
INTRODUCTION
Lung cancer is a major cause of cancer-related deaths worldwide, occurring in more than a million new patients annually [1]. Genetic factors, along with exposure to environmental carcinogens contribute significantly to the risk of developing lung cancer [2]. Non-Small Cell Lung Cancer (NSCLC) is the most common type of lung cancer, comprising 80–85% of all cases. Lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) are the two major subtypes of NSCLC, each comprising about 30% of lung cancer diagnosis. The tumor genomics profiles between LUAD and LUSC differed significantly and high heterogeneity was observed within each cancer type [3–5]. Therapies for LUAD are often ineffective for LUSC [6]. LUSC is a highly heterogeneous disease that develops via multiple complex steps [7, 8]. Genetic etiology of LUSC has been studied extensively [9–11], however, more research is needed to increase the knowledge base of LUSC and design more effective prevention and therapeutic strategies.
A mouse model of LUSC in which the tumors are induced by the carcinogen, N-nitroso-tris-chloroethylurea (NTCU), was developed and widely used in chemoprevention studies of LUSC [12]. However, the underlying genetic profiles of this murine model and its resemblance to human LUSCs have not been well characterized. Because carcinogen-induced mouse models have become important for studies of oncoimmunology [13], it is necessary to systematically analyze the mutation and transcriptome profiles of the NTCU-induced murine LUSC model and compare them to the corresponding profiles of human LUSCs [14, 15] to identify the genes with potential clinical value for further study. In the present study, we assessed the mutational and transcriptional characteristics of mice LUSCs. We identified the the novel patterns of clonal evolution, mutational landscapes and expression signatures of LUSC, which could contribute to the development of new LUSC therapeutic strategies.
RESULTS
Whole-exome sequencing (WES) of mouse lung squamous cell carcinoma (LUSC) tumors
To characterize the genetic alterations that occur in mouse LUSCs, we performed WES in 16 surgically resected NTCU-induced NIH Swiss mouse LUSC tumors, along with 8 normal mouse lung tissues. LUSC tumors and normal lung tissues were sequenced to an average read depth of 129 X (range: 98 X to 165 X) and 137 X (range: 112 X to 179 X), respectively (Supplementary Table 1). The 16 mouse LUSCs harbored 5,664 somatic coding mutations (mean = 354) that consisted of 2,885 missense, 106 nonsense, 2,426 silent mutations, 167 small insertions and deletions (indels) and 80 alterations residing in exon-exon boundaries (Supplementary Table 2 [Excel File]). The most common substitutions were C>T transversions (Supplementary Figure 1), indicative of oxidative DNA damage [16].
WES identified recurrently mutated cancer genes and clonal mutations in mouse LUSC
Identification of cancer genes that are recurrently mutated in mouse LUSC tumors (mutated in at least two tumors) may reveal the novel genetic mechanisms of carcinogenesis and progression of LUSC. Therefore, we analyzed the mutation profiles of the mouse LUSCs focusing on the comprehensive list of 2,027 cancer related genes (from the file “allOnco_Feb2017.tsv” available at http://www.bushmanlab.org/links/genelists). It was found that 59 cancer genes were recurrently mutated in our mouse LUSC tumors (Figure 1). The cancer genes most frequently mutated in the mouse LUSC tumors (≥25%, i.e., mutated in at least 4 samples of 16 tumors) were as follows: Muc4, Prg4, Igf2r, Ctsll3, Dlgap1, Hspa9, Armcx3, Cdk1, Pcdhb15, Fus, Gga1, Il2rb, Kmt2d (Mll2), Mapk6, Myh1, Ncoa3 (Src3), Obscn, Runx2, Zmynd8, Ido1, Nkain2, Pyy, Stil, Tcl1b4, Tfeb, and Trpv1 (Figure 1).
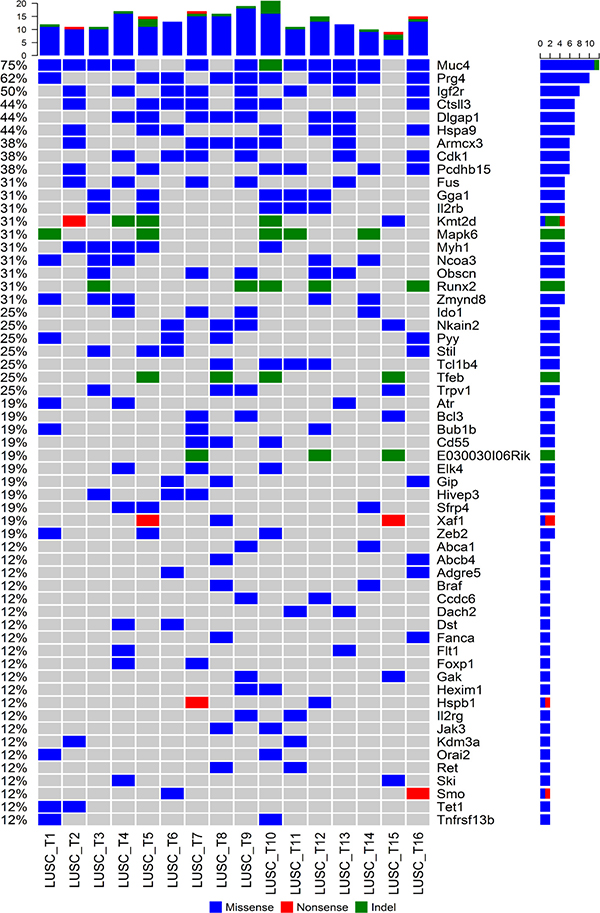
Figure 1: Profiles of recurrently mutated cancer genes in mouse LUSC tumors. The non-silent somatic mutation spectrum of the 59 cancer genes that were recurrently mutated in the 16 mice LUSC tumors was plotted. The order of genes was sorted by the mutation frequency decreasingly. The top bar plot showed the total number of mutations in any of the 59 genes per sample, the right-side and the left-side legends showed the number and frequency of mutations within each gene across the 16 mouse LUSC tumors.
To identify whether the 59 recurrently mutated cancer genes in mouse LUSC tumors were also mutated in human LUSC tumors, we downloaded the somatic mutation profiles of 191 human LUSC tumors that are accessible through COSMIC database (http://cancer.sanger.ac.uk/cosmic) and performed cross-species comparisons. We found that 47 of the 59 cancer genes recurrently mutated in the mouse LUSCs were also mutated in the human LUSCs (Figure 2).The mutation profiles of most of these 47 genes were mutually exclusive across the human LUSC tumors and they comprehensively characterized the human LUSC tumors (Figure 2). Among the 47 commonly mutated cancer genes shared between mouse and human LUSCs, the frequently mutated ones in human LUSCs include KMT2D (MLL2), MYH1, OBSCN, ZEB2, BRAF, IGF2R, FLT1, HIVEP3, PRG4, ABCA1, ATR, DACH2, ABCB4, DST, and MUC4 (Figure 2). The proportion of the cancer genes recurrently mutated in mice LUSCs that were also mutated in human LUSCs was as high as 80% (= 47/59), suggesting that the NTCU-induced mouse LUSC model is an excellent model to study the genetic mechanisms of human LUSC. However, about 20% (= 12/59) of recurrently mutated cancer genes of mouse LUSCs were not found to be mutated in human LUSCs, suggesting either the unique mechanisms of LUSC carcinogenesis in mice or the necessity to sequence more human LUSC tumors to enlarge the mutation spectrum of human LUSCs.
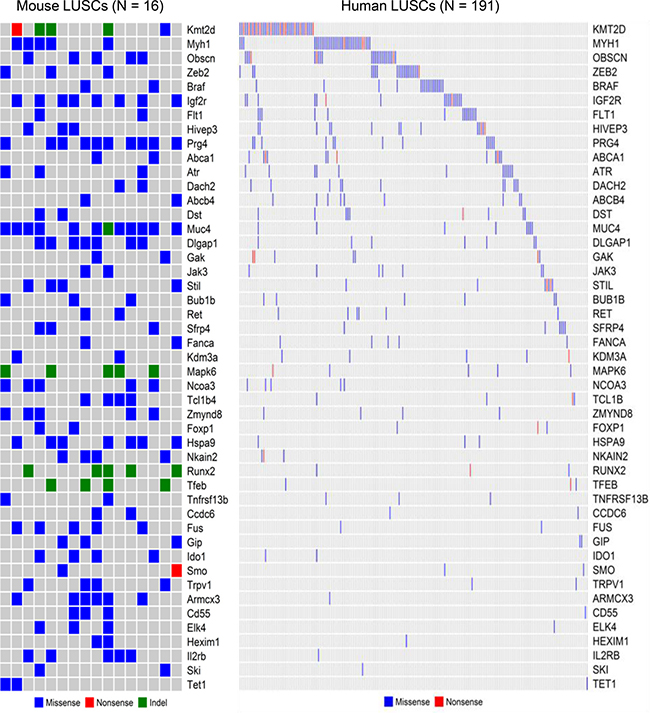
Figure 2: Comparison study of mouse and human LUSC genes. In terms of non-silent somatic mutations, 47 of the 59 cancer genes recurrently mutated in the mouse LUSCs were also mutated in the human LUSCs archived in the COSMIC database. The waterfall plots showed the side-by-side comparison of the 47 mutated LUSC genes shared between mouse and human.
In addition, our analysis revealed that each LUSC tumor had a sample-specific mutational landscape consisting of a mixture of recurrent and private clonal mutations (Supplementary Figure 2). The clonal mutation spectra across the 16 mouse LUSCs were shown in Supplementary Figure 3 and Supplementary Table 3. There were nine most frequent clonal mutations identified in at least 4 of 16 LUSC tumors, including Hspa9: A651S (7), Cdk1: S39I (6), Pcdhb15: R461C (6), Ctsll3: P329S (5), Gga1: D358N (5), Il2rb: R475S (5), Dlgap1: A329T (4), Nkain2: V67L (4), and Pyy: P42L (4).
scRNA-seq (single-cell RNA-sequencing) identified clonal mutations in the two mice LUSC tumors
Single-cell analysis by scRNA-seq was used to characterize the nonsilent somatic mutations in two mouse LUSC tumors, with a specific focus on the known cancer genes or the mutated genes identified by our exome-seq of mouse LUSC tumors. Single tumor and normal cells were differentiated from one another based on whether any mutations in the above genes can be identified. With respect to the first mouse LUSC tumor (LUSC1), 36 single cells from LUSC1 were classified into 28 tumor cells and 8 normal cells according to the mutation status of the cancer genes (Figure 3A). The 33 single cells from the second mouse LUSC tumor (LUSC2) were classified into 20 tumor cells and 13 normal cells according to the mutation status of the cancer genes (Figure 3B). The details of the cancer gene mutations of the two mouse LUSC tumors can be seen in Supplementary Table 4.
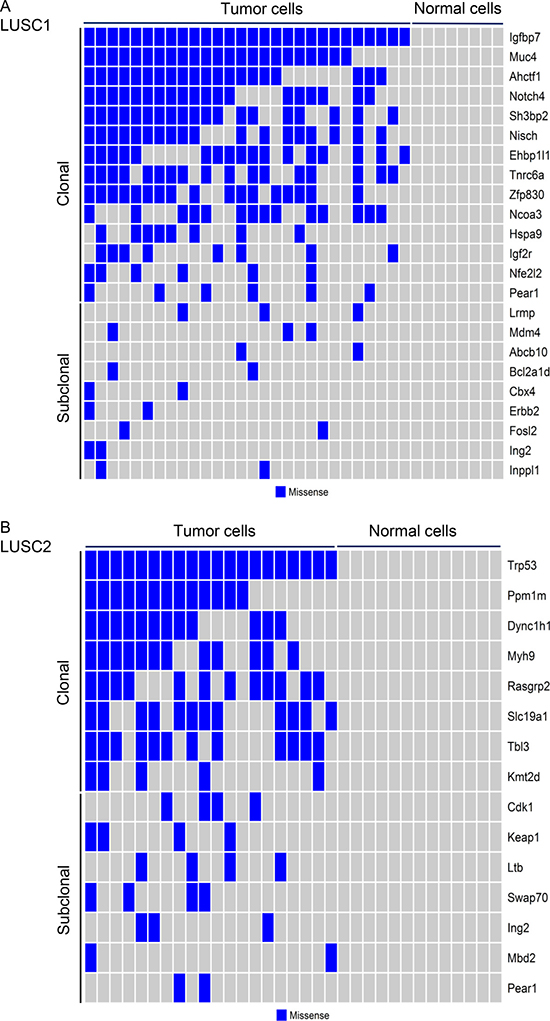
Figure 3: Clonal and subclonal mutations across the tumor cells from the two mouse LUSC tumors. The somatic missense mutations in the cancer related genes of single tumor cells from LUSC1 (A) and LUSC2 (B) were plotted. The corresponding normal cells did not carry any mutations.
Next, we inferred the clonal evolutionary history of the two mouse LUSC tumors (Supplementary Figures 4 and 5). For LUSC1, all the single tumor cells had the clonal mutation (R45P) in Igfbp7 and a subset of single tumor cells had the mutation (R2457S) in Igf2r (Figure 3A). IGFBPs participate in a complex signaling axis called IGF-IGFR-IGFBP. In addition, the genes having somatic mutations in LUSC1 involve a number of transcription regulators such as Ahctf1, Notch4, Ncoa3, Nfe2l2 (Figure 3A). The scRNA-seq of LUSC2 identified a Trp53 somatic missense mutation (Q97L) in all the tumor cells (Figure 3B). Somatic mutations of the LUSC2 tumor were also found in a number of novel driver genes such as Myh9, Kmt2d and Keap1 (Figure 3B).
The activation of a common cancer gene expression module in the two mouse LUSC tumors
Based on the mutation spectra of the two LUSC tumors, differential expression (DE) analysis was performed between tumor and normal cells in each tumor. Interestingly, DE analysis and comparison study of the two LUSC tumors identified a common set of 80 cancer genes termed ‘G80’ that were activated in both the LUSC1 and LUSC2 tumor cells although they had a different set of key cancer gene mutations (Figures 4 and 5). The G80 geneset was further tested in the mouse and human LUSC tumors at the bulk sample level. Analysis of the 16 mice LUSC tumors and 8 control normal lungs showed that the G80 module expression could differentiate the tumor samples from the normal lung samples (Figure 6). We also analyzed the G80 module cancer gene expression in the 502 human LUSCs and 51 control normal lung samples from TCGA. Interestingly, the G80 gene expression signature clearly differentiated LUSC tumors from normal samples again (Figure 7). These data suggested that the G80 module can serve as a classifier for LUSC tumors in both mouse and human samples.
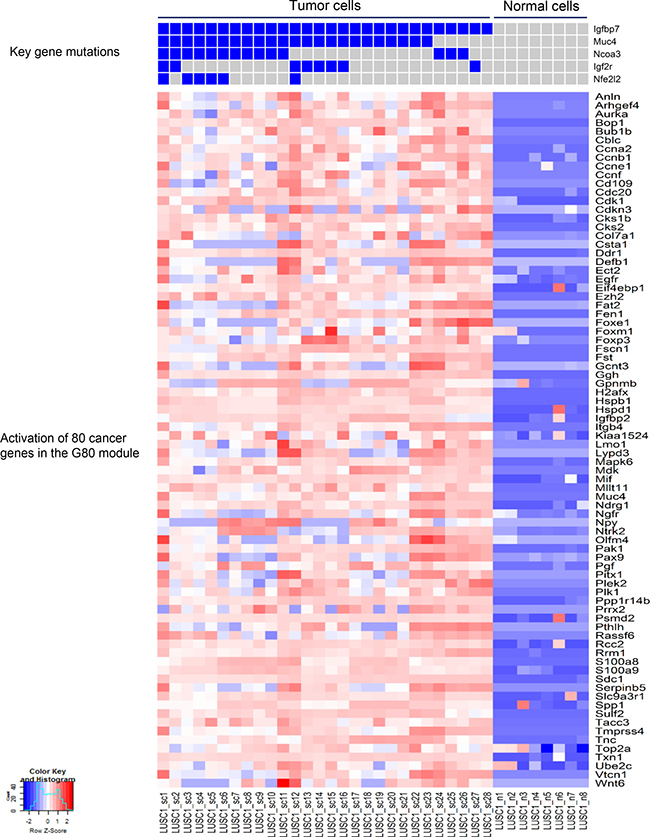
Figure 4: Mutation profiles of the key cancer genes and the corresponding up-regulated cancer genes in LUSC1 single tumor cells. The nonsilent mutations in the key genes correlated with significant upregulation of the expression of 80 cancer genes in the G80 module in the single tumor cells from the mouse LUSC1 tumor.
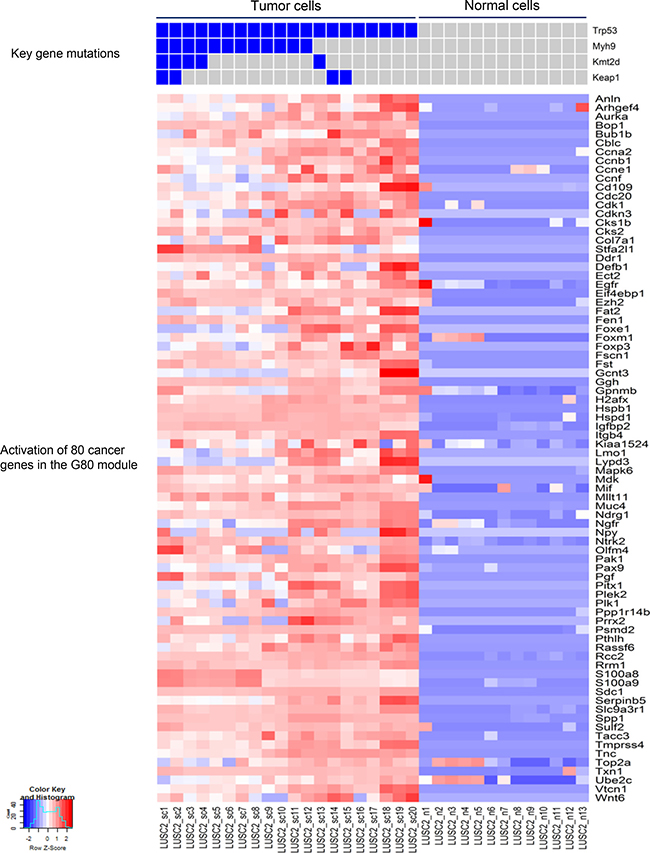
Figure 5: Mutation profiles of the key cancer genes and the corresponding up-regulated cancer genes in LUSC2 single tumor cells. The nonsilent mutations in the key genes correlated with the significant upregulation of the expression of 80 cancer genes in the G80 module in the single tumor cells from the mouse LUSC2 tumor.
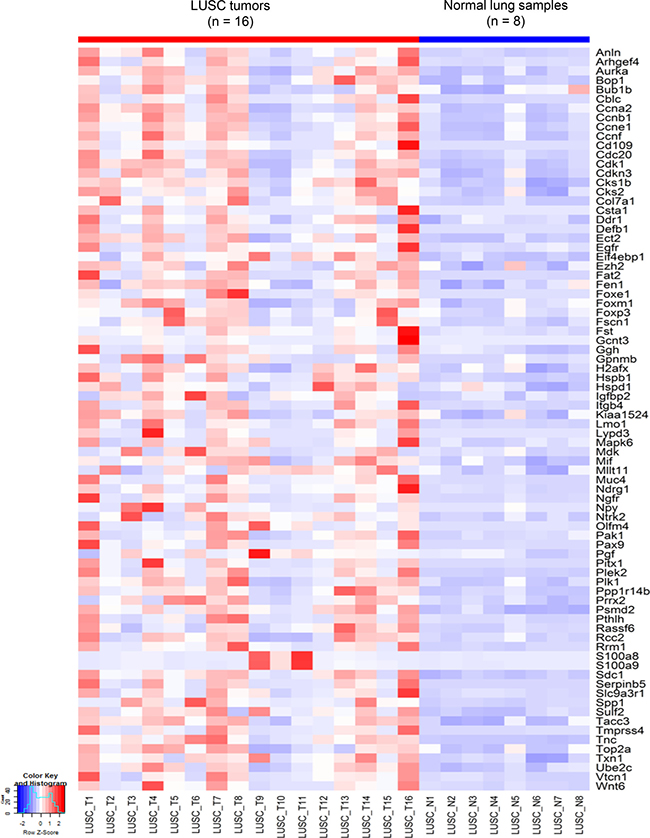
Figure 6: Heatmap of G80 expression in the 16 mouse LUSC tumors versus 8 control normal lung samples. At the bulk inter-tumoral level, the expression patterns of the 80 cancer genes in the G80 module still clearly separated the mouse LUSC tumors from the control normal lung tissues.
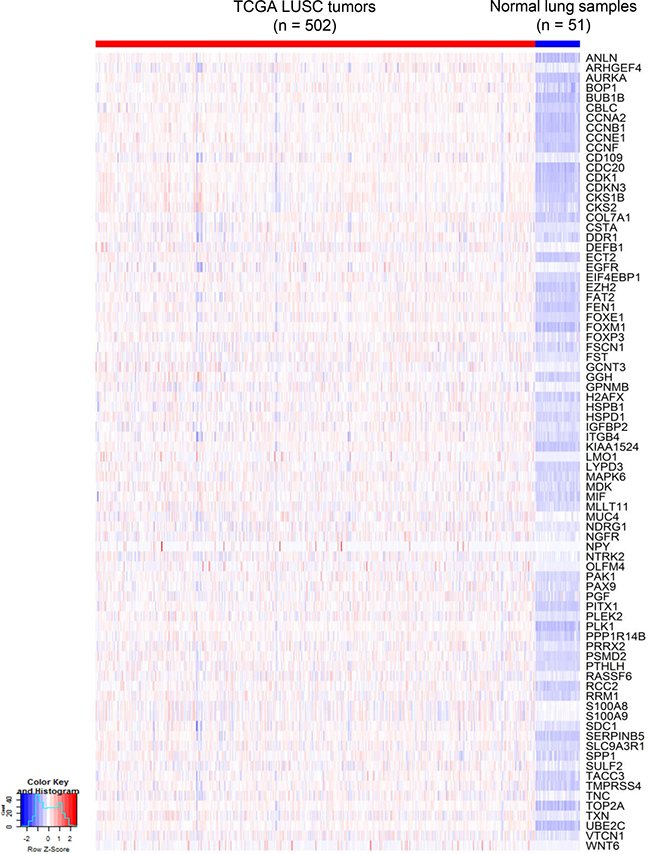
Figure 7: Heatmap of G80 expression in 502 TCGA LUSC tumors versus 51 control normal lung samples. At the bulk inter-tumoral level, the G80 gene expression signature that distinguished between single cells in mouse LUSC model also clearly differentiated the same sub-groups for the TCGA LUSC samples.
Next, we sought to explore whether the expression signature of the G80 cancer gene module was associated with survival outcome in lung cancer patients. The TCGA LUSC cohort and the other three independent human NSCLC gene expression data sets were analyzed [17–20]. The high-risk gene expression signature of the G80 cancer gene module was significantly associated with poor overall survival outcome across these four large and representative human lung cancer datasets (Figure 8). The risk group hazard ratio (HR) based on G80 module expression in the TCGA LUSC cohort was 10.2 (P = 1.7E–14), which was much greater than the HR values of the other three NSCLC cohorts (HR = 3.3, 3.5, 4.4, Figure 8). This reflected that the G80 module was developed from LUSC samples so its performance was better in the LUSC cohort than in the NSCLC cohort which also contains LUAD (lung adenocarcinoma) samples. Our data suggested that the overall expression of the G80 module genes can be used as a biomarker to assess the survival outcomes of lung cancer patients, especially patients with LUSC.
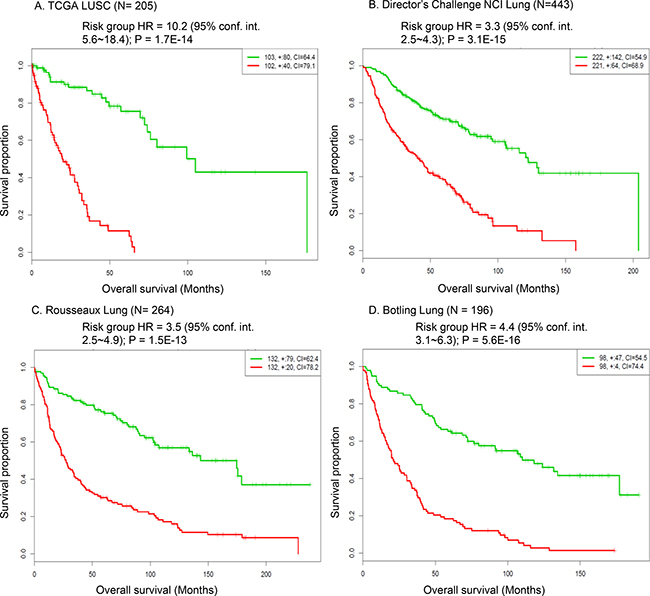
Figure 8: Survival analysis of the G80 module in human LUSC and NSCLC patient cohorts. Kaplan-Meier analysis showing that the expression signature of the G80 cancer gene module was significantly associated with the overall survival outcome in the TCGA LUSC cohort and the other three NSCLC patient cohorts. Red curve denoted high risk group based on the G80 module expression, green curve denoted low risk group based on the G80 module expression.
Associations of mutant LUSC genes with the immune checkpoint gene expression in human LUSCs
Our comprehensive analysis yielded a list of cancer genes with recurrent or clonal mutations for LUSC (Figures 1–3, Supplementary Figure 3, and totaling 106 genes). In order to test whether these mutated genes have potential clinical application, the associations between mutational status of the identified LUSC genes and gene expression of immune checkpoint genes were analyzed by using a set of 176 TCGA LUSC tumor samples. We examined the expression of inhibitory immune checkpoint genes, including CTLA4, PD-1 (PDCD1), LAG3, TIM3 (HAVCR2), PD-L1 (CD274), and VISTA (C10orf54). The suppressed tumoral PD-L1 expression was significantly associated with mutations in eight genes (Figure 9, Supplementary Table 5): HIVEP3 (P = 2.8E-11), NKAIN2 (P = 2.9E-11), RUNX2 (P = 2.5E-09), MUC4(P = 2.3E-06), CUX1 (P = 9.0E-06), NIPBL (P = 6.1E-05), PLAGL2 (P = 0.0001), and NFE2L2 (P = 0.019). The mutant NFE2L2 was associated with increased tumoral PD-L1 expression while the other seven mutant genes associated with decreased PD-L1 expression. The tumoral VISTA expression was significantly decreased in the LUSCs with mutations in the KEAP1 (P = 0.0005), FANCA (P = 0.0076), and AFF3 (P = 0.008) genes while increased in the LUSCs with the mutant FLT1 gene (P = 0.02). Moreover, tumoral TIM3 was significantly decreased in the mutant RET (P = 4.2E-10), FANCA (P = 0.0007), or ZMYND8 (P = 0.028) LUSC tumors while increased in the mutant DYNC1H1 (P = 0.027) LUSC tumors. The suppressed tumoral LAG3 expression was significantly associated with mutations in CUX1 (P = 6.9E-07), FANCA (P = 0.0005) or NOTCH4 (P = 0.028) genes.
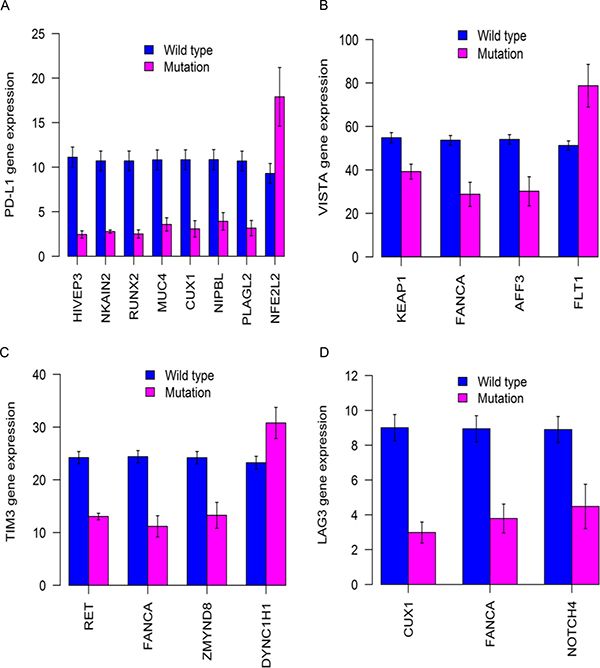
Figure 9: Differential expression of immune markers in 176 TCGA LUSCs with significantly mutated cancer genes. Different mutant genes identified from sequencing of mouse LUSC tumors were associated with significantly decreased expression of tumoral PD-L1 (A), VISTA (B), TIM3 (C), and LAG3 (D). All the P values < 0.05 after multiple testing adjustment.
In addition, the LUSC patients with overexpressed PD-L1 or VISTA had significantly worse overall survival outcome compared to the patients with decreased expression of PD-L1 or VISTA (Figure 10). The median overall survival time of high PD-L1 vs. low PD-L1expression group was 1189 days vs. 2945 days (P = 0.04) and of high VISTA vs. low VISTA expression group was 1640 days vs. 2945 days (P = 0.01) (Figure 10). These data suggested that aberrant immune checkpoint gene expression could have significant impact on the survival outcome of LUSC patients.
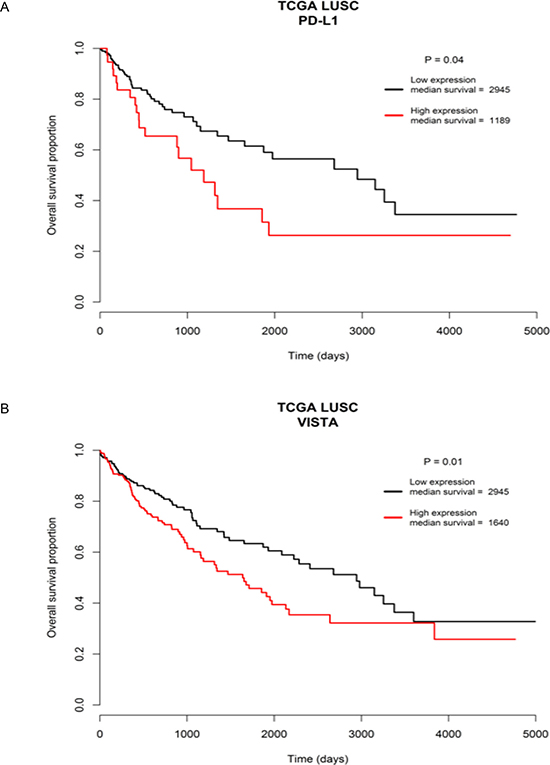
Figure 10: Survival outcome analysis based on immune checkpoint gene expression. Worse overall survival outcome was significantly associated with overexpressed tumoral PD-L1 gene expression group (A), and overexpressed tumoral VISTA gene expression group (B), relative to the down-regulated expression group of the two genes, respectively.
DISCUSSION
We identified 59 cancer genes that are recurrently mutated in mice LUSC tumors (Figure 1), 47 of which (80%) were also mutated in the human LUSCs (Figure 2). Most of the commonly mutated genes in both mouse and human LUSCs have genetic and/or functional significance to lung cancer, such as Kmt2d (Mll2), Zeb2, Braf, Igf2r, Flt1, Atr, Muc4, Ncoa3 (Src3), and Ido1. KMT2D, the key enzyme governing histone modification, is a well-known driver gene with a high somatic mutation rate in LUSC tumors [14, 21]. The mutant KMT2D significantly associated with poor prognosis of LUSC [14]. ZEB2 expression increased in NSCLC [22] and it can induce epithelial-mesenchymal transition (EMT) to facilitate the metastasis of cancer cells [23, 24]. BRAF mutated lung cancer is a genetically distinct subtype that accounts for about 5% of NSCLC [25]. IGF2R is mutated frequently in LUSC and it suppresses cancer cell growth [26]. NSCLC patients with low IGF2R expression had a poorer prognosis than those with high IGF2R expression [27]. IGF2R inhibition in NSCLC cell lines resulted in increased proliferation, migration and invasion abilities and a reduced apoptosis rate of the cancer cells [27]. The functional FLT1 variant and FLT1 mRNA expression are prognostic determinants of patient survival and recurrence in stage I-III NSCLC [28, 29]. ATR encoded protein kinase is a master regulator of the DNA-damage response [30] and its genetic alteration was associated with lung cancer risk [31]. MUC4 has been shown to play a tumor-suppressor role in NSCLC by altering p53 expression [32]. A reduced expression of MUC4 was observed in LUSC tumors, especially in advanced tumor stages [32, 33]. A high somatic mutation rate in MUC4 was found in smokers having NSCLC, suggesting that the MUC4 gene is a potential target of nicotine in causing NSCLC [34]. This is supported by our data revealing Muc4 as one of the most recurrently mutated genes in mice LUSC. NCOA3 may be an oncogene for lung cancer due to its role in promoting lung cancer cell invasion [35, 36]. IDO1 is an immune checkpoint gene involved in lung cancer progression and metastasis [37, 38] and thus a promising target for lung cancer immunotherapy.
Furthermore, we detected clonal mutations in the 16 mice LUSC tumors. Each tumor had a unique set of clonal mutations (Supplementary Figures 2 and 3), supporting previous findings of distinct clonal mutation structures across the same type of NSCLC tumors [39]. Although the overall clonal mutation spectra were different across the 16 mice LUSC tumors, a number of recurrent clonal mutations were identified (Supplementary Figure 3, Supplementary Table 3). The most frequent clonal mutations were found in the following cancer genes: Hspa9, Cdk1, Pcdhb15, Ctsll3, Gga1, Igf2r, Il2rb, Dlgap1, Nkain2, and Pyy. HSPA9 (mortalin/GRP75/PBP74), was overexpressed in different tumor types and detected in different subcellular compartments of cancer cells, indicating its functional role in cancer [40, 41]. HSPA9 may play an important role in the progression of lung carcinoma by regulating the expressions of p53 and bcl-2 [42]. CDK1 is essential for cell cycle and its overexpression is directly correlated with the clinical features such as tumor stage and therapy outcome [43]. CTSL (Ctsll3 in mice), a lysosomal acid cysteine protease, is known to play important roles in tumor metastasis and resistance to chemotherapy [44]. IL2RB gene variants have been associated with human lung cancer risk in a large patient population [45]. NKAIN2 is a novel tumor suppressor gene [46–49]. The recurrent somatic clonal mutations of these genes in LUSC tumors suggest that they could be the therapeutic targets of LUSC treatment.
Tumor progression is a dynamic evolutionary process acting at the level of individual cells. A tumor typically arises from a single progenitor cell whose founder mutation gives it a growth advantage over the surrounding cells and helps it to evade the immune response. Consequently, the clone arising from this cell expands and, over time, the descendant cells develop further into subclones by acquiring additional somatic mutations [50]. The mouse LUSC1 tumor was initiated from clonal cells having a mutant Igfbp7 gene, which may lead to an aberrant Igf-Igfr-Igfbp axis that is an important driver of cancer [51]. Igfbp7 is a tumor suppressor gene inactivated in lung cancer by DNA hypermethylation and it is regulated by p53 [52]. Aberrant methylation and downregulation of IGFBP7 were frequently observed in NSCLCs [53, 54]. Our findings provided evidence indicating that non-silent somatic mutations in IGFBP7 could be the important driver alteration leading to LUSCs. Another important driver gene for LUSC1 is Nfe2l2 in the Keap1-Nrf2 pathway. Mutations in NFE2L2 could cause the excessive intracellular accumulation of NFE2L2 protein and the subsequent activation of downstream oncogenes resulting in tumor growth promotion [55].
For LUSC2, the founder clone was driven by mutant Trp53, the mouse homolog of the human TP53 gene that is a well known driver gene frequently mutated in the human LUSCs [14, 56]. In addition, a number of driver genes, such as Myh9, Kmt2d and Keap1, accumulated mutations in the subclones of LUSC2. The mutations or abnormal expression of these genes have been linked to aberrant molecular events leading to cancer, such as the interruption of p53 stabilization (MYH9) [57], abnormal chromatin/histone modification (KMT2D) [58], and aberrant Keap1-Nrf2 pathway (KEAP1) activity [55, 59]. Myh9, which encodes nonmuscle myosin IIa, has been identified as tumor suppressor of squamous cell carcinomas (SCCs) [57]. Myh9 knockout triggers invasive formation of SCCs. Myh9 plays a role in regulating posttranscriptional p53 stabilization and it is frequently mutated and diminished in human SCCs [57]. Kmt2d serves as the key enzyme in histone lysine methyltransfer and thus is one of the important epigenetic regulators whose mutations could lead to the development of tumors with abnormal histone modifications [58]. Human studies revealed a high frequency of non-silent somatic mutations within KMT2D for LUSC tumors [14, 21]. Moreover, mutant KMT2D was the alteration most significantly associated with poor prognosis of LUSC [14]. In human cancers, similar to NFE2L2, KEAP1 mutations would disrupt the normal combination of NFE2L2 and KEAP1 in tumors, resulting in accumulation of excessive intracellular NFE2L2 and activation of its downstream genes and the eventual promotion of tumor growth [55]. The Keap1-Nrf2 pathway is important for cytoprotection from oxidative stress. The discrepancy from normal in the Keap1-Nrf2 pathway may lead to promotion of tumor [59].
In addition, scRNA-seq identified a set of cancer genes; i.e., the G80 module that is an effective classifier for LUSC tumors in both mice and humans and an effective biomarker to assess the survival outcomes of lung cancer patients. Pathway enrichment analysis of the G80 module cancer genes showed that these genes are significantly enriched in six cancer related molecular pathways, including Cell cycle, PI3K-Akt, p53 and ErbB signaling, Focal adhesion and ECM-receptor interaction pathways (Supplementary Figure 6, Supplementary Table 6). Alteration of transcripts in these pathways may be a common mechanism involved in LUSC tumorigenesis and progression. The G80 module was activated in both LUSC1 and LUSC2 tumor cells although the two tumors had different clonal mutations. This is not surprising because the major driver genes in the two tumors functionally interacted with each other and played similar roles in the same pathway. For example, IGFBP7 might be regulated by TP53 in lung cancer cells [52] and mutant NFE2L2 or KEAP1 led to similar functional consequences in terms of causing NFE2L2 accumulation and aberrant Keap1-Nrf2 pathway activity in cancer cells [59].
Lastly, we found that the mutant LUSC genes could be associated with the significantly altered tumoral expression of inhibitory immune checkpoint genes such as PD-L1, VISTA, TIM3 and LAG3. These findings suggest that immune checkpoint signaling activity could be significantly altered by mutations in LUSC genes identified in this study. We observed both up- and down-regulated immune checkpoint gene expression patterns according to different mutant genes in LUSC tumors (Figure 9). Notably, suppressed tumoral expression of immune checkpoint genes has been associated with mutated oncogenes in lung cancer [14, 60]. Particularly, the increased tumoral PD-L1 gene expression was significantly associated with the mutant NFE2L2 gene (Figure 9). This supported the finding that PD-L1 expression in LUSC tumor cells was associated with NFE2L2 mutations [61] and indicated that LUSC patients carrying NFE2L2 mutations may be more responsive to anti-PD-L1 immunotherapy. Similarly, the mutant FLT1 and DYNC1H1 genes associated with higher tumoral gene expression of VISTA and TIM3, respectively, suggesting that they may serve as biomarkers to predict the effectiveness of VISTA-targeted and TIM3-targeted antitumor immunotherapy for LUSC patients. Moreover, we detected that the tumoral overexpression of PD-L1 and VISTA were significantly associated with worse survival outcome (Figure 10). This suggested that the mutant genes such as NFE2L2 and FLT1 associating with higher expression of PD-L1 and VISTA may be correlated with worse survival outcome. Interestingly, we found the trends of the associations with worse overall survival outcome for the NFE2L2 and FLT1 mutations (Supplementary Figure 7). The association of NFE2L2, FLT1 and DYNC1H1 mutations with higher expression of PD-L1, VISTA and TIM3 might reflect the necessity of the neoplastic cells to compensate the high immunologic visibility through the mechanism of aberrant activation of immune checkpoint genes that counteracts the cytotoxic effects of the immune response [61].
In summary, we identified the new candidate genes for LUSC and a cancer gene expression signature that proved to be an effective classifier of LUSC tumors and a biomarker for predicting survival outcome of LUSC patients. Overall, the identified novel patterns of clonal evolution, mutational landscapes and expression signatures of LUSC could contribute to the development of new LUSC therapeutic strategies.
MATERIALS AND METHODS
Mouse model and lung tissue collection
In this study, the NTCU induced NIH Swiss mouse lung squamous cell carcinoma (LUSC) model was used as previously reported [12, 62]. NTCU was purchased from Toronto Research Chemicals, Inc. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (MCW). For bulk sequencing, lung tumor tissues were obtained from NTCU induced LUSC tumor-bearing NIH Swiss mice (n = 16) and normal lung tissues were obtained from control healthy NIH Swiss mice without NTCU treatment (n = 8). For single-cell RNA-sequencing (scRNA-seq), two LUSC tumors from another two NIH Swiss mice independent of the 16 mice subjected to the bulk RNA-seq were used. NTCU treatment caused the development of LUSC tumors within 24 to 26 weeks [63]. In this study, mice LUSC tumor tissues were obtained 28 weeks after the initial treatment of NTCU when mice were euthanized by CO2 asphyxiation.
Bulk DNA/RNA extraction and sequencing
For the bulk exome-sequencing (exome-seq) and RNA-sequencing (RNA-seq), both the DNA and RNA samples from each lung tissue sample (SCC tumor or normal) were isolated simultaneously using the AllPrep DNA/RNA Mini Kit (Qiagen Inc., CA). Whole exome sequencing (WES) was conducted on 16 lung SCC tumors and 8 normal lung tissue samples. Detailed WES procedures can be seen in our previous publication [64]. Briefly, whole exome capture was carried out using the protocol for Agilent’s SureSelectXT Mouse All Exon Kit. The exome-seq libraries were sequenced for 100 bp paired-end reads by the MCW Human and Molecular Genetics Center (HMGC) Sequencing Core using HiSeq 2500 platforms (Illumina, San Diego, CA). Each sample was sequenced at a mean depth of about 133 X. Sequence short reads were aligned to a reference genome (NCBI mouse genome assembly mm9) using the BWA (Burrows-Wheeler Aligner) program [65]. Each alignment was assigned a mapping quality score by BWA [65], which is the Phred-scaled probability that the alignment is incorrect. Reads with low mapping quality scores (<5) were removed to reduce false positive rate. The PCR duplicates were detected and removed by the Picard program. We then performed local realignment of the BWA-aligned reads using the Genome Analysis Toolkit (GATK) [66]. VarScan 2 [67] was used to call somatic variants based on the local realignment results comparing each tumor with the eight normal lung samples. Default parameters in VarScan 2 were used. The lists of shared SNVs/indels were then annotated using ANNOVAR [68]. Single nucleotide polymorphisms (SNPs) were filtered using the mouse dbSNP VCF file (mm9_snp128). For RNA-seq, the quality of the total RNA samples obtained was very high, with RIN (RNA integrity number) values in the range of 9–10. The TruSeq RNA Library Preparation Kit v2 (Illumina Inc., CA) was used to construct the RNA-seq libraries. The sequencing of the RNA-seq library samples was performed in MCW-HMGC Sequencing Core using HiSeq 2500 platforms. The reads generated were pair-end with 100 nucleotides in length. The qualities of the RNA-seq reads were analyzed using the FastQC program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The coverage ranged from 15 million to 32 million reads per RNA-seq sample. The quality scores of >95.3% of all the bases of each sample are >30, averaging around 40. The pre-processed sample RNA-seq reads were aligned to the mm9 mouse genome using Bowtie-TopHat [version 2.0.4 [69, 70]]. Gene-level read counts were obtained using the htseq-count Python script (http://www-huber.embl.de/users/anders/HTSeq/) in the “union” mode. Further differential gene expression analysis was conducted using edgeR software [71]. FDR (False discovery rate) corrected P-values of less than 0.05 were used as criteria for significantly regulated genes.
Single-cell isolation and RNA sequencing
Single-cell suspensions were prepared from two mouse LUSC tumor tissue samples using a mouse Tumor Dissociation kit (Miltenyi Biotec Inc., CA). For single-cell analysis cell capture, lysis, reverse transcription, and cDNA amplification were performed on the C1 integrated fluidic circuit (IFC) for mRNA-seq on a Fluidigm C1 Single-Cell Auto Prep System following the manufacturer’s protocol (Fluidigm Corporation, CA). Medium-sized C1 mRNA-Seq chips (10–17 μm) were used to capture single cells from each of the two tumors. The C1 Auto Prep System captures the dissociated single cells and the whole-transcriptome amplified cDNA was prepared on chip using the SMARTer Ultra Low RNA kit from Illumina (Clontech). Cells captured are manually inspected as a quality control measure to remove empty well, doublets, or debris-containing wells. cDNA from cells are checked by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Single-cell libraries were constructed with the use of the Illumina Nextera XT DNA Sample Preparation kit with 96 dual barcoded indices and were multiplexed and sequenced to a depth of 2–4 million reads (HiSeq 2500; Illumina) using 50-bp single-end sequencing. The program RSEM [72] was utilized to quantify transcript expression. Differential expression analysis was conducted on RSEM derived TPM (Transcripts Per Kilobase Million) values using the software AltAnalyzer [73]. FDR (False discovery rate) corrected P-values of less than 0.05 were used as criteria for significantly regulated genes.
Clonal mutation, pathway enrichment, survival and association analyses
The bioinformatics tools – SciClone [74] and clonevol package (https://github.com/hdng/clonevol) were used to identify the clonal structures of each of the 16 mouse LUSC tumors. Single-cell gene mutations were analyzed using the rnaseqmut program (https://github.com/davidliwei/rnaseqmut/blob/master/README) based on scRNA-seq data. The software SCITE [75] was used to analyze the orders of the sequential mutational events in the two LUSC tumors subjected to scRNA-seq. Pathway enrichment analysis of the G80 module cancer genes was performed by using ConsensusPathDB software (http://cpdb.molgen.mpg.de/). Four published lung cancer gene expression data sets [18–20, 56] were analyzed using the program SurvExpress [76] to test whether the G80 cancer gene module was associated with overall survival of the patients. In order to test whether these mutated genes have potential clinical application, the associations between mutational status of the identified LUSC genes and markers of immune response were analyzed by using the set of 176 TCGA LUSC tumor samples with gene mutation data and expression data available through NCI GDC Data Portal (https://portal.gdc.cancer.gov/). The association test between mutation genotypes and immune checkpoint gene expression in LUSC was performed using the t test implemented in the computing environment R (R Development Core Team, 2005). FDR (False discovery rate) corrected p-values of less than 0.05 were used as criteria for significant association. Plots of mutations were generated using the “oncoPrint” function provided by the R package – ComplexHeatmap [77] and gene expression heatmaps were generated using the R package – heatmap3 (https://cran.r-project.org/web/packages/heatmap3/).
ACKNOWLEDGMENTS AND FUNDING
We thank the outside reviewers for the suggestions and revisions of this manuscript. This work was supported in part by National Institutes of Health (N01CN201200015 and R01CA134682).
CONFLICTS OF INTEREST
The authors have declared that no conflicts of interest exists.
REFERENCES
1. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566.
2. Gariboldi M, Spinola M, Milani S, Pignatiello C, Kadota K, Bono H, Hayashizaki Y, Dragani TA, Okazaki Y. Gene expression profile of normal lungs predicts genetic predisposition to lung cancer in mice. Carcinogenesis. 2003; 24:1819–1826.
3. Chang JT, Lee YM, Huang RS. The impact of the Cancer Genome Atlas on lung cancer. Transl Res. 2015; 166:568–585.
4. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–525.
5. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550.
6. Rekhtman N, Paik PK, Arcila ME, Tafe LJ, Oxnard GR, Moreira AL, Travis WD, Zakowski MF, Kris MG, Ladanyi M. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res. 2012; 18:1167–1176.
7. Colby TV, Wistuba II, Gazdar A. Precursors to pulmonary neoplasia. Adv Anat Pathol. 1998; 5:205–215.
8. Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002; 1:49–52.
9. Sy SM, Wong N, Lee TW, Tse G, Mok TS, Fan B, Pang E, Johnson PJ, Yim A. Distinct patterns of genetic alterations in adenocarcinoma and squamous cell carcinoma of the lung. Eur J Cancer. 2004; 40:1082–1094.
10. Yakut T, Schulten HJ, Demir A, Frank D, Danner B, Egeli U, Gebitekin C, Kahler E, Gunawan B, Urer N, Ozturk H, Fuzesi L. Assessment of molecular events in squamous and non-squamous cell lung carcinoma. Lung Cancer. 2006; 54:293–301.
11. Zochbauer-Muller S, Minna JD. The biology of lung cancer including potential clinical applications. Chest Surg Clin N Am. 2000; 10:691–708.
12. Wang Y, Zhang Z, Yan Y, Lemon WJ, LaRegina M, Morrison C, Lubet R, You M. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res. 2004; 64:1647–1654.
13. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016; 16:759–773.
14. Choi M, Kadara H, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, Kim K, Kalhor N, Moran C, et al. Mutation profiles in early-stage lung squamous cell carcinoma with clinical follow-up and correlation with markers of immune function. Ann Oncol. 2017; 28:83–89.
15. Lee HY, Lee SH, Won JK, Lee DS, Kwon NJ, Choi SM, Lee J, Lee CH, Lee SM, Yim JJ, Yoo CG, Kim YW, Han SK, et al. Analysis of Fifty Hotspot Mutations of Lung Squamous Cell Carcinoma in Never-smokers. J Korean Med Sci. 2017; 32:415–420.
16. Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc Natl Acad Sci USA. 1998; 95:3578–3582.
17. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–525.
18. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008; 14:822–827.
19. Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, Laffaire J, de Reynies A, Beer DG, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013; 5:186ra166.
20. Botling J, Edlund K, Lohr M, Hellwig B, Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Ponten F, Konig A, Fernandes O, Karlsson M, et al. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin Cancer Res. 2013; 19:194–204.
21. Kim Y, Hammerman PS, Kim J, Yoon JA, Lee Y, Sun JM, Wilkerson MD, Pedamallu CS, Cibulskis K, Yoo YK, Lawrence MS, Stojanov P, Carter SL, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol. 2014; 32:121–128.
22. Lin X, Yang Z, Zhang P, Liu Y, Shao G. miR-154 inhibits migration and invasion of human non-small cell lung cancer by targeting ZEB2. Oncol Lett. 2016; 12:301–306.
23. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001; 7:1267–1278.
24. Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005; 33:6566–6578.
25. McEvoy SH, Halpenny DF, Viteri-Jusue A, Hayes SA, Plodkowski AJ, Riely GJ, Ginsberg MS. Investigation of patterns of nodal metastases in BRAF mutant lung cancer. Lung Cancer. 2017; 108:62–65.
26. Kong FM, Anscher MS, Washington MK, Killian JK, Jirtle RL. M6P/IGF2R is mutated in squamous cell carcinoma of the lung. Oncogene. 2000; 19:1572–1578.
27. Tian Z, Yao G, Song H, Zhou Y, Geng J. IGF2R expression is associated with the chemotherapy response and prognosis of patients with advanced NSCLC. Cell Physiol Biochem. 2014; 34:1578–1588.
28. Glubb DM, Pare-Brunet L, Jantus-Lewintre E, Jiang C, Crona D, Etheridge AS, Mirza O, Zhang W, Seiser EL, Rzyman W, Jassem J, Auman T, Hirsch FR, et al. Functional FLT1 Genetic Variation is a Prognostic Factor for Recurrence in Stage I-III Non-Small-Cell Lung Cancer. J Thorac Oncol. 2015; 10:1067–1075.
29. Zhang SD, McCrudden CM, Kwok HF. Prognostic significance of combining VEGFA, FLT1 and KDR mRNA expression in lung cancer. Oncol Lett. 2015; 10:1893–1901.
30. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008; 9:616–627.
31. Zhang Y, Zhang L, Li R, Chang DW, Ye Y, Minna JD, Roth JA, Han B, Wu X. Genetic variations in cancer-related significantly mutated genes and lung cancer susceptibility. Ann Oncol. 2017; 28:1625–1630.
32. Majhi PD, Lakshmanan I, Ponnusamy MP, Jain M, Das S, Kaur S, Shimizu ST, West WW, Johansson SL, Smith LM, Yu F, Rolle CE, Sharma P, et al. Pathobiological implications of MUC4 in non-small-cell lung cancer. J Thorac Oncol. 2013; 8:398–407.
33. Lopez-Ferrer A, Curull V, Barranco C, Garrido M, Lloreta J, Real FX, de Bolos C. Mucins as differentiation markers in bronchial epithelium. Squamous cell carcinoma and adenocarcinoma display similar expression patterns. Am J Respir Cell Mol Biol. 2001; 24:22–29.
34. Bavarva JH, Tae H, McIver L, Garner HR. Nicotine and oxidative stress induced exomic variations are concordant and overrepresented in cancer-associated genes. Oncotarget. 2014; 5:4788–4798. https://doi.org/10.18632/oncotarget.2033.
35. Long W, Foulds CE, Qin J, Liu J, Ding C, Lonard DM, Solis LM, Wistuba II, Tsai SY, Tsai MJ, O’Malley BW. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J Clin Invest. 2012; 122:1869–1880.
36. Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, Girard L, Sullivan JP, Gao B, Peyton M, Nanjundan M, Byers L, Heymach J, et al. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res. 2010; 70:6477–6485.
37. Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, Gao W. P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget. 2017; 8:54548–54557. https://doi.org/10.18632/oncotarget.17408.
38. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, Boulden J, Sutanto-Ward E, Soler AP, Laury-Kleintop LD, Mandik-Nayak L, Metz R, Ostrand-Rosenberg S, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2012; 2:722–735.
39. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012; 150:1121–1134.
40. Kaul SC, Deocaris CC, Wadhwa R. Three faces of mortalin: a housekeeper, guardian and killer. Exp Gerontol. 2007; 42:263–274.
41. Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, Kaul SC. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer. 2006; 118:2973–2980.
42. Shi S, He Z, Cai J, Qiu J. [RNA Interference targeting GRP75 decreases cisplatin resistance in human lung adenocarcinoma cell]. [Article in Chinese]. Zhongguo Fei Ai Za Zhi. 2011; 14:305–310.
43. Shi YX, Zhu T, Zou T, Zhuo W, Chen YX, Huang MS, Zheng W, Wang CJ, Li X, Mao XY, Zhang W, Zhou HH, Yin JY, et al. Prognostic and predictive values of CDK1 and MAD2L1 in lung adenocarcinoma. Oncotarget. 2016; 7:85235–85243. https://doi.org/10.18632/oncotarget.13252.
44. Han ML, Zhao YF, Tan CH, Xiong YJ, Wang WJ, Wu F, Fei Y, Wang L, Liang ZQ. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol Sin. 2016; 37:1606–1622.
45. Spitz MR, Gorlov IP, Dong Q, Wu X, Chen W, Chang DW, Etzel CJ, Caporaso NE, Zhao Y, Christiani DC, Brennan P, Albanes D, Shi J, et al. Multistage analysis of variants in the inflammation pathway and lung cancer risk in smokers. Cancer Epidemiol Biomarkers Prev. 2012; 21:1213–1221.
46. Mao X, Luo F, Boyd LK, Zhou B, Zhang Y, Stankiewicz E, Marzec J, Vasiljevic N, Yu Y, Feng N, Xu J, Lorincz A, Jiang Y, et al. NKAIN2 functions as a novel tumor suppressor in prostate cancer. Oncotarget. 2016; 7:63793–63803. https://doi.org/10.18632/oncotarget.11690.
47. Zhao SC, Zhou BW, Luo F, Mao X, Lu YJ. The structure and function of NKAIN2-a candidate tumor suppressor. Int J Clin Exp Med. 2015; 8:17072–17079.
48. Zhang G, Liu D, Long G, Shi L, Qiu H, Hu G, Liu S. Downregulation of microRNA-181d had suppressive effect on pancreatic cancer development through inverse regulation of KNAIN2. Tumour Biol. 2017; 39:1010428317698364.
49. Romania P, Castellano A, Surace C, Citti A, De Ioris MA, Sirleto P, De Mariano M, Longo L, Boldrini R, Angioni A, Locatelli F, Fruci D. High-resolution array CGH profiling identifies Na/K transporting ATPase interacting 2 (NKAIN2) as a predisposing candidate gene in neuroblastoma. PLoS One. 2013; 8:e78481.
50. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, et al. The life history of 21 breast cancers. Cell. 2012; 149:994–1007.
51. Kashyap MK. Role of insulin-like growth factor-binding proteins in the pathophysiology and tumorigenesis of gastroesophageal cancers. Tumour Biol. 2015; 36:8247–8257.
52. Chen Y, Cui T, Knosel T, Yang L, Zoller K, Petersen I. IGFBP7 is a p53 target gene inactivated in human lung cancer by DNA hypermethylation. Lung Cancer. 2011; 73:38–44.
53. Suzuki M, Shiraishi K, Eguchi A, Ikeda K, Mori T, Yoshimoto K, Ohba Y, Yamada T, Ito T, Baba Y, Baba H. Aberrant methylation of LINE-1, SLIT2, MAL and IGFBP7 in non-small cell lung cancer. Oncol Rep. 2013; 29:1308–1314.
54. Okamura J, Huang Y, Moon D, Brait M, Chang X, Kim MS. Downregulation of insulin-like growth factor-binding protein 7 in cisplatin-resistant non-small cell lung cancer. Cancer Biol Ther. 2012; 13:148–155.
55. Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009; 34:176–188.
56. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–525.
57. Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, Fuchs E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014; 343:309–313.
58. Li X. Emerging role of mutations in epigenetic regulators including MLL2 derived from The Cancer Genome Atlas for cervical cancer. BMC Cancer. 2017; 17:252.
59. Pandey P, Singh AK, Singh M, Tewari M, Shukla HS, Gambhir IS. The see-saw of Keap1-Nrf2 pathway in cancer. Crit Rev Oncol Hematol. 2017; 116:89–98.
60. Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, Yoo Y, Kalhor N, Moran C, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. 2017; 28:75–82.
61. Scheel AH, Ansen S, Schultheis AM, Scheffler M, Fischer RN, Michels S, Hellmich M, George J, Zander T, Brockmann M, Stoelben E, Groen H, Timens W, et al. PD-L1 expression in non-small cell lung cancer: Correlations with genetic alterations. Oncoimmunology. 2016; 5:e1131379.
62. Rehm S, Lijinsky W, Singh G, Katyal SL. Mouse bronchiolar cell carcinogenesis. Histologic characterization and expression of Clara cell antigen in lesions induced by N-nitrosobis-(2-chloroethyl) ureas. Am J Pathol. 1991; 139:413–422.
63. Pan J, Zhang Q, Liu Q, Komas SM, Kalyanaraman B, Lubet RA, Wang Y, You M. Honokiol inhibits lung tumorigenesis through inhibition of mitochondrial function. Cancer Prev Res (Phila). 2014; 7:1149–1159.
64. Xiong D, Li G, Li K, Xu Q, Pan Z, Ding F, Vedell P, Liu P, Cui P, Hua X, Jiang H, Yin Y, Zhu Z, et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis. 2012; 33:1797–1805.
65. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760.
66. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303.
67. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–576.
68. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164.
69. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25.
70. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111.
71. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140.
72. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323.
73. Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, Grimes HL. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016; 537:698–702.
74. Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, Vij R, Tomasson MH, Graubert TA, Walter MJ, Ellis MJ, Schierding W, DiPersio JF, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014; 10:e1003665.
75. Jahn K, Kuipers J, Beerenwinkel N. Tree inference for single-cell data. Genome Biol. 2016; 17:86.
76. Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, Tamez-Pena JG, Trevino V. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 16:e74250.
77. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849.