
INTRODUCTION
Epilepsy is a common serious neurological disease, which does latent harm to human health [1]. Treatments of epilepsy are currently limited to drugs that provide only symptomatic control of seizures. Current treatments have adverse effects and are ineffective in up to 40% of patients [2]. However, the pathogenesis of epilepsy, and the resistance to available antiepileptic drugs remains poorly understood [1]. Though a better understanding of the genetic targets and mechanisms of anti-seizure drug activity offers trusty direction in drug selection for individual patients, the experiments in need are ethically and technically difficult. For this reason, animal models are useful in anti-seizure drug research [3]. One such model organism is the dog, because both humans and companion animals suffer from epilepsy, which is a chronic neurologic disorder affecting nearly 50 million people and 0.5–5% of the canine population worldwide [4]. Conversely, models of anti-seizure medication targets and activities that are derived from humans are useful in epilepsy research in a canine context.
Domestic dogs share many parallel syndromes with humans [4]. Evolutionarily speaking, similarities in medical genetics between dogs and humans can be found throughout history. Dogs evolved from the gray wolf in East Asia at least 15,000 years ago, co-operationally integrated with mankind, and developed a mutually beneficial relationship over a long period of time. Many dog breeds have shown a high prevalence of specific diseases, such as blindness, cancer, heart disease, and epilepsy [5], most of which are also common in humans. Furthermore, the clinical manifestations of the two species are often similar [6]. High-quality draft genome sequencing, has verified a degree of similarity of 96% between dog and human genes [7]. Therefore, the domestic dog is treated as a valuable model species for human diseases [4], and canine epilepsy pathogenesis is regarded as the same as that of human epilepsy. Thus, human anti-epilepsy drugs are promising agents for canine therapy.
Even prior to the problems of selection of anti-seizure medications, diagnosis of epilepsy is complicated. Generally, at least two unprovoked seizures, or one seizure combined with additional symptoms, e.g., adverse head injury, previous stroke or an abnormality on an electroencephalogram (EEG) brain scan, are required for diagnosis [8]. However, it is more difficult than that in some cases, the cause of most cases of epilepsy is unknown [9]. Generally speaking, canine epilepsy can be divided into reactive seizures, structural epilepsy, and idiopathic epilepsy according to etiology [10]. The mechanism of reactive seizures is the response of the brain to abnormal systemic metabolism, such as hypoglycemia, hypocalcemia, uremia, poisoning and so on. Structural epilepsy is mainly due to abnormal brain structure, including brain abnormalities caused by hydrocephalus, craniocerebral tumor causing compression of brain parenchyma, trauma caused by brain injury or damage, infectious diseases such as toxoplasmosis, rabies, tetanus and other pathogens into the brain tissue, or destruction of the normal structure of brain tissue. Idiopathic epilepsy or primarily genetic epilepsy, which is associated with congenital or genetic factors, is the most common neurological disorder in dogs [11]. However, the mechanism of idiopathic epilepsy remains unclear.
Anti-epileptic drugs (AEDs) have long been administered to humans and dogs. Early in 1857, bromide was introduced to treat epilepsy [12]. It was combined with potassium or sodium to form a crystal powder. Currently, there are over 20 anti-epileptic drugs on the market for treating humans. These include carbamazepine (also known as carbatrol, equetro, tegretol), gabapentin (neurontin), levetiracetam (keppra), lamotrigine (lamictal), oxcarbazepine (trileptal), pregabalin (lyrica), tiagabine (gabitril), topiramate (topamax), and valproate (depakote, depakene). The first three types of drugs are also widely used in dogs. Furthermore, there are also AEDs for dogs, such as clorazepate, diazepam, felbamate, phenobarbital, phenytoin, primidone, valproic acid, and zonisamide (http://www.canine-epilepsy.com/AEDs.html). Though many new drugs have been developed that work well in humans, their efficacies decrease in canine treatment because dogs metabolize these drugs much more quickly, making new drug discovery for dogs challenging.
The recent availability of a large number of canine genetic samples presents an opportunity to realize the promise of using understanding of the genetic targets and mechanisms of anti-seizure drug activity to help in drug selection for individual patients. A promising approach to finding these genetic things is the Genome-Wide Association Study (GWAS) [13]. The first GWAS on focal epilepsy was carried out with 3,445 patients and 6,935 controls from Northern Europe. No significant genetic markers were found, as none of the single nucleotide polymorphisms (SNPs) corresponded to P-values that reached significance after Bonferroni correction [14]. Flank expansion of 95 and 116 kb from the top SNP only led to a predicted pseudogene. A second GWAS study on 1,000 patients from China identified two highly correlated variants in CAMSAP1L1 (CAMSAP1-Like 1), a calmodulin-regulated spectrin-associated protein expressed in neurons and astrocytes in the mammalian nervous system [15]. Although each GWAS study thus far has offered a list of candidate genes, few of these genes have been functionally validated. For most patients, epilepsy is regarded as a complex disorder associated with multiple genes and external environmental factors, which also makes it difficult to rely on single-gene associations. Therefore, it is hoped that earlier findings can be enhanced by additional GWAS results and next-generation DNA sequencing (NGS) [16].
Prior to the GWAS, 10 genes had been reported as underlying canine epilepsy [4]. Among these, 9 genes are associated with progressive myoclonic epilepsy in several dog breeds, i.e., EPM2B [17], CLN8 [18], CLN5 [19], CTSD [20], TPP1 [21], PPT1 [22], ARSG [23], CLN6 [24], and ATP13A2 [25], and one gene, LGI2, is associated with genetic epilepsy [26]. In 2015, using GWAS, Hayward et al. [27] collected the largest set of canine GWAS results. Among these results, the authors found additional candidate genes related to epilepsy in Irish Wolfhounds using 34 cases and 168 controls. However, very little is currently known about the genetic factors that are involved here. Several research efforts are currently focused on the discovery of key genes associated with epilepsy, although the results remain far from unveiling the entire mechanism. This concern leads to our current research.
Among published idiopathic epilepsy studies that have investigated inheritance patterns, various breeds show evidence of autosomal recessive inheritance. Many of these studies also demonstrated polygenic inheritance, indicating that the genetic basis of idiopathic epilepsy is complex in dogs [4]. In this paper, we used a hybrid method combining GWAS and HotNet2 with Hayward et al.’s data [27] and found 26 significant subnetworks correlated with canine epilepsy. Combined with gene ontology (GO) enrichment analysis, our method not only identified PPFIA1, ASB5, SLC1A2 and MCU to correspond to significant P-values in the GWAS results but also identified the pathogenic genes PTPRM, PTPRD and SGCE, which were omitted by the GWAS analysis. Among the above seven genes, PPFIA1 [28], SLC1A2 [29], PTPRM [30] and SGCE [31] were reported to be associated with epilepsy, and the others were associated with mental illness [32–34]. Moreover, drug enrichment analysis showed that the subnetworks we identified are capable of enriching anti-epilepsy drugs. In view of the combination therapy in cancer enhances efficacy and reduces drug resistance compared to the mono-therapy approach [35], we believe that this strategy can work on epilepsy. When targets of different anti-epilepsy drugs are enriched in the same subnetwork, their combinations are also more likely to effectively treat epilepsy. Ultimately, 22 drug combinations were obtained, one of which was confirmed in the Drug Combination database (DCDB, https://omictools.com/drug-combination-database-tool). The results of this study showed the effectiveness of our method in exploring the pathogenesis of canine epilepsy, and this approach increases cost efficiency for anti-epilepsy drug discovery.
RESULTS AND DISCUSSION
Replication of the GWAS experiment
MLogit [36] was used to analyze the GWAS data. Our experiment completely replicated the results of the previous GWAS and achieved high consistency.
By using the NCBI SNP database (ftp://ftp.ncbi.nih.gov/snp/organisms/dog_9615/chr_rpts/), 44,542 SNPs were mapped to dog genes. Among them, the top 82 SNPs passed the Bonferroni thresholded P-value of 1.12e-6, and the top 45 SNPs passed the significance test with the empirical thresholded P-value of 5e-8. These results are shown in Figure 1 and Table 1, and the full dataset is shown in Supplementary Table 1.
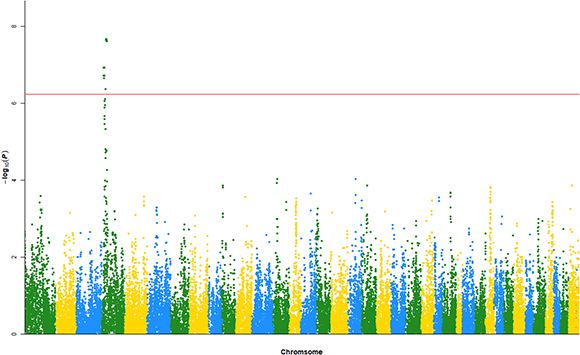
Figure 1: Manhattan plot of the replicated GWAS experiment.
Table 1: Results that reached genome-wide significance in Hayward et al.’s study [27] and our replicated GWAS experiment
Disease |
Case/Control |
Name of breeds |
Hayward et al. [27] |
Our replication |
|---|---|---|---|---|
Idiopathic epilepsy |
34/168 |
Irish Wolfhound |
4:7.5–21 |
4:7.53–20.824 |
Identification of significant subnetworks by HotNet2 and subsequent gene ontology analysis
Using automated procedures, HotNet2 identified 26 significant subnetworks (P = 0.02, Supplementary Table 2), which contained a total of 370 genes. A GO enrichment analysis of the genes was performed for each subnetwork (http://www.geneontology.org). Intriguingly, the results of the GO analysis from 4 of the 26 subnetworks were associated with epilepsy, as shown in Table 2.
Table 2: Results of the subnetwork GO analyses
Subnetwork index |
Gene ontology analysis |
Genes |
P-value |
|---|---|---|---|
1 |
GO molecular function complete |
PTPRD, PTPRK, |
0.0126 |
4 |
amino acid transmembrane transport |
SLC36A3, SLC1A2, |
6.06E-06 |
11 |
GO molecular function complete |
CREB3L1, CREB3L2 |
0.0316 |
17 |
sarcoglycan complex |
SGCD, SGCZ, SGCE |
1.77E-06 |
The first subnetwork was the largest, containing 50 genes. The enrichment term used was “protein tyrosine phosphatase activity,” which is a significant molecular function, as previous studies have shown that mutations in protein tyrosine phosphatase can cause progressive myoclonus epilepsy [37–39]. Specifically, the genes involved in protein tyrosine phosphatase activity in this subnetwork are PTPRD, PTPRK, PTPRM, PTPRT and EYA3. Among these five genes, PTPRM is associated with mesial temporal lobe epilepsy [30], and PTPRD is implicated in the neurological disorder restless legs syndrome (RLS) [32].
PTPRM was reported to be involved in mesial temporal lobe epilepsy [30]. PTPRM encodes a cell surface receptor that is a homophilic cell-cell adhesion molecule expressed in neuronal, glial and endothelial cells [40]. PTPRM is part of the network associated with cell growth and differentiation mechanisms [41] and is also associated with neurite outgrowth [42]. Santos et al quantified PTPRM transcripts in hippocampal samples from patients and controls and observed up-regulated PTPRM transcript levels in the patient group compared with the control group [30]. Their results indicated involvement of PTPRM in the predisposition to mesial temporal lobe epilepsy.
Some evidence has shown that variants in the 5′ UTR of PTPRD are implicated in the neurological disorder RLS. These RLS-associated SNPs consist of ten noncoding exons contained in two known long splice variants expressed predominantly in fetal and adult brain tissue [32]. PTPRD encodes protein tyrosine phosphatase receptor type D, and it is associated with the regulation of synapse development and function [43]. RLS is the same neurological disease as epilepsy, and therefore, PTPRD may play a role in epilepsy.
Although the P-values of PTPRM and PTPRD were 0.0816 and 0.0723, respectively, which were not significant in the GWAS, they were highlighted by HotNet2. The other three protein tyrosine phosphatases were not associated with any relevant reports but may be involved in epilepsy etiology and deserve further study. Along with the other four genes, PTPRD was associated with two other latent vital genes discovered by HotNet2, PPFIA1 and ASB5, which are marked as two dashed circles in Figure 2. PPFIA1 encodes the Liprin-a1 protein, which contains 1202 amino acid residues with a molecular weight of approximately 136 kDa. Yin et al first reported that Liprin-a1 protein exists in the temporal neocortex of intractable epilepsy patients. They found that the expression level of Liprin-a1 was significantly higher in intractable temporal lobe epilepsy patients than in controls. Their results suggested that increased expression of Liprin-a1 in the brain is associated with human intractable epilepsy [28]. In addition, ASB5 encodes a protein in the ankyrin repeat and SOCS box-containing (ASB) family of proteins. Novel copy number variations (CNVs) in ASB5 have been identified in children with autism [33]. Davis et al found a 166-kb deletion on chromosome 4q34.2 in two of three trizygotic triplets, and this deletion included ASB5. Another study reported that deletions in this chromosome region are associated with several features including mild mental retardation, velo-cardio-facial (VCF) syndrome-like features, and finger clinodactyly [33]. Considering that autism and epilepsy are both neurological diseases, these results demonstrate the effectiveness of this method.
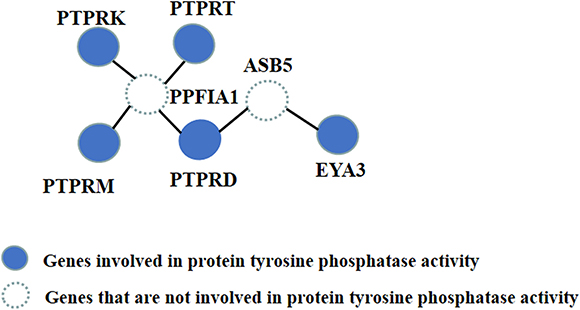
Figure 2: The topologies of genes involved in protein tyrosine phosphatase activity.
The fourth subnetwork contained 31 genes. The GO biological process results are shown in Table 2. The amino acid transmembrane transport function gene results yielded SLC36A3, SLC1A2, SLC38A2, SLC7A8, SLC38A10, and SLC38A11. All these genes transcribe proteins in the solute carrier family. Mutations in SLC1A2 are critical causes of epileptic encephalopathies [29]. SLC1A2 encodes one of the major glutamate transporters; mutations in SLC1A2 in mice cause impaired glutamate uptake, and the resulting excess glutamate leads to subsequent excitotoxicity. The previous study defined a type of SLC1A2 encephalopathy and highlighted the importance of SLC1A2 as a glutamatergic gene associated with epileptic encephalopathies [29]. The other genes, SLC36A3, SLC38A2, and SLC7A8, are associated with the transport of anti-epilepsy drugs [44–46].
HotNet2 also identified a subnetwork containing eleven proteins. The GO molecular functions are presented in Table 2. Studies have shown that inhibition of cAMP response element binding protein (CREB) transcription can shorten the duration of status epilepticus [47]. In this subnetwork, CREB3L1 and CREB3L2 were classified as belonging to the CREB protein family, and therefore, their mutations may regulate CRE transcription and appear to play a role in epilepsy.
The seventeenth subnetwork included eight genes, and the GO cellular components are listed in Table 2. The three genes involved in the sarcoglycan complex were SGCD, SGCZ, and SGCE. Interestingly, though SGCE was not significantly associated with epilepsy based on the GWAS method alone (P = 0.0517), it was highlighted in our method. In fact, it has been reported that epsilon-sarcoglycan (SGCE) mutations can lead to seizures in myoclonus-dystonia patients [31]. This gene encodes the epsilon member of the sarcoglycan family. Sarcoglycans are transmembrane proteins that are components of the dystrophin-glycoprotein complex, which link the actin cytoskeleton to the extracellular matrix. Studies have indicated that loss-of-function mutations in the SGCE gene are found in approximately 50% (range 20–80%) of epsilon-sarcoglycan cases [34, 48]. The mitochondrial calcium uniporter (MCU), which is associated with the uniplex complex, is also involved in many neurological diseases [49]. There is evidence demonstrating that mitochondrial dysfunction contributes to the pathophysiology of epilepsy [50, 51]. The MCU localizes to the inner membrane and is widely accepted to be responsible for calcium uptake by mitochondria [52]. Wang et al investigated the role of MCU in the rat pilocarpine model of epilepsy, which replicates the key features of temporal lobe epilepsy, and they found that MCU inhibition exerted a neuroprotective effect on seizure-induced brain injury through the mitochondria/ROS/CytC pathway [49].
In summary, the systems genetics analysis performed here broadened our understanding of mutations in epilepsy. Some gene mutations in these subnetworks have been reported to be significant in epilepsy. These results illustrate the effectiveness of the systems genetics method in determining the pathogenesis of epilepsy.
Drug enrichment analysis of HotNet2 subnetworks
To test the effectiveness of the HotNet2 method in drug discovery, we counted the enrichment of the drugs in these subnetworks. We collected and sorted 5,452 active drugs corresponding to 2,440 targets in three databases: DrugBank (https://www.drugbank.ca), Therapeutic Target Database, (TTD, http://bidd.nus.edu.sg/group/cjttd/), and ClinicalTrials (https://www.clinicaltrials.gov). Among them, there were 79 drugs for the treatment of epilepsy, generating 226 targets. The HotNet2 results based on the relationships of drugs and their targets included 370 genes. We were able to identify 221 active drugs, of which 22 are used for the treatment of epilepsy (Table 3). The results of a hypergeometric test were significant (P = 2.130943e-13), indicating that the HotNet2 approach can markedly enrich anti-epilepsy drugs.
Table 3: Drug enrichment of subnetworks
Subnetwork index |
Drugs |
Targets |
|---|---|---|
2 |
Adinazolam |
GABRP |
Clobazam |
GABRP |
|
Clonazepam |
GABRP |
|
Diazepam |
GABRP |
|
Lorazepam |
GABRP |
|
Meprobamate |
GABRP |
|
Metharbital |
GABRP |
|
Nitrazepam |
GABRP |
|
Primidone |
GABRP |
|
Propofol |
GABRP |
|
Topiramate |
GABRP |
|
Lacosamide |
SCN10A |
|
Valproic Acid |
SCN10A |
|
4 |
Felbamate |
GRIN3A |
Gabapentin |
GRIN3A |
|
Ketamine |
GRIN3A |
|
Phenobarbital |
GRIN3A |
|
14 |
CPP-15 |
ABAT |
DP-VPA |
ABAT |
|
Tiagabine |
ABAT |
|
Valproic acid |
ABAT |
|
Vigabatrin |
ABAT |
|
19 |
Zonisamide |
CA13 |
We also examined the drug enrichment of a single genetic locus in the GWAS results. We selected the most significant P-value of the first 370 genes and obtained 248 active agents, including 15 epilepsy agents. A P-value of 1.912487e-06 was obtained using the hypergeometric test. By comparison, the capacity of the HotNet2 method to enrich anti-epilepsy drugs was dramatically higher than that of the GWAS method.
Potential combinatorial anti-epilepsy drugs
If drugs that treat epilepsy are enriched in the same subnetwork and correspond to different targets, these drugs may be more effective in combination. Through drug enrichment, the second subnetwork contained 13 drugs, of which 11 drugs corresponded to the target GABRP, and the other two drugs corresponded to the target SCN10A. Therefore, we considered 22 potential combinations of anti-epilepsy drugs (Table 4). In PubMed, we counted the number of co-occurrences of these combinations of drugs and the keyword “epilepsy” in abstracts. As shown in Table 4, six of the drug combinations were found in a total of more than 100 abstracts. In addition, in the Drug Combination database (DCDB, https://omictools.com/drug-combination-database-tool), we found that the combination of topiramate and valproic acid (Drug Combination ID: DC000445) has been reported to play a role in anti-epileptic treatment, indicating that this method for identifying drug combinations is reliable.
Table 4: Potential drug combinations for epilepsy
Index |
Potential combinations of drugs |
Count |
|---|---|---|
1 |
Adinazolam + Lacosamide |
0 |
2 |
Clobazam + Lacosamide |
17 |
3 |
Clonazepam +Lacosamide |
10 |
4 |
Diazepam + Lacosamide |
13 |
5 |
Lorazepam + Lacosamide |
12 |
6 |
Meprobamate + Lacosamide |
0 |
7 |
Metharbital + Lacosamide |
0 |
8 |
Nitrazepam + Lacosamide |
2 |
9 |
Primidone + Lacosamide |
7 |
10 |
Propofol + Lacosamide |
9 |
11 |
Topiramate + Lacosamide |
58 |
12 |
Adinazolam + Valproic Acid |
0 |
13 |
Clobazam + Valproic Acid |
142 |
14 |
Clonazepam + Valproic Acid |
457 |
15 |
Diazepam + Valproic Acid |
407 |
16 |
Lorazepam + Valproic Acid |
115 |
17 |
Meprobamate + Valproic Acid |
9 |
18 |
Metharbital + Valproic Acid |
1 |
19 |
Nitrazepam + Valproic Acid |
35 |
20 |
Primidone + Valproic Acid |
272 |
21 |
Propofol + Valproic Acid |
41 |
22 |
Topiramate + Valproic Acid |
521 |
MATERIALS AND METHODS
Data & materials
The data were from a study by Hayward et al. [27], an extensive dog genotyping dataset with 4,224 samples genotyped on a semicustom 180,000 SNP array for several complex traits. Among the most common recorded diseases collected in these data, there were 7 across-breed phenotypes (canine hip dysplasia, elbow dysplasia, cranial cruciate ligament disease, mast cell tumor, lymphoma, portosystemic vascular anomalies and mitral valve degeneration) and 5 within-breed phenotypes (idiopathic epilepsy in Irish Wolfhounds, granulomatous colitis in Boxers and Bulldogs, lymphoma in Golden Retrievers, mast cell tumor in Labrador Retrievers and portosystemic vascular anomalies in Yorkshire Terriers). To avoid the diversity between across-breed individuals, we retained the disease idiopathic epilepsy (IE) in Irish Wolfhounds, which consisted of 200 individuals (34 cases and 168 controls) and 160,727 SNPs. Case dogs were diagnosed by metabolic screening, magnetic resonance imaging and EEG, while control dogs were older than 5 years and without a history of seizures.
The raw GWAS data were preprocessed by PLINK v1.9 (http://zzz.bwh.harvard.edu/plink/tutorial.shtml). Only SNPs with a minor allele frequency (MAF) > 0.05 and Hardy–Weinberg test (HWE) < 0.001 in these individuals were included. After quality control, 89,085 SNPs remained.
Replication of the GWAS experiment using multiple logistic regression
Logistic regression (LR) is a suitable algorithm for associating phenotypes with different genotypes. Assuming Y is the binary genotype variable, and x is the phenotype variable (continuous or discrete), we can obtain the association of y and x by LR. The goal of LR is to model p(x) ≡ P(Y = 1 | x). In LR, the probability p(x) is modeled by 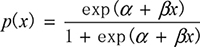 , which can be conveniently converted to
, which can be conveniently converted to 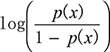 . To test H0 : β = 0 vs. H1 : β ≠ 0, the test statistic
. To test H0 : β = 0 vs. H1 : β ≠ 0, the test statistic 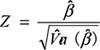 is used.
is used.
To match the multinomial nature of the genotype data, we use multinomial logistic regression (MLogit, or multiclass logistic regression [36]) to calculate the association of y and x. We denote the genotype variable y = 0, 1, and 2, and the MLogit model is as follows:
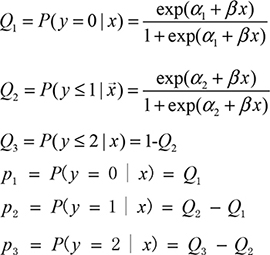
We can estimate the parameters α, β1, β2 using the maximum likelihood method and obtain the probabilities p1, p2 and p3. In this study, we used the R package “MLogit” [36] to obtain the estimators.
Using the HotNet2 algorithm to find subnetworks related to epilepsy
HotNet2 (HotNet diffusion-oriented subnetworks) performs well for finding significant subnetworks, which consist of genes with a higher mutation tendency than expected by chance in a large gene interaction network. HotNet2 is based on a heat diffusion kernel algorithm by considering random walks with restarts [53].
The main inputs to HotNet2 include a heat vector, which contains the score of each gene and the interactions among the corresponding gene pairs. First, to compute the heat vector for each gene, the sorted P-values of SNPs in the GWAS were used after removing redundancy. Specifically, the minimum GWAS P-value of the SNPs in the gene was set as the final heat value of the gene. Thus, 12,355 genes with their corresponding P-values were obtained. To avoid information loss of the amount of gene mutations, Spearman testing was performed to indicate the positive correlations between the number of gene mutations and the negative logarithm of the minimum P-value (i.e., credibility). Here, the correlation value for Spearman testing was 0.294, and the credibility was 9.0459e-245, i.e., the minimum P-value contains information on the number of mutations. Afterwards, the top 20% of genes (n = 2471) were selected as the input, and the negative log base 10 of each P-value was used for the computation formula. Second, to obtain the interactions used for HotNet2, canis lupus familiaris protein-protein interaction networks were derived from the STRING database (STRING, http://www.string-db.org). As the default score threshold is usually 400 [54], interactions with scores lower than 400 were discarded.
The parameter in HotNet2 was set as the default, β = 0.4, as cited by Leiserson et al [53]. Finally, the most significant (P < 0.05) subnetworks were identified.
Study flowchart
A flowchart of this study is shown in Figure 3.
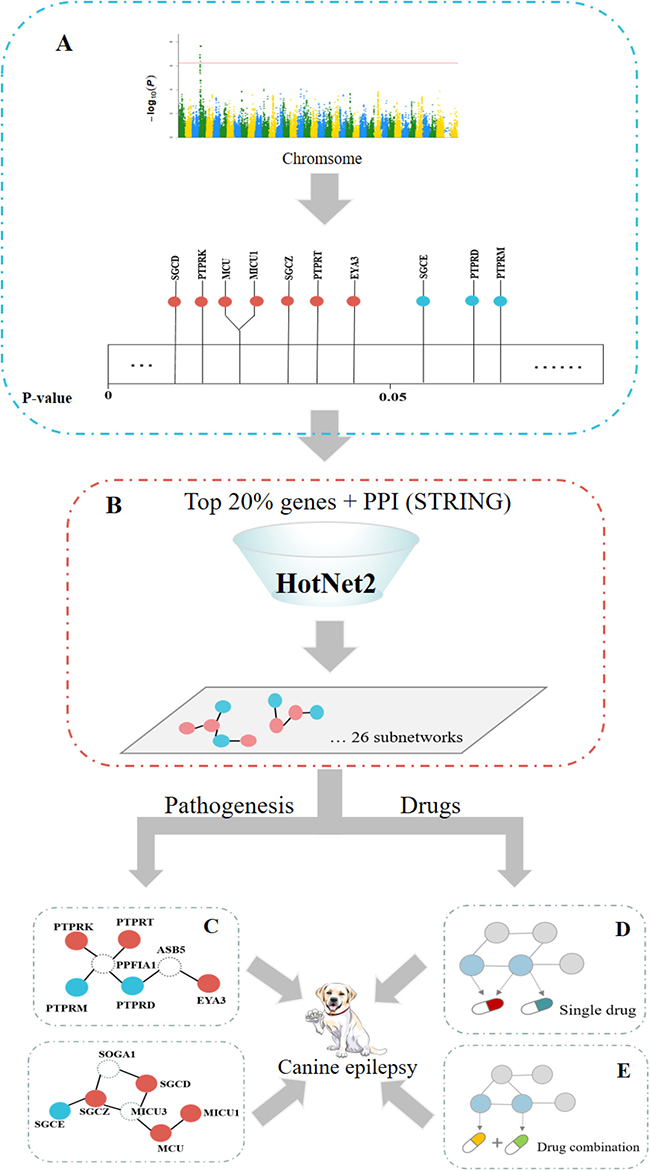
Figure 3: Study flowchart. (A) The associations of genes with canine epilepsy were calculated by the GWAS method. Some genes were significantly associated with canine epilepsy (P < 0.05, marked with orange), and some were not (marked by blue in the figure). (B) The significant subnetworks related to canine epilepsy were found using the HotNet2 method. (C) GO enrichment analysis of each subnetwork led to enhancement of the GWAS results and allowed exploration of the pathogenesis of canine epilepsy. (D) Relationship statistics of single drug-gene pairs that passed the enrichment testing of the anti-epilepsy drugs in these subnetworks. (E) These drug combinations are likely to produce synergistic effects if separate target genes of different drugs co-occur in a vital HotNet2 subnetwork.
CONCLUSIONS
The domestic dog (Canis familiaris) is not only a faithful partner of humans but also an excellent model animal for studying human diseases, as the dog genome and clinical manifestations are extremely similar to those of humans [7]. In this study, the genotype and epilepsy phenotype data of Hayward et al. [27] were used to explore the pathogenesis of canine epilepsy in a combined method using GWAS and HotNet2.
Using the combination of GWAS and HotNet2, we ultimately identified 26 subnetworks associated with canine epilepsy. Through GO enrichment analysis and a literature review, we focused on the relationship between gene mutations and epilepsy mechanisms in four subnetworks. We automatically identified PTPRD, PPFIA1, ASB5, SLC1A2, and SGCE from a large-scale interaction network. PTPRM, PTPRD and SGCE were of particular interest, as they were not significant results of the GWAS and, thus, demonstrated the power of this method. Moreover, PTPRM, PPFIA1, SLC1A2, and SGCE were confirmed to be associated with epilepsy, while PTPRD, ASB5, and MCU were involved in neurological disorders. This evidence suggests that the method for key gene discovery is reliable.
In addition, we performed drug enrichment on each subnetwork, resulting in 221 active drugs, of which 22 were drugs used for the treatment of epilepsy. The hypergeometric test was extremely significant and was stronger than the single-site GWAS method of drug enrichment. If targets of agents correspond to different target genes in the same subnetwork, their combinations are likely to induce synergistic effects. We found 22 potential combinations of anti-epilepsy drug treatments. One of these drug combinations, topiramate and valproic acid (DrugCombination_ID: DC000445), have been confirmed by the DCDB database for the treatment of epilepsy.
In summary, this study demonstrated that the combined GWAS and HotNet2 methods can effectively exploit the pathogenic factors of epilepsy and be used to identify potential drug combinations. Moreover, not only is the present approach helpful for epilepsy, but it will also be useful for exploring the pathogenic mechanisms of other diseases and potential drug combinations.
Author contributions
Ze-Jia Cui and Ye-Mao Liu generated the data, Ze-Jia Cui analyzed the data, Qiang Zhu discussed the algorithm, Jingbo Xia and Ze-Jia Cui drafted the manuscript, Hong-Yu Zhang designed the entire pipeline. All authors approved the final manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the National Natural Science Foundation of China (Grant No. 61202305).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Xia L, Li D, Lin C, Ou S, Li X, Pan S. Comparative study of joint bioinformatics analysis of underlying potential of ‘neurimmiR’, miR-212-3P/miR-132-3P, being involved in epilepsy and its emerging role in human cancer. Oncotarget. 2017; 8:40668–40682. https://doi.org/10.18632/oncotarget.16541.
2. Iori V, Aronica E, Vezzani A. Epigenetic control of epileptogenesis by miR-146a. Oncotarget. 2017; 8:45040–45041. https://doi.org/10.18632/oncotarget.18364.
3. Löscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002; 50:105–23.
4. Ekenstedt KJ, Oberbauer AM. Inherited Epilepsy in Dogs. Top Companion Anim Med. 2013; 28:51–8.
5. Patterson DF. Companion animal medicine in the age of medical genetics. J Vet Intern Med. 2000; 14:1–9.
6. Sargan DR. IDID: inherited diseases in dogs: web-based information for canine inherited disease genetics. Mamm Genome. 2004; 15:503–6.
7. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ 3rd, Zody MC, Mauceli E, Xie X, Breen M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005; 438:803–19.
8. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, Hesdorffer DC, Lee BI, Mathern GW, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014; 55:475–82.
9. World Health Organization (WHO). Epilepsy Fact sheet. 2016; URL: http://www.who.int/mediacentre/factsheets/fs999/en/.
10. De Risio L, Bhatti S, Muñana K, Penderis J, Stein V, Tipold A, Berendt M, Farqhuar R, Fischer A, Long S, Mandigers PJ, Matiasek K, Packer RM, et al. International veterinary epilepsy task force consensus proposal: diagnostic approach to epilepsy in dogs. BMC Vet Res. 2015; 11:148.
11. Kearsley-Fleet L, O’Neill DG, Volk HA, Church DB, Brodbelt DC. Prevalence and risk factors for canine epilepsy of unknown origin in the UK. Vet Rec. 2013; 172:338.
12. Johannessen Landmark C, Johannessen SI. Pharmacological management of epilepsy: recent advances and future prospects. Drugs. 2008; 68:1925–39.
13. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308:385–9.
14. Kasperaviciūte D, Catarino CB, Heinzen EL, Depondt C, Cavalleri GL, Caboclo LO, Tate SK, Jamnadas-Khoda J, Chinthapalli K, Clayton LM, Shianna KV, Radtke RA, Mikati MA, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. 2010; 133:2136–47.
15. Guo Y, Baum LW, Sham PC, Wong V, Ng PW, Lui CH, Sin NC, Tsoi TH, Tang CS, Kwan JS, Yip BH, Xiao SM, Thomas GN, et al. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum Mol Genet. 2012; 21:1184–9.
16. Buono RJ. Genome wide association studies (GWAS) and common forms of human epilepsy. Epilepsy Behav. 2013; 28:S63–5.
17. Lohi H, Young EJ, Fitzmaurice SN, Rusbridge C, Chan EM, Vervoort M, Turnbull J, Zhao XC, Ianzano L, Paterson AD, Sutter NB, Ostrander EA, André C, et al. Expanded Repeat in Canine Epilepsy. Science. 2005; 307:81.
18. Katz ML, Khan S, Awano T, Shahid SA, Siakotos AN, Johnson GS. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid-lipofuscinosis. Biochem Biophys Res Commun. 2005; 327:541–7.
19. Melville SA, Wilson CL, Chiang CS, Studdert VP, Lingaas F, Wilton AN. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. Genomics. 2005; 86:287–94.
20. Awano T, Katz ML, O’Brien DP, Taylor JF, Evans J, Khan S, Sohar I, Lobel P, Johnson GS. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006; 87:341–8.
21. Awano T, Katz ML, O’Brien DP, Sohar I, Lobel P, Coates JR, Khan S, Johnson GC, Giger U, Johnson GS. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006; 89:254–60.
22. Sanders DN, Farias FH, Johnson GS, Chiang V, Cook JR, O’Brien DP, Hofmann SL, Lu JY, Katz ML. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a Dachshund. Mol Genet Metab. 2010; 100:349–56.
23. Abitbol M, Thibaud JL, Olby NJ, Hitte C, Puech JP, Maurer M, Pilot-Storck F, Hédan B, Dréano S, Brahimi S, Delattre D, André C, Gray F, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci USA. 2010; 107:14775–80.
24. Katz ML, Farias FH, Sanders DN, Zeng R, Khan S, Johnson GS, O’Brien DP. A missense mutation in canine CLN6 in an Australian shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotechnol. 2011; 2011:198042.
25. Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppälä EH, Wade CM, Lindblad-Toh K, O’Brien DP, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. 2011; 42:468–74.
26. Seppälä EH, Jokinen TS, Fukata M, Fukata Y, Webster MT, Karlsson EK, Kilpinen SK, Steffen F, Dietschi E, Leeb T, Eklund R, Zhao X, Rilstone JJ, et al. LGI2 truncation causes a remitting focal epilepsy in dogs. Plos Genetics. PLoS Genet. 2011; 7:e1002194.
27. Hayward JJ, Castelhano MG, Oliveira KC, Corey E, Balkman C, Baxter TL, Casal ML, Center SA, Fang M, Garrison SJ, Kalla SE, Korniliev P, Kotlikoff MI, et al. Complex disease and phenotype mapping in the domestic dog. Nat Commun. 2016; 7:10460.
28. Yin H, Wang L, Xiao F, Huang Z, Huang Y, Zhou C, Han Y, Tao S, Yang H, Wang X. Upregulation of liprin-α1 protein in the temporal neocortex of intractable epileptic patients and experimental rats. Synapse. 2011; 65:742–50.
29. Myers CT, McMahon JM, Schneider AL, Petrovski S, Allen AS, Carvill GL, Zemel M, Saykally JE, LaCroix AJ, Heinzen EL, Hollingsworth G, Nikanorova M, Corbett M, et al, and Epi4K Consortium. De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am J Hum Genet. 2016; 99:287–98.
30. Santos RO. Investigating candidate genes in mesial temporal lobe epilepsy: PTPRM and IL1B genes.2015. http://repositorio.unicamp.br/bitstream/REPOSIP/312709/1/Santos_RenatoOliveirados_D.pdf.
31. Haugarvoll K, Tzoulis C, Tran GT, Karlsen B, Engelsen BA, Knappskog PM, Bindoff LA. Myoclonus-dystonia and epilepsy in a family with a novel epsilon-sarcoglycan mutation. J Neurol. 2014; 261:358–62.
32. Schormair B, Kemlink D, Roeske D, Eckstein G, Xiong L, Lichtner P, Ripke S, Trenkwalder C, Zimprich A, Stiasny-Kolster K, Oertel W, Bachmann CG, Paulus W, et al. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat Genet. 2008; 40:946–8.
33. Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH. Novel copy number variants in children with autism and additional developmental anomalies. J Neurodev Disord. 2009; 1:292–301.
34. Ritz K, Gerrits MC, Foncke EM, van Ruissen F, van der Linden C, Vergouwen MD, Bloem BR, Vandenberghe W, Crols R, Speelman JD, Baas F, Tijssen MA. Myoclonus-dystonia: clinical and genetic evaluation of a large cohort. J Neurol Neurosurg Psychiatry. 2009; 80:653–58.
35. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017; 8:38022–38043. https://doi.org/10.18632/oncotarget.16723.
36. Croissant Y. Estimation of multinomial logit models in R: The mlogit Packages. R package version 02–2. 2012. https://cran.r-project.org/web/packages/mlogit/mlogit.pdf.
37. Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, Jardim L, Satishchandra P, Andermann E, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998; 20:171–4.
38. Serratosa JM, Gómez-Garre P, Gallardo ME, Anta B, de Bernabé DB, Lindhout D, Augustijn PB, Tassinari CA, Malafosse RM, Topcu M, Grid D, Dravet C, Berkovic SF, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet. 1999; 8:345–52.
39. Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004; 117:699–711.
40. Navis AC, van den Eijnden M, Schepens JT, Hooft van Huijsduijnen R, Wesseling P, Hendriks WJ. Protein tyrosine phosphatases in glioma biology. Acta Neuropathol. 2010; 119:157–75.
41. Zondag GC, Reynolds AB, Moolenaar WH. Receptor protein-tyrosine phosphatase RPTPmu binds to and dephosphorylates the catenin p120(ctn). J Biol Chem. 2000; 275:11264–9.
42. Burden-Gulley SM, Brady-Kalnay SM. PTPmu regulates N-cadherin-dependent neurite outgrowth. J Cell Biol. 1999; 144:1323–36.
43. Ackley BD, Harrington RJ, Hudson ML, Williams L, Kenyon CJ, Chisholm AD, Jin Y. The two isoforms of the Caenorhabditis elegans leukocyte-common antigen related receptor tyrosine phosphatase PTP-3 function independently in axon guidance and synapse formation. J Neurosci. 2005; 25:7517–28.
44. Thwaites DT, Anderson CM. The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. Br J Pharmacol. 2011; 164:1802–16.
45. Tani H, Bandrowski AE, Parada I, Wynn M, Huguenard JR, Prince DA, Reimer RJ. Modulation of epileptiform activity by glutamine and system A transport in a model of post-traumatic epilepsy. Neurobiol Dis. 2007; 25:230–8.
46. Dickens D, Webb SD, Antonyuk S, Giannoudis A, Owen A, Rädisch S, Hasnain SS, Pirmohamed M. Transport of gabapentin by LAT1 (SLC7A5). Biochem Pharmacol. 2013; 85:1672–83.
47. Zhu X, Dubey D, Bermudez C, Porter BE. Suppressing cAMP response element-binding protein transcription shortens the duration of status epilepticus and decreases the number of spontaneous seizures in the pilocarpine model of epilepsy. Epilepsia. 2015; 56:1870–8.
48. Tezenas du Montcel S, Clot F, Vidailhet M, Roze E, Damier P, Jedynak CP, Camuzat A, Lagueny A, Vercueil L, Doummar D, Guyant-Maréchal L, Houeto JL, Ponsot G, et al, and French Dystonia Network. Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes. J Med Genet. 2006; 43:394–400.
49. Wang C, Xie N, Wang Y, Li Y, Ge X, Wang M. Role of the Mitochondrial Calcium Uniporter in Rat Hippocampal Neuronal Death After Pilocarpine-Induced Status Epilepticus. Neurochem Res. 2015; 40:1739–46.
50. Acharya MM, Katyare SS. Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp Neurol. 2005; 192:79–88.
51. Chuang YC, Chang AY, Lin JW, Hsu SP, Chan SH. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia. 2004; 45:1202–9.
52. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011; 476:341–5.
53. Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, Lawrence MS, Gonzalez-Perez A, Tamborero D, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015; 47:106–14.
54. Franceschini A. STRINGdb Package Vignette. 2015. http://www.bioconductor.org/packages/3.0/bioc/vignettes/STRINGdb/inst/doc/STRINGdb.pdf.