
INTRODUCTION
Autophagy is a cellular process that engulfs, digests, and recycles long-lived or aggregated proteins, defective organelles, and various soluble molecules to sustain cellular metabolism [1]. Previous studies have shown that autophagy is activated in many neuronal diseases such as cerebral hemorrhage, cerebral ischemia, Alzheimer’s disease and Parkinson’s disease. [2–4]. Autophagy has different biological effects under different conditions. For example, stimulation of autophagy during cerebral hemorrhage is a cell-defense mechanism [2], while excessive activation of autophagy in ischemia-reperfusion injury which occurs during cerebral ischemia can aggravate cellular injury [5]. Recently, it was also reported that the effects of autophagy were various in different hypoxia environments. In chronic and moderate continuous hypoxia, autophagy plays a protective role by removal of damaged organelles and proteins. Contrary to the protective role of autophagy, during rapid and severe oxygen fluctuations, autophagy may be detrimental and induce cell death [6]. This new evidence suggests the effect of autophagy might be varied under different forms of hypoxia.
Intermittent hypoxia is a characteristic pathophysiological change of obstructive sleep apnea (OSA), a commonly-diagnosed chronic sleep disorder. With the progression of OSA, OSA patients often suffer from nervous system damage and multiple cognitive dysfunctions [7]. Intermittent hypoxia-caused hippocampal neuronal apoptosis may be the main cause of nervous system damage induced by OSA [8]. Intermittent hypoxia, as a special form of hypoxia, can also lead to the release of ROS, inflammation, and organelle injury which can promote autophagy activation [9]. It is noteworthy that intermittent hypoxia, different from continuous hypoxia, is a process of repetitive oxygen desaturation-reoxygenation which can lead to severe cellular damage [10], and the relationship between intermittent hypoxia and autophagy in hippocampal neurons is unclear. Therefore, in this article, we investigated the effects and molecular mechanism of autophagy in intermittent hypoxia-induced hippocampal neuronal injury.
RESULTS
Intermittent hypoxia increases the expression of markers for autophagy in hippocampal neurons
In this study, we first examined the expressions of LC3-II and Beclin1, two key markers of autophagy in hippocampal neurons by western blot assay. Results revealed that the levels of LC3-II and Beclin1 increased with the duration of intermittent hypoxia, especially in the 12 h intermittent hypoxic group (Figure 1A and 1B). We stained hippocampal neurons to detect LC3-II expression. As shown in Figure 1C, compared with the control group, there were significantly more LC3-II positive dots in the intermittent hypoxic group, especially in the 12 h intermittent hypoxic group. We then evaluated the autophagic vacuoles by transmission electron microscopy (TEM), the gold standard technique for detecting autophagy. Similar to the results of western blot and immunofluorescence, the number of autolysosomes (AL, formerly called the degradative/late autophagic vacuoles) was increased in the intermittent hypoxic group compared with the control group (Figure 1D).
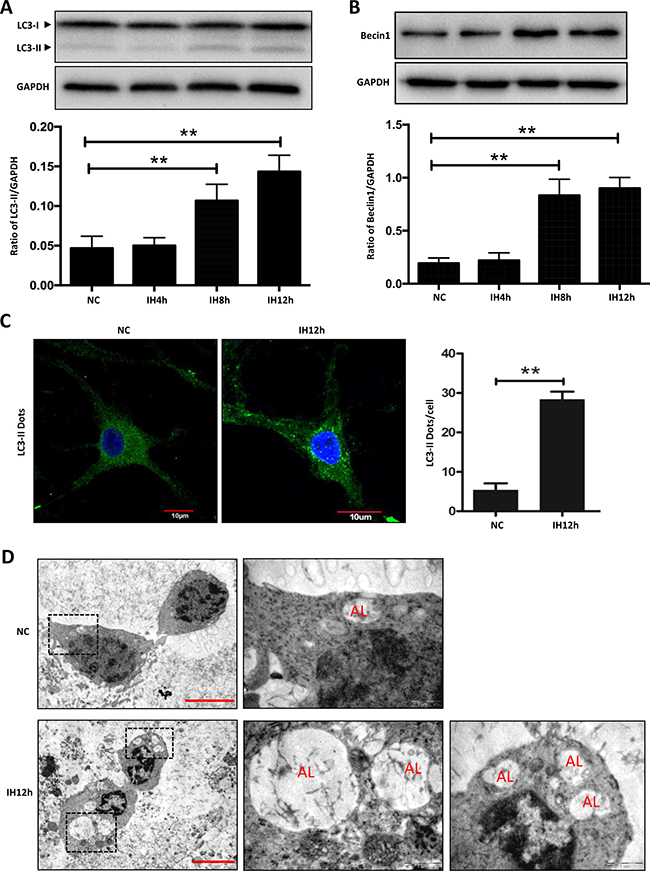
Figure 1: Intermittent hypoxia increases the expression of markers for autophagy in hippocampal neurons. (A and B) The expressions of LC3-II and Beclin1 in hippocampal neurons were assayed by western blot in NC, IH4h, IH8h and IH12h group respectively. (C) Measurement of LC3-II in hippocampal neurons with the indicated treatments by immunofluorescence microscopy assay. The cell nucleus are seen as blue fluorescence and LC3-II dots are shown in green fluorescence. Scale bar: 10 μm. (D) Ultrastructural images of autophagic vacuoles in IH12h cells and the controls. TEM images showed representative ALs. NC, nomoxia control; IH, intermittent hypoxia; Scale bar: 5 μm. *P < 0.05, **P < 0.01.
Intermittent hypoxia activates autophagy in hippocampal neurons
The dynamic process of autophagy includes initiation, elongation, maturation and degradation, also called autophagy flux. Next, we investigated the effects of intermittent hypoxia on autophagy flux in hippocampal neurons. LC3-I to LC3-II turnover and SQSTM1/p62 protein levels were regarded as the principal markers to monitor autophagy flux [11]. Results of western blot showed that the level of LC3-II and LC3 turnover were increased, while the level of SQSTM1/p62 was decreased in hippocampal neurons exposed to 12 h intermittent hypoxia (Figure 2A). As is known, Earle’s balanced salt solution (EBSS) starvation can initiate autophagy. Results showed that the expression level of LC3-II and LC3 turnover in EBSS groups were significantly increased, while SQSTM1/p62 expression was significantly decreased when compared with the control group. However, there was no significant difference in LC3 turnover and SQSTM1/p62 levels between EBSS and the intermittent hypoxic group (Figure 2A). We then used the autophagy activator rapamycin, and the autophagy inhibitor chloroquine, to further detect the autophagy flux. Rapamycin increased LC3-II lipidation, and intermittent hypoxia plus rapamycin further upregulated LC3-II/LC3-I levels and downregulated SQSTM1/p62 levels, but intermittent hypoxia plus chloroquine did not increase expression of SQSTM1/p62 (Figure 2B), suggesting that intermittent hypoxia increased autophagy flux rather than blocked the fusion of autophagosomes with lysosomes. In addition, there was no difference in LC3-II levels between DMSO (the dissolution agent of rapamycin) and the nomoxia group (Supplementary Figure 1). All of the above changes indicate that intermittent hypoxia can activate autophagy in hippocampal neurons.
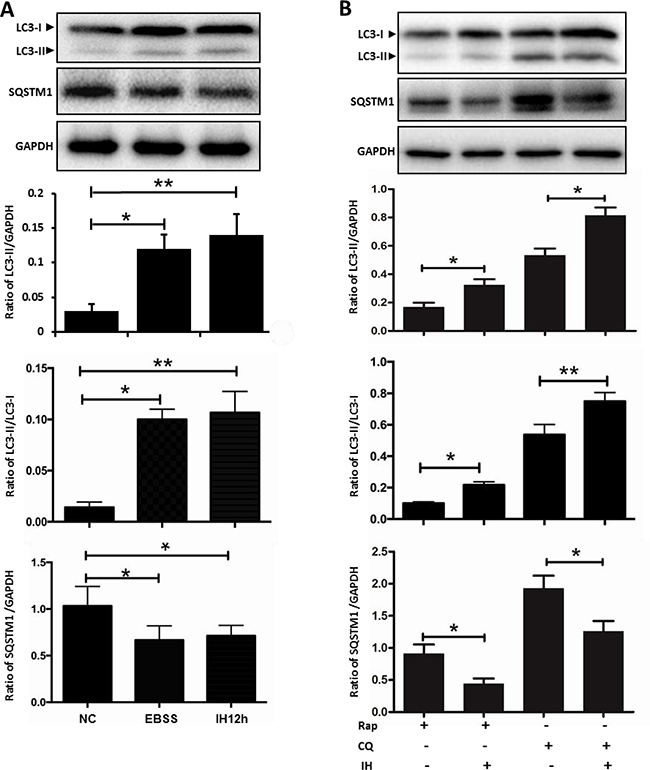
Figure 2: The effect of intermittent hypoxia on autophagy flux in hippocampal neurons. (A) The cultured hippocampal neurons were treated with IH for 12 h or were starved in EBSS for 3 h. The ratio of LC3II/GAPDH, LC3II/LC3I and SQSTM1/GAPDH was determined by western blot. (B) Employing autophagy activator Rap and autophagy inhibitor CQ to further detect autophagy activation in IH12h. The ratio of LC3II/GAPDH, LC3II/LC3I and SQSTM1/GAPDH was checked by western blot. Rap, rapamycin, 20 μM; CQ, chloroquine, 30 μM; NC, normoxia control; IH, intermittent hypoxia; *P < 0.05, **P < 0.01.
Intermittent hypoxia activates autophagy through the HIF-1α/BNIP3/Beclin1 signaling pathway
HIF-1α is a sensitive to hypoxia and is thought to induce the general autophagy pathway [12]. We therefore aimed to characterize its contribution in intermittent hypoxia. Western blot results showed that a time-dependent increase of HIF-1α was seen when hippocampal neurons were treated with intermittent hypoxia, with the greatest increase seen at 12 h during intermittent hypoxia treatment (Figure 3A). Similar patterns of HIF-1α were observed by immunofluorescence microscopy assay (Figure 3D). Next, we used the HIF-1α inhibitor YC-1, a novel specific HIF-1-targeting drug [13], to further explore the autophagy activation. As shown in Figure 3B, the expression of HIF-1α was significantly decreased in cells treated with YC-1 compared to those treated with intermittent hypoxia alone. Consistently, the addition of YC-1 suppressed intermittent hypoxia-induced upregulation of LC3-II (Figure 3C). We further demonstrated the critical role of HIF-1α on autophagy after exposure to intermittent hypoxia by knocking down HIF-1α expression. As expected, transfection with gene-specific small interfering RNAs successfully reduced HIF-1α protein levels and suppressed the biosynthesis of LC3-II and the degradation of SQSTM1/p62 induced by intermittent hypoxia (Figure 3E and 3F). Consistent with these results, the levels of the apoptosis marker, cleaved caspase 3, also decreased with decreasing levels of HIF-1α (Figure 3G). These data suggested that intermittent hypoxia-induced autophagy was related to HIF-1α.
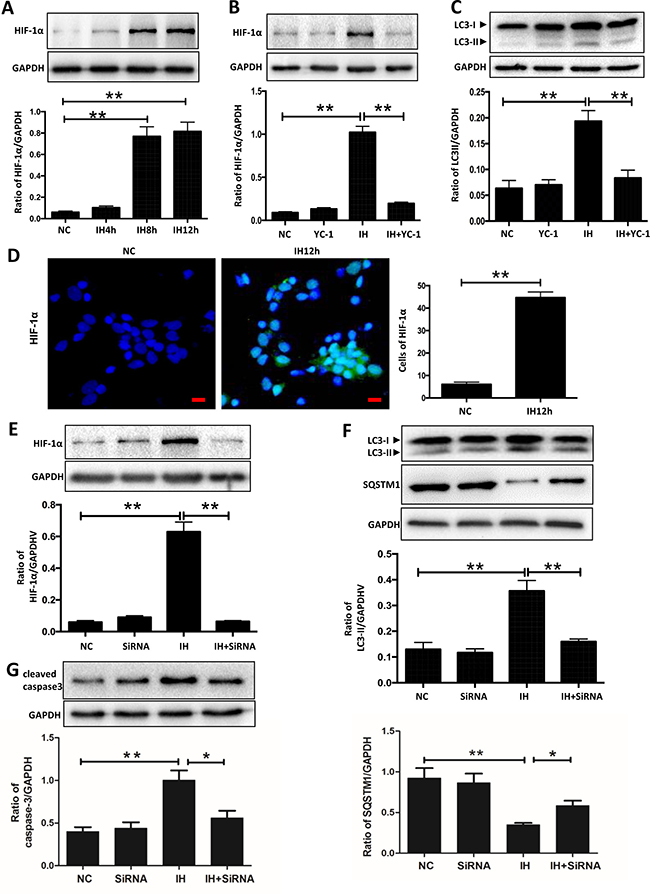
Figure 3: HIF-1α is related to intermittent hypoxia-induced autophagy activation in hippocampal neurons. (A) The hippocampal neurons were treated with NC, IH4h, IH8h and IH12h. The expression of HIF-1α in hippocampal neurons was detected by western blot. (B and C) The hippocampal neurons were treated with 10 mM YC-1 and then cultured with intermittent hypoxia for 12 h. The expression of HIF-1α and LC3-II in hippocampal neurons was assayed by western blot. (D) Measurement of HIF-1α in hippocampal neurons with the indicated treatments by immunofluorescence microscopy assay. Scale bar: 10 μm (E) The hippocampal neurons were treated with HIF-1α siRNA and then cultured with intermittent hypoxia for 12 h. The expression of HIF-1α in hippocampal neurons was assayed by western blot. (F) The expression of LC3-II and SQSTM1/p62 in hippocampal neurons were assayed by western blot with HIF-1α siRNA treatment. (G) The expression of cleaved caspase 3 in hippocampal neurons was assayed by western blot with HIF-1α siRNA treatment. NC, normoxia control; IH, intermittent hypoxia; *P < 0.05, **P < 0.01.
However, the specific mechanism through which HIF-1α mediates intermittent hypoxia-induced autophagy is still not clear. To unmask the precise underlying mechanisms behind the above demonstrated activation of autophagy in hippocampal neurons, we further tested the alteration of BNIP3, an important hypoxia-inducible protein downstream of HIF-1α, which can mediate autophagy by disruption of Beclin1 [14]. Our results showed that intermittent hypoxic incubation time dependently upregulated the expression of HIF-1α, which was consistent with its effect on BNIP3 and Beclin1 (Figure 4A). These findings initially indicated the involvement of a HIF-1α/BNIP3/Beclin1 signaling axis. To further determine the functional significance of the HIF-1α/BNIP3/Beclin1 pathway in the promotive effects of intermittent hypoxia, we then evaluated the influence of YC-1 on intermittent hypoxia- induced upregulation of BNIP3 in hippocampal neuronal cells. As shown in Figure 4B, treatment with YC-1 significantly prevented the occurrence of LC3-II biosynthesis as well as intermittent hypoxia-mediated over-expression of BNIP3 and Beclin1. Also, transfection with HIF-1α siRNA successfully reduced LC3-II biosynthesis and the increased expression of BNIP3 and Beclin1 caused by intermittent hypoxia (Figure 4C).
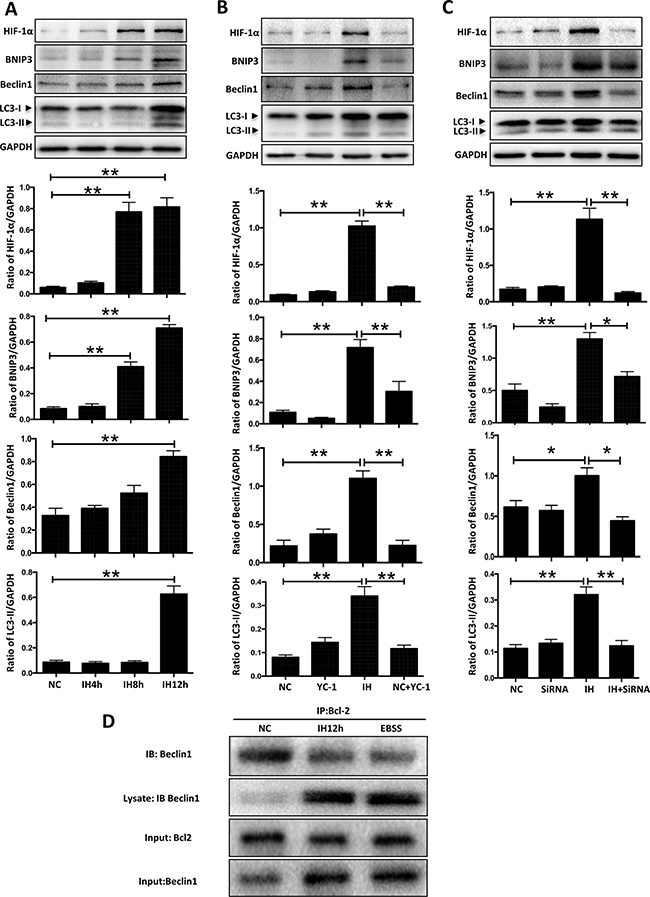
Figure 4: HIF-1α-BNIP3-Beclin1 mediates the autophagy activation induced by intermittent hypoxia. (A) The hippocampal neurons were treated with NC, IH4h, IH8h and IH12h. The expressions of LC3-II and the HIF-1α/BNIP3/Beclin1 signaling pathway related proteins in hippocampal neurons were assayed by western blot. (B) The hippocampal neurons were treated with 10 mM YC-1, and the cells were cultured with intermittent hypoxia for 12 h. The expression of LC3-II and the HIF-1α /BNIP3/Beclin1 signaling pathway related protein in hippocampal neurons was assayed by western blot. (C) The hippocampal neurons were treated with HIF-1α siRNA and the cells were cultured with intermittent hypoxia for 12 h. The expression of LC3-II and the HIF-1α /BNIP3/Beclin1 signaling pathway related proteins in hippocampal neurons were assayed by western blot. (D) Co-immunoprecipitation of endogenous Beclin1 with Bcl2. The hippocampal neurons were subjected to intermittent hypoxia for 12 hours, or EBSS for 3 hours, and cell lysates were immunoprecipitated with an anti-Beclin1 antibody resin. The presence of Beclin1 protein in the lysates and the immunoprecipitates is shown. IB, immunoblot; IP, immunoprecipitate. NC, normoxia control; IH, intermittent hypoxia; *P < 0.05 , **P < 0.01.
As noted, Beclin1 was originally isolated as a Bcl2-interacting protein, and several anti-apoptotic members of the Bcl2 family, such as Bcl2 and Bcl-XL, bind to Beclin1 and are negative regulators of its autophagy function. Studies found that starvation and hypoxia could induce autophagy through disruption of the Bcl2/Beclin1 complex [14–15]. In keeping with this notion, we detected the combination of Beclin1 and Bcl2 with a co-immunoprecipitation assay. Results showed that the endogenous Bcl2/Beclin1 complex, formed under normoxic conditions in hippocampal neuron cells, was partially dissociated following 12 h intermittent hypoxia or 3 h EBSS (Figure 4D). Taking all the above results into consideration, we concluded that the HIF-1α/BNIP3/Beclin1 pathway regulates autophagy activation in hippocampal neurons under intermittent hypoxia conditions.
Oxidative stress is involved in intermittent hypoxia-induced autophagy activation
Oxidative stress is not only considered to be one of the pathological mechanisms of intermittent hypoxia-induced multisystem damage, but it is also an important mechanism for inducing autophagy under cellular stresses [16–17]. Therefore, we hypothesized that oxidative stress may mediate the changes of autophagy in intermittent hypoxia-induced hippocampal neuronal injury. As shown in Supplementary Figure 2A, a fluorescence microscopy assay revealed that exposing hippocampal neurons to intermittent hypoxia induced an obvious ROS response in a time-dependent manner. To indicate the role of ROS, we used N-acetyl-L-cysteine (NAC), an antioxidant which can inhibit the expression of ROS induced by intermittent hypoxia (Figure 5A). The results of our western blot showed that treatment with NAC suppressed intermittent hypoxia-induced upregulation of LC3-II (Figure 5B). Similar patterns of LC3-II biosynthesis were observed by immunofluorescence assay (Supplementary Figure 2B). Consistent with these results, levels of the apoptosis marker cleaved caspase 3, also decreased with the decreasing levels of ROS and LC3-II (Figure 5C). All of these results demonstrated that ROS may be involved in the changes observed in autophagy in hippocampal neurons induced by intermittent hypoxia.
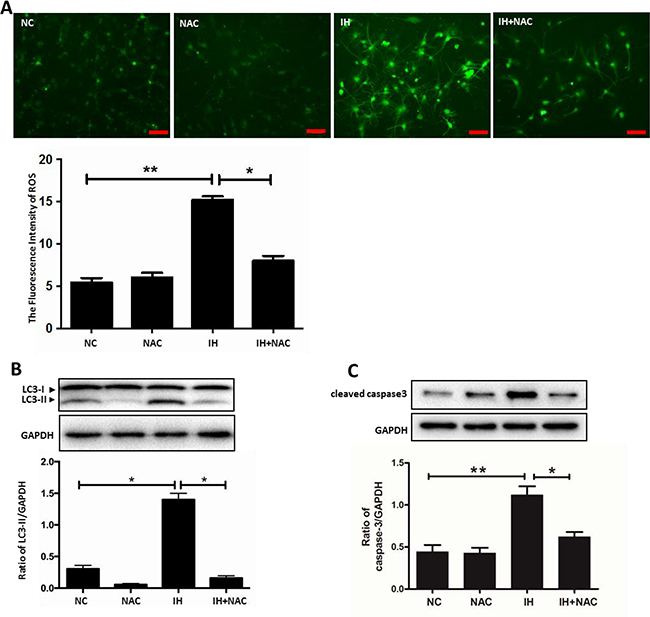
Figure 5: ROS is involved in intermittent hypoxia-induced autophagy activation. (A) The hippocampal neurons were treated with IH or NAC (10 mM) for 12 h. Measurement of ROS in hippocampal neurons with the indicated treatments seen with fluorescence microscopy. Scale bar: 50 μm. (B) The hippocampal neurons were treated with IH and NAC (10 mM). (C) The level of LC3-II and cleaved caspase 3 were determined by western blot assay. NC, normoxia control; IH, intermittent hypoxia. *P < 0.05, **P < 0.01.
Autophagy activation aggravates hippocampal neuronal apoptosis in intermittent hypoxia
Autophagy plays different roles under different conditions, and the effect of autophagy under intermittent hypoxia is still ambiguous. As shown in Figure 6A, a time-dependent increase of cleaved caspase 3, an apoptosis protein, was evident when hippocampal neurons were exposed to intermittent hypoxia, and the highest increase was seen at 12 h of intermittent hypoxia. In order to further detect the effects of intermittent hypoxia on hippocampal neuronal apoptosis, we ran a TUNEL assay to check the rate of apoptosis. Our results showed that the levels of apoptosis in hippocampal neurons for 12 h intermittent hypoxic group were significantly higher than the normal control group (Figure 6B).
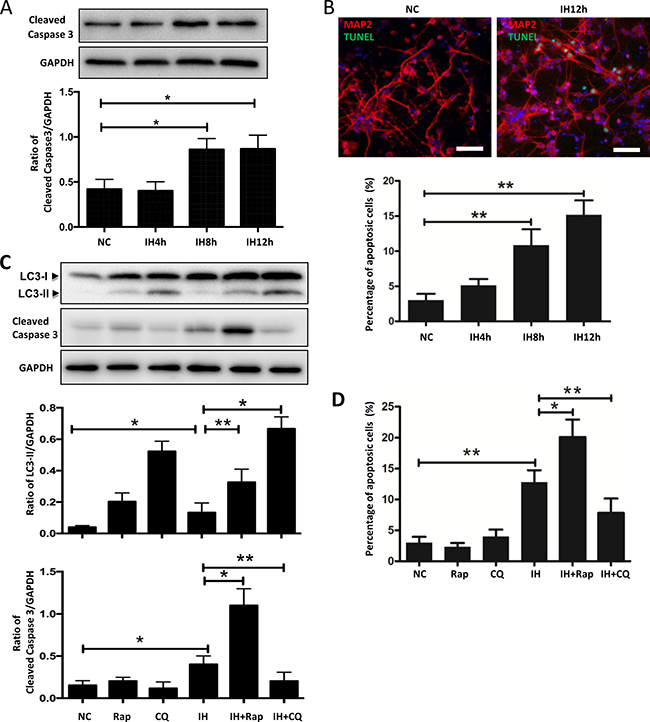
Figure 6: Autophagy activation aggravates hippocampal neuronal apoptosis in intermittent hypoxia. (A) The hippocampal neurons were treated with NC, IH4h, IH8h and IH12h. The expression of cleaved caspase 3 in hippocampal neurons was assayed by western blot. (B) The hippocampal neurons were treated with NC, IH4h, IH8h and IH12h. Detection of apoptotic cells was determined by TUNEL assay. Figure B only has photos for NC and IH12h groups. Scale bar: 100 μm. (C) Western blot analysis the expression of LC3-II or cleaved caspase 3 in hippocampal neurons after exposure to IH12h, with or without Rap or CQ for 12 h. (D) Quantitative data of the percentage of apoptotic cells in hippocampal neurons, determined by TUNEL assay, treatments the same as in (C). Rap, rapamycin, 20 μM; CQ, chloroquine, 30 μM; NC, normoxia control; IH, intermittent hypoxia; *P < 0.05, **P < 0.01.
It is thought that rapamycin increases autophagy flux by promoting autophagosome formation, while chloroquine impairs autophagic flux by blocking autophagic degradation [11, 18]. Thus, we used these 2 drugs to determine the functional role of autophagy activation in intermittent hypoxia-induced cell injury. The addition of rapamycin or chloroquine to hippocampal neurons further increased intermittent hypoxia-induced LC3-II expression compared to intermittent hypoxia alone (Figure 6C). The data suggested that rapamycin and chloroquine functioned by modulating the autophagy response induced by intermittent hypoxia exposure. We then examined whether rapamycin or chloroquine affected intermittent hypoxia-induced hippocampal neuronal apoptosis. Exposure to intermittent hypoxia significantly enhanced the level of cleaved caspase 3, such effects were further upregulated by rapamycin and attenuated by chloroquine (Figure 6C). Similar to the results shown in Figure 6C, a TUNEL assay showed that the upregulation of apoptotic cells caused by intermittent hypoxia was increased after exposure to rapamycin and decreased by chloroquine (Figure 6D and Supplementary Figure 3).
We further demonstrated the critical role of autophagy on hippocampal neuronal apoptosis after exposure to intermittent hypoxia by knocking down autophagy-related genes (Beclin1, Atg5 and Atg7). As expected, transfection with gene-specific small interfering RNAs successfully reduced autophagy-related proteins (Beclin1, ATG5 and ATG7) levels and suppressed intermittent hypoxia-induced upregulation of LC3-II biosynthesis. Similar to the effects induced by chloroquine, siRNAs against autophagy-related genes also decreased hippocampal neuronal apoptosis (Figure 7A–7C). These results indicated that autophagy activation might be an injurious mechanism in hippocampal neurons exposed to intermittent hypoxia.
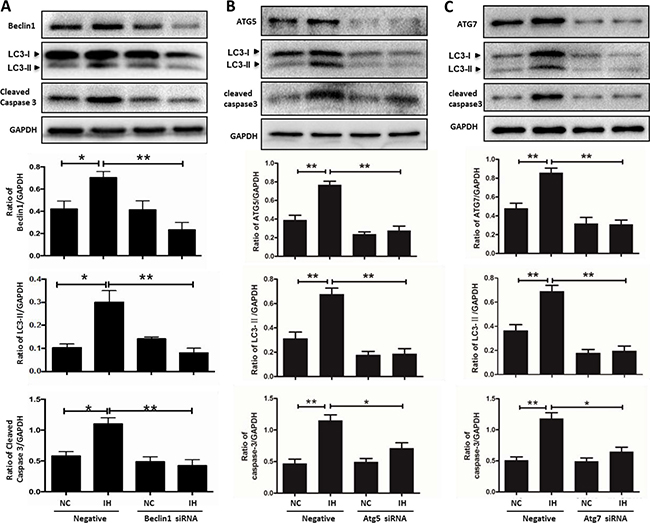
Figure 7: siRNA targeting autophagy-related genes alleviates hippocampal neuronal apoptosis in intermittent hypoxia. The hippocampal neurons were treated with Beclin1, Atg5 and Atg7 siRNA respectively, and then cultured with intermittent hypoxia for 12 h. (A–C) The expressions of autophagy- related proteins and cleaved caspase 3 were assayed by western blot. NC, normoxia control; IH, intermittent hypoxia; *P < 0.05, **P < 0.01.
The decrease of XIAP might be the reason for hippocampal neuronal apoptosis caused by autophagy activation in intermittent hypoxia
XIAP is an inhibitor of apoptosis protein, which is closely related to many causes of apoptosis, including autophagy activation [19]. In order to detect the expression of XIAP and to clarify the mechanism by which autophagy aggravates apoptosis, we investigated the expression of XIAP in intermittent hypoxia. As shown in Figure 8A, compared with the control group, the expression of XIAP decreased, especially in the 12 h intermittent hypoxic group. And we further investigated the effects of XIAP using autophagy activators or inhibitors. As shown in Figure 8B, the addition of rapamycin to hippocampal neurons further decreased intermittent hypoxia-induced XIAP expression compared to intermittent hypoxia alone, but when autophagy was inhibited by chloroquine, the effects were attenuated. Similar to the results shown in Figure 8B, Beclin1 siRNA also reversed the decrease observed in XIAP levels induced by intermittent hypoxia in hippocampal neurons (Figure 8C). These results indicate that the decreasing levels of XIAP may be the reason for hippocampal neuronal apoptosis caused by autophagy activation in intermittent hypoxia.
DISCUSSION
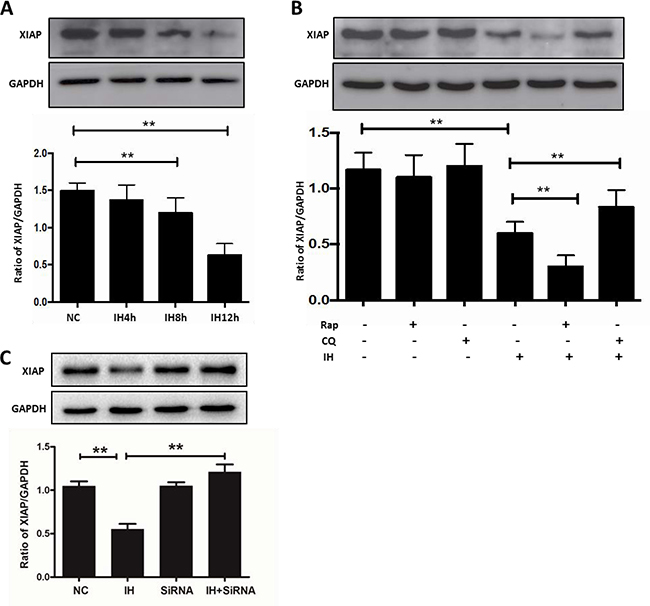
Figure 8: The inhibition of XIAP might be the reason for apoptosis caused by autophagy in intermittent hypoxia. (A) The hippocampal neurons were treated with NC, IH4h, IH8h and IH12h. Detection of XIAP was assayed by western blot. (B) Western blot analysis of XIAP expression in hippocampal neurons after exposure to IH12h, with or without Rap or CQ for 12 h. (C) The hippocampal neurons were treated with Beclin1 siRNA and then cultured with intermittent hypoxia for 12 h. The expression of XIAP was assayed by western blot. (the effect of Beclin1 siRNA on Beclin1 as shown in Figure 7A) Rap, rapamycin, 20 μM; CQ, chloroquine, 30 μM; NC, normoxia control; IH, intermittent hypoxia; *P < 0.05, **P < 0.01.
In our study, we found that the expression of autophagy marker proteins LC3-II and Beclin1 in hippocampal neurons were increased after intermittent hypoxia treatment. In addition, the increase of autophagosomes in hippocampal neurons was observed with electron microscopy. These results suggest that autophagy may be induced by intermittent hypoxia in hippocampal neurons. However, autophagy is a dynamic process including the initiation, formation, maturation and degradation of autophagosomes -- a dynamic flow defined as autophagic flux [20]. Increased expression of LC3-II and Beclin1 or visualization of autophagosomes may result from either an enhancement of autophagosomal formation or inhibition of autophagosomal degradation [21]. Therefore, we examined autophagic flux after exposure to intermittent hypoxia in the following three perspectives. First, we detected the turnover of LC3-I to LC3-II after intermittent hypoxic exposure. Our results showed that the ratio of LC3-II/LC3-I was significantly increased after intermittent hypoxic exposure. At the same time, when EBSS starvation treatment was used as a positive control group in vitro study, the same results were found in hippocampal neurons. Second, we also determined that the expression level of SQSTM1/p62, a well-known autophagic substrate [22], was decreased by intermittent hypoxia treatment. Third, we showed that intermittent hypoxia plus rapamycin enhanced LC3-II/LC3-I turnover compared to rapamycin alone, and the level of SQSTM1/p62 was dramatically reduced. Furthermore, the level of SQSTM1/p62 in hippocampal neurons treated with chloroquine and intermittent hypoxia together was less than that treated by chloroquine alone. These suggest that intermittent hypoxia increases the formation/maturation of autophagosomes rather than blocks the fusion of the autophagosomes and lysosomes. All these data together provide strong evidence that intermittent hypoxia promotes autophagic initiation and maturation.
HIF-1α is a key regulator in hypoxia [23], which can mediate autophagy activation induced by hypoxia and ischemia [24–25]. We found that the expression of HIF-1α is increased in hippocampal neurons exposed to intermittent hypoxia. In addition, we exposed hippocampal neurons to YC-1 (a HIF-1α inhibitor) [26–27], and found that the level of autophagy decreased significantly. What’s more, hippocampal neurons treated with HIF-1α siRNA showed the same results as above. Zhao Yang et al. also found that the activation of autophagy induced by hypoxia could be inhibited by HIF-1α siRNA in microglia cells [28]. These suggest that HIF-1α is necessary for activation of autophagy induced by intermittent hypoxia in hippocampal neurons.
BNIP3 is one of the downstream target proteins of HIF-1α. Recent studies have confirmed that BNIP3 induced autophagy by disrupting Bcl2-Beclin1 interaction under hypoxia conditions [29–30]. Simultaneously, we found that the expression of HIF-1α, BNIP3 and Beclin1 proteins were all increased in hippocampal neurons after intermittent hypoxia exposure. Further results demonstrated that HIF-1α inhibition could significantly decrease the expression of HIF-1α, BNIP3 and Beclin1 induced by intermittent hypoxia. These further suggest that the HIF-1α-BNIP3-Beclin1 signaling pathway is involved in hippocampal neuron autophagy induced by intermittent hypoxia. In addition, some studies have shown that BNIP3 could compete for binding to Bcl2 or phosphorylation of Bcl2 to disrupt the Bcl2-Beclin1 interaction and thus promote the release of potent activated Beclin1 [14, 31], which initiates autophagy. Our results showed that intermittent hypoxia induced the isolation of Bcl2 and Beclin1, resulting in autophagy activation. These findings suggest that intermittent hypoxia induces autophagy activation mainly through HIF-1α activating its downstream effector BNIP3. BNIP3 may then disrupt the interaction of Bcl2-Beclin1 and release Beclin1, thus initiating the autophagy process.
Oxidative stress is not only considered to be one of the pathological mechanisms of intermittent hypoxia-induced multisystem damage, but also an important mechanism that induces autophagy under cellular stresses [16–17]. Our results found that the expression of ROS in the intermittent hypoxia group was remarkably higher than in the control group. However, the expression of ROS and autophagy was significantly decreased by antioxidant intervention. These results indicate that the HIF-1α-BNIP3-Beclin1 signaling pathway mediates autophagy activation, and that ROS also play an important role in the activation. Additionally, studies showed that the activation of autophagy mediated by ROS was closely related to HIF-1α expression under hypoxic conditions [32]. In well oxygenated environments, HIF-1α subunits are hydroxylated at conserved proline residues. These modifications are mediated by PHDs, whose activities are regulated by O2 availability [33]. In conditions of hypoxic stress, PHDs activity is diminished by ROS, and stabilized HIF-1α proteins can induce gene transcription [34]. However, the potential mechanisms of ROS-mediated autophagy activation are still unclear, and require further research.
Autophagy and apoptosis may act independently in parallel pathways or may influence one another. Studies have shown that autophagy could alleviate cell apoptosis under continuous hypoxia, starvation and ischemic conditions [35–38]. Autophagy activation can aggravate neuronal apoptosis injury in ischemia reperfusion [39]. Our results showed inhibition of autophagy by chloroquine significantly decreased the percentage of cell apoptosis or cleaved caspase 3 expression caused by intermittent hypoxia. However, rapamycin increased the injury to hippocampal neurons due to intermittent hypoxia. In addition, we found that apoptosis was also significantly decreased after knockdown of autophagy-related genes. These results suggest that autophagy promotes apoptosis in hippocampal neurons exposed to intermittent hypoxia. Moreover, some studies have shown that autophagy induced by intermittent hypoxia increased apoptosis in the endothelium [40]. However, other studies have shown that intermittent hypoxia-induced autophagy attenuated contractile dysfunction and myocardial injury in rat heart [41]. We speculate that the reasons for these differences may be related to the different duration of intermittent hypoxia and the different objects of study. In addition, many studies have shown that activation of autophagy reduced apoptosis and played a protective role under continuous hypoxia [42]. By contrast, our results demonstrated that autophagy activation induced by intermittent hypoxia played a pro-apoptotic role in hippocampal neuronal cells. As for continuous hypoxia, Carloni S et al. found that autophagy played a protective role in neonatal hypoxic-ischemia induced brain injury. When autophagy acts as a cellular cleaner, it plays a protective role, removing damaged organelles and intracellular proteins [43–44]. However, intermittent hypoxia, as a repeated hypoxia/re-oxygenation process, plays a pro-apoptotic role, which is similar to ischemia and reperfusion. Unfortunately, the pro-apoptotic mechanism of autophagy due to ischemia-reperfusion and intermittent hypoxia is not clear.
A previous study has shown that autophagy can promote apoptosis by clearing the XIAP from nurse cells in late Drosophila melanogaster [19]. Our results showed that decreased expression of XIAP was followed by an increase of hippocampal neuronal apoptosis, which is consistent with the previous study that XIAP is an apoptosis inhibitor. In 1997, Deveraux, Q.L. et al. have found that IAP can suppress caspase 3 and 7, and prevent apoptosis [45]. As for the potential mechanism behind autophagy aggravated the apoptosis, study has shown that inhibition of autophagy could lead to the accumulation of XIAP, which inhibits apoptosis in nurse cells and hepatocellular carcinoma cells [19, 46]. In addition, study has shown that XIAP inhibits autophagy via XIAP-Mdm2-p53 signaling, which promotes tumorigenesis [47]. This suggests that XIAP might interact with autophagy to mediate the apoptosis process. We found that apoptosis was increased and the expression level of XIAP was decreased when autophagy was activated, while these results were reversed when autophagy was inhibited. These results show that the decrease of XIAP may be one of the potential mechanism that autophagy uses to promote apoptosis of hippocampal neurons in intermittent hypoxia. But the mechanism of how XIAP interact with autophagy is still unclear.
In conclusion, we have demonstrated that HIF-1α-BNIP3-Beclin1 signaling pathway and oxidative stress-dependent autophagy activation promotes hippocampal neuronal apoptosis caused by intermittent hypoxia, which may be associated with a decrease of XIAP. Our study suggested that inhibition of autophagy may be a possible intervention strategy for clinical management of cognitive impairment caused by OSA.
MATERIALS AND METHODS
Ethics statement
Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health, and approved by the Animal Care and Use Committee of Tianjin Medical University. All animal procedures were in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. The study was reviewed and approved by the Ethics Committee of Tianjin Medical University. All methods were performed in accordance with the relevant guidelines and regulations of the Animal Care and Use Committee and the Ethics Committee of Tianjin Medical University.
Materials
The following materials were used: chloroquine (Sigma, C6628), rapamycin (Selleck, s1039), YC-1 (Sigma, Y102), NAC (Sigma, A7250–5G), anti-GAPDH (CST, 2118S), anti-SQSTM1 antibody (CST, 5114S), anti-HIF-1α (CST, 14179S), anti-BNIP3 (CST, 3769S), anti-Beclin1 (CST, 3495S), anti-cleaved caspase 3 (CST, 9664S), anti-LC3B (CST, 2775S), anti-XIAP (CST, 287635S), anti-MAP2 (Boster, BM1243), anti-Bcl2 antibody (Santa Cruz, sc-7382), anti-ATG5 (CST, 12994), anti-ATG7 (CST, 8558), secondary antibodies for goat anti-rabbit IgG (Beyotime, A0208), secondary antibodies for goat anti-mouse IgG (Beyotime, A0216), secondary antibodies for goat anti-mouse IgG antibodies (Zhongshan Goldenbridge Biotechnology, ZF-0313) and secondary antibodies for goat anti-rabbit (Zhongshan Goldenbridge Biotechnology, ZF-0311).
Hippocampal neurons culture
Primary hippocampal neuron cultures were obtained from Sprague-Dawley rat pups (< 24 h), which were purchased from the Chinese Academy of Military Science (Beijing, China), and cultured according to the procedure described previously [48–49]. Briefly, the brain was dissected and the hippocampus was removed. The meninges and blood vessels were carefully removed and the hippocampus was minced into 1 mm3 fragments. Then, the hippocampal pieces were dissociated by 0.25% Trypsin-EDTA (Gibco, 25200056) and 100 ng/ml DNase (Promega, M6101) at 37ºC for 30 min. The isolated cells were resuspended in DMEM medium (Gibco, 8116058) with 10% FBS (Gibco, 10099–141) and seeded into 6-well plates or 22*22mm2 glass coverslips in 35-mm Petri dishes pre-coated with 10 μg/ml poly-D-lysine (Beyotime, ST508) at a density of 5*105 per well. After 4 h, the neurons were cultured in Neurobasal-A medium with 2% B27, 1% glutamine and HEPES (Gibco, 15630080) for 7 days at 37ºC in 5% CO2 and 95% O2 atmosphere. Next, the purity of hippocampal neurons were identified by morphology and fluorescence staining of MAP2 (a neuron-specific cytoskeletal protein). (Supplementary Figure 4)
Exposure of hippocampal neurons to intermittent hypoxia
Intermittent hypoxia exposure for hippocampal neurons was conducted as described previously [49]. Briefly, hippocampal neurons were maintained in the cycle of 1.5% O2 for 5 min and 21% O2 for 10 min and sustained normoxia gas mixture. The chamber was equipped with a humidifier, thermostat and molecular sieve to maintain an inner temperature of 37ºC, humidity of 45% and a relative germfree environment. Change of oxygen concentrations in the chamber was monitored by an O2 analyzer. O2 levels in the culture media, 1mm above the cell layer, were sampled and monitored by an oxygen electrode (Lazar, USA) and a blood gas analyzer (AVL OMNI, Swiss), 6 samples per gas environment (data not shown).
Western blot analysis
Hippocampal neuronal protein was isolated following standard protocol for the radioimmunoprecipitation assay (RIPA) buffer (Beyotime, P0013B). Protein concentrations were measured using a BCA assay kit (Thermo, 23227). Equal amounts of the protein were resolved by 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes (Merck Millipore). The membranes were incubated overnight at 4°C with diluted primary antibody against LC3, Beclin1, SQSTM1/p62, HIF-1α, cleaved caspase 3, BNIP3, Bcl2, ATG5, ATTG7, XIAP MAP2 and GAPDH (1:1000), followed by a horseradish peroxidase (HRP) conjugated secondary antibody against rabbit IgG (1: 1000). The blots were treated with enhanced chemiluminescence, and the signals were detected by a ChemiDoc™ Imaging Systems (BIO-RAD). The relative signal intensity was quantified by densitometry with Image J software. All western blots were shown are from a single experiment that is representative of at least three biological replicates.
Immunofluorescence microscopy
The primary hippocampal neurons, grown on coverslips in 3.5-cm dishes, were observed and photographed by an Optical microscope after being treated with intermittent hypoxia or normoxia. The cells were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 15min. Then they were permeabilized with 0.1% Triton X-100 for 10 min and blocked with BSA buffer for 30min at room temperature. Subsequently, cells were incubated with the anti-LC3B (1:50), anti-HIF-1α (1:400), anti-MAP2 (1:100) at 4ºC overnight. After being rinsed with PBS, the sections were incubated for 1 h at room temperature with an appropriate secondary antibody. Finally, 4, 6-diamidino-2-phenylindole (DAPI) (Beyotime, C1002) was used as a counterstain. Images were taken under an OLYMPUS confocal microscope (FV-ASW) and OLYMPUS microscope (cellSens system). Apoptosis was assessed using a TUNEL apoptosis assay kit (Roche, 11684817910) according to the manufacturer’s instructions. LC3-II dots were counted in individual hippocampal neurons and the average of dots in at least 30 cells was presented in the Figures. HIF-1α and TUNEL positive cells were counted in individual views.
Transmission electron microscopy
The primary hippocampal neurons collected by centrifugation were fixed, embedded and sectioned as described previously [50]. Ultrathin sections were observed using a HITACHI H-7500 electron microscope. The AL was defined as single-membrane structures containing cytoplasmic components at various stages of degradation.
Transfection of small interfering RNAs
The primary hippocampal neurons were transfected with siRNA-HIF-1α, siRNA-Beclin1, siRNA-Atg5, and siRNA-Atg7 via Lipofectamine 3000 (Invitrogen, 1734497) according to the manufacturer’s instructions. The negative control, HIF-1α, Beclin1, Atg5 and Atg7 siRNA were purchased from Gene Pharma. The primary hippocampal neurons transfected with HIF-1α and autophagy-related genes siRNA were then incubated with intermittent hypoxia pretreatment. The results of negative control experiments were shown in the Supplementary Figure 5.
Immunoprecipitate assay
An acellular assay of immunoprecipitate checking the disruption of the Bcl2–Beclin1 complex by intermittent hypoxia was performed. Briefly, the hippocampal neurons were subjected to intermittent hypoxia for 12 h or starvation for 3 hours. Then, cells were washed three times in phosphate-buffered saline (PBS) and the cells were lysed in cold RIPA buffer for 30 min on ice before being centrifuged at 15,000g for 15 min at 4°C. The immunoprecipitate assay was performed with 500 ug of a whole-cell lysate. The samples were incubated overnight with 10 ug of anti-Bcl2 (Santa Cruz, sc-7382) and then incubated with 20 ug Protein A/G PLUS-Agarose (Santa Cruz, sc-2003) for 5 hours. The immunoprecipitates then were analyzed by immunoblot with anti-Beclin antibody (CST, 3495S).
Detection of intracellular ROS
Intracellular ROS levels were measured using the Reactive Oxygen Species Assay Kit (Beyotime, S0033) according to the manufacturer’s instructions. Cells were treated with intermittent hypoxia for the indicated hours in 6-well plates. After treatment, the media was removed, 1ml pre-warmed serum-free medium containing 1 μM DCFH-DA was added to each well and incubated for 20min at room temperature. Cells were then washed with serum-free medium three times and the intracellular ROS levels (DCF signals) were measured using an OLYMPUS microscope (cellSens system). The fluorescence intensity of ROS was quantified by densitometry with Image J software.
Statistical analysis
All statistical tests were performed with SPSS 16.0. All data are expressed as the means ± standard error of the mean (S.E.M.). Two group comparisons were carried out using our independent-sample t test. Multiple group comparison was performed using ANOVA, followed by Bonferroni or Dunnett post-hoc tests. The P value was considered as statistically significant if it was less than 0.05.
ACKNOWLEDGMENTS AND FUNDING
We thank Dr Zuoming Sun (Beckman Research Institute of the City of Hope) for technical assistance. This work was supported by National Natural Science Foundation of China (No. 81670086 and 81370183), Tianjin Natural Science Foundation (No. 14JCYBJC27800 and 16JCQNJC11000) and International S&T Cooperation Program of China (ISTCP, 2015DFA50310).
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
1. Mizushima N. Autophagy: process and function. Genes Dev. 2007; 21:2861–2873.
2. Hu S, Xi G, Jin H, He Y, Keep RF, Hua Y. Thrombin-induced autophagy: a potential role in intracerebral hemorrhage. Brain Res. 2011; 1424:60–66.
3. Descloux C, Ginet V, Clarke PG, Puyal J, Truttmann AC. Neuronal death after perinatal cerebral hypoxia-ischemia: Focus on autophagy-mediated cell death. Int J Dev Neurosci. 2015; 45:75–85.
4. Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008; 13:718–736.
5. Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012; 18:250–260.
6. Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. 2015; 39:891–898.
7. Aaronson JA, van Bennekom CA, Hofman WF, van Bezeij T, van den Aardweg JG, Groet E, Kylstra WA, Schmand B. Obstructive Sleep Apnea is Related to Impaired Cognitive and Functional Status after Stroke. Sleep. 2015; 38:1431–1437.
8. Dayyat EA, Zhang SX, Wang Y, Cheng ZJ, Gozal D. Exogenous erythropoietin administration attenuates intermittent hypoxia-induced cognitive deficits in a murine model of sleep apnea. BMC Neurosci. 2012; 13:77.
9. Wu J, Stefaniak J, Hafner C, Schramel JP, Kaun C, Wojta J, Ullrich R, Tretter VE, Markstaller K, Klein KU. Intermittent Hypoxia Causes Inflammation and Injury to Human Adult Cardiac Myocytes. Anesth Analg. 2016; 122:373–380.
10. Torres M, Palomer X, Montserrat JM, Vázquez-Carrera M, Farré R. Effect of ovariectomy on inflammation induced by intermittent hypoxia in a mouse model of sleep apnea. Respir Physiol Neurobiol. 2014; 202:71–74.
11. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010; 140:313–326.
12. Sun Y, Xing X, Liu Q, Wang Z, Xin Y, Zhang P, Hu C, Liu Y. Hypoxia-induced autophagy reduces radiosensitivity by the HIF-1α/miR-210/Bcl-2 pathway in colon cancer cells. Int J Oncol. 2015; 46:750–756.
13. Na JI, Na JY, Choi WY, Lee MC, Park MS, Choi KH, Lee JK, Kim KT, Park JT, Kim HS. The HIF-1 inhibitor YC-1 decreases reactive astrocyte formation in a rodent ischemia model. Am J Transl Res. 2015; 7:751–760.
14. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009; 29:2570–2581.
15. Li D, Wang J, Hou J, Fu J, Chang D, Bensoussan A, Liu J. Ginsenoside Rg1protects starving H9c2 cells by dissociation of Bcl-2-Beclin1 complex. BMC Complement Altern Med. 2016; 16:146.
16. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007; 26:1749–1760.
17. Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011; 14:2179–2190.
18. Kumamoto T, Ueyama H, Watanabe S, Murakami T, Araki S. Effect of denervation on overdevelopment of chloroquine-induced autophagic vacuoles in skeletal muscles. Muscle Nerve. 1993; 16:819–826.
19. Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010; 190:523–531.
20. Esteban-Martinez L, Boya P. Autophagic flux determination in vivo and ex vivo. Methods. 2015; 75:79–86.
21. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8:445–544.
22. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015; 282:4672–4678.
23. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92:5510–5514.
24. Han ZB, Ren H, Zhao H, Chi Y, Chen K, Zhou B, Liu YJ, Zhang L, Xu B, Liu B, Yang R, Han ZC. Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis. 2008; 29:1853–1861.
25. Shi H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med Chem. 2009; 16:4593–4600.
26. Tsui L, Fong TH, Wang IJ. The effect of 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1) on cell viability under hypoxia. Mol Vis. 2013; 19:2260–73.
27. Kim HL, Yeo EJ, Chun YS, Park JW. A domain responsible for HIF-1alpha degradation by YC-1, a novel anticancer agent. Int J Oncol. 2006; 29:255–60.
28. Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H. Hypoxia Induces Autophagic Cell Death through Hypoxia-I nducible Factor 1 a in Microglia. PLoS One. 2014; 9:e96509.
29. Zhao Y, Chen G, Zhang W, Xu N, Zhu JY, Jia J, Sun ZJ, Wang YN, Zhao YF. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1α/BNIP3 signaling pathway. J Cell Physiol. 2012; 227:639–648.
30. Zhao ST, Chen M, Li SJ, Zhang MH, Li BX, Das M, Bean JC, Kong JM, Zhu XH, Gao TM. Mitochondrial BNIP3 upregulation precedes endonuclease G translocation in hippocampal neuronal death following oxygen-glucose deprivation. BMC Neurosci. 2009; 10:113.
31. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates StarvationInduced Autophagy. Mol Cell. 2008; 30:678–688.
32. Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, Lee D, Yang JS, Kim S, Mair WB, Lee C, Lee SS, Lee SJ. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2014; 111:E4458-E4467.
33. Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012; 31:2448–2460.
34. Chowdhury R, Godoy LC, Thiantanawat A, Trudel LJ, Deen WM, Wogan GN. Nitric oxide produced endogenously is responsible for hypoxia-induced HIF-1αstabilization in colon carcinoma cells. Chem Res Toxicol. 2012; 25:2194–2202.
35. Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008; 172:454–469.
36. Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014; 10:846–60.
37. Xia DY, Li W, Qian HR, Yao S, Liu JG, Qi XK. Ischemia preconditioning is neuroprotective in a rat cerebral ischemic injury model through autophagy activation and apoptosis inhibition. Braz J Med Biol Res. 2013; 46:580–588.
38. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004; 432:1032–1036.
39. Chen W, Sun Y, Liu K, Sun X. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014; 9: 1210–1216.
40. Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, Lee PJ. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol. 2015; 35:1166–1178.
41. Maeda H, Nagai H, Takemura G, Shintani-Ishida K, Komatsu M, Ogura S, Aki T, Shirai M, Kuwahira I, Yoshida K. Intermittent-hypoxia induced autophagy attenuates contractile dysfunction and myocardial injury in rat heart. Biochim Biophys Acta. 2013; 1832: 1159–1166.
42. Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy. 2009; 5:221–223.
43. Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008; 32:329–339.
44. Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010; 115:68–78.
45. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997; 388:300–304.
46. Wang N, Feng Y, Zhu M, Siu FM, Ng KM, Che CM. A novel mechanism of XIAP degradation induced by timosaponin AIII in hepatocellular carcinoma. Biochim Biophys Acta. 2013; 1833:2890–2899.
47. Huang X, Wu Z, Mei Y, Wu M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signaling. EMBO J. 2013; 32: 2204–2216.
48. Pont-Lezica L, Colasse S, Bessis A. Depletion of microglia from primary cellular cultures. Methods Mol Biol. 2013; 1041:55–61.
49. Song S, Tan J, Miao Y, Zhang Q. Effect of different levels of intermittent hypoxia on autophagy of hippocampal neurons. Sleep Breath. 2017; 21:791–98. https://doi.org/10.1007/s11325-017-1512-7.
50. Fukunaga Y, Hirase A, Kim H, Wada N, Nishino Y, Miyazawa A. Electron microscopic analysis of a fusion protein of postsynaptic density-95 and metallothionein in cultured hippocampal neurons. J Electron Microsc (Tokyo). 2007; 56: 119–129.