding exogenous Cu [139], Cu gluconate of CUSP9 has been deleted in CUSP9*.
In CUSP9 [1] we gave the basic rationale for adding disulfiram to glioblastoma treatment. Since then the rationale has been further elaborated and details added [35, 143, 144]. We give here in CUSP9* a short background and a little update on disulfiram developments since end of 2012.
The original suggestion to use disulfiram in treatment of glioblastoma came from dozens of reports associating high ALDH expression in individual cells with those cells having stem cell attributes [142, 143]. This held true in various cancers including glioblastoma [141, 142, 146]. There are currently 30 open studies on disulfiram as adjuvant treatment in various cancers [clinicaltrials.gov].
Some recent data on disulfiram: Mesothelioma cells’ growth inhibition by disulfiram/copper was associated with weaker NFkB activation, accumulation of ubiquitinated proteins and cleavage of vimentin [145 ]. A study in glioblastoma also showed disulfiram cytotoxicity was related to inhibition of NFkB activation [146- 148]. Cytotoxicity by disulfiram/copper to non-small cell lung cancer cells resulted in cell cycle arrest at G2/M [147]. In hepatocellular cancer disulfiram/copper mediated cytotoxicity was shown to be mediated through an ROS increasing step [148], findings concordant with our findings on disulfiram/copper cytotoxicity to epithelial ovarian cancer cells being mediated in part by increased ROS [35, 149]. Likewise, pancreatic cancer cell growth inhibition in a murine xenotransplant model was also ROS dependent [150]. A compound screen including several thousand drugs showed prostate cancer cells to be sensitive to disulfiram which was partially reversible by adding ROS scavenging agents [151] again indicating that disulfiram-mediated cytotoxicity at least in part is due to the increased generation of ROS. ROS increase as a primary mode of disulfiram-mediated cell death was already indicated in early works with melanoma cells over ten years ago [152, 153].
In the course of replicating disulfiram’s increased ROS cytotoxicity to breast and colon cancer cells, and quite happily for our intended use, hypoxia and lower pH – conditions prevailing within glioblastoma tissue - both enhance disulfiram’s cytotoxicity [154]. Disulfiram alone, but particularly so in combination with auranofin, results in large increases in intracellular ROS [35].
In a genomic study examining a panel of ovarian cancer cells ALDH expression as a feature of stemness was again confirmed suggesting disulfiram as a stemness defeating drug for that type of cancer [155]. As for other cancers, the subset of high ALDH expressing cells was associated with stem cell attributes and disulfiram again, as predicted, defeated much of the stem cell functions, both in vitro and in a xenograft model [156]. Breast cancer cells expressing ALDH were shown to have decreased sensitivity to paclitaxel and cisplatin compared to ALDH non-expressing cells. Decreased sensitivity in these ALDH expressing cells reverses after exposure to disulfiram [157]. Triple negative breast cancer cells showed synergy in cytotoxicity when disulfiram is added to doxorubicin [158]. Gemcitabine cytotoxicity is enhanced by disulfiram in glioblastoma cells [159], in pancreatic cancer cells [156, 160] and in breast and colon cancer cells [161].
However, disulfiram related cytotoxicity seems to be exerted by at least two main paths- one Cu++ dependent, one not [35, 162, 163]. Importantly, for our intended use cancer cell specific cytotoxicity peaks when disulfiram and copper are present in a 1:1 molar ratio [162]. By an unclear mechanism, cytotoxicity is reduced by excess disulfiram [162]. Data from early 2014 indicate that disulfiram is a partial inhibitor of the main temozolomide damage repair enzyme, O6-methylguanine-DNA methyltransferase, MGMT [163].
However, a study of disulfiram 250 mg/day or 500 mg/day as isolated treatment failed to significantly affect progression in men with localized recurrent post-resection prostate cancer as measured by PSA slope [164]. We interpret the discrepancy with in vitro data to mean a] in human disease there are compensatory paths engaged when ALDH is inhibited, and b] these compensatory growth paths must also be blocked simultaneously with ALDH. Certainly, there are also other interpretations for the failure of disulfiram to change prostate cancer growth slope.
It still remains a matter of debate whether disulfiram’s anti-cancer mechanism of action is based on ALDH inhibition, MGMT inhibition, NFkB activation inhibition, proteasome inhibition, or increased intracellular ROS generation. Is one event primary and the others secondary to that one? Or is stem cell selectivity achieved by a combination of actions? Answers to these important questions are currently unknown.
Itraconazole
General
Itraconazole is a 706 Da broad spectrum anti-fungal agent used clinically since the late 1980’s [165, 166]. It is commonly used today for onychomycosis and as anti-fungal prophylaxis, or as an empirical treatment for fever, in neutropenic patients [167]. As with many other highly lipophilic drugs, brain tissue concentration is greater than CSF [165]. Although 5% of itraconazole treated patients develop some elevation of LFTs, this has lead to overt liver damage in < o.o2% of treated patients [165].
In a study initiated on the basis of preclinical evidence for itraconazole’s inhibition of angiogenesis, Rudin et al recently showed prominent lengthening of OS in itraconazole plus pemetrexed treated metastatic nonsquamous non-small cell lung cancer patients when compared to those receiving pemetrexed alone [168]. Based on positive murine prostate cancer xenograft studies, Antonarakis et al in a clinical study found clear evidence for itraconazole’s anti-tumor effect- reduced PSA levels [of note without effect on androgen levels], shallower PSA slope, and fewer circulating cancer cells in metastatic prostate cancer patient [169]. Skin biopsies derived from these patients showed diminished Hedgehog, Hh, signaling, [vide infra], a finding concordant with studies on mesothelioma cells exposed to itraconazole [170] and murine Hh signaling studies with itraconazole [171].
Proton pump inhibitors, H2 inhibitors, and other stomach acid reducing agents must be avoided during itraconazole due to decreased absorption at higher pH [172]. In HIV patients receiving ritonavir 400 mg twice daily, the related azole antifungal drug ketoconazole increased cerebrospinal fluid ritonavir (from 2.4 to 6.6 ng/mL) to disproportionately greater degree than it raised blood ritonavir levels [173]. This was a major reason for adding ketoconazole to the original CUSP9 regimen [1]. We believe itraconazole will achieve similar increased ritonavir concentrations based on itraconazole’s similar efflux pump inhibition as that seen with ketoconazole [174, 175].
From the outset of our work constructing a non-cytotoxic adjuvant protocol for cytotoxic treatment of recurrent glioblastoma we considered 5-LO inhibition as an important component. In a screen of hundreds of FDA approved drugs across multiple classes, none of which were traditional cancer chemotherapeutic drugs, Chong et al found itraconazole to be a specific inhibitor of human umbilical cord endothelial cell proliferation while surprisingly having little or weak inhibitory effect on normal fibroblasts, Jurkat T cells or HeLa cells [176]. In support of our substitution of ketoconazole in CUSP9 with itraconazole in CUSP9*, ketoconazole was shown to provide less anti-angiogenesis activity in this model. Chong et al’s premise was to “uncover novel biological activity among existing drugs,” [176] - precisely one of our main driving principles also in CUSP9*. Another group showed diminished nascent capillary sprouting as well as endothelial proliferation inhibition during exposure to itraconazole [177].
Easily achievable levels of itraconazole will block vascular endothelial growth factor, VEGF, binding to VEGF receptor-2 [178]. This was traced to defective receptor trafficking, which in turn was secondary to itraconazole mediated defective glycosylation of VEGF receptor-2 [178]. Another interesting and helpful aspect of this trafficking inhibition by itraconazole is its interference with mTOR activation [179].
mTOR/AKT signaling in glioblastoma cells was inhibited during exposure to itraconazole with autophagic cell death to follow [180]. Since “angiogenesis is dependent on multiple growth factors and a broad signaling network in vivo” [180] it seems reasonable that we must block multiple growth factors with multiple drugs, until and unless we can find a crucial single sine qua non element in this process. So far, it seems there is none such. Hence, at least for now, CUSP9*.
The pharmacokinetics of itraconazole oral solution was measured in seven patients receiving chemotherapy followed by autologous bone marrow transplantation for leukaemia or lymphoma. Patients receiving itraconazole 5 mg/kg/day [70 kg = 350 mg/day] gave pre-dose serum level of ~800 ng/mL at steady state [about two weeks] [181]. A retrospective analysis of patients with acute lymphoblastic and acute myelogenous leukemia receiving daunorubicin where itraconazole was used as antifungal prophylaxis during the neutropenic nadir showed better remission rates than those not receiving itraconazole [182]. The effect was not large but was statistically significant. In vitro, intracellular levels of daunorubicin increased proportionately as itraconazole levels went from o.5 to 5.0 microg/ml [183].
Special position of 5-lipoxygenase
We now place greater emphasis on 5-LO in CUSP9* than we did in CUSP9. Leukotrienes are arachidonate derived signaling molecules of importance that are overexpressed in many cancers [184] and demonstrated to be so specifically in glioblastoma [185-189]. Given that glioblastomas express excess 5-LO and that 5-LO product leukotrienes are major mediators of glioblastoma-related brain edema [185] we predict therefore that itraconazole will lower brain edema and the consequent need for dexamethasone. Steroid-sparing action alone would be expected to improve QOL and OS [82, 190].
Itraconazole at low micromolar concentrations inhibits synthesis of leukotriene LTB4 in activated neutrophils even more deeply than does ketoconazole or miconazole [191-193]. “Changes across metabolic networks” [194] that are not malignant by themselves become an integral part of malignant growth when combined with the suite of so deranged networks and genomic changes driving them. We aim to dismantle or block enough of these networks to hobble glioblastoma growth. Morin et al thinking along these lines found that zileuton, a specific drug marketed as an 5-LO inhibitor [to treat allergy symptoms] interferes with glioblastoma cell line growth [194] as previously hypothesized by Omahen in 2011 [195]. Herbal-derived 5-LO inhibitors [201c;Nordy201d; and others] have also shown anti-glioma growth activity, activity in xenograft model, cytotoxicity that exceeded that of the classical alkylating cytotoxic drug carmustine (BCNU) [196]. Itraconazole blocks 5-LO activity without affecting activity of COX in vitro in low micromolar concentrations [192], levels easily achieved in clinical use.
Much of our aim in CUSP9* is inhibiting or blocking normal cell processes pathologically employed by cancer cells to grow or evade senescence, apoptosis or cytotoxicity. Others are thinking along these lines also [196, 194, 196, 197].
Some glioblastoma patients currently use cannabidiol, based on preclinical studies [198-200], internet chatroom discussions, and anecdotal reports of benefit. In a formal study of cannabidiol, much of the in vitro anti-cancer and specifically anti-glioma effects derive from its 5-LO inhibition. Cannabidiol is one of the many biologically active but not psychoactive molecules present in marijuana. It is sold openly, legally, and without prescription in most jurisdictions on the herbal market.
Again within the principle of ¨ganging up¨ disulfiram as well inhibited rat neutrophil soluble 5-LO with IC50 <1 micromol [201]. Diethyldithiocarbamate, the prominent circulating metabolite of disulfiram, still had 5-LO inhibiting properties but was less potent than its parent compund [201]. Concordant with that work, disulfiram inhibited LTB4 release from isolated human neutrophils at IC50 <5 micromol and in an in vivo rat model but less effectively so [202]. Thinking along these lines of “ganging up”, similar to our CUSP9 protocol, Jiang et al reported earlier this year [196] results from a drug screen empirically looking at a thousand non-cytotoxic, non-cancer chemotherapy-related FDA approved drugs for cytotoxicity against glioma cell lines [196]. Two of the CUSP9* drugs, itraconazole and sertraline happened to have- as predicted in CUSP9 last year- to have good anti-glioma cell activity (at least in vitro) [196].
Itraconazole and p-gp
Itraconazole significantly inhibited breast cancer resistance protein, BCRP, efflux pump, thereby lowering cytotoxicity resistance to topotecan [203] and leukemia cell line resistance to doxorubicin and etoposide [204].
P-glycoprotein [p-gp] is a 170 kDa, ATP consuming, outer cell membrane drug [xenobiotic molecule]efflux pump synonomous with MDR-1 [205, 206]. It preferentially exports lipophilic molecules, a veritable intracellular “hydrophobic vacuum cleaner” [205] particularly active at the blood-brain barrier (BBB) endothelium [206]. Itraconazole is both an efflux substrate and partial inhibitor of p-gp [206-208].
Recognizing itraconazole’s remarkably useful attributes of both Hh signaling inhibition and p-gp inhibition, a retrospective chart review showed that clear cell ovarian cancer patients receiving itraconazole 2 days before and during treatment with platinum and taxane had over twice the overall survival compared to those receiving the same chemotherapy regimen but without itraconazole and had even greater effect in prolonging survival in other forms of epithelial ovarian cancer [208].
Ritonavir
Ritonavir is a 721 Da protease inhibitor, the first such approved for use in humans to treat HIV [209]. As mentioned above, ketaconazole more than doubles CSF ritonavir levels (2.4 to 6.6 ng/mL) in HIV positive people [173]. Concomitant administration of ritonavir (400 mg twice daily) plus ketoconazole [200 mg twice daily] was well tolerated and resulted a in disproportionate increase in CSF ritonavir level compared to a small increase in plasma level [173]. Both drugs, ritonavir and ketoconazole, as well as itraconazole, are substrates for and inhibitors of p-gp and MRP1.
A trial of ritonavir 100mg with lopinavir 400 mg twice daily reporting in 2011 showed little or no benefit in prolonging OS of recurrent glioblastoma [210]. Of uncertain significance, there was one complete radiological remission that lasted 11 months before progression [210]. Parenthetically, note that in this report the published doses of ritonavir and lopinavir were reversed from the doses actually given. The authors have assured us that he standard commercial capsules of KlatraTM (ritonavir 100mg with lopinavir 400 mg) were used.
Ritonavir inhibited in vitro proliferation of a glioma cell line [211]. Ritonavir in vitro exposure increased apoptosis and decreased proliferation of pancreatic ductal adenocarcinoma cell lines which led the authors to conclude that using ritonavir in pancreatic duct cell cancer by “drug repositioning...would limit the costs and reduce risks” [212]. Our point exactly times nine.
Sato et al demonstrated renal cell carcinoma proliferation inhibition by ritonavir in 2012 [213]. Cervical carcinoma in situ cells synthesize both MMP-2 and MMP-9. Activity of both was reduced by ritonavir with corresponding in vitro invasion inhibition [214]. Non-small cell lung cancer cell lines’ growth was inhibited by ritonavir as well [215].
The eleven members of the ABC efflux pump group are associated with cancer chemotherapeutic drug efflux from cells and therefore become an element of chemotherapy resistance. Breast cancer resistance protein (BCRP), p-gp, or various multidrug resistance proteins are better known members of this group and all are inhibited to varying degrees by ritonavir [216]. Kumar et al showed that ritonavir at 20 microM generated G1 cell cycle arrest and apoptosis in ovarian cancer cell lines MDH-2774 and SKOV-3 [217]. Kumar et al conclude that HIV protease inhibitors like ritonavir “are efficient blockers of MDR1 (P-gp), MRP1 and BCRP” [217]. Ritonavir induced cell-cycle arrest at G1-phase and apoptosis in EBV-positive lymphoblastoid B cells in vitro [218].
Decreased AKT phosphorylation is a basic mode of action in ritonavir cytotoxicity [212, 217, 219]. Again the refrain, “[ritonavir] repositioning for ovarian cancer... would reduce risks, limit the costs and decrease the time needed to bring the drug from bench to bedside” [219]. We concur.
Ritonavir increases etoposide cytotoxicity in MRP-1 over-expressing cells [220]. Long-term exposure to ritonavir seems to upregulate p-gp expression [221] but at the same time ritonavir inhibits the efflux of paclitaxel and vinblastine in p-gp-positive cell lines [222]. Ritonavir inhibited the xenobiotic export pump BCRP in low micromolar concentrations but was not a substrate for BCRP [223, 224]. A rat glioblastoma cell line’s in vitro proliferation was suppressed by ritonavir, and direct inhibition of proteasomal chymotrypsin-like activity by ritonavir could be demonstrated. However, in an in vivo rat model ritonavir had no effect on glioma growth [225].
To what extent the many paths of ritonavir-mediated cell death in cancer cell lines and relative resistance to such death in non-malignant cell lines [226 ] is fundamentally secondary to AKT inhibition or not, is unknown.
Like itraconazole, ritonavir is both an efflux substrate and partial inhibitor of p-gp [227-229]. Ritonavir penetration into normal brain tissue is poor but as mentioned above adding ketoconazole increased cerebrospinal fluid concentrations of ritonavir (from 2.4 to 6.6 ng/mL) in those receiving 400 mg ritonavir twice daily [183]. We expect itraconazole to similarly assist ritonavir penetration of the BBB.
Several multiple myeloma cell lines responded to ritonavir with decreasing proliferation and increasing apoptosis [230]. Ritonavir is a strong inhibitor of hepatic P450 3A4 and a clear but relatively weak inhibitor of p-gp efflux pump, but together these two attributes ended up increasing orally administered circulating docetaxel 50 fold [231]. Human endothelium growing in vitro show mitochondrial DNA damage and reduced proliferation after exposure to ritonavir [232.]. Colon carcinoma cell apoptosis was increased after exposure to ritonavir [233]. Ritonavir is a strong hepatic P450 3A4 inhibitor that lowered affinity of activated NFkB with its DNA target sequence [233]. Ritonavir enhanced radiation-induced apoptosis in a murine model of head and neck squamous cell carcinoma [234].
Sertraline
Introduction
Sertraline is a 306 Da antidepressant of the selective serotonin re-uptake (SSRI) class with a minimal side effect profile [235]. Sertraline is widely used worldwide. It is a mainstay in current treatment of excessive anxiety states as well. Side effects are usually well-tolerated consisting of reduced libido (occurs in one third of treated patients) and some loosening of bowel movements (in one tenth of those treated). Other side effects are rare [235]. Sertraline is also commonly used in metastatic cancer with co-morbid depression where it is a safe and effective antidepressant/anti-anxiety agent with side effects not different from that seen when used in non-cancer settings [236].
Sertraline was included in the original CUSP9 protocol partly based on the empirical observation that glioblastoma patients on SSRI class antidepressants had a longer OS than those not on SSRIs [237]. The authors of that study reported on a 1.6 month longer OS in those treated with SSRIs which was not statistically significant but we considered the possibility that if a larger cohort continued to show similarly longer OS this would become statistically significant [1]. The eminent safety and tolerability of sertraline made the risk/benefit skewed enough to include sertraline in CUSP9*.
Sertraline effects on cancer cells
Sertraline was shown to induce cytotoxicity in a human osteosarcoma cell line [238], a human prostate cancer cell line [239] and a squamous oral cancer cell line [240] by mediating phospholipase C -dependent Ca++ release from the endoplasmic reticulum, ER, and Ca++ cell entry by L-type Ca++ channels. Inhibition of proliferation by sertraline of colon cancer cell lines, both in vitro and when xenografted, was traced to Bcl-2 inhibition [241]. In a drug pair study of sertraline in five glioma cell lines (U87MG, U343MG, U373MG, A172, T98G) robust inhibitory activity was seen across these cell lines [242].
Translationally controlled tumor protein, TCTP, is an ancient ~20 kDa intracellular chaperone protein dysregulated in mammalian cancers [243]. TCTP is seemingly closely related to heat shock proteins [244]. Sertraline inhibits TCTP [245-247].
An important role of TCTP relating to glioblastoma’s treatment-resistance is TCTP’s non-covalent bonding to p-53 preventing p-53’s function [248]. p-53 is a 44 kDa protein active in both normal and malignant cells that, although a multi-functional protein, i.a. is a transcription factor leading to G1-arrest and/or apoptosis during cytotoxic chemotherapy. Thereby p-53 becomes most important in cancer research and hence in CUSP9* [249]. In glioblastoma, TCTP overexpression destabilizes p-53, preventing p-53’s pro-apoptosis signaling during chemotherapy [250]. By sertraline’s inhibition of TCTP [245-247] we intend to restore some p-53 function.
Glioblastoma biopsy with TCTP overexpression indicates a more aggressive clinical trajectory, have a higher proliferation rate, and were associated with shorter OS [250, 251]. Stemming from observations that revertant subclones [sub-clones with reduced malignant characteristics]of cancer cell lines had reduced TCTP [247], sertraline was noted to both reduce TCTP and enhance the reversion process [247]. An inverse reciprocal quantitative relationship exists between TCTP and p-53. TCTP enhanced p-53 degradation while p-53 repressed TCTP transcription [245, 246]. The repressive function of TCTP is inhibited by sertraline in a breast cancer cell line, thereby de-repressing p-53, allowing resumption of p-53 mediated apoptosis [245].
Empirically, sertraline inhibited glioma cell line U87 proliferation at even lower concentrations than did temozolomide [252]. Proliferation inhibition at low micromolar in vitro concentrations was also seen in a breast cancer cell line that was traced to mTOR inhibition [253]. Jurkat cell line proliferation was inhibited by sertraline at lower concentration than by vincristine or cyclophosphamide and cytotoxicity of vincristine and doxorubicin was enhanced by clinically achievable sertraline levels [254]. Melanoma xenograft growth was inhibited likewise by sertraline at clinically achievable doses and correlated with inhibition of AKT phosphorylation[255].
The sertraline dose has been increased from 50 mg twice daily to 100 mg twice daily in CUSP9* based on early reports of good tolerability, increased fatigue has been the main side effect noted from the increase, in those who have taken it as part of compassionate use in CUSP9.
Hedgehog signaling pathway
General
The hedgehog signaling pathway, Hh, [256] is one of the growth driving signaling systems in cancer generally [257, 258] and in glioblastoma specifically [259, 260]. Autocrine, juxtacrine and paracrine Hh activation modes are recognized. Hh signaling is a multi-stage process starting at the base of cells’ cilia going to the cilia tip, then back to cells’ nucleus. Glioblastoma cells bear cilia and they are faulty [261, 262]. Hh is a particularly important signaling system in that it branches out to transactivate numerous other signaling systems [257], particularly so and well documented in the case of glioblastoma [260, 263].
Vismodegib is a small molecule inhibitor of Hh that, by binding to SMO, tends to limit efficiency of Hh signaling [264, 265]. Vismodegib is FDA approved and marketed in USA/Canada, and several countries of the EU for treatment of metastatic or advanced basal cell carcinoma [265]. Basal cell carcinoma gives us an important lesson in understanding cancer in general and with specific reference principles that drove our CUSP9*.
In basal cell carcinoma, Hh plays a central role and acts in the absence of important enough cross-covering growth signaling pathways such that effective tumor growth suppression can be achieved with pharmacologic inhibition using the small molecule drug vismodegib [264, 265, 266], inhibiting a single pathway. An easy enemy to defeat.
Many other cancers where Hh has been shown to have important growth stimulating role cannot be so easily suppressed. In these cases, single agent inhibition of Hh is not efficient. The lesson: basal cell cancer is an indolent disease of low degree malignancy. Metastases are rare or a late occurrence in disease course. Bulk tumor invasion is clear but not very cancer-like (cf. cancer as crab). Simple surgical resection commonly results in cure. The microscopic invading tentacles common to other cancers and responsible for local recurrence and metastases are not usually seen in basal cell cancer. Corresponding to this it seems that simple blocking of a single signaling path - Hh - stops growth. The fact that vismodegib doesn’t cure other cancers we take as evidence for multiple cross-covering growth signaling paths being generally active in cancers and specifically in glioblastoma. Hh has well documented partially cross-covering intersections with four other growth promoting pathways-1) RAS/RAF/MEK/ERK, 2) PI3K/AKT/mTOR, 3) EGFR [257]. Again we face the Nile Distributary Problem and aim to address it with a multi-drug regimen- CUSP9*.
Hh and Itraconazole
As mentioned itraconazole inhibits Hh signaling [170, 171]. Murine medulloblastoma growth inhibition by itraconazole was shown to act via Hh, specifically by blocking SMO translocation [171, 267]. In accord with previously documented Hh inhibition, a clinical study showed that itraconazole at 100 mg twice daily inhibited Hh signaling and proliferation in basal cell carcinomas [267]. Of particular interest, confirming our understanding of basal cell carcinoma, is the observation that basal cell carcinomas previously exposed and resistant to vismodegib showed no effect subsequent to itraconazole treatment [267]. If these cancers were resistant to one Hh inhibitor they were resistant to the other.
Mesothelioma cells were killed by Hh inhibition by itraconazole to an equivalent degree as did Hh RNA knockdown [170]. Itraconazole has significant anti-myeloma cell activity via Hh inhibition, particularly so in the ALDH co-expressing sub-population indicating Hh as a stem cell feature [268].
Based on strong in vitro and in vivo (xenograft) inhibition of endothelial cell proliferation, migration, and tubule formation in response to vascular endothelial growth factor (VEGF) and basic fibroblast growth factor in non-small cell cancer models [269], a phase II study compared standard cytotoxic therapy with or without daily oral itraconazole in recurrent metastatic non-small cell lung cancer [270]. The authors reported “trends suggestive of improved disease control” in the cohort treated with itraconazole 200mg/day [270].
In considering these data showing itraconazole as both p-gp efflux inhibitor and an Hh inhibitor, Tsubamoto et al found evidence for clinical activity in a 2014 clinical trial of itraconazole in advanced clear cell epithelial ovarian cancer [208].
High dose itraconazole [600 mg/day] given to men with advanced prostate cancer resulted in halving of the PSA level without changes in androgen [testosterone and dehydroepiandrosterone] levels [271]. Intriguingly and without current explanation, patients’ aldosterone dropped to 25% of pre-itraconazole levels [271]. This latter finding is particularly felicitous in that a study from 1994 showed increased CSF aldosterone in brain tumor patients of diverse pathologies including glioblastoma, which was correlated to brain edema [272]. Since brain [273], and glioblastoma tissue itself have autonomous functioning renin-angiotensin systems [80, 274, 275], our expectation is that much of itraconazole’s brain edema prevention derives from this diminished aldosterone synthesis during itraconazole treatment and will be augmented by our use of captopril.
Reciprocal 5-LO and COX shunting
Recent findings of a reciprocal shunting between COX-2 and 5-LO [195, 276, 277] referred to earlier, has necessitated adding COX inhibition (with celecoxib in CUSP9*) along with the 5-LO inhibitor itraconazole. This would be then a specifically delineated case of addressing the Nile Distributary Problem. D.A. Omahen, noting increased expression of both COX-2 and 5-LO in gliomas, suggested in 2011 treating gliomas with simultaneous COX and 5-LO inhibitors ¨using readily available, well-tolerated medications¨, ¨thereby priming glioma cells for treatment-induced apoptotic cell death¨ [195].
Examples of COX/5-LO reciprocal relationship tendency: Smokers with elevated urinary PGE metabolite levels showed decreased urinary PGE metabolite levels after celecoxib administration but increased urinary levels of LTB4 [278]. Exposure of human chondrocytes to naproxen, a COX-1 and 2 inhibitor, increased LTB4 secretion and 5-LO mRNA [279].
Celecoxib had some 5-LO inhibition in a single in vivo study [280] but COX-2 inhibition predominated. Both COX-2 and 5-LO were up-regulated in head/neck squamous cancer cell lines [281]. COX-2 inhibition had little effect on proliferation but COX-2 inhibition did increase LTB4 [281]. Knockdown of both COX-2 and 5-LO gave significantly impaired proliferation and, parenthetically, VEGF production [281]. COX inhibitors suppressed PGE2 production but enhanced LTB4 secretion in COX-2 expressing colon cancer cell lines [278].
The reverse process can be seen as well. Some patients given the marketed pharmaceutical 5-LO inhibitor zileuton develop increased prostaglandin levels [282]. In a cardiomyocyte cell line zileuton upregulated COX-2 expression [283]. We have therefore potential for a bound bilateral see-saw [reciprocal] relationship between 5-LO and COX-2, with either cross-covering for the other. A specific example of the Nile Distributary Problem.
Dosing and practical considations
Dose suggestions
Specific addition and uptitration schedules are available from the authors. The CUSP9* drugs are generally forgiving and have been well-tolerated in the handful of cases given CUSP9 drugs on a compassionate-use basis that have been reported to the authors to date. No remarkable toxicities have been reported. This benign side effect profile was expected based on wide experience over decades of use with these drugs.
Artesunate and auranofin are the only two drugs that might be unfamiliar to standard general medical practioners. Artesunate, although widely used to treat malaria around the world, is distinctly uncommonly used in developed countries due to the rarity of malaria there. Auranofin, although in use for several decades to treat rheumatoid arthritis, is uncommonly used. Captopril, celecoxib, ritonavir, and sertraline are common drugs, not rarely used together in day-to-day medical practice. Aprepitant is an unusually silent drug side effect wise when used in chemotherapy related nausea and vomiting control.
The doses for CUSP9* drugs are those commonly used doses when given for their approved indications- auranofin in rheumatoid arthritis, captopril for hypertension, celecoxib for joint pain, itraconazole for fungal infections. Doses for disulfiram and sertraline are slightly higher than average dosing in alcoholism or depression respectively, but CUSP9* doses below are at the higher end of the dose range given clinically. CUSP9* dose for artesunate, nominally ~ 1.4 mg/kg/day, is lower than that used in malaria treatment, ~ 4 mg/kg/day [284] but we will be using artesunate longer than is usual during malaria treatment and chose a conservative dose. Aprepitant is given at standard anti-emetic dose but for longer duration than is current clinical practice.
Weekly evaluations with complete blood count are essential given the potential for unforeseen drug-drug interactions in a medical regimen as complex as CUSP9*. Monthly MRI’s will be done in the upcoming clinical trial. Both theoretical considerations [starting with the drugs least likely to give side effects [1] ] and limited initial clinical experience suggest the following drug addition schedule: The initial four drugs and doses are aprepitant 80 mg once daily, auranofin 3 mg once daily, celecoxib 200 mg once daily, disulfiram 250 mg once daily. These can be started on day one. After seven days artesunate 50 mg once daily, captopril 50 mg once daily, itraconazole 200 mg once daily, ritonavir 400 mg once daily, and sertraline 100 mg once daily are started. On the three week check [day 21] drugs are brought to full doses, given below.
Aprepitant 80 mg twice daily.
Artesunate 50 mg twice daily.
Auranofin 3 mg twice daily.
Captopril 50 mg twice daily.
Celecoxib 400 mg twice daily
Disulfiram 250 mg twice daily.
Itraconazole 200 mg twice daily.
Ritonavir 400 mg twice daily.
Sertraline 100 mg twice daily.
Several practical recommendations
As part of CUSP9* we specify first-choice medicines for four common problems arising in the course of glioblastoma treatment generally. We specify these drugs to treat ancillary problems due to the high potential for drug-drug interactions when so many drugs are used at once as in CUSP9*. The four selected drugs are unlikely to interact pharmacokinetically or pharmacodynamically with the CUSP9* drugs. No other medicine, herbal preparation, over-the-counter medicine or nutritional supplement will be allowed, again to minimize the risk of drug-drug interaction.
i. Diarrhea can be treated with loperamide 2 mg ii bid. This dose can be increased or decresed as needed. Loperamide is not absorbed systemically, yet by stimulating gut opiate receptors effectively reduces the loose bowel movements some of the CUSP9* drugs can give.
ii. Anxiety or poor sleep should be treated with lorazepam 1 mg or 2 mg at hour of sleep. This can be increased or decreased as needed. Half this dose can be used several times during the day should daytime anxiety become a problem.
iii. If seizures occur levetiracetam 1 gram twice daily can be used, with down titration if side effects are encountered.
iv. Headache treatment with low-dose hydromorphone is allowed.
An overview of pharmacodynamic and pharmacokinetic data and interaction analysis of the CUSP9* drugs is given in Table 4. Ritonavir and aprepitant are also inducers of 3A4. CUSP9* uses some of the most potent hepatic [and small intestine] P-450 3A4 inhibitors, itraconazole and riotonavir [see Table 4.]. This necessitates particular strictness in avoidance of any herbal preparations as well as any drug treatment beyond those specified by the CUSP9* regimen and the four ancillary drugs allowed- loperamide for diarrhea, lorazepam for anxiety or insomnia, levetiracetam for seizures, low-dose hydromorphone for headache.
Strictest avoidance of any source of ethanol, even in such sources as wine vinegar or casseroles and grapefruit juice avoidance [a 3A4 inhibitor] will be the only two dietary restrictions. Coffee consumption is allowed but caution advised, as caffeine effects can be magnified by 1A2 inhibitors like disulfiram.
Severe alcohol intolerance is not listed as a disulfiram side effect in that severe intolerance of even slight amounts of alcohol is universal and the main effect of disulfiram.
Edema, dexamethasone, & survival
The use of dexamethasone is common during the course of glioblastoma particularly in the perioperative reduction of brain edema, but greater dexamethasone use is a negative prognostic sign [285]. Dexamethasone or other related corticosteroid use contributes to disease morbidity and rarely, mortality. It is immunosuppressive [286], exacerbating the inherent, unmedicated, glioblastoma-related immunosuppression [287], Dexamethasone reduced in vitro temozolomide cytotoxicity in U87 [288] and T98G glioma cells [289]. Although dexamethasone has some anti-cancer activities in other cancers, and may have such at some stages of glioblastoma, we consider the sum of data indicates probable benefit from less, or ideally no, dexamethasone use. We have outlined how several of the CUSP9* medicines are expected to act together to reduce glioblastoma related edema and thereby reduce or eliminate the need for dexamethasone use in the course of glioblastoma treatment.
Conclusion
We presented six themes important to cancer therapy generally that we apply to recurrent glioblastoma treatment and attempt to address by CUSP9*:
1. Nile Distributary Problem where cancer uses multiple cross-covering growth enhancing pathways to grow and avoid cytotoxic interventions.
2. Intratumoral heterogeneity in space and over time as general feature of cancer that must be accounted for in treatment.
3. Individual mutually supporting sub-populations existing within a tumor, working together to enhance growth, generating vigor of the malignant state.
4. Ganging up. We see the need for multiple attacks against the same growth-enhancing subsystem, again in the effort to defeat compensating reactions by tumors.
5. We must fight today with the weapons we have today. We therefore use ancillary attributes of nine already-marketed drugs to condition glioblastoma cells to be less fit to thrive.
6. “Changes across metabolic networks” that are not malignant by themselves become an integral part of malignant growth when combined with the suite of so deranged networks and genomic changes driving them. We aim to dismantle or block these pathologically employed but not inherently pathological processes enough to hobble glioblastoma growth.
We have shown how past research indicates how the nine drugs of CUSP9* have a good chance of inhibiting the growth enhancing functions of 17 different systems used by glioblastoma to grow, migrate, and avoid cell death. These growth systems are 1) AKT phosphorylation, 2) ALDH, 3) ACE, 4) carbonic anhydrase 2, 9, and 12, 5) COX-2, 6) cathepsin B, 7) Hh, 8) interleukin-6, 9] 5-LO, 10) MMP-2 and -9, 11) mTOR, 12) NK-1, 12) p-gp efflux pump, 14) thioredoxin reductase, 15) TF, 16) TCTP, and 17) VEGF.
Given the stalled progress in glioblastoma treatment since the last advance, introduction of temozolomide with the Stupp Protocol in 2005, we suggest a conceptual departure from the usual cytotoxic efforts that have so far been futile in prolonging survival or QOL. A formal clinical trial of CUSP9* in patients on first recurrence is in advanced stage planning. Initial experience on the handful of patients as reported to us who were given CUSP9 without remarkable side effects leads us to be optimistic that this will hold for the closely related CUSP9*.
To break the impasse we propose using nine commonly used drugs marketed for non-cancer indications, drugs that show minimal cytotoxicity to normal cells and minimal changes to organ systems when used singly. We use the nine drugs of CUSP9* to “shape the battlefield”. By this we mean if we can inhibit or block a pathological protective system engaged or used by glioblastoma cells to grow, migrate, or avoid cell death and senescence, our directly cell-killing drugs like temozolomide will be more effective. The nine drugs of CUSP9* each have data from animal and human study showing they can individually inhibit or block one or another identified cellular pathway to enhance or stimulate their growth, helping CUSP9* drugs acting in concert to set the stage for more effective cell killing.
Abbreviations
angiotensin receptor blockers, ARB; angiotensin converting enzyme, ACE; ATR1, angiotensin II receptor 1; blood-brain barrier, BBB; breast cancer resistance protein BCRP; cyclooxygenase, COX; CUSP9, Coordinated Undermining of Survival Paths; DMARDs, disease modifying anti-rheumatic drugs; European Medicines Agency, EMA; inducible nitric oxide synthase, iNOS; 5-lipoxygenase, 5-LO; MCP-1, macrophage chemotactic protein-1, synonymous with CCL2; matrix metalloproteinase, MMP; O6-methylguanine-DNA methyltransferase, MGMT; neurokinin-1 receptors, NK-1; OS, overall survival; P-glycoprotein, p-gp, synonymous with MDR-1; prostaglandin E2, PGE2; renin-angiotensin system RAS; quality of life, QOL; ROS, reactive oxygen species; renal cell carcinoma, RCC; Translationally controlled tumor protein , TCTP; Tissue Factor, TF, also termed thromboplastin; TNF-alpha, tumor necrosis factor-alpha; vascular endothelial growth factor, VEGF
References
1. Kast RE, Boockvar JA, Brüning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, Ketola K, Khoshnevisan A, Keizman D, Magné N, Marosi C, McDonald K, Muñoz M, Paranjpe A, Pourgholami MH, Sardi I, Sella A, Srivenugopal KS, Tuccori M, Wang W, Wirtz CR, Halatsch ME. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013;4(4):502-30.
2. Chang L, Su J, Jia X, Ren H. Treating malignant glioma in Chinese patients: update on temozolomide. Onco Targets Ther. 2014;7:235-244.
3. Gilbert MR. Recurrent glioblastoma: a fresh look at current therapies and emerging novel approaches. Semin Oncol. 2011;38 Suppl 4:S21-33. doi:10.1053/j.seminoncol.2011.09.008.
4. Vartanian A, Singh SK, Agnihotri S, Jalali S, Burrell K, Aldape KD, Zadeh G. GBM’s multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro Oncol. 2014 Mar 18. [Epub ahead of print] PMID:24642524.
5. Costa ET, Barnabé GF, Li M, Dias AA, Machado TR, Asprino PF, Cavalher FP, Ferreira EN, Del Mar Inda M, Nagai MH, Malnic B, Duarte ML, Leite KR, de Barros AC, Carraro DM, Chammas R, Armelin HA, Cavenee W, Furnari F, Camargo AA. Intratumoral heterogeneity of ADAM23 promotes tumor growth and metastasis through LGI4 and nitric oxide signals. Oncogene. 2014;0. doi:10.1038/onc.2014.70.
6. Zapletalova D, André N, Deak L, Kyr M, Bajciova V, Mudry P, Dubska L, Demlova R, Pavelka Z, Zitterbart K, Skotakova J, Husek K, Martincekova A, Mazanek P, Kepak T, Doubek M, Kutnikova L, Valik D, Sterba J. Metronomic chemotherapy with the COMBAT regimen in advanced pediatric malignancies: a multicenter experience. Oncology. 2012;82(5):249-60. doi: 10.1159/000336483.
7. Omuro A, Chan TA, Abrey LE, Khasraw M, Reiner AS, Kaley TJ, Deangelis LM, Lassman AB, Nolan CP, Gavrilovic IT, Hormigo A, Salvant C, Heguy A, Kaufman A, Huse JT, Panageas KS, Hottinger AF, Mellinghoff I. Phase II trial of continuous low-dose temozolomide for patients with recurrent malignant glioma. Neuro Oncol. 2013;15(2):242-50. doi: 10.1093/neuonc/nos295.
8. Stockhammer F, Misch M, Koch A, Czabanka M, Plotkin M, Blechschmidt C, Tuettenberg J, Vajkoczy P. Continuous low-dose temozolomide and celecoxib in recurrent glioblastoma. J Neurooncol. 2010;100(3):407-15. doi: 10.1007/s11060-010-0192-y.
9. Aapro MS, Schmoll HJ, Jahn F, Carides AD, Webb RT. Review of the efficacy of aprepitant for the prevention of chemotherapy-induced nausea and vomiting in a range of tumor types. Cancer Treat Rev. 2013;39(1):113-7. doi:10.1016/j.ctrv.2012.09.002.
10. dos Santos LV, Souza FH, Brunetto AT, Sasse AD, da Silveira Nogueira Lima JP. Neurokinin-1 receptor antagonists for chemotherapy-induced nausea and vomiting: a systematic review. J Natl Cancer Inst. 2012 5;104(17):1280-92. doi:10.1093/jnci/djs335.
11. Coveñas R, Muñoz M. Cancer progression and substance P. Histol Histopathol. 2014 Feb 18. [Epub ahead of print] PMID: 24535838.
12. Muñoz M, González-Ortega A, Rosso M, Robles-Frias MJ, Carranza A, Salinas-Martín MV, Coveñas R. The substance P/neurokinin-1 receptor system in lung cancer: focus on the antitumor action of neurokinin-1 receptor antagonists. Peptides. 2012;38(2):318-25. doi: 10.1016/j.peptides.2012.09.024.
13. Muñoz M, Martinez-Armesto J, Coveñas R. NK-1 receptor antagonists as antitumor drugs: a survey of the literature from 2000 to 2011. Expert Opin Ther Pat. 2012;22(7):735-46. doi: 10.1517/13543776.2012.697153.
14. Muñoz M, Rosso M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Invest New Drugs. 2010;28(2):187-93. doi:10.1007/s10637-009-9218-8.
15. Kast RE. Why cerebellar glioblastoma is rare and how that indicates adjunctive use of the FDA-approved anti-emetic aprepitant might retard cerebral glioblastoma growth: a new hypothesis to an old question. Clin Transl Oncol. 2009;11(7):408-10.
16. Kast RE. Glioblastoma: synergy of growth promotion between CCL5 and NK-1R can be thwarted by blocking CCL5 with miraviroc, an FDA approved anti-HIV drug and blocking NK-1R with aprepitant, an FDA approved anti-nausea drug. J Clin Pharm Ther. 2010;35(6):657-63. doi: 10.1111/j.1365-2710.2009.01148.x.
17. Lewis KM, Harford-Wright E, Vink R, Ghabriel MN. NK1 receptor antagonists and dexamethasone as anticancer agents in vitro and in a model of brain tumours secondary to breast cancer. Anticancer Drugs. 2013;24(4):344-54. doi:10.1097/CAD.0b013e32835ef440.
18. Berger M, Neth O, Ilmer M, Garnier A, Salinas-Martín MV, de Agustín Asencio JC, von Schweinitz D, Kappler R, Muñoz M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J Hepatol. 2014 May;60(5):985-94. doi: 10.1016/j.jhep.2013.12.024.
19. Muñoz M, Coveñas R. Involvement of substance P and the NK-1 receptor in pancreatic cancer. World J Gastroenterol. 2014;20(9):2321-34. doi: 10.3748/wjg.v20.i9.2321.
20. Gogtay N, Kannan S, Thatte UM, Olliaro PL, Sinclair D. Artemisinin-based combination therapy for treating uncomplicated Plasmodium vivax malaria. Cochrane Database Syst Rev. 2013;10:CD008492. doi:10.1002/14651858.CD008492.pub3.
21. Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin CS, Fleckenstein L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar J. 2011;10:263. doi: 10.1186/1475-2875-10-263.
22. Ho WE, Peh HY, Chan TK, Wong WS. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol Ther. 2014;142(1):126-139. doi: 10.1016/j.pharmthera.2013.12.001.
23. Cai H, Kapoor A, He R, Venkatadri R, Forman M, Posner GH, Arav-Boger R. In Vitro Combination of Anti-Cytomegalovirus Compounds Acting through Different Targets: Role of the Slope Parameter and Insights into Mechanisms of Action. Antimicrob Agents Chemother. 2014;58(2):986-94. doi: 10.1128/AAC.01972-13.
24. Schreiber A, Härter G, Schubert A, Bunjes D, Mertens T, Michel D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin Pharmacother. 2009;10(2):191-209. doi: 10.1517/14656560802678138.
25. Holien T, Olsen OE, Misund K, Hella H, Waage A, Rø TB, Sundan A. Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. Eur J Haematol. 2013;91(4):339-46. doi: 10.1111/ejh.12176.
26. Liu Y, Cui YF. Synergism of cytotoxicity effects of triptolide and artesunate combination treatment in pancreatic cancer cell lines. Asian Pac J Cancer Prev. 2013;14(9):5243-8.
27. Jin M, Shen X, Zhao C, Qin X, Liu H, Huang L, Qiu Z, Liu Y. In vivo study of effects of artesunate nanoliposomes on human hepatocellular carcinoma xenografts in nude mice. Drug Deliv. 2013;20(3-4):127-33. doi: 10.3109/10717544.2013.801047.
28. Jiang W, Huang Y, Wang JP, Yu XY, Zhang LY. The synergistic anticancer effect of artesunate combined with allicin in osteosarcoma cell line in vitro and in vivo. Asian Pac J Cancer Prev. 2013;14(8):4615-9.
29. Zhou X, Sun WJ, Wang WM, Chen K, Zheng JH, Lu MD, Li PH, Zheng ZQ. Artesunate inhibits the growth of gastric cancer cells through the mechanism of promoting oncosis both in vitro and in vivo. Anticancer Drugs. 2013;24(9):920-7. doi: 10.1097/CAD.0b013e328364a109.
30. Zhou HJ, Wang WQ, Wu GD, Lee J, Li A. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol. 2007;47(2-3):131-8.
31. Li Y, Feng L, Li Y, Jiang W, Shan N, Wang X. Artesunate possesses anti-leukemia properties that can be enhanced by arsenic trioxide. Leuk Lymphoma. 2014;55(6):1366-72. doi: 10.3109/10428194.2013.829573.
32. Karpel-Massler G, Westhoff MA, Kast RE, Dwucet A, Nonnenmacher L, Wirtz CR, Debatin KM, Halatsch ME. Artesunate Enhances the Antiproliferative Effect of Temozolomide on U87MG and A172 Glioblastoma Cell Lines. Anticancer Agents Med Chem. 2014;14(2):313-8.
33. Miller RS, Li Q, Cantilena LR, Leary KJ, Saviolakis GA, Melendez V, Smith B, Weina PJ. Pharmacokinetic profiles of artesunate following multiple intravenous doses of 2, 4, and 8 mg/kg in healthy volunteers: phase 1b study. Malar J. 2012;11:255. doi: 10.1186/1475-2875-11-255.
34. Aquino I, Tsuboy MS, Marcarini JC, Mantovani MS, Perazzo FF, Maistro EL. Genotoxic evaluation of the antimalarial drugs artemisinin and artesunate in human HepG2 cells and effects on CASP3 and SOD1 gene expressions. Genet Mol Res. 2013;12(3):2517-27. doi: 10.4238/2013.July.24.6.
35. Brüning A, Kast RE. Oxidizing to death: disulfiram for cancer cell killing. Cell Cycle. 2014;13(10):1513-4. doi: 10.4161/cc.28959.
36. Efferth T, Giaisi M, Merling A, Krammer PH, Li-Weber M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One. 2007;2(8):e693.
37. Cheng R, Li C, Li C, Wei L, Li L, Zhang Y, Yao Y, Gu X, Cai W, Yang Z, Ma J, Yang X, Gao G. The artemisinin derivative artesunate inhibits corneal neovascularization by inducing ROS-dependent apoptosis in vascular endothelial cells.Invest Ophthalmol Vis Sci. 2013;54(5):3400-9. doi: 10.1167/iovs.12-11068.
38. Antoine T, Fisher N, Amewu R, O’Neill PM, Ward SA, Biagini GA. Rapid kill of malaria parasites by artemisinin and semi-synthetic endoperoxides involves ROS-dependent depolarization of the membrane potential. J Antimicrob Chemother. 2014;69(4):1005-16. doi: 10.1093/jac/dkt486.
39. Krusche B, Arend J, Efferth T. Synergistic inhibition of angiogenesis by artesunate and captopril in vitro and in vivo. Evid Based Complement Alternat Med. 2013;2013:454783. doi: 10.1155/2013/454783.
40. Chen K, Shou LM, Lin F, Duan WM, Wu MY, Xie X, Xie YF, Li W, Tao M. Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs. 2014;25(6):652-62. doi:10.1097/CAD.0000000000000089.
41. Reichert S, Reinboldt V, Hehlgans S, Efferth T, Rödel C, Rödel F. A radiosensitizing effect of artesunate in glioblastoma cells is associated with a diminished expression of the inhibitor of apoptosis protein survivin. Radiother Oncol. 2012;103(3):394-401. doi: 10.1016/j.radonc.2012.03.018.
42. Hooft van Huijsduijnen R, Guy RK, Chibale K, Haynes RK, Peitz I, Kelter G, Phillips MA, Vennerstrom JL, Yuthavong Y, Wells TN. Anticancer properties of distinct antimalarial drug classes. PLoS One. 2013;8(12):e82962. doi: 10.1371/journal.pone.0082962. ECollection 2013.
43. Abdin AA, Ashour DS, Shoheib ZS. Artesunate effect on schistosome thioredoxin glutathione reductase and cytochrome c peroxidase as new molecular targets in schistosoma mansoni-infected mice. Biomed Environ Sci. 2013;26(12):953-61. doi: 10.3967/bes2013.030.
44. Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology. 2012;20(6):297-306. doi: 10.1007/s10787-012-0149-1.
45. Debnath A, Ndao M, Reed SL. Reprofiled drug targets ancient protozoans: drug discovery for parasitic diarrheal diseases. Gut Microbes. 2013;4(1):66-71. doi: 10.4161/gmic.22596.
46. Casini A, Messori L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr Top Med Chem. 2011;11(21):2647-60.
47. Becker K, Gromer S, Schirmer RH, Müller S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur J Biochem. 2000;267(20):6118-25.
48. Messori L, Marcon G. Gold complexes in the treatment of rheumatoid arthritis. Met Ions Biol Syst. 2004;41:279-304.
49. Kim NH, Park HJ, Oh MK, Kim IS. Antiproliferative effect of gold(I) compound auranofin through inhibition of STAT3 and telomerase activity in MDA-MB 231 human breast cancer cells. BMB Rep. 2013;46(1):59-64.
50. Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD, Chauhan L, Rao R, Gunewardena S, Schorno K, Austin CP, Maddocks K, Byrd J, Melnick A, Huang P, Wiestner A, Bhalla KN. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014;74(9):2520-32. doi:10.1158/0008-5472.CAN-13-2033.
51. Luo Z, Yu L, Yang F, Zhao Z, Yu B, Lai H, Wong KH, Ngai SM, Zheng W, Chen T. Ruthenium polypyridyl complexes as inducer of ROS-mediated apoptosis in cancer cells by targeting thioredoxin reductase. Metallomics. 2014 May 14. [Epub ahead of print] PubMed PMID: 24823440.
52. Fan C, Zheng W, Fu X, Li X, Wong YS, Chen T. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014;5:e1191. doi:10.1038/cddis.2014.132.
53. Gamberi T, Massai L, Magherini F, Landini I, Fiaschi T, Scaletti F, Gabbiani C, Bianchi L, Bini L, Nobili S, Perrone G, Mini E, Messori L, Modesti A. Proteomic analysis of A2780/S ovarian cancer cell response to the cytotoxic organogold(III) compound Aubipy(c.). J Proteomics. 2014;103C:103-120. doi: 10.1016/j.jprot.2014.03.032.
54. Liu C, Liu Z, Li M, Li X, Wong YS, Ngai SM, Zheng W, Zhang Y, Chen T. Enhancement of auranofin-induced apoptosis in MCF-7 human breast cells by selenocystine, a synergistic inhibitor of thioredoxin reductase. PLoS One. 2013;8(1):e53945. doi:10.1371/journal.pone.0053945.
55. Shen SC, Wu MS, Lin HY, Yang LY, Chen YH, Chen YC. Reactive Oxygen Species-Dependent Nitric Oxide Production in Reciprocal Interactions of Glioma and Microglial Cells. J Cell Physiol. 2014 Apr 29.doi: 10.1002/jcp.24659. [Epub ahead of print
56. Schruefer R, Lutze N, Schymeinsky J, Walzog B. Human neutrophils promote angiogenesis by a paracrine feedforward mechanism involving endothelial interleukin-8. Am J Physiol Heart Circ Physiol. 2005;288(3):H1186-92.
57. da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol. 2013;2013:264124. doi: 10.1155/2013/264124.
58. Vasco C, Canazza A, Rizzo A, Mossa A, Corsini E, Silvani A, Fariselli L, Salmaggi A, Ciusani E. Circulating T regulatory cells migration and phenotype in glioblastoma patients: an in vitro study. J Neurooncol. 2013;115(3):353-63.doi: 10.1007/s11060-013-1236-x.
59. Lin Y, Zhang G, Zhang J, Gao G, Li M, Chen Y, Wang J, Li G, Song SW, Qiu X, Wang Y, Jiang T. A panel of four cytokines predicts the prognosis of patients with malignant gliomas. J Neurooncol. 2013;114(2):199-208. doi:10.1007/s11060-013-1171-x.
60. Betts WH, Hurst NP, Murphy GA, Cleland LG. Auranofin stimulates LTA hydrolase and inhibits 5-lipoxygenase/LTA synthase activity of isolated human neutrophils. Biochem Pharmacol. 1990;39(7):1233-7.
61. Elmgreen J, Ahnfelt-Rønne I, Nielsen OH. Inhibition of human neutrophils by auranofin: chemotaxis and metabolism of arachidonate via the 5-lipoxygenase pathway. Ann Rheum Dis. 1989;48(2):134-8.
62. Herlin T, Fogh K, Christiansen NO, Kragballe K. Effect of auranofin on eicosanoids and protein kinase C in human neutrophils. Agents Actions. 1989;28(1-2):121-9.
63. Peters-Golden M, Shelly C. Inhibition of alveolar macrophage 5-lipoxygenase metabolism by auranofin. Biochem Pharmacol. 1989;38(10):1589-95.
64. James MJ, Cleland LG, Gibson RA. Inhibition of human neutrophil leukotriene B4 synthesis by combination auranofin and eicosapentaenoic acid. Biochem Pharmacol. 1992;43(4):695-700.
65. Hafström I. The effect of auranofin on polymorphonuclear granulocytes. Scand J Rheumatol Suppl. 1983;51:36-41.
66. Hafström I, Ringertz B, Palmblad J, Malmsten C. Effects of auranofin on leukotriene production and leukotriene stimulated neutrophil function. Agents Actions. 1984;15(5-6):551-5.
67. Hafström I, Seligmann BE, Friedman MM, Gallin JI. Auranofin affects early events in human polymorphonuclear neutrophil activation by receptor-mediated stimuli. J Immunol. 1984;132(4):2007-14.
68. Formolo CA, Williams R, Gordish-Dressman H, MacDonald TJ, Lee NH, Hathout Y. Secretome signature of invasive glioblastoma multiforme. J Proteome Res. 2011;10(7):3149-59. doi: 10.1021/pr200210w.
69. Malla R, Gopinath S, Alapati K, Gondi CS, Gujrati M, Dinh DH, Mohanam S, Rao JS. Downregulation of uPAR and cathepsin B induces apoptosis via regulation of Bcl-2 and Bax and inhibition of the PI3K/Akt pathway in gliomas. PLoS One. 2010;5(10):e13731. doi: 10.1371/journal.pone.0013731.
70. Colin C, Voutsinos-Porche B, Nanni I, Fina F, Metellus P, Intagliata D, Baeza N, Bouvier C, Delfino C, Loundou A, Chinot O, Lah T, Kos J, Martin PM, Ouafik L, Figarella-Branger D. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol. 2009;118(6):745-54. doi: 10.1007/s00401-009-0592-2.
71. Kenig S, Alonso MB, Mueller MM, Lah TT. Glioblastoma and endothelial cells cross-talk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010;289(1):53-61. doi: 10.1016/j.canlet.2009.07.014.
72. Kast RE. Profound blockage of CXCR4 signaling at multiple points using the synergy between plerixafor, mirtazapine, and clotrimazole as a new glioblastoma treatment adjunct. Turk Neurosurg. 2010;20(4):425-9. doi:10.5137/1019-5149.JTN.3334-10.0.
73. Würth R, Bajetto A, Harrison JK, Barbieri F, Florio T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front Cell Neurosci. 2014;8:144. doi:10.3389/fncel.2014.00144.
74. Gunatilleke SS, Barrios AM. Inhibition of lysosomal cysteine proteases by a series of Au(I) complexes: a detailed mechanistic investigation. J Med Chem. 2006;49(13):3933-7.
75. Migdalof BH, Antonaccio MJ, McKinstry DN, Singhvi SM, Lan SJ, Egli P, Kripalani KJ. Captopril: pharmacology, metabolism and disposition. Drug Metab Rev. 1984;15(4):841-69.
76. Brogden RN, Todd PA, Sorkin EM. Captopril. An update of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension and congestive heart failure. Drugs. 1988;36(5):540-600.
77. Nakagawa T, Kubota T, Kabuto M, Kodera T. Captopril inhibits glioma cell invasion in vitro: involvement of matrix metalloproteinases. Anticancer Res. 1995;15(5B):1985-9.
78. Rooprai HK, Kandanearatachi A, Rucklidge G, Pilkington GJ. Influence of putative antiinvasive agents on matrix metalloproteinase secretion by human neoplastic glia in vitro. Ann N Y Acad Sci. 1999;878:654-7.
79. Rooprai HK, Kandanearatchi A, Maidment SL, Christidou M, Trillo-Pazos G, Dexter DT, Rucklidge GJ, Widmer W, Pilkington GJ. Evaluation of the effects of swainsonine, captopril, tangeretin and nobiletin on the biological behaviour of brain tumour cells in vitro. Neuropathol Appl Neurobiol. 2001;27(1):29-39.
80. Kast RE, Halatsch ME. Matrix metalloproteinase-2 and -9 in glioblastoma: a trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch Med Res. 2012;43(3):243-7. doi:10.1016/j.arcmed.2012.04.005.
81. Takada Y, Hiwada K, Kokubu T. Characterization and immunohistologic demonstration of angiotensin-converting enzyme in human renal cell carcinoma. Oncodev Biol Med. 1983;4(5):327-34.
82. Carpentier AF, Ferrari D, Bailon O, Ursu R, Banissi C, Dubessy AL, Belin C, Levy C. Steroid-sparing effects of angiotensin-II inhibitors in glioblastoma patients. Eur J Neurol. 2012;19(10):1337-42. doi:10.1111/j.1468-1331.2012.03766.x.
83. Ronquist G, Frithz G, Wang YH, Lindeborg T. Captopril may reduce biochemical PSA failure following radical prostatectomy for clinically localized prostate cancer. Scand J Urol Nephrol. 2009;43(1):32-6. doi: 10.1080/00365590802468875.
84. Jones PH, Christodoulos K, Dobbs N, Thavasu P, Balkwill F, Blann AD, Caine GJ, Kumar S, Kakkar AJ, Gompertz N, Talbot DC, Ganesan TS, Harris AL. Combination antiangiogenesis therapy with marimastat, captopril and fragmin in patients with advanced cancer. Br J Cancer. 2004;91(1):30-6.
85. Ronquist G, Rodríguez LA, Ruigómez A, Johansson S, Wallander MA, Frithz G, Svärdsudd K. Association between captopril, other antihypertensive drugs and risk of prostate cancer. Prostate. 2004;58(1):50-6.
86. Wilson MJ, Mack MS, Woodson M, Sinha AA. Angiontensin-converting enzyme activity in dunning rat prostate tumor. Arch Androl. 2003;49(6):457-61.
87. Ager EI, Wen SW, Chan J, Chong WW, Neo JH, Christophi C. Altered efficacy of AT1R-targeted treatment after spontaneous cancer cell-AT1R upregulation. BMC Cancer. 2011;11:274. doi: 10.1186/1471-2407-11-274.
88. Wang L, Cai SR, Zhang CH, He YL, Zhan WH, Wu H, Peng JJ. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor blockers on lymphangiogenesis of gastric cancer in a nude mouse model. Chin Med J (Engl). 2008;121(21):2167-71.
89. De la Iglesia Iñigo S, López-Jorge CE, Gómez-Casares MT, Lemes Castellano A, Martín Cabrera P, López Brito J, Suárez Cabrera A, Molero Labarta T. Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: a new role in the treatment of leukaemia for these agents. Leuk Res. 2009;33(6):810-6. doi: 10.1016/leukres.2008.09.029.
90. Arafat HA, Gong Q, Chipitsyna G, Rizvi A, Saa CT, Yeo CJ. Antihypertensives as novel antineoplastics: angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptor blockers in pancreatic ductal adenocarcinoma. J Am Coll Surg. 2007;204(5):996-1005.
91. Hii SI, Nicol DL, Gotley DC, Thompson LC, Green MK, Jonsson JR. Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br J Cancer. 1998;77(6):880-3.
92. Small W Jr, Molteni A, Kim YT, Taylor JM, Chen Z, Ward WF. Captopril modulates hormone receptor concentration and inhibits proliferation of human mammary ductal carcinoma cells in culture. Breast Cancer Res Treat. 1997;44(3):217-24.
93. Small W Jr, Molteni A, Kim YT, Taylor JM, Ts’ao CH, Ward WF. Mechanism of captopril toxicity to a human mammary ductal carcinoma cell line in the presence of copper. Breast Cancer Res Treat. 1999;55(3):223-9.
94. Volpert OV, Ward WF, Lingen MW, Chesler L, Solt DB, Johnson MD, Molteni A, Polverini PJ, Bouck NP. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest. 1996;98(3):671-9.
95. Kowalski J, Herman ZS. Captopril augments antitumor activity of cyclophosphamide in mice. Pol J Pharmacol. 1996;48(3):281-5.
96. Ward WF, Molteni A, Ts’ao C, Hinz JM. The effect of Captopril on benign and malignant reactions in irradiated rat skin. Br J Radiol. 1990;63(749):349-54.
97. de Groot-Besseling RR, Ruers TJ, van Kraats AA, Poelen GJ, Ruiter DJ, de Waal RM, Westphal JR. Anti-tumor activity of a combination of plasminogen activator and captopril in a human melanoma xenograft model. Int J Cancer. 2004;112(2):329-34.
98. Stanojkovic TP, Zizak Z, Mihailovic-Stanojevic N, Petrovic T, Juranic Z. Inhibition of proliferation on some neoplastic cell lines-act of carvedilol and captopril. J Exp Clin Cancer Res. 2005;24(3):387-95.
99. Chisi JE, Briscoe CV, Ezan E, Genet R, Riches AC, Wdzieczak-Bakala J. Captopril inhibits in vitro and in vivo the proliferation of primitive haematopoietic cells induced into cell cycle by cytotoxic drug administration or irradiation but has no effect on myeloid leukaemia cell proliferation. Br J Haematol. 2000;109(3):563-70
100. Williams RN, Parsons SL, Morris TM, Rowlands BJ, Watson SA. Inhibition of matrix metalloproteinase activity and growth of gastric adenocarcinoma cells by an angiotensin converting enzyme inhibitor in in vitro and murine models. Eur J Surg Oncol. 2005;31(9):1042-50.
101. Prontera C, Mariani B, Rossi C, Poggi A, Rotilio D. Inhibition of gelatinase A (MMP-2) by batimastat and captopril reduces tumor growth and lung metastases in mice bearing Lewis lung carcinoma. Int J Cancer. 1999;81(5):761-6.
102. Brown RE, Lun M, Prichard JW, Blasick TM, Zhang PL. Morphoproteomic and pharmacoproteomic correlates in hormone-receptor-negative breast carcinoma cell lines. Ann Clin Lab Sci. 2004;34(3):251-62.
103. Tao Z, Sakurada C, Yokosawa H. Evidence for thiol-dependent metallo-endopeptidase involved in degradation of luteinizing hormone-releasing hormone in glioma cells. Neuropeptides. 1991;20(2):125-31.
104. Koh SL, Ager EI, Costa PL, Malcontenti-Wilson C, Muralidharan V, Christophi C. Blockade of the renin-angiotensin system inhibits growth of colorectal cancer liver metastases in the regenerating liver. Clin Exp Metastasis. 2014 Jan 18. [Epub ahead of print] PMID: 24442969
105. Attoub S, Gaben AM, Al-Salam S, Al Sultan MA, John A, Nicholls MG, Mester J, Petroianu G. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann N Y Acad Sci. 2008;1138:65-72. doi: 10.1196/annals.1414.011.
106. Kocatürk B, Versteeg HH. Tissue factor-integrin interactions in cancer and thrombosis: every Jack has his Jill. J Thromb Haemost. 2013;11 Suppl 1:285-93. doi: 10.1111/jth.12222.
107. Perry JR. Thromboembolic disease in patients with high-grade glioma. Neuro Oncol. 2012;14 Suppl 4:iv73-80. doi: 10.1093/neuonc/nos197.
108. Jenkins EO, Schiff D, Mackman N, Key NS. Venous thromboembolism in malignant gliomas. J Thromb Haemost. 2010;8(2):221-7. doi:10.1111/j.1538-7836.2009.03690.x.
109. Rak J, Milsom C, Magnus N, Yu J. Tissue factor in tumour progression. Best Pract Res Clin Haematol. 2009;22(1):71-83. doi: 10.1016/j.beha.2008.12.008.
110. Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood. 2010;116(5):815-8. doi: 10.1182/blood-2009-10-250639.
111. Napoleone E, Cutrone A, Cugino D, Amore C, Di Santo A, Iacoviello L, de Gaetano G, Donati MB, Lorenzet R. Inhibition of the renin-angiotensin system downregulates tissue factor and vascular endothelial growth factor in human breast carcinoma cells. Thromb Res. 2012;129(6):736-42. doi: 10.1016/j.thromres.2011.11.047.
112. Brat DJ, Van Meir EG. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest. 2004;84(4):397-405.
113. Ornstein DL, Meehan KR, Zacharski LR. The coagulation system as a target for the treatment of human gliomas. Semin Thromb Hemost. 2002;28(1):19-28.
114. Milsom C, Magnus N, Meehan B, Al-Nedawi K, Garnier D, Rak J. Tissue factor and cancer stem cells: is there a linkage? Arterioscler Thromb Vasc Biol. 2009;29(12):2005-14. doi: 10.1161/ATVBAHA.108.177444.
115. Garnier D, Milsom C, Magnus N, Meehan B, Weitz J, Yu J, Rak J. Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb Res. 2010;125 Suppl 2:S44-50. doi: 10.1016/S0049-3848(10)70012-8.
116. Jo J, Schiff D, Purow B. Angiogenic inhibition in high-grade gliomas: past, present and future. Expert Rev Neurother. 2012;12(6):733-47. doi:10.1586/ern.12.53.
117. McCormack PL. Celecoxib: a review of its use for symptomatic relief in the treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. Drugs. 2011;71(18):2457-89. doi: 10.2165/11208240-000000000-00000. 118. Tive L. Celecoxib clinical profile. Rheumatology (Oxford). 2000;39 Suppl 2:21-8;
119. Scott WW, Levy M, Rickert KL, Madden CJ, Beshay JE, Sarode R. Assessment of Common Non-Steroidal Anti-inflammatory Medications by Whole Blood Aggregometry: A Clinical Evaluation for the Perioperative Setting. World Neurosurg. 2014 Mar 31.pii: S1878-8750(14)00371-4. doi: 10.1016/j.wneu.2014.03.043.
120. Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D, Zhou Y. Effect of celecoxib on cyclooxygenase-1-mediated prostacyclin synthesis and endothelium-dependent contraction in mouse arteries. Eur J Pharmacol. 2013;698(1-3):354-61. doi: 10.1016/j.ejphar.2012.10.040.
121. Sud’ina GF, Pushkareva MA, Shephard P, Klein T. Cyclooxygenase (COX) and 5-lipoxygenase (5-LOX) selectivity of COX inhibitors. Prostaglandins Leukot Essent Fatty Acids. 2008;78(2):99-108. doi: 10.1016/j.plefa.2007.12.006.
122. Germann B, Neuhaus W, Hofer-Warbinek R, Noe CR. Applying blood-brain barrier in vitro models to study the influence of drugs on endothelial cells--effects of selected COX-inhibitors. Pharmazie. 2008;63(4):303-7.
123. Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82(1-4):85-94.
124. Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9(2 Suppl):28-35.
125. Robison NJ, Campigotto F, Chi SN, Manley PE, Turner CD, Zimmerman MA, Chordas CA, Werger AM, Allen JC, Goldman S, Rubin JB, Isakoff MS, Pan WJ, Khatib ZA, Comito MA, Bendel AE, Pietrantonio JB, Kondrat L, Hubbs SM, Neuberg DS, Kieran MW. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr Blood Cancer. 2014;61(4):636-42. doi: 10.1002/pbc.24794.
126. Perroud HA, Rico MJ, Alasino CM, Queralt F, Mainetti LE, Pezzotto SM, Rozados VR, Scharovsky OG. Safety and therapeutic effect of metronomic chemotherapy with cyclophosphamide and celecoxib in advanced breast cancer patients. Future Oncol. 2013;9(3):451-62. doi: 10.2217/fon.12.196.
127. Chow LW, Tung SY, Ng TY, Im SA, Lee MH, Yip AY, Toi M, Glück S. Concurrent celecoxib with 5 fluorouracil/epirubicin/cyclophosphamide followed by docetaxel for stages II - III invasive breast cancer: the OOTR-N001 study. Expert Opin Investig Drugs. 2013;22(3):299-307. doi: 10.1517/13543784.2013.766715.
128. Sörenson S, Fohlin H, Lindgren A, Lindskog M, Bergman B, Sederholm C, Ek L, Lamberg K, Clinchy B. Predictive role of plasma vascular endothelial growth factor for the effect of celecoxib in advanced non-small cell lung cancer treated with chemotherapy. Eur J Cancer. 2013;49(1):115-20. doi:10.1016/j.ejca.2012.07.032.
129. Reyners AK, de Munck L, Erdkamp FL, Smit WM, Hoekman K, Lalisang RI, de Graaf H, Wymenga AN, Polee M, Hollema H, van Vugt MA, Schaapveld M, Willemse PH; DoCaCel Study Group. A randomized phase II study investigating the addition of the specific COX-2 inhibitor celecoxib to docetaxel plus carboplatin as first-line chemotherapy for stage IC to IV epithelial ovarian cancer, Fallopian tube or primary peritoneal carcinomas: the DoCaCel study. Ann Oncol. 2012;23(11):2896-902. doi: 10.1093/annonc/mds107.
130. Iyer R, Barrese AA 3rd, Parakh S, Parker CN, Tripp BC. Inhibition profiling of human carbonic anhydrase II by high-throughput screening of structurally diverse, biologically active compounds. J Biomol Screen. 2006;11(7):782-91.
131. Ivanov S, Liao SY, Ivanova A, Danilkovitch-Miagkova A, Tarasova N, Weirich G, Merrill MJ, Proescholdt MA, Oldfield EH, Lee J, Zavada J, Waheed A, Sly W, Lerman MI, Stanbridge EJ. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol. 2001;158(3):905-19.
132. Haapasalo J, Nordfors K, Järvelä S, Bragge H, Rantala I, Parkkila AK, Haapasalo H, Parkkila S. Carbonic anhydrase II in the endothelium of glial tumors: a potential target for therapy. Neuro Oncol. 2007;9(3):308-13.
133. Haapasalo J, Hilvo M, Nordfors K, Haapasalo H, Parkkila S, Hyrskyluoto A, Rantala I, Waheed A, Sly WS, Pastorekova S, Pastorek J, Parkkila AK. Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro Oncol. 2008;10(2):131-8. doi: 10.1215/15228517-2007-065.
134. Proescholdt MA, Merrill MJ, Stoerr EM, Lohmeier A, Pohl F, Brawanski A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol. 2012;14(11):1357-66. doi: 10.1093/neuonc/nos216.
135. Sathornsumetee S, Cao Y, Marcello JE, Herndon JE 2nd, McLendon RE, Desjardins A, Friedman HS, Dewhirst MW, Vredenburgh JJ, Rich JN. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 2008;26(2):271-8. doi: 10.1200/JCO.2007.13.3652.
136. Flynn JR, Wang L, Gillespie DL, Stoddard GJ, Reid JK, Owens J, Ellsworth GB, Salzman KL, Kinney AY, Jensen RL. Hypoxia-regulated protein expression, patient characteristics, and preoperative imaging as predictors of survival in adults with glioblastoma multiforme. Cancer. 2008;113(5):1032-42. doi: 10.1002/cncr.23678.
137. Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J, Kopacek J, Pastorekova S. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol. 2006;29(4):1025-33.
138. Das A, Banik NL, Ray SK. Modulatory effects of acetazolomide and dexamethasone on temozolomide-mediated apoptosis in human glioblastoma T98G and U87MG cells. Cancer Invest. 2008;26(4):352-8. doi: 10.1080/07357900701788080.
139. Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl. 1992;369:15-26.
140. Helander A, Johansson B. Inhibition of human erythrocyte and leukocyte aldehyde dehydrogenase activities by diethylthiocarbamic acid methyl ester. An in vivo metabolite of disulfiram. Biochem Pharmacol. 1989;38(13):2195-8.
141. Kast RE, Belda-Iniesta C. Suppressing glioblastoma stem cell function by aldehyde dehydrogenase inhibition with chloramphenicol or disulfiram as a new treatment adjunct: an hypothesis. Curr Stem Cell Res Ther. 2009;4(4):314-7.
142. Wang W, Darling JL. How could a drug used to treat alcoholism also be effective against glioblastoma? Expert Rev Anticancer Ther. 2013;13(3):239-41. doi: 10.1586/era.12.169.
143. Xia P. Surface markers of cancer stem cells in solid tumors. Curr Stem Cell Res Ther. 2014;9(2):102-11.
144. Singh S, Brocker C, Koppaka V, Chen Y, Jackson BC, Matsumoto A, Thompson DC, Vasiliou V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med. 2013;56:89-101. doi:10.1016/j.freeradbiomed.2012.11.010.
145. Cheriyan VT, Wang Y, Muthu M, Jamal S, Chen D, Yang H, Polin LA, Tarca AL, Pass HI, Dou QP, Sharma S, Wali A, Rishi AK. Disulfiram suppresses growth of the malignant pleural mesothelioma cells in part by inducing apoptosis. PLoS One.2014;9(4):e93711. doi: 10.1371/journal.pone.0093711.
146. Westhoff MA, Zhou S, Nonnenmacher L, Karpel-Massler G, Jennewein C, Schneider M, Halatsch ME, Carragher NO, Baumann B, Krause A, Simmet T, Bachem MG, Wirtz CR, Debatin KM. Inhibition of NF-κB signaling ablates the invasive phenotype of glioblastoma. Mol Cancer Res. 2013;11(12):1611-23. doi:10.1158/1541-7786.MCR-13-0435-T.
147. Duan L, Shen H, Zhao G, Yang R, Cai X, Zhang L, Jin C, Huang Y. Inhibitory effect of Disulfiram/copper complex on non-small cell lung cancer cells. Biochem Biophys Res Commun. 2014 Mar 19. pii: S0006-291X(14)00489-6. doi:10.1016/j.bbrc.2014.03.047.
148. Chiba T, Suzuki E, Yuki K, Zen Y, Oshima M, Miyagi S, Saraya A, Koide S, Motoyama T, Ogasawara S, Ooka Y, Tawada A, Nakatsura T, Hayashi T, Yamashita T, Kaneko S, Miyazaki M, Iwama A, Yokosuka O. Disulfiram eradicates tumor-initiating hepatocellular carcinoma cells in ROS-p38 MAPK pathway-dependent and –independent manners. PLoS One. 2014;9(1):e84807. doi: 10.1371/journal.pone.0084807.
149. Papaioannou M, Mylonas I, Kast RE, Brüning A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience 2014;1.1:21-29.
150. Dinnen RD, Mao Y, Qiu W, Cassai N, Slavkovich VN, Nichols G, Su GH, Brandt-Rauf P, Fine RL. Redirecting apoptosis to aponecrosis induces selective cytotoxicity to pancreatic cancer cells through increased ROS, decline in ATP levels, and VDAC. Mol Cancer Ther. 2013;12(12):2792-803. doi:10.1158/1535-7163.MCT-13-0234.
151. Ketola K, Kallioniemi O, Iljin K. Chemical biology drug sensitivity screen identifies sunitinib as synergistic agent with disulfiram in prostate cancer cells. PLoS One. 2012;7(12):e51470. doi: 10.1371/journal.pone.0051470.
152. Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL Jr. Disulfiram induces apoptosis in human melanoma cells: a redox-related process. Mol Cancer Ther. 2002;1(3):197-204.
153. Morrison BW, Doudican NA, Patel KR, Orlow SJ. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010;20(1):11-20. doi: 10.1097/CMR.0b013e328334131d.
154. Navrátilová J, Hankeová T, Beneš P, Šmarda J. Acidic pH of tumor microenvironment enhances cytotoxicity of the disulfiram/Cu2+ complex to breast and colon cancer cells. Chemotherapy. 2013;59(2):112-20. doi: 10.1159/000353915.
155. Huang Y, Ju B, Tian J, Liu F, Yu H, Xiao H, Liu X, Liu W, Yao Z, Hao Q. Ovarian cancer stem cell-specific gene expression profiling and targeted drug prescreening. Oncol Rep. 2014;31(3):1235-48. doi: 10.3892/or.2014.2976.
156. Kim SK, Kim H, Lee DH, Kim TS, Kim T, Chung C, Koh GY, Kim H, Lim DS. Reversing the intractable nature of pancreatic cancer by selectively targeting ALDH-high, therapy-resistant cancer cells. PLoS One. 2013 Oct 23;8(10):e78130. doi: 10.1371/journal.pone.0078130.
157. Liu P, Kumar IS, Brown S, Kannappan V, Tawari PE, Tang JZ, Jiang W, Armesilla AL, Darling JL, Wang W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer. 2013;109(7):1876-85. doi:10.1038/bjc.2013.534.
158. Robinson TJ, Pai M, Liu JC, Vizeacoumar F, Sun T, Egan SE, Datti A, Huang J, Zacksenhaus E. High-throughput screen identifies disulfiram as a potential therapeutic for triple-negative breast cancer cells: interaction with IQ motif-containing factors. Cell Cycle. 2013;12(18):3013-24. doi:10.4161/cc.26063.
159. Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, Guichet PO, Bian X, Armesilla AL, Darling JL, Wang W. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer. 2012;107(9):1488-97. doi:10.1038/bjc.2012.442.
160. Dalla Pozza E, Donadelli M, Costanzo C, Zaniboni T, Dando I, Franchini M, Arpicco S, Scarpa A, Palmieri M. Gemcitabine response in pancreatic adenocarcinoma cells is synergistically enhanced by dithiocarbamate derivatives. Free Radic Biol Med. 2011;50 (8):926-33. doi:10.1016/j.freeradbiomed.2011.01.001.
161. Guo X, Xu B, Pandey S, Goessl E, Brown J, Armesilla AL, Darling JL, Wang W. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010;290(1):104-13. doi: 10.1016/j.canlet.2009.09.002.
162. Rae C, Tesson M, Babich JW, Boyd M, Sorensen A, Mairs RJ. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. J Nucl Med. 2013;54(6):953-60. doi: 10.2967/jnumed.112.113324.
163. Paranjpe A, Zhang R, Ali-Osman F, Bobustuc GC, Srivenugopal KS. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis. 2014;35(3):692-702. doi: 10.1093/carcin/bgt366.
164. Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA,Yegnasubramanian S, Carducci MA. Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis. 2013;16(4):357-61. doi: 10.1038/pcan.2013.28.
165. Lestner J, Hope WW. Itraconazole: an update on pharmacology and clinical use for treatment of invasive and allergic fungal infections. Expert Opin Drug Metab Toxicol. 2013;9(7):911-26. doi:10.1517/17425255.2013.794785.
166. Caputo R. Itraconazole (Sporanox) in superficial and systemic fungal infections. Expert Rev Anti Infect Ther. 2003;1(4):531-42.
167. Kim JS, Cheong JW, Kim YK, Park J, Mun YC, Kang HJ, Yi HG, Lee JH, Kim YS, Ryoo HM, Kim SH, Kim HY, Kim JY, Lee DG, Kim HG, Kim H, Joo YD, Min YH. The relationship between the success rate of empirical antifungal therapy with intravenous itraconazole and clinical parameters, including plasma levels of itraconazole, in immunocompromised patients receiving itraconazole oral solution as prophylaxis: a multicenter, prospective, open-label, observational study in Korea. Ann Hematol. 2014;93(1):33-42. doi: 10.1007/s00277-013-1826-x.
168. Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R, Smith R, Aftab BT, Huang P, Liu JO. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J Thorac Oncol. 2013;8(5):619-23. doi: 10.1097/JTO.0b013e31828c3950.
169. Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC, King S, Frost A, Ajiboye AS, Zhao M, Mendonca J, Kachhap SK, Rudek MA, Carducci MA. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist. 2013;18(2):163-73. doi: 10.1634/theoncologist.2012-314.
170. You M, Varona-Santos J, Singh S, Robbins DJ, Savaraj N, Nguyen DM. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg. 2014;147(1):508-16. doi:10.1016/j.jtcvs.2013.08.035.
171. Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, Gardner D, Reya T, Liu JO, Epstein EH, Stevens DA, Beachy PA. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388-99. doi: 10.1016/j.ccr.2010.02.027.
172. Kanda Y, Kami M, Matsuyama T, Mitani K, Chiba S, Yazaki Y, Hirai H. Plasma concentration of itraconazole in patients receiving chemotherapy for hematological malignancies: the effect of famotidine on the absorption of itraconazole. Hematol Oncol. 1998;16(1):33-7.
173. Khaliq Y, Gallicano K, Venance S, Kravcik S, Cameron DW. Effect of ketoconazole on ritonavir and saquinavir concentrations in plasma and cerebrospinal fluid from patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 2000;68(6):637-46.
174. Wang EJ, Lew K, Casciano CN, Clement RP, Johnson WW. Interaction of common azole antifungals with P glycoprotein. Antimicrob Agents Chemother. 2002;46(1):160-5.
175. Zolnerciks JK, Booth-Genthe CL, Gupta A, Harris J, Unadkat JD. Substrate- and species-dependent inhibition of P-glycoprotein-mediated transport: implications for predicting in vivo drug interactions. J Pharm Sci. 2011;100(8):3055-61. doi:10.1002/jps.22566.
176. Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ Jr, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2(4):263-70.
177. Nacev BA, Liu JO. Synergistic inhibition of endothelial cell proliferation, tube formation, and sprouting by cyclosporin A and itraconazole. PLoS One. 2011;6(9):e24793. doi: 10.1371/journal.pone.0024793.
178. Nacev BA, Grassi P, Dell A, Haslam SM, Liu JO. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J Biol Chem. 2011;286(51):44045-56. doi: 10.1074/jbc.M111.278754.
179. Xu J, Dang Y, Ren YR, Liu JO. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci U S A. 2010;107(10):4764-9. doi: 10.1073/pnas.0910872107.
180. Liu R, Li J, Zhang T, Zou L, Chen Y, Wang K, Lei Y, Yuan K, Li Y, Lan J, Cheng L, Xie N, Xiang R, Nice EC, Huang C, Wei Y. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy. 2014 May 15;10(7). [Epub ahead of print] PubMed PMID:24905460.
181. Prentice AG, Warnock DW, Johnson SA, Phillips MJ, Oliver DA. Multiple dose pharmacokinetics of an oral solution of itraconazole in autologous bone marrow transplant recipients. J Antimicrob Chemother. 1994;34(2):247-52.
182. Vreugdenhil G, Raemaekers JM, van Dijke BJ, de Pauw BE. Itraconazole and multidrug resistance: possible effects on remission rate and disease-free survival in acute leukemia. Ann Hematol. 1993;67(3):107-9.
183. Gupta S, Kim J, Gollapudi S. Reversal of daunorubicin resistance in P388/ADR cells by itraconazole. J Clin Invest. 1991;87(4):1467-9.
184. Comba A, Pasqualini ME. Primers on molecular pathways - lipoxygenases: their role as an oncogenic pathway in pancreatic cancer. Pancreatology. 2009;9(6):724-8. doi: 10.1159/000235623.
185. Boado RJ, Pardridge WM, Vinters HV, Black KL. Differential expression of arachidonate 5-lipoxygenase transcripts in human brain tumors: evidence for the expression of a multitranscript family. Proc Natl Acad Sci U S A. 1992;89(19):9044-8
186. Lim JY, Oh JH, Jung JR, Kim SM, Ryu CH, Kim HT, Jeun SS. MK886-induced apoptosis depends on the 5-LO expression level in human malignant glioma cells. J Neurooncol. 2010;97(3):339-46. doi: 10.1007/s11060-009-0036-9.
187. Nathoo N, Prayson RA, Bondar J, Vargo L, Arrigain S, Mascha EJ, Suh JH, Barnett GH, Golubic M. Increased expression of 5-lipoxygenase in high-grade astrocytomas. Neurosurgery. 2006;58(2):347-54.
188. Zhang L, Zhang WP, Hu H, Wang ML, Sheng WW, Yao HT, Ding W, Chen Z, Wei EQ. Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma. Neuropathology. 2006;26(2):99-106.
189. Ishii K, Zaitsu M, Yonemitsu N, Kan Y, Hamasaki Y, Matsuo M. 5-lipoxygenase pathway promotes cell proliferation in human glioma cell lines. Clin Neuropathol. 2009;28(6):445-52.
190. Gorlia T, Stupp R, Brandes AA, Rampling RR, Fumoleau P, Dittrich C, Campone MM, Twelves CC, Raymond E, Hegi ME, Lacombe D, van den Bent MJ. New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer. 2012;48(8):1176-84. doi:10.1016/j.ejca.2012.02.004.
192. Steinhilber D, Jaschonek K, Knospe J, Morof O, Roth HJ. Effects of novel antifungal azole derivatives on the 5-lipoxygenase and cyclooxygenase pathway. Arzneimittelforschung. 1990;40(11):1260-3.
193. Jaschonek K, Steinhilber D, Einsele H, Ehninger G, Roth HJ. 5-Lipoxygenase inhibition by antifungal azole derivatives: new tools for immunosuppression? Eicosanoids. 1989;2(3):189-90.
194. Morin P Jr, Ferguson D, LeBlanc LM, Hébert MJ, Paré AF, Jean-François J, Surette nvbnvbnME, Touaibia M, Cuperlovic-Culf M. NMR metabolomics analysis of the effects of 5-lipoxygenase inhibitors on metabolism in glioblastomas. J Proteome Res. 2013;12(5):2165-76. doi: 10.1021/pr400026q.
195. Omahen DA. Augmentation of chemotherapy-triggered glioma cell apoptosis by blockade of arachidonic acid metabolism--the potential role of ceramide accumulation. Med Hypotheses. 2011;77(5):726-33. doi:10.1016/j.mehy.2011.07.025.
196. Jiang P, Mukthavavam R, Chao Y, Bharati IS, Fogal V, Pastorino S, Cong X, Nomura N, Gallagher M, Abbasi T, Vali S, Pingle SC, Makale M, Kesari S. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J Transl Med. 2014;12:13. doi:10.1186/1479-5876-12-13.
197. Gottlieb A, Altman RB. Integrating Systems Biology Sources Illuminates Drug Action. Clin Pharmacol Ther. 2014 Feb 27. doi: 10.1038/clpt.2014.51. [Epub ahead of print] PMID: 24577151.
198. Hernán Pérez de la Ossa D, Lorente M, Gil-Alegre ME, Torres S, García-Taboada E, Aberturas Mdel R, Molpeceres J, Velasco G, Torres-Suárez AI. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS One. 2013;8(1):e54795. doi:10.1371/journal.pone.0054795.
199. Torres S, Lorente M, Rodríguez-Fornés F, Hernández-Tiedra S, Salazar M, García-Taboada E, Barcia J, Guzmán M, Velasco G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther. 2011;10(1):90-103. doi: 10.1158/1535-7163.MCT-10-0688.
200. Massi P, Valenti M, Solinas M, Parolaro D. Molecular mechanisms involved in the antitumor activity of cannabinoids on gliomas: role for oxidative stress. Cancers (Basel). 2010;2(2):1013-26. doi: 10.3390/cancers2021013.
201. Choo LY, Riendeau D. Disulfiram is a potent inhibitor of rat 5-lipoxygenase activity. Can J Physiol Pharmacol. 1987;65(12):2503-6.
202. Riendeau D, Guay J, Foster A, Wolfe S, Chan CC. Inhibition of leukotriene B4 biosynthesis by disulfiram and A-64077 during carrageenan-induced pleurisy in the rat. Gen Pharmacol. 1991;22(2):371-4.
203. Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50(1):161-78. doi: 10.1042/bse0500161.
204. Kurosawa M, Okabe M, Hara N, Kawamura K, Suzuki S, Sakurada K, Asaka M. Reversal effect of itraconazole on adriamycin and etoposide resistance in human leukemia cells. Ann Hematol. 1996;72(1):17-21.
205. Bendayan R, Ronaldson PT, Gingras D, Bendayan M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54(10):1159-67.
206. Tapaninen T, Backman JT, Kurkinen KJ, Neuvonen PJ, Niemi M. Itraconazole, a P-glycoprotein and CYP3A4 inhibitor, markedly raises the plasma concentrations and enhances the renin-inhibiting effect of aliskiren. J Clin Pharmacol. 2011;51(3):359-67. doi: 10.1177/0091270010365885.
207. Ni L, Yu X, Yu Q, Chen X, Jia L. Effects of cyclosporine A and itraconazole on permeability, biliary excretion and pharmacokinetics of amlodipine. Drug Metab Lett. 2008;2(3):163-8.
208. Tsubamoto H, Sonoda T, Yamasaki M, Inoue K. Impact of combination chemotherapy with itraconazole on survival of patients with refractory ovarian cancer. Anticancer Res. 2014;34(5):2481-7.
209. Hoetelmans RM, Meenhorst PL, Mulder JW, Burger DM, Koks CH, Beijnen JH. Clinical pharmacology of HIV protease inhibitors: focus on saquinavir, indinavir, and ritonavir. Pharm World Sci. 1997;19(4):159-75.
210. Ahluwalia MS, Patton C, Stevens G, Tekautz T, Angelov L, Vogelbaum MA, Weil RJ, Chao S, Elson P, Suh JH, Barnett GH, Peereboom DM. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J Neurooncol. 2011;102(2):317-21. doi: 10.1007/s11060-010-0325-3.
211. Moawad EY. Identifying the optimal dose of ritonavir in the treatment of malignancies. Metab Brain Dis. 2014;29(2):533-40. doi:10.1007/s11011-013-9448-5.
212. Batchu RB, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, Kung ST(7), Sanka RS, Puttagunta US, Weaver DW, Gruber SA. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals (Basel). 2014;7(1):46-57. doi: 10.3390/ph7010046.
213. Sato A, Asano T, Ito K, Asano T. 17-Allylamino-17-demethoxygeldanamycin and ritonavir inhibit renal cancer growth by inhibiting the expression of heat shock factor-1. Int J Oncol. 2012;41(1):46-52. doi: 10.3892/ijo.2012.1419.
214. Barillari G, Iovane A, Bacigalupo I, Palladino C, Bellino S, Leone P, Monini P, Ensoli B. Ritonavir or saquinavir impairs the invasion of cervical intraepithelial neoplasia cells via a reduction of MMP expression and activity. AIDS. 2012;26(8):909-19. doi: 10.1097/QAD.0b013e328351f7a5.
215. Srirangam A, Milani M, Mitra R, Guo Z, Rodriguez M, Kathuria H, Fukuda S, Rizzardi A, Schmechel S, Skalnik DG, Pelus LM, Potter DA. The human immunodeficiency virus protease inhibitor ritonavir inhibits lung cancer cells, in part, by inhibition of survivin. J Thorac Oncol. 2011;6(4):661-70. doi: 10.1097/JTO.0b013e31820c9e3c.
216. Bierman WF, Scheffer GL, Schoonderwoerd A, Jansen G, van Agtmael MA, Danner SA, Scheper RJ. Protease inhibitors atazanavir, lopinavir and ritonavir are potent blockers, but poor substrates, of ABC transporters in a broad panel of ABC transporter-overexpressing cell lines. J Antimicrob Chemother. 2010;65(8):1672-80. doi: 10.1093/jac/dkq209.
217. Kumar S, Bryant CS, Chamala S, Qazi A, Seward S, Pal J, Steffes CP, Weaver DW, Morris R, Malone JM, Shammas MA, Prasad M, Batchu RB. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. 2009;8:26. doi: 10.1186/1476-4598-8-26.
218. Kraus M, Müller-Ide H, Rückrich T, Bader J, Overkleeft H, Driessen C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk Res. 2014;38(3):383-92.doi: 10.1016/j.leukres.2013.12.017.
219. Srirangam A, Mitra R, Wang M, Gorski JC, Badve S, Baldridge L, Hamilton J, Kishimoto H, Hawes J, Li L, Orschell CM, Srour EF, Blum JS, Donner D, Sledge GW, Nakshatri H, Potter DA. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12(6):1883-96
220. Olson DP, Scadden DT, D’Aquila RT, De Pasquale MP. The protease inhibitor ritonavir inhibits the functional activity of the multidrug resistance related-protein 1 (MRP-1). AIDS. 2002;16(13):1743-7.
221. Perloff MD, Von Moltke LL, Marchand JE, Greenblatt DJ. Ritonavir induces P-glycoprotein expression, multidrug resistance-associated protein (MRP1) expression, and drug transporter-mediated activity in a human intestinal cell line. J Pharm Sci. 2001;90(11):1829-37.
222. Washington CB, Duran GE, Man MC, Sikic BI, Blaschke TF. Interaction of anti-HIV protease inhibitors with the multidrug transporter P-glycoprotein (P-gp) in human cultured cells. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19(3):203-9.
223. Hashiguchi Y, Hamada A, Shinohara T, Tsuchiya K, Jono H, Saito H. Role of P-glycoprotein in the efflux of raltegravir from human intestinal cells and CD4+ T-cells as an interaction target for anti-HIV agents. Biochem Biophys Res Commun. 2013;439(2):221-7. doi: 10.1016/j.bbrc.2013.08.054.
224. Gupta A, Zhang Y, Unadkat JD, Mao Q. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther. 2004;310(1):334-41.
225. Laurent N, de Boüard S, Guillamo JS, Christov C, Zini R, Jouault H, Andre P, Lotteau V, Peschanski M. Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther. 2004;3(2):129-36.
226. Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M, Galanos C, Niedermann G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002;62(23):6901-8.
227. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M, Hollander MC, Kawabata S, Tsokos M, Figg WD, Steeg PS, Dennis PA. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183-94.
228. Zha W, Wang G, Xu W, Liu X, Wang Y, Zha BS, Shi J, Zhao Q, Gerk PM, Studer E, Hylemon PB, Pandak WM Jr, Zhou H. Inhibition of P-glycoprotein by HIV protease inhibitors increases intracellular accumulation of berberine in murine and human macrophages. PLoS One. 2013;8(1):e54349. doi: 10.1371/journal.pone.0054349.
229. Fukushima K, Kobuchi S, Mizuhara K, Aoyama H, Takada K, Sugioka N. Time-dependent interaction of ritonavir in chronic use: the power balance between inhibition and induction of P-glycoprotein and cytochrome P450 3A. J Pharm Sci. 2013;102(6):2044-55. doi: 10.1002/jps.23545.
230. Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol Cancer Ther. 2004;3(4):473-9.
231. Bardelmeijer HA, Ouwehand M, Buckle T, Huisman MT, Schellens JH, Beijnen JH, van Tellingen O. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002;62(21):6158-64.
232. Zhong DS, Lu XH, Conklin BS, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir induces cytotoxicity of human endothelial cells. Arterioscler Thromb Vasc Biol. 2002;22(10):1560-6.
233. Mühl H, Paulukat J, Höfler S, Hellmuth M, Franzen R, Pfeilschifter J. The HIV protease inhibitor ritonavir synergizes with butyrate for induction of apoptotic cell death and mediates expression of heme oxygenase-1 in DLD-1 colon carcinoma cells. Br J Pharmacol. 2004;143(7):890-8.
234. Maggiorella L, Wen B, Frascogna V, Opolon P, Bourhis J, Deutsch E. Combined radiation sensitizing and anti-angiogenic effects of ionizing radiation and the protease inhibitor ritonavir in a head and neck carcinoma model. Anticancer Res. 2005;25(6B):4357-62.
235. Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol. 2009;24(2):43-60.
236. Torta R, Siri I, Caldera P. Sertraline effectiveness and safety in depressed oncological patients. Support Care Cancer. 2008;16(1):83-91.
237. Caudill JS, Brown PD, Cerhan JH, Rummans TA. Selective serotonin reuptake inhibitors, glioblastoma multiforme, and impact on toxicities and overall survival: the mayo clinic experience. Am J Clin Oncol. 2011;34(4):385-7. doi:10.1097/COC.0b013e3181e8461a.
238. Lin KL, Chi CC, Lu T, Tseng LL, Wang JL, Lu YC, Jan CR. Effect of sertraline on Ca2+ and viability of human MG63 osteosarcoma cells. Drug Chem Toxicol. 2013;36(2):231-40. doi:10.3109/01480545.2012.710625.
239. Huang JK, Chang HT, Chou CT, Shu SS, Kuo CC, Tsai JY, Liao WC, Wang JL, Lin KL, Lu YC, Chen IS, Liu SI, Ho CM, Jan CR. The mechanism of sertraline-induced Ca(2+ rise in human PC3 prostate cancer cells. Basic Clin Pharmacol Toxicol. 2011;109(2):103-10. doi:10.1111/j.1742-7843.2011.00690.x.
240. Chien JM, Chou CT, Pan CC, Kuo CC, Tsai JY, Liao WC, Kuo DH, Shieh P, Ho CM, Chu ST, Su HH, Chi CC, Jan CR. The mechanism of sertraline-induced [Ca2+]i rise in human OC2 oral cancer cells. Hum Exp Toxicol. 2011;30(10):1635-43. doi:10.1177/0960327110396523.
241. Gil-Ad I, Zolokov A, Lomnitski L, Taler M, Bar M, Luria D, Ram E, Weizman A. Evaluation of the potential anti-cancer activity of the antidepressant sertraline in human colon cancer cell lines and in colorectal cancer-xenografted mice. Int J Oncol. 2008;33(2):277-86.
242. Schmidt L, Kling T, Monsefi N, Olsson M, Hansson C, Baskaran S, Lundgren B, Martens U, Häggblad M, Westermark B, Forsberg Nilsson K, Uhrbom L, Karlsson-Lindahl L, Gerlee P, Nelander S. Comparative drug pair screening across multiple glioblastoma cell lines reveals novel drug-drug interactions. Neuro Oncol. 2013;15(11):1469-78. doi: 10.1093/neuonc/not111.
243. Acunzo J, Baylot V, So A, Rocchi P. TCTP as therapeutic target in cancers. Cancer Treat Rev. 2014 Mar 2. pii: S0305-7372(14)00027-9. doi:10.1016/j.ctrv.2014.02.007.
244. Gnanasekar M, Dakshinamoorthy G, Ramaswamy K. Translationally controlled tumor protein is a novel heat shock protein with chaperone-like activity. Biochem Biophys Res Commun. 2009;386(2):333-7. doi:10.1016/j.bbrc.2009.06.028.
244. Gnanasekar M, Dakshinamoorthy G, Ramaswamy K. Translationally controlled tumor protein is a novel heat shock protein with chaperone-like activity. Biochem Biophys Res Commun. 2009;386(2):333-7. doi:10.1016/j.bbrc.2009.06.028.
245. Amson R, Karp JE, Telerman A. Lessons from tumor reversion for cancer treatment. Curr Opin Oncol. 2013;25(1):59-65. doi:10.1097/CCO.0b013e32835b7d21.
246. Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, Tosoni D, Colaluca I, Viale G, Rodrigues-Ferreira S, Wynendaele J, Chaloin O, Hoebeke J, Marine JC, Di Fiore PP, Telerman A. Reciprocal repression between P53 and TCTP. Nat Med. 2011;18(1):91-9. doi: 10.1038/nm.2546.
247. Tuynder M, Fiucci G, Prieur S, Lespagnol A, Géant A, Beaucourt S, Duflaut D, Besse S, Susini L, Cavarelli J, Moras D, Amson R, Telerman A. Translationally controlled tumor protein is a target of tumor reversion. Proc Natl Acad Sci U SA. 2004;101(43):15364-9.
248. Rho SB, Lee JH, Park MS, Byun HJ, Kang S, Seo SS, Kim JY, Park SY. Anti-apoptotic protein TCTP controls the stability of the tumor suppressor p53. FEBS Lett. 2011;585(1):29-35. doi: 10.1016/j.febslet.2010.11.014.
249. Brambilla E, Brambilla C. p53 and lung cancer. Pathol Biol (Paris). 1997;45(10):852-63.
250. Gu X, Yao L, Ma G, Cui L, Li Y, Liang W, Zhao B, Li K. TCTP promotes glioma cell proliferation in vitro and in vivo via enhanced β-catenin/TCF-4 transcription. Neuro Oncol. 2014;16(2):217-27. doi:10.1093/neuonc/not194.
251. Miao X, Chen YB, Xu SL, Zhao T, Liu JY, Li YR, Wang J, Zhang J, Guo GZ. TCTP overexpression is associated with the development and progression of glioma. Tumour Biol. 2013;34(6):3357-61. doi:10.1007/s13277-013-0906-9.
252. Tzadok S, Beery E, Israeli M, Uziel O, Lahav M, Fenig E, Gil-Ad I, Weizman A, Nordenberg J. In vitro novel combinations of psychotropics and anti-cancer modalities in U87 human glioblastoma cells. Int J Oncol. 2010;37(4):1043-51.
253. Lin CJ, Robert F, Sukarieh R, Michnick S, Pelletier J. The antidepressant sertraline inhibits translation initiation by curtailing mammalian target of rapamycin signaling. Cancer Res. 2010;70(8):3199-208. doi:10.1158/0008-5472.CAN-09-4072.
254. Amit BH, Gil-Ad I, Taler M, Bar M, Zolokov A, Weizman A. Proapoptotic and chemosensitizing effects of selective serotonin reuptake inhibitors on T cell lymphoma/leukemia (Jurkat) in vitro. Eur Neuropsychopharmacol. 2009;19(10):726-34. doi:10.1016/j.euroneuro.2009.06.003.
255. Reddy KK, Lefkove B, Chen LB, Govindarajan B, Carracedo A, Velasco G, Carrillo CO, Bhandarkar SS, Owens MJ, Mechta-Grigoriou F, Arbiser JL. The antidepressant sertraline downregulates Akt and has activity against melanoma cells. Pigment Cell Melanoma Res. 2008;21(4):451-6. doi:10.1111/j.1755-148X.2008.00481.x.
256. Mukhopadhyay S, Rohatgi R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin Cell Dev Biol. 2014 May 17. pii: S1084-9521(14)00129-3. doi:10.1016/j.semcdb.2014.05.002.
257. Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014 Feb 24. pii: S0305-7372(14)00023-1. doi:10.1016/j.ctrv.2014.02.003.
258. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19(11):1410-22. doi:10.1038/nm.3389.
259, Natsume A, Kinjo S, Yuki K, Kato T, Ohno M, Motomura K, Iwami K, Wakabayashi T. Glioma-initiating cells and molecular pathology: implications for therapy. Brain Tumor Pathol. 2011;28(1):1-12. doi: 10.1007/s10014-010-0011-3.
260. Takezaki T, Hide T, Takanaga H, Nakamura H, Kuratsu J, Kondo T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011;102(7):1306-12. doi: 10.1111/j.1349-7006.2011.01943.x.
261. Sarkisian MR, Siebzehnrubl D, Hoang-Minh L, Deleyrolle L, Silver DJ, Siebzehnrubl FA, Guadiana SM, Srivinasan G, Semple-Rowland S, Harrison JK, Steindler DA, Reynolds BA. Detection of primary cilia in human glioblastoma. J Neurooncol. 2014;117(1):15-24. doi: 10.1007/s11060-013-1340-y.
262. Moser JJ, Fritzler MJ, Rattner JB. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer. 2009;9:448. doi:10.1186/1471-2407-9-448.
263. Mimeault M, Batra SK. Complex oncogenic signaling networks regulate brain tumor-initiating cells and their progenies: pivotal roles of wild-type EGFR, EGFRvIII mutant and hedgehog cascades and novel multitargeted therapies. Brain Pathol. 2011;21 (5):479-500. doi: 10.1111/j.1750-3639.2011.00505.x.
264. Meiss F, Zeiser R. Vismodegib. Recent Results Cancer Res. 2014;201:405-17. doi: 10.1007/978-3-642-54490-3_25.
265. Amin SH, Motamedi KK, Ochsner MC, Song TE, Hybarger CP. Mechanisms and efficacy of vismodegib in the treatment of basal cell carcinoma. Discov Med. 2013;16(89):229-32.
266. Xie J, Bartels CM, Barton SW, Gu D. Targeting hedgehog signaling in cancer: research and clinical developments. Onco Targets Ther. 2013;6:1425-1435.
267. Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K, Fu T, Gilliam A, Molgo M, Beachy PA, Tang JY. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. 2014;32(8):745-51. doi: 10.1200/JCO.2013.49.9525.
268. Yang Y, Shi J, Tolomelli G, Xu H, Xia J, Wang H, Zhou W, Zhou Y, Das S, Gu Z, Levasseur D, Zhan F, Tricot G. RARα2 expression confers myeloma stem cell features. Blood. 2013;122(8):1437-47. doi: 10.1182/blood-2013-02-482919.
269. Aftab BT, Dobromilskaya I, Liu JO, Rudin CM. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011;71(21):6764-72. doi:10.1158/0008-5472.CAN-11-0691.
270. Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R, Smith R, Aftab BT, Huang P, Liu JO. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J Thorac Oncol. 2013;8(5):619-23.doi: 10.1097/JTO.0b013e31828c3950.
271. Suzman DL, Antonarakis ES. High-dose itraconazole as a noncastrating therapy for a patient with biochemically recurrent prostate cancer. Clin Genitourin Cancer. 2014;12(2):e51-3. doi: 10.1016/j.clgc.2013.11.015.
272. Tenedieva VD, Kulikovsky VP, Lyamin PV, Nepomnyaschi VP. The CSF aldosterone in brain tumors with brain edema. Acta Neurochir Suppl (Wien). 1994;60:390-4.
273. Grossmann C, Gekle M. New aspects of rapid aldosterone signaling. Mol Cell Endocrinol. 2009;308(1-2):53-62. doi: 10.1016/j.mce.2009.02.005.
274. Juillerat-Jeanneret L, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P, Gasc JM. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer. 2004;90(5):1059-68.
277. Ganesh R, Marks DJ, Sales K, Winslet MC, Seifalian M. Cyclooxygenase/lipoxygenase shunting lowers the anti-cancer effect of cyclooxygenase-2 inhibition in colorectal cancer cells. World J Surg Oncol. 2012;10:200. doi: 10.1186/1477-7819-10-200.
278. Duffield-Lillico AJ, Boyle JO, Zhou XK, Ghosh A, Butala GS, Subbaramaiah K, Newman RA, Morrow JD, Milne GL, Dannenberg AJ. Levels of prostaglandin E metabolite and leukotriene E(4) are increased in the urine of smokers: evidence that celecoxib shunts arachidonic acid into the 5-lipoxygenase pathway. Cancer Prev Res (Phila). 2009;2(4):322-9. doi: 10.1158/1940-6207.CAPR-09-0005.
279. Martel-Pelletier J, Mineau F, Fahmi H, Laufer S, Reboul P, Boileau C, Lavigne M, Pelletier JP. Regulation of the expression of 5-lipoxygenase-activating protein/5-lipoxygenase and the synthesis of leukotriene B(4) in osteoarthritic chondrocytes: role of transforming growth factor beta and eicosanoids. Arthritis Rheum. 2004;50(12):3925-33.
280. Maier TJ, Tausch L, Hoernig M, Coste O, Schmidt R, Angioni C, Metzner J, Groesch S, Pergola C, Steinhilber D, Werz O, Geisslinger G. Celecoxib inhibits 5-lipoxygenase. Biochem Pharmacol. 2008;76(7):862-72. doi: 10.1016/j.bcp.2008.07.009.
281. Park SW, Heo DS, Sung MW. The shunting of arachidonic acid metabolism to 5-lipoxygenase and cytochrome p450 epoxygenase antagonizes the anti-cancer effect of cyclooxygenase-2 inhibition in head and neck cancer cells. Cell Oncol (Dordr). 2012;35 (1):1-8. doi: 10.1007/s13402-011-0051-7.
282. Mohebati A, Milne GL, Zhou XK, Duffield-Lillico AJ, Boyle JO, Knutson A, Bosworth BP, Kingsley PJ, Marnett LJ, Brown PH, Akpa EG, Szabo E, Dannenberg AJ. Effect of zileuton and celecoxib on urinary LTE4 and PGE-M levels in smokers. Cancer Prev Res (Phila). 2013;6(7):646-55. doi:10.1158/1940-6207.CAPR-13-0083.
283. Kwak HJ, Park KM, Choi HE, Lim HJ, Park JH, Park HY. The cardioprotective effects of zileuton, a 5-lipoxygenase inhibitor, are mediated by COX-2 via activation of PKC delta. Cell Signal. 2010;22(1):80-7. doi:10.1016/j.cellsig.2009.09.014.
284. Angus BJ, Thaiaporn I, Chanthapadith K, Suputtamongkol Y, White NJ. Oral artesunate dose-response relationship in acute falciparum malaria. Antimicrob Agents Chemother. 2002;46:778–82.
285. Gundersen S, Lote K, Hannisdal E. Prognostic factors for glioblastoma multiforme--development of a prognostic index. Acta Oncol. 1996;35 Suppl 8:123-7.
286. Löwenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol Med. 2007 ;13(4):158-63.
287. Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, Dietz AB. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12(7):631-44. doi: 10.1093/neuonc/noq001.
288. Das A, Banik NL, Patel SJ, Ray SK. Dexamethasone protected human glioblastoma U87MG cells from temozolomide induced apoptosis by maintaining Bax:Bcl-2 ratio and preventing proteolytic activities.Mol Cancer. 2004;3(1):36.
289. Sur P, Sribnick EA, Patel SJ, Ray SK, Banik NL. Dexamethasone decreases temozolomide-induced apoptosis in human gliobastoma T98G cells. Glia. 2005;50(2):160-7.
290. Di Cristofori A, Carrabba G, Lanfranchi G, Menghetti C, Rampini P, Caroli M. Continuous tamoxifen and dose-dense temozolomide in recurrent glioblastoma. Anticancer Res. 2013;33(8):3383-9.
291. Mizumoto M, Okumura T, Ishikawa E, Yamamoto T, Takano S, Matsumura A, Oshiro Y, Ishikawa H, Sakurai H, Tsuboi K. Reirradiation for recurrent malignant brain tumor with radiotherapy or proton beam therapy. Technical considerations based on experience at a single institution. Strahlenther Onkol. 2013;189(8):656-63. doi: 10.1007/s00066-013-0390-6.
292. Lassen U, Sorensen M, Gaziel TB, Hasselbalch B, Poulsen HS. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer Res. 2013;33(4):1657-60.
293. Sloan AE, Ahluwalia MS, Valerio-Pascua J, Manjila S, Torchia MG, Jones SE, Sunshine JL, Phillips M, Griswold MA, Clampitt M, Brewer C, Jochum J, McGraw MV, Diorio D, Ditz G, Barnett GH. Results of the NeuroBlate System first-in-humans Phase I clinical trial for recurrent glioblastoma: clinical article. J Neurosurg. 2013;118(6):1202-19. doi: 10.3171/2013.1.JNS1291.
294. Norden AD, Lesser GJ, Drappatz J, Ligon KL, Hammond SN, Lee EQ, Reardon DR, Fadul CE, Plotkin SR, Batchelor TT, Zhu JJ, Beroukhim R, Muzikansky A, Doherty L, Lafrankie D, Smith K, Tafoya V, Lis R, Stack EC, Rosenfeld MR, Wen PY. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro Oncol. 2013;15(7):930-5. doi: 10.1093/neuonc/not040.
295. Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, Nabors LB, Rosenfeld MR, Mikkelsen T, Grossman SA; New Approaches to Brain Tumor Therapy Consortium. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013;15(4):490-6. doi: 10.1093/neuonc/nos322.
296. Karavasilis V, Kotoula V, Pentheroudakis G, Televantou D, Lambaki S, Chrisafi S, Bobos M, Fountzilas G. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. J Neurol. 2013;260(6):1469-80. doi: 10.1007/s00415-012-6812-z.
297. Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neurooncol. 2013;111(2):205-12. doi: 10.1007/s11060-012-1009-y.
298. D’Alessandris QG, Montano N, Cenci T, Martini M, Lauretti L, Bianchi F, Larocca LM, Maira G, Fernandez E, Pallini R. Targeted therapy with bevacizumab and erlotinib tailored to the molecular profile of patients with recurrent glioblastoma. Preliminary experience. Acta Neurochir (Wien). 2013;155(1):33-40. doi: 10.1007/s00701-012-1536-5.
299. Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, Fine HA. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol. 2013;111(1):41-8. doi: 10.1007/s11060-012-0988-z.
300. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, Butowski N, Chang SM, Clarke J, Berger MS, McDermott MW, Prados MD, Parsa AT. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19(1):205-14. doi: 10.1158/1078-0432.CCR-11-3358.
301. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883-92. doi: 10.1056/NEJMoa1113205.
302. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72(19):4875-82. doi: 10.1158/0008-5472.CAN-12-2217.
303. Zhang XC, Xu C, Mitchell RM, Zhang B, Zhao D, Li Y, Huang X, Fan W, Wang H, Lerma LA, Upton MP, Hay A, Méndez E, Zhao LP. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia. 2013;15(12):1371-8.
304. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59(1):168-79. doi: 10.1373/clinchem.2012.184655.
305. Yang T, Rycaj K, Liu ZM, Tang DG. Cancer Stem Cells: Constantly Evolving and Functionally Heterogeneous Therapeutic Targets. Cancer Res. 2014 May 2. [Epub ahead of print] PubMed PMID: 24713433.
306. Joy A, Ramesh A, Smirnov I, Reiser M, Misra A, Shapiro WR, Mills GB, Kim S, Feuerstein BG. AKT Pathway Genes Define 5 Prognostic Subgroups in Glioblastoma. PLoS One. 2014;9(7):e100827. doi: 10.1371/journal.pone.0100827
307. Asimus S, Elsherbiny D, Hai TN, Jansson B, Huong NV, Petzold MG, Simonsson US, Ashton M. Artemisinin antimalarials moderately affect cytochrome P450 enzyme activity in healthy subjects. Fundam Clin Pharmacol. 2007;21(3):307-16.
308. Chaffman M, Brogden RN, Heel RC, Speight TM, Avery GS. Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs. 1984;27(5):378-424.
309. Dando TM, Perry CM. Aprepitant: a review of its use in the prevention of chemotherapy-induced nausea and vomiting. Drugs. 2004;64(7):777-94.
310. Davis P. Auranofin. Clin Rheum Dis. 1984;10(2):369-83.
311. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38(1):41-57.
312. Frye RF, Branch RA. Effect of chronic disulfiram administration on the activities of CYP1A2, CYP2C19, CYP2D6, CYP2E1, and N-acetyltransferase in healthy human subjects. Br J Clin Pharmacol. 2002;53(2):155-62.
313. Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of HIV protease inhibitors 1: inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39(6):1070-8.
314. Kivitz A, Fairfax M, Sheldon EA, Xiang Q, Jones BA, Gammaitoni AR, Gould EM. Comparison of the effectiveness and tolerability of lidocaine patch 5% versus celecoxib for osteoarthritis-related knee pain: post hoc analysis of a 12 week, prospective, randomized, active-controlled, open-label, parallel-group trial in adults. Clin Ther. 2008;30(12):2366-77.
315. Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277(1):423-31.
316. Massaro AM, Lenz KL. Aprepitant: a novel antiemetic for chemotherapy-induced nausea and vomiting. Ann Pharmacother. 2005;39(1):77-85.
317. Poirier JM, Cheymol G. Optimisation of itraconazole therapy using target drug concentrations. Clin Pharmacokinet. 1998;35(6):461-73.
318. Preskorn SH. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin Pharmacokinet. 1997;32 Suppl 1:1-21.
319. Sandberg M, Yasar U, Strömberg P, Höög JO, Eliasson E. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol. 2002;54(4):423-9.
320. Tang C, Shou M, Mei Q, Rushmore TH, Rodrigues AD. Major role of human liver microsomal cytochrome P450 2C9 (CYP2C9) in the oxidative metabolism of celecoxib, a novel cyclooxygenase-II inhibitor. J Pharmacol Exp Ther. 2000;293(2):453-9.
321. Warrington SJ. Clinical implications of the pharmacology of sertraline. Int Clin Psychopharmacol. 1991;6 Suppl 2:11-21.
322. White CM. Pharmacologic, pharmacokinetic, and therapeutic differences among ACE inhibitors. Pharmacotherapy. 1998;18(3):588-99.
323. Petersen EN. The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr Scand Suppl. 1992;369:7-13.