
INTRODUCTION
Medullary thyroid carcinoma (MTC) is a rare endocrine tumor originating from parafollicular C cells of the thyroid. This neoplasia is inherited as an autosomal dominant trait in 25% of patients [1]. In these cases, other organs besides the thyroid (e.g., the parathyroid and adrenal glands) can be involved, thus giving rise to the multiple endocrine neoplasia type 2 (MEN 2) syndromes, which are categorized into three different subtypes (e.g., MEN 2A, MEN 2B and familial medullary thyroid carcinoma or FMTC) according to their phenotype [2]. Activating germline point mutations in the RET proto-oncogene have been shown to cause approximately 95–98% of MEN 2 cases [2, 3]. In the other 75% of cases, MTC is a sporadic tumor, and with exception of RAS alterations that have been found in approximately 10% of cases [4, 5], somatic mutations in the RET proto-oncogene appear to be the most common genetic alteration in MTC tumorigenesis [3]. The most common alterations in the RET proto-oncogene are missense gain of function mutations mainly located in the extracellular domain of RET (exons 10 or 11) and in the RET tyrosine kinase domain (exons 13, 14, 15 and 16).
These mutations are able to cause the constitutive activation of the ret onco-protein [6].
In particular, we demonstrated that somatic RET mutation prevalence increases with increasing tumor size [7], reaching a prevalence of approximately 90% in advanced cases [8].
The predominant role of a single initiating mutation in MEN 2 was proposed several years ago [9]. Recently, studies performed with deep sequencing technologies, either whole exome sequencing (WES) [10] or targeted sequencing [11–13], have shown that, with a few exceptions, RET is the only oncogene altered in MTC.
Over the years, spatial and temporal intratumor heterogeneity have been demonstrated in several human cancers such as clear cell renal cancer, glioblastoma, pancreatic cancer and breast cancer [14–17]. In addition to a different mutation profile, DNA copy number alterations represents an additional feature of intratumor heterogeneity [18]. As far as thyroid cancer genetic heterogeneity is concerned, little is known about the frequency of different mutations in metastases of papillary thyroid histotypes [19–21]. Even less evidence is available on the genetic heterogeneity of MTC. A single study performed on 28 sporadic MTC cases showed an intratumoral heterogenic RET mutation profile in 50% of cases [22].
Taking into consideration that RET is almost the only oncogene altered in MTC, it is conceivable that, if tumoral heterogeneity exists, different RET mutations might be present in different tumoral specimens (i.e., primary and metastatic tissues). The aim of the present study was to investigate the RET somatic mutation profiles in a large series of primary MTC cases and corresponding synchronous or metachronous metastatic tissues.
RESULTS
RET mutations
Among the 56 metastatic MTC cases included in the study, 7 cases were found to CARRY a RET germline mutation (Table 1). However case n. 36 was considered sporadic since it was positive for an A883T mutation in exon 15 that was previously demonstrated, both in vitro and in vivo, to have a very low or null transforming ability [23, 24]. The remaining 49 patients were found to be negative for the presence of a RET germline mutation; thus, a total of 50 cases were considered truly sporadic.
Table 1: Available tumoral tissues from the 56 patients with metastatic MTC included in the study
num |
Primary tumor |
local recurrence (LC)/metastases |
RET germline mutation |
num |
primary tumor |
local recurrence (LC)/metastases |
RET germline mutation |
|---|---|---|---|---|---|---|---|
1 |
n.a. |
2 LNF |
Neg |
29 |
n.a. |
7 liver |
Neg |
2 |
1 |
1 LNF |
Neg |
30 |
1 |
1 LNM |
Neg |
3 |
1 |
1 LC/5 LNF |
Neg |
31 |
n.a. |
3 LNF |
Neg |
4 |
1 |
1 LNF |
Neg |
32 |
1 |
4 LNF |
Neg |
5 |
1 |
1 LNF |
Neg |
33 |
1 |
1 LNF CC |
Neg |
6 |
n.a. |
6 LNF |
Neg |
34 |
2 |
2 LNF |
C634S |
7 |
1 |
2 LNF |
Neg |
35 |
1 |
1 LNF |
Neg |
8 |
1 |
2 LNF |
Neg |
36* |
3 |
11 LNF 1 |
A883T |
9 |
1 |
1 LNF |
Neg |
37 |
n.a. |
2 LNF |
Neg |
10 |
1 |
1 LC |
Neg |
38 |
1 |
1 LNF |
Neg |
11 |
1 |
1 LNF |
Neg |
39 |
n.a. |
3 LNF, 2 brain, 2 liver, 1 kidney, 1 adrenal |
C634R |
12 |
1 |
3 LNF |
Neg |
40 |
1 |
1 LNF |
Neg |
13 |
n.a. |
2 LNF |
Neg |
41 |
n.a. |
2 LNF |
Neg |
14 |
1 |
5 LNF, 2 liver |
Neg |
42 |
1 |
1 liver |
Neg |
15 |
1 |
4 LNF |
Neg |
43 |
1 |
1 LNF |
Neg |
16 |
1 |
1 LC/1 LNF |
Neg |
44 |
2 |
1 LNF |
Neg |
17 |
1 |
1 LNF |
Neg |
45 |
n.a. |
2 LNF |
Neg |
18 |
1 |
1 LNF |
Neg |
46 |
n.a. |
5 LNF |
Neg |
19 |
1 |
1 LNF |
V804M |
47 |
1 |
2 |
M918T |
20 |
1 |
1 LNF |
Neg |
48 |
1 |
1 LNF |
Neg |
21 |
1 |
1 LNF |
Neg |
49 |
1 |
4 LNF, 2 PHEO |
M918T |
22 |
1 |
1 LNF |
Neg |
50 |
1 |
1 trachea |
Neg |
23 |
1 |
12 LNF |
Neg |
51 |
1 |
1 LNF |
M918T |
24 |
1 |
8 LNF |
Neg |
52 |
1 |
1 LNF |
Neg |
25 |
1 |
2 LNF |
Neg |
53 |
2 |
7 LNF |
Neg |
26 |
1 |
2 LNF |
Neg |
54 |
1 |
1 kidney |
Neg |
27 |
2 |
1 LNF |
Neg |
55 |
3 |
3 LNF |
Neg |
28 |
2 |
2 LNF |
Neg |
56 |
1 |
2 LNF |
Neg |
n.a. not available; * this patient was considered as sporadic since A883T is not transforming (ref 24) and had the somatic deletion p.D898_E901del, c.2694_2705del12 reported in Table 2. PHEO: pheochromocitoma.
As reported in Table 2, 45/50 (90%) cases were positive for a RET somatic mutation, while 5 were RET mutation negative. The most frequent RET somatic mutation was the M918T mutation in exon 16 that was found in 34/50 (68%) sporadic cases. Other RET somatic point mutations or in-frame deletions were found in 5/50 (10%) and 6/50 (12%) cases, respectively. As reported in Table 2, 4/50 (8%) cases were found to carry 2 different somatic mutations, either 2 point mutations or 2 deletions, in the same tissue.
Table 2: RET somatic mutations in sporadic cases
Sporadic cases (n = 50) RET somatic mutations (45/50 = 90%) |
|
|---|---|
mutation |
n. of cases/50 (%) |
M918T* |
34 (70.4) |
C620A |
2 (4) |
C634G |
1 (2) |
C634R |
1 (2) |
A883F |
1 (2) |
p.D899_E902del, c.2694_2705 |
2 (4) |
p.D898_E901del, c.2692_2703del12 |
1 (2) |
p.Glu632fs c.1894_1904del11 |
1 (2) |
p.Ile638fs c.1912_1918del7** |
|
p.E632_L633del, c.1894_1899del6 + p.D898_E901del, c.2692_2703del12 ** |
1 (2) |
p.E632_L633del, c.1894_1899del6 |
1 (2) |
NM |
5 (11.5) |
*in 2 cases M918T mutation was associated with an additional point mutation in the same tissue (S891A or C620F); ** in 2 cases two different deletions in exon 11 and in both exon 11 and 15, respectively, were found in the same tissue.
Comparison of RET somatic mutations in different tumoral tissues of the same patient
In the whole series of 56 metastatic MTC cases, we compared the presence of RET somatic mutations in different tumoral tissues of the same patient. In the majority of cases (n = 45), the comparison was made between primary and metastatic tissues, while in the others (n = 11), the RET genetic profile was compared in different metastases. The comparison showed that in 45/56 (80.4%) cases, the RET mutation profile, either positive or negative, was the same in all the specimens of the same patient. In this group of concordant cases, no double mutants of RET were observed. Moreover, in 7 cases (Table 1: n. 27, n. 28, n. 34, n. 36, n. 44, n. 53, and n. 55) of which 2 or 3 different sections of the same primary tissue were available, no differences in the RET genetic profile, either positive or negative, were observed.
The other 11/56 (19.6%) cases showed a heterogenic RET mutation profile. As shown in Table 3 (panel A), in 5 cases (cases n. 3, n. 18, n. 36, n. 53 and n. 54), a RET heterozygous somatic mutation, either point or complex, was present in the primary tumor. This subgroup contained some metastatic lesions that showed the same RET mutation as found in the primary tumor, while others were RET negative (n. 3, n. 18, n. 36, n. 53). Moreover, case n. 53 showed a lymph node metastases with the same mutation as the primary tumor plus an additional 12 bp deletion of exon 15. Finally, the kidney metastases of case n. 54 that was characterized by a double somatic mutation in the primary tumor showed only one of the two mutations.
Table 3: Cases with a different RET mutation profile in different samples
PANEL A |
|||
|---|---|---|---|
patient |
Primary |
LNF |
Distant met |
3 |
M918T (1/1) |
M918T (2/5) NM (3/5) |
n.a. |
18 |
M918T (1/1) |
NM (1/1) |
n.a. |
36 |
p.D898_E901del, c.2694_2705del12 (3/3) |
p.D898_E901del, c.2694_2705del12 (10/11) NM (1/11) |
n.a |
53 |
M918T (2/2) |
M918T (1/7) p.D898_E901del, c.2694_2705del12 + M918T (1/7) NM (5/7) |
n.a |
54 |
M918T/S891A (1/1) |
Not present |
only M918T (1/1) |
PANEL B |
|||
6 |
n.a. |
M918T (5/6) NM (1/6) |
n.a. |
|---|---|---|---|
10 |
NM (1/1) |
n.a. |
p.E632_L633del, c.1894_1899del6 p.D898_E901del, c.2692_2703del12 (1/1) |
23 |
NM (1/1) |
p.D898_E901del, c.2692_2703del12 (12/12) |
n.a. |
24 |
NM (1/1) |
C620F+M918T (2/8) M918T (5/8) NM (1/8) |
n.a. |
PANEL C |
|||
9 |
M918T homo/hemizygous (1/1) |
M918T heterozygous (1/1) |
n.a. |
|---|---|---|---|
14 |
p.E632_L633del, c.1894_1899del6 heterozygous (1/1) |
p.E632_L633del, c.1894_1899del6 heterozygous (4/5) p.E632_L633del, c.1894_1899del6 homo/hemizygous(1/5) |
p.E632_L633del, c.1894_1899del6 heterozygous (1/2) p.E632_L633del, c.1894_1899del6 homo/hemizygous (1/2) |
n.a. not available
Another subgroup of discordant cases (Table 3, panel B) was characterized by different patterns: a) different metastases of the same patient were either positive or negative for a specific RET mutation (n. 6); b) two different RET mutations, none of them found in the primary, were present in the same metastatic lesion (n. 10); c) the same RET mutation, not found in the primary, was present in all lymphnode metastases (n. 23); d) two different RET mutations or just one of them or even none, as it happened in the primary, were found in different metastatic lesions (n 24).
The last two cases (Table 3, panel C) showed a peculiar RET genetic profile since in some lesions of the same patient the RET mutation was apparently homozygous, while in others it was clearly heterozygous (n. 9 and 14).
In order to verify whether the RET negative tumoral tissues belonging to cases with other RET positive tissues were false negative due to a low percentage of cancer cells in the sample, we evaluated the prevalence of tumoral cells in the tissue samples. As shown in Table 4, when the percentage of tumoral cells was at least 20% RET mutation was detected. Among cases negative for RET mutations, some cases had a high percentage of tumoral cells and could be considered real negative instead some cases had low percentage of tumoral cells (10–15%) and they could be potentially false negative. Nevertheless, if we exclude these latter cases, the number of heterogenic cases would not change and affect the final results of the present study.
Table 4: Comparison between RET mutation status and percentage of tumoral cells in the analysed tumoral tissue
patient |
Type of tissue |
% of tumoral cells |
RET mutation |
|---|---|---|---|
3 |
LNF |
n.a. |
NM |
LNF1 |
80 |
NM |
|
LNF3 |
70 |
NM |
|
primary tumor |
20 |
M918T |
|
53 |
LNF1 |
5 |
NM |
LNF2 |
5 |
NM |
|
LNF3 |
20 |
p.D898_E901del,c.2694_2705del12 |
|
LNF4 |
60 |
NM |
|
LNF5 |
30 |
NM |
|
LNF6 |
n.a. |
NM |
|
6 |
LNF4 |
20 |
NM |
24 |
LNF6 |
30 |
M918T/C620F |
LNF7 |
10 |
NM |
|
LNF8 |
10 |
NM |
|
primary tumor |
20 |
NM |
|
53 |
LNF1 |
5 |
NM |
LNF2 |
5 |
NM |
|
LNF3 |
20 |
p.D898_E901del,c.2694_2705del12 |
|
LNF4 |
60 |
NM |
|
LNF5 |
30 |
NM |
|
LNF6 |
n.a. |
NM |
|
LNF LC SIN |
50 |
M918T |
Multiplex ligation-dependent probe amplification (MLPA) assay
As previously stated, some tumoral tissues of patient n. 14 showed a heterozygous somatic deletion in exon 11 encompassing codons 632–634 (Figure 1, panel A). Very interestingly, the same deletion was found as homozygous in other metastatic lesions (Figure 1, panel B). No RET alterations were found at the germline level (Figure 1, panel C).
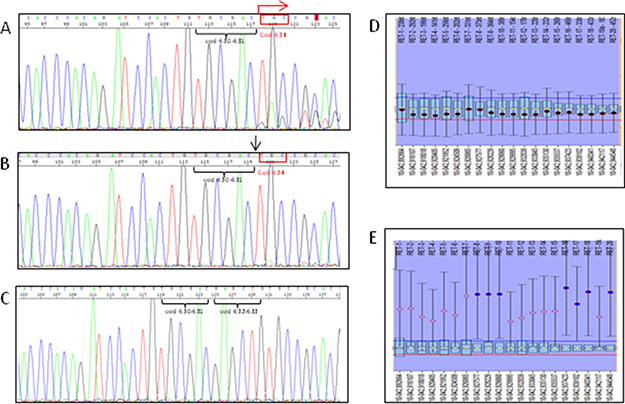
Figure 1: Sanger sequencing pherograms and MLPA graphics of the case n 14. Heterozygous deletion in exon 11 encompassing codons 632–634 (panel A) is revealed by the presence of double peaks in the pherogram starting from codon 634 (see red arrow); homo/hemizygous RET somatic deletion in exon 11 encompassing codons 632–634 is shown in (panel B) and revealed by the absence of both codons 632 and 633 (see black arrow). No RET deletion was found at the germline level (panel C) as demonstrated by the wild type sequence of RET oncogene. MLPA showed that no copy number variation was found within the RET gene in the tissues with the heterozygous somatic 6 bp deletion of exon 11 (panel D) suggesting a balance between mutated and not mutated alleles. At variance, an amplification of one RET allele was observed in the tissue with apparent homozygous 6 bp deletion of exon 11 (panel E) suggesting that the amplified allele should be the mutated one.
MLPA was performed on all tumoral tissues of case n. 14 to determine if the “homozygosity” of the deletion was caused by a RET copy number variation. As shown in Figure 1, panel D, no copy number variation was found within the RET gene in the tumor that was characterized by the heterozygous somatic 6 bp deletion of exon 11. However, one of the two RET alleles was highly amplified with respect to the other in the tissue with the apparent homozygous somatic 6 bp deletion of exon 11 (Figure 1, panel E). Based on these MLPA results, we can reasonably assume that the amplified allele was the mutated one, thus leading to the apparently homozygous pattern observed with the sequencing analysis.
DISCUSSION
Several studies have demonstrated that RET is the most prevalent oncogene involved in MTC tumorigenesis. From the study of the International RET consortium [25], the prevalence of RET mutations in sporadic MTC cases was found to be approximately 40%, with a high rate of M918T mutations. Over the years, the overall RET mutation prevalence was essentially confirmed, but RET mutation levels were demonstrated to be significantly lower in small size tumors and much higher in advanced metastatic cases [7, 8, 26, 27]. This latter finding has been confirmed by the results of the present study, since the prevalence of somatic RET mutations was 90% in this advanced metastatic MTC series, with M918T mutations being the most frequent. Among the RET mutations other than M918T, we found several complex somatic alterations, almost exclusively deletions, affecting exons 11 and/or 15. The presence of these type of RET mutations, although more rare than point mutations, has been previously reported in other series, and they have been demonstrated to have high transforming capabilities [28, 29].
Only few cases of sporadic MTC with multiple RET mutations in the same tumoral tissue have been reported so far [30]. In our series, we found 4 cases with intratumoral heterogeneity characterized by the presence of 2 RET alterations, either point mutations or deletions, in the same tumoral specimen. Although the prevalence of these mixed cases (8% of our series) is rather low, this finding is something new with respect to the results of Eng et al. [22], who found a high prevalence of mixed subpopulations, but only mixed in regards to RET positive or negative mutation status. However, it must be noted that the methodology used in the Eng et al study was rather restrictive and did not allow for the identification of all RET mutations and in particular could not detect complex mutations. The pathogenic role of a double mutant in the same tissue is still not clear, but Nowell’s evolutionary theory of cancer, which describes how mutations can accumulate in cell subpopulations, remains the most likely explanation [31, 32].
Recently, some studies have described the genetic heterogeneity in different human cancers [14–17]. These findings are rather expected in those human tumors (i.e., lung and melanoma) in which multiple oncogenes (up to 163 and 147 different oncogenes, respectively) have been found to be altered in the same tumor [33–35]. Differentiated thyroid cancers have been shown to have a low number of altered oncogenes, which are usually mutually exclusive [36]. This is particularly true for sporadic MTC, which has a high prevalence of RET mutations and a low prevalence of RAS mutations, with no other driver oncogenes found so far [3, 5]. For this reason, we concentrated our attention on the RET oncogene alone and we showed that 20% of MTC cases were characterized by a different RET mutation profile in primary and metastatic tissues. As reported in Table 2, three different patterns of heterogeneity were found, and different hypotheses can be proposed to explain them. The first pattern was characterized by the presence of a RET heterozygous somatic mutation in the primary tumor that was not necessarily found in all the corresponding metastases or an additional unique mutation (from that found in the primary tumor) that was found in one metastatic lymph node. To understand this finding, we might hypothesize that the primary tumor is composed of RET-positive and RET-negative cell subpopulations. Although we cannot be sure that different cells within a tumor can carry different mutations, the study of Eng et al [22] clearly demonstrated that it was possible. As far as the case with the additional mutation is concerned, the most plausible explanation is the evolutionary theory of cancer according to which new mutations can occur in already mutated, developing tumors [31, 32].
In the second pattern, the primary tumor was RET negative, and different metastases of the same patient were either positive or negative for a specific RET mutation. It is conceivable that a few cells in the primary tumor were positive for the mutations found in the lymphnodes which received a growth advantage during the selective pressures of the metastatic process and produced RET-positive metastases [32]. This explanation and the possibility that during the growth of mutated cells other mutations can be added could also justify those cases in which different metastases are genetically different.
The third pattern of heterogeneity showed a peculiar RET genetic profile: In some lesions, the RET mutation was apparently homozygous, while in others, it was clearly heterozygous. In these cases, a loss of heterozygosity (LOH) (as reported also by Dvorakova et al in their series [30, 37]) or a copy number alteration should be responsible for the observed pattern. The MLPA experiment suggested that an increased number of RET mutated alleles were present in our sample. This result agrees with our previous findings, which showed that RET gene amplifications were present in RET-positive tumors [38].
A limitation of the present study can be due the sensitivity of the Sanger method that has been estimated to be between 10–20% [7, 39, 40]. This implies the possibility to have a technical bias due to the presence of micro metastases, with a low number of metastatic tumoral cells surrounded by normal cells and for these reason not detectable by Sanger sequencing. We tried to rule out this problem and effectively we found that when the tumoral cells were > 20% RET mutation was detectable but we also had cases with > 20% of tumoral cells that were RET negative. These latter can be considered as true negative cases. For those cases with < 20% of tumoral cells we cannot exclude the possibility they were false negative cases. Nevertheless, the number of heterogenic cases would not change if we exclude these potential false negative cases.
In conclusion, this study showed that a different RET mutation profile between primary and metastatic tissues of the same MTC was present in 20% of cases, and in 8% of these, RET intratumor heterogeneity was observed. These results, together with the evidence that MTC has a low rate of mutations other than RET, explains the relatively moderate level of aggressiveness of the disease with respect to other, more aggressive tumors (lung adenocarcinoma or melanoma) that are characterized by a high rate of mutation and heterogeneity.
MATERIALS AND METHODS
Patients
We studied the tumoral tissues of 56 patients (24 females and 32 males) affected with metastatic sporadic or familial MTC. The histological diagnosis and classification of tumoral tissues were performed by an experienced team of local pathologists. MTC cases were included in the study only if at least two tumoral tissues obtained from different lesions were available, either from primary and metastatic tissues or from several metastases. A total of 209 tumoral specimens were analyzed. In particular, 54 primary tumoral tissues, 2 local recurrences, 132 lymph node metastases and 21 distant metastases (i.e., lung, liver, kidney, adrenal gland, brain) were included in the study. The details of the analyzed tumoral tissues and their corresponding patients’ statistics are reported in Table 1.
Tissues were either collected at surgery, immediately frozen in liquid nitrogen and kept at -80°C, or recovered from paraffin-embedded tissue blocks. A peripheral blood sample was available for both sporadic and familial cases.
All patients provided written informed consent. This investigation was approved by our institutional review board and by the local Ethic Committee (protocol number 469, approved 29/1/2015).
Methods
Pathologic diagnosis
Patients underwent total or subtotal thyroidectomy at the Department of Surgery of the University of Pisa, Italy. The presence of typical histological (i.e., tumoral cells arranged in trabecular, insular or sheet-like growth patterns) and immunohistochemical (cells positive for calcitonin and chromogranin) features defined an MTC histological diagnosis. In some cases, the percentage of tumoral cells in the sample was recorded. High levels of serum calcitonin were confirmatory of the C cell origin of the cases.
Genotyping
DNA was prepared from blood, fresh and paraffin-embedded tumoral tissues according to a previously described protocol [8]. RET exons 10, 11, 13, 14, 15 and 16 were analyzed by direct sequencing [41]. To exclude false negative cases, primary tumors and metastases found to be discordant for the presence/absence of the M918T RET mutation were further analyzed using a more sensitive TaqMan SNP genotyping assay (ThermoFisher Scientific, Waltham, Massachusetts, USA). The experiments were run according to the manufacturer’s guidelines.
Multiplex ligation-dependent probe amplification (MLPA) assay
MLPA was used to detect potential large deletions in RET gene. Experiments were performed on primary and metastatic tumor samples using the SALSA MLPA P169 HIRSCHSPRUNG PROBEMIX (MRC-Holland, Amsterdam, the Netherlands). Coffalyser.Net software (MRC-Holland, Amsterdam, the Netherlands) was used to identify copy number variations. Experiments were repeated at least twice. Three reference DNAs from the blood of healthy subjects and a negative control (a sample without DNA) were included in all experiments.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest that could affect the impartiality of the reported research.
FUNDING
This work was supported by the Associazione Italiana Ricerca sul Cancro (AIRC, Investigator grant 2014, project code 15431).
REFERENCES
1. Raue F, Frank-Raue K. Epidemiology and Clinical Presentation of Medullary Thyroid Carcinoma. Recent Results Cancer Res. 2015; 204:61–90. https://doi.org/10.1007/978-3-319-22542-5_3.
2. Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015; 204:139–56. https://doi.org/10.1007/978-3-319-22542-5_6.
3. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016; 12:192–202. Available from http://www.ncbi.nlm.nih.gov/pubmed/26868437.
4. Ciampi R, Mian C, Fugazzola L, Cosci B, Romei C, Barollo S, Cirello V, Bottici V, Marconcini G, Rosa PM, Borrello MG, Basolo F, Ugolini C, et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid. 2013; 23:50–7. https://doi.org/10.1089/thy.2012.0207.
5. Moura MM, Cavaco BM, Leite V. RAS proto-oncogene in medullary thyroid carcinoma. Endocr Relat Cancer. 2015; 22:R235-52. https://doi.org/10.1530/ERC-15-0070.
6. Hedayati M, Zarif Yeganeh M, Sheikholeslami S, Afsari F. Diversity of mutations in the RET proto-oncogene and its oncogenic mechanism in medullary thyroid cancer. Crit Rev Clin Lab Sci. 2016; 53:217–27. https://doi.org/10.3109/10408363.2015.1129529.
7. Romei C, Ugolini C, Cosci B, Torregrossa L, Vivaldi A, Ciampi R, Tacito A, Basolo F, Materazzi G, Miccoli P, Vitti P, Pinchera A, Elisei R. Low prevalence of the somatic M918T RET mutation in micro-medullary thyroid cancer. Thyroid. 2012; 22:476–81. https://doi.org/10.1089/thy.2011.0358.
8. Romei C, Casella F, Tacito A, Bottici V, Valerio L, Viola D, Cappagli V, Matrone A, Ciampi R, Piaggi P, Ugolini C, Torregrossa L, Basolo F, et al. New insights in the molecular signature of advanced medullary thyroid cancer: evidence of a bad outcome of cases with double RET mutations. J Med Genet. 2016. https://doi.org/10.1136/jmedgenet-2016-103833.
9. Mulligan LM, Gardner E, Smith BA, Mathew CG, Ponder BA. Genetic events in tumour initiation and progression in multiple endocrine neoplasia type 2. Genes Chromosomes Cancer. 1993; 6:166–77.
10. Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, Bhan S, Ho AS, Khan Z, Bishop J, Westra WH, Wood LD, Hruban RH, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013; 98:E364-9. https://doi.org/10.1210/jc.2012-2703.
11. Simbolo M, Mian C, Barollo S, Fassan M, Mafficini A, Neves D, Scardoni M, Pennelli G, Rugge M, Pelizzo MR, Cavedon E, Fugazzola L, Scarpa A. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014; 465:73–8. https://doi.org/10.1007/s00428-014-1589-3.
12. Sherman SI, Clary DO, Elisei R, Schlumberger MJ, Cohen EEW, Schoffski P, Wirth LJ, Mangeshkar M, Aftab DT, Brose MS. Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer. 2016; 122:3856–64. https://doi.org/10.1002/cncr.30252.
13. Wei S, LiVolsi VA, Montone KT, Morrissette JJD, Baloch ZW. Detection of Molecular Alterations in Medullary Thyroid Carcinoma Using Next-Generation Sequencing: an Institutional Experience. Endocr Pathol. 2016; 27:359–62. https://doi.org/10.1007/s12022-016-9446-3.
14. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–92. https://doi.org/10.1056/NEJMoa1113205.
15. Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013; 110:4009–14. https://doi.org/10.1073/pnas.1219747110.
16. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114–7. https://doi.org/10.1038/nature09515.
17. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109–13. https://doi.org/10.1038/nature09460.
18. Martinez P, Birkbak NJ, Gerlinger M, McGranahan N, Burrell RA, Rowan AJ, Joshi T, Fisher R, Larkin J, Szallasi Z, Swanton C. Parallel evolution of tumour subclones mimics diversity between tumours. J Pathol. 2013; 230:356–64. https://doi.org/10.1002/path.4214.
19. Yang X, Li J, Li X, Liang Z, Gao W, Liang J, Cheng S, Lin Y. TERT Promoter Mutation Predicts Radioiodine-Refractory Character in Distant Metastatic Differentiated Thyroid Cancer. J Nucl Med. 2017; 58:258–65. https://doi.org/10.2967/jnumed.116.180240.
20. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, Janakiraman M, Solit D, Knauf JA, Tuttle RM, Ghossein RA, Fagin JA. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009; 69:4885–93. https://doi.org/10.1158/0008-5472.CAN-09-0727.
21. Melo M, Gaspar da Rocha A, Batista R, Vinagre J, Martins MJ, Costa G, Ribeiro C, Carrilho F, Leite V, Lobo C, Cameselle-Teijeiro JM, Cavadas B, Pereira L, et al. TERT, BRAF, and NRAS in Primary Thyroid Cancer and Metastatic Disease. J Clin Endocrinol Metab. 2017; 102:1898–907. https://doi.org/10.1210/jc.2016-2785.
22. Eng C, Mulligan LM, Healey CS, Houghton C, Frilling A, Raue F, Thomas GA, Ponder BA. Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res. 1996; 56:2167–70.
23. Cosci B, Vivaldi A, Romei C, Gemignani F, Landi S, Ciampi R, Tacito A, Molinaro E, Agate L, Bottici V, Cappagli V, Viola D, Piaggi P, et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr Relat Cancer. 2011; 18:603–12. https://doi.org/10.1530/ERC-11-0117.
24. Elisei R, Cosci B, Romei C, Agate L, Piampiani P, Miccoli P, Berti P, Basolo F, Ugolini C, Ciampi R, Nikiforov Y, Pinchera A. Identification of a novel point mutation in the RET gene (Ala883Thr), which is associated with medullary thyroid carcinoma phenotype only in homozygous condition. J Clin Endocrinol Metab. 2004; 89:5823–7. https://doi.org/10.1210/jc.2004-0312.
25. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996; 276:1575–9.
26. Wells SAJ, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015; 25:567–610. https://doi.org/10.1089/thy.2014.0335.
27. Schlumberger M, Jarzab B, Cabanillas ME, Robinson B, Pacini F, Ball DW, McCaffrey J, Newbold K, Allison R, Martins RG, Licitra LF, Shah MH, Bodenner D, et al. A Phase II Trial of the Multitargeted Tyrosine Kinase Inhibitor Lenvatinib (E7080) in Advanced Medullary Thyroid Cancer. Clin Cancer Res. 2016; 22:44–53. https://doi.org/10.1158/1078-0432.CCR-15-1127.
28. Uchino S, Noguchi S, Yamashita H, Sato M, Adachi M, Yamashita H, Watanabe S, Ohshima A, Mitsuyama S, Iwashita T, Takahashi M. Somatic mutations in RET exons 12 and 15 in sporadic medullary thyroid carcinomas: different spectrum of mutations in sporadic type from hereditary type. Jpn J Cancer Res. 1999; 90:1231–7.
29. Ceccherini I, Pasini B, Pacini F, Gullo M, Bongarzone I, Romei C, Santamaria G, Matera I, Mondellini P, Scopsi L, Pinchera A, Pierotti MA, Romeo G. Somatic in frame deletions not involving juxtamembranous cysteine residues strongly activate the RET proto-oncogene. Oncogene. 1997; 14:2609–12. https://doi.org/10.1038/sj.onc.1201079.
30. Dvorakova S, Vaclavikova E, Sykorova V, Duskova J, Vlcek P, Ryska A, Novak Z, Bendlova B. New multiple somatic mutations in the RET proto-oncogene associated with a sporadic medullary thyroid carcinoma. Thyroid. 2006; 16:311–6. https://doi.org/10.1089/thy.2006.16.311.
31. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–8.
32. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481:306–13. https://doi.org/10.1038/nature10762.
33. Hong SM, Park JY, Hruban RH, Goggins M. Molecular signatures of pancreatic cancer. Arch Pathol Lab Med. 2011; 135:716–27. https://doi.org/10.1043/2010-0566-RA.1.
34. Brugarolas J. Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol. 2014; 32:1968–76. https://doi.org/10.1200/JCO.2012.45.2003.
35. Riesco-Eizaguirre G, Santisteban P. Endocrine Tumours: Advances in the molecular pathogenesis of thyroid cancer: lessons from the cancer genome. Eur J Endocrinol. 2016; 175:R203–17. https://doi.org/10.1530/EJE-16-0202.
36. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676–90. https://doi.org/10.1016/j.cell.2014.09.050.
37. Dvorakova S, Vaclavikova E, Sykorova V, Vcelak J, Novak Z, Duskova J, Ryska A, Laco J, Cap J, Kodetova D, Kodet R, Krskova L, Vlcek P, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Mol Cell Endocrinol. 2008; 284:21–7. https://doi.org/10.1016/j.mce.2007.12.016.
38. Ciampi R, Romei C, Cosci B, Vivaldi A, Bottici V, Renzini G, Ugolini C, Tacito A, Basolo F, Pinchera A, Elisei R. Chromosome 10 and RET gene copy number alterations in hereditary and sporadic Medullary Thyroid Carcinoma. Mol Cell Endocrinol. 2012; 348:176–82. https://doi.org/10.1016/j.mce.2011.08.004.
39. Monzon FA, Ogino S, Hammond MEH, Halling KC, Bloom KJ, Nikiforova MN. The role of KRAS mutation testing in the management of patients with metastatic colorectal cancer. Arch Pathol Lab Med. 2009; 133:1600–6. https://doi.org/10.1043/1543-2165-133.10.1600.
40. Ihle MA, Fassunke J, Konig K, Grunewald I, Schlaak M, Kreuzberg N, Tietze L, Schildhaus HU, Buttner R, Merkelbach-Bruse S. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014; 14:13. https://doi.org/10.1186/1471-2407-14-13.
41. Romei C, Cosci B, Renzini G, Bottici V, Molinaro E, Agate L, Passannanti P, Viola D, Biagini A, Basolo F, Ugolini C, Materazzi G, Pinchera A, et al. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin Endocrinol (Oxf). 2011; 74:241–7. https://doi.org/10.1111/j.1365-2265.2010.03900.x.