
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide, despite a reduced incidence in western countries and a remarkable improvement in its therapeutic approach. Among lung tumors, Non Small Cell Lung Cancer (NSCLC) is the most common diagnosis and the adenocarcinoma is the predominant subtype [1]. Several driver mutations have been described, in the recent years, in lung adenocarcinomas including those affecting KRAS (15-25%) and EGFR (10-35%). Less commonly AKT1, PIK3CA, HER2, MAPK1, MEK1 and MET mutations have been reported. Moreover, rearrangements involving ALK, ROS1 or RET locus have been identified [2].
The majority of these alterations are targetable by the EGFR tyrosine kinase inhibitors (TKIs) (i.e. gefitinib, erlotinib, afatinib in EGFR-mutant; crizotinib, ceritinib, brigatinib, and alectinib in ALK rearranged tumors) [3–8].
However, there are no specific approved drugs for patients with KRAS mutant tumors and all the anti-KRAS evaluated compounds have failed to demonstrate any clinical activity. Indeed, because of the high frequency of KRAS mutations in NSCLC, several preclinical and clinical investigations have been conducted including inhibition of KRAS protein expression via RNA interference (RNAi), blocking post-translational modification with farnesyl-transferase inhibitors (FTIs) or blocking KRAS localization at the cellular membrane [9–11]. Different strategies evaluated an epigenetic approach, using cyclin-dependent kinases, heat shock proteins or focal adhesion inhibitors [12–14]. Several researchers wagered on inhibitors of downstream effectors of the KRAS signaling pathways (PI3K/AKT/mTOR and RAF/MEK/ERK) without significant success [15–17].
This review focuses on the molecular pathways of KRAS in order to point out possible targets for an anti-KRAS approach reporting success and failures of compounds developed to date.
MATERIALS AND METHODS
A bibliographic search of the NCBI PubMed database was conducted for articles published in English, using the following keywords: KRAS, KRAS mutations in non-small cell lung cancer, KRAS and tumorigenesis, KRAS and TKIs, KRAS and chemotherapy, KRAS and monoclonal antibody, KRAS and immunotherapy, KRAS and drugs, KRAS and drug resistance.
RESULTS
KRAS and its signaling pathways
KRAS belongs to a group of small GTP-binding proteins called the RAS superfamily or RAS-like GTP-ases. This group includes Harvey-Ras (H-RAS), neuroblastoma-Ras (N-RAS) and two splice variants of KRAS: KRAS4A and KRAS4B [18]. Following to the binding of growth factors with the respective receptors (i.e. EGFR), KRAS protein bounds to GTP and become able to activate intracellular pathways between a GTP-bound active and inactive state. GTPase activating proteins (GAPs) facilitate GTP hydrolysis, amplifying the intrinsic GTPase activity of KRAS. The interaction with guanine-exchanging/releasing factors (GEFs) promotes the exchange of the GDP with GTP [19]. In the GTP-bound state, KRAS interacts with multiple downstream effectors, including PI3K/AKT/mTOR, RAF/MEK/ERK and Ral-GEF pathways, and regulates cell proliferation, survival, motility, differentiation, endocytosis, angiogenesis, and apoptosis (Figure 1) [20]. Many other signaling pathways are involved in the feedback regulation and crosstalk, which contribute to the complexity of the KRAS signaling network. These pathways include the downstream effector protein kinase Cι (PKCι) involved in tumor-initiating cell phenotype through the PKCι/ELF3/NOTCH3 axis [21]; the EphA2 a receptor tyrosine kinase (RTK) that negatively regulates the KRAS/MEK/ERK pathway after the binding of its ligand ephrin A1 [22]; or the CUB domain–containing protein 1 (CDCP1) and AXL that are two RTKs regulated by many KRAS effectors, like RAF/MEK/ERK and PI3K/AKT/mTOR [23, 24].
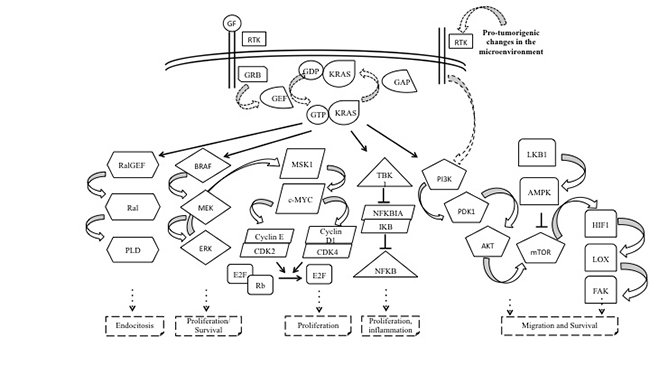
Figure 1: Complexity of KRAS signalling pathways. KRAS belongs to the Ras superfamily, a group of small GTP-binding proteins, and serves as a signal transducer from its tyrosine kinase receptors, that are engaged by autocrine and paracrine stimuli. In the active GTP-bound conformation, KRAS activates several effector molecules resulting in endocytosis, cell growth and proliferation, apoptosis, migration, and survival. Abbreviations: GF: growth factor; RTK: receptor tyrosine kinase; GRB: growth factor receptor-binding protein; GEF: guanine nucleotide exchange factor; GTP: guanosine triphosphate; GDP: guanosine diphosphate; KRAS: Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; GAP: GTPase activating protein; RalGEF: RAL-guanine nucleotide exchange factor; PLD: phospholipase D; BRAF: v-Raf murine sarcoma viral oncogene homolog B; Mek: Mitogen-activated protein kinase kinase; Erk: extracellular signal-related kinase; MSK1: mitogen- and stress-activated protein kinase; C-MYC: v-Myc avian myelocytomatosis viral oncogene homolog; CDK2/4: cyclin-dependent kinase 2/4; E2F: E2 transcription factor; Rb: retinoblastoma protein; TBK1: TANK Binding Kinase 1; NFKB: nuclear factor kappa-light-chain-enhancer of activated B cells; NFKBIA: NFKB inhibitor alpha; FAK: focal adhesion kinase; PI3K: phosphoinositide-3-kinase; AKT: v-Akt murine thymoma viral oncogene homolog 1; mTOR: mammalian target of rapamycin; LKB1: serine/threonine kinase 11; AMPK: AMP-activated protein kinase; HIF1: hypoxia-inducible factor 1; LOX: lysyl oxidase; FAK: focal adhesion kinase.
KRAS mutations and NSCLC
KRAS is mutated in 15-25% of NSCLC, mostly adenocarcinoma and occasionally squamous cell carcinoma [25], and is more frequent in white than in Asian populations (25–50% vs 5–15%, respectively) [26]. There is a linkage between smoking habits and KRAS mutations, with an incidence of 25–35% in smokers and only 5% in never-smokers [27–29]. Approximately, 97% of KRAS mutations interest codons 12 and 13 in exon 1, less frequently, mutations occur at codon 61 (Table 1) [30–32]. While these mutations are located near to the GTP binding site, the intrinsic GTP-ase activity is impaired and KRAS accumulates in GTP-bound constitutively active form, which sustains the activation of the KRAS downstream signalling [33].
Table 1: Frequency of KRAS mutations in NSCLC
KRAS genotype |
Mutation |
Frequency (%) |
Reference |
|---|---|---|---|
p.G12C |
c.34G>T |
42 |
[25] |
p.G12V |
c.35G>T |
21 |
|
p.G12D |
c.35G>A |
17 |
|
p.G12A |
c.35G>C |
7 |
|
p.G13D |
c.38G>A |
2 |
KRAS and EGFR mutations are usually mutually exclusive and, therefore, KRAS mutations can be considered as innate resistance factor for anti-EGFR TKI. However, recent published data, obtained with high sensible technologies, suggest that some tumors can share EGFR and KRAS mutation in a heterogeneous tumor cell population, as well as the co-occurrence of genomic alteration in LKB1 and TP53 [34–39].
Therapeutic approaches for KRAS-mutant NSCLC patients
Inhibition of the KRAS gene expression
One effect of KRAS activation is the induction of telomerase (TERT) transcription [40]. Recently, Liu and colleagues confirmed an increased mRNA expression, telomerase activity and telomere length in lung adenocarcinoma cells with KRAS mutations. However, BIBR1532, a telomerase inhibitor, hampered KRAS-induced cells’ proliferation suggesting that telomerase could represent a promising target in KRAS-mutated NSCLCs [41]. In fact, in a randomized phase II trial, the maintenance treatment with the telomerase inhibitor imetelstat did not improve progression-free survival (PFS) in advanced NSCLC patients [42].
Zhang and colleagues evaluated the anti-tumor effect of an anti-KRAS ribozyme adenoviral vector (KRbz-ADV) in NSCLC cell lines with or without KRAS mutation, finding that KRbz-ADV inhibits significantly the growth of KRAS mutant than wild type cells [43].
Using an RNA interference (RNAi), Sunaga and colleagues in 2011 investigated if the knock down mutant KRAS transcript may revert the malignant phenotype of NSCLC. KRAS expression was inhibited and cell proliferation was reduced alongside with a down regulation of MAPK pathway, however, tumorigenicity was not abolished. These findings remarked the complexity of mutant KRAS oncogenic signaling and cell capability to overcome the KRAS “targeted inhibition” [9]. Despite recent researches encouraged the clinical application of the targeted silencing for NSCLC patients harboring a KRAS mutation [44, 45], further investigations are still needed in the gene therapy field.
HDAC inhibitors
The histone deacetylase inhibitors (HDACi) block gene transcription, inhibit proliferation and induce apoptosis in tumor cells with promising results for the treatment of some neoplastic proliferative diseases [46]. Kurtze and colleagues evaluated if the treatment of KRAS-mutant NSCLC A549 cell line with vorinostat, an HDACi, could overcome the resistance to the EGFR TKIs [47]. The combination of vorinostat with gefitinib or erlotinib was found to induce apoptosis, revering the TKI-resistance status of A549 cells [47]. Similar results were observed when three NSCLC cell lines, including the KRAS-mutant and EGFR wild type A549, were tested with panobinostat, an HDACi, and erlotinib. As expected, while proliferation of A549 cell line was not inhibited by erlotinib alone, it was impaired by panobinostat treatment, as well as by the synergistic combination of both drugs [48]. In an exploratory biomarker analysis of phase I/II trial investigating the efficacy of gefitinib plus vorinostat in NSCLC, the presence of sensitive EGFR mutations was predictive of higher response rate (RR), longer PFS and overall survival (OS) compared to KRAS mutations [14]. However, these results could be simply related to the high activity of gefitinib in EGFR-mutant patients, and do not clarify the role of HDAC inhibition.
Inhibition of the KRAS trafficking
Post-translational modifications of the KRAS protein is another target for KRAS inhibition. After transduction, KRAS undergoes multi-stage post-translational modifications to become active. First, the protein undergoes prenylation by the addition of a farnesyl tail to its carboxyl-terminal by the farnesyl-transferase (FT-ase) [49]. Several studies have been conducted to determine whether the farnesyltransferase inhibitors (FTIs) have clinical activity in NSCLC patients [10, 50, 51]. Tipifarnib and lonafarnib showed activity in vitro and in chemically-induced KRAS-mutant lung tumors in mice [52, 53]. In clinical trials FTIs did not show activity in NSCLC, and they have never been tested in a defined KRAS mutant population [10, 50]. A possible explanation for the FTIs failure may be the presence of an alternative modification, the geranylgeranylation, that is another process to localize protein to the membrane (Figure 2) [54].
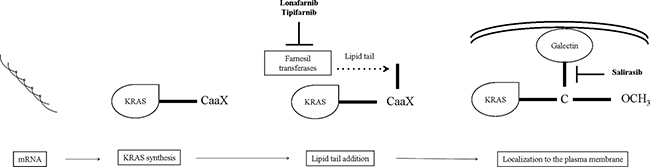
Figure 2: Steps towards KRAS membrane trafficking and localization. After KRAS synthesis in the cytoplasm, farnesyl transferases add a lipid tail at a CaaX tetrapeptide motif (C: amminoacid cysteine; aa: two aliphatic residues; X: a variable residue) on the C-terminus of inactive KRAS protein. Lonafarnib and tipifarnib may inhibit this step, interfering with KRAS membrane trafficking. On the other hand, KRAS signaling could be block by salirasib, that targets the localization of KRAS to the membrane. Abbreviations: KRAS: Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; CaaX: carboxyl-terminal.
Inhibition of KRAS localization
Other attempts to block the KRAS signalling is to interfere with its localization in cellular membranes using RAS farnesyl cysteine mimetic drugs, like salirasib (farnesylthiosalicylic acid). Mimetic drugs dislodge KRAS from its membrane-anchoring sites and prevent activation of the signaling cascades [54]. Despite promising preclinical data [55], early-phase clinical trials were not successful. Riely and colleagues enrolled 33 patients with stage IIIb/IV lung adenocarcinoma, of which 30 had a KRAS mutation, however, none of the patients raised a radiographic partial response (PR). Despite moderate toxicity (diarrhea, nausea, and fatigue), this phase II trial testing salirasib failed to show any clinical benefit for NSCLC patients harboring KRAS mutations. Interestingly, this was the first trial to examine a targeted therapy specifically in KRAS-mutant NSCLC (Figure 2) [11].
The failure of this trial emphasized the challenges in targeting challenges KRAS prenylation and its membrane localization. First, it is known that an alternative process that could prenylate KRAS proteins exists (geranyl-geranylation). In addition, several signaling molecules are farnesylated (Rho-B, Rho-E, Lamin A, Lamin B, PTP-CAAX1/2), supporting a pleiotropic biological effect, even if KRAS were significantly inhibited by FTIs [49].
Targeting the downstream effectors of oncogenic KRAS
PI3K/AKT/mTOR inhibitors
The PI3K/AKT/mTOR pathway is frequently activated in cancer and maintains tumor growth [56]. In lung cancer, mTOR phosphorylation was found in 51% of NSCLC patients [57]. PI3K/AKT/mTOR pathway is a downstream effector of KRAS and its inhibition could have a role in KRAS mutant NSCLC [58]. Castellano and colleagues reported that PI3K inhibitors cause the regression of KRAS p.G12D-induced benign lung tumors in genetically engineered mouse models [59]. Instead, in mice with malignant lung cancer harboring the KRAS p.G12D, PI3K p.H1047R mutations, and TP53-null, Green et colleagues showed a modest growth inhibition using PI3K inhibitors and little or no survival benefit [60]. Moreover, these results are in line with several clinical observations suggesting a limited activity of PI3K/AKT/mTOR inhibitors in NSCLC. The BASALT-1 trial, evaluating the combination of buparlisib, a PIK3CA inhibitor, with chemotherapy was closed for futility at first interim analysis. The study included 12 patients with KRAS mutation, which had a trend for a better PFS [61].
mTOR inhibitors seem to be able to stop the malignant progression in mice and in preclinical models of NSCLC with a KRAS mutation [62]. However, in the randomized clinical trial, 79 patients with KRAS mutant NSCLC treated with ridaforolimus, only achieved an overall response rate of 1% (Figure 3) [63].
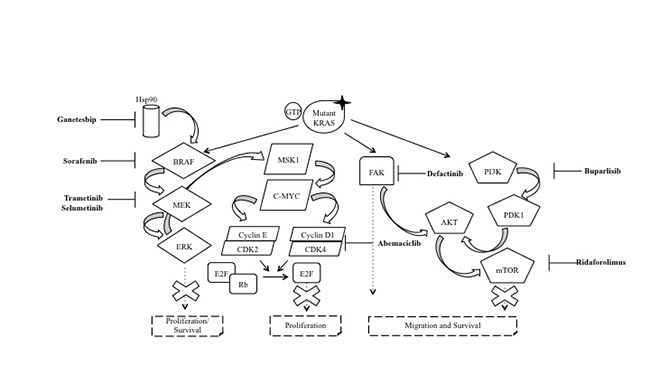
Figure 3: Targeting downstream effectors of oncogenic KRAS. In NSCLC, the KRAS protein is often mutated (mutant KRAS) leading to the inactivation of its GTPase activity. The result is the constitutive activation of KRAS and, therefore, of the several effector pathways that are activated downstream of KRAS, with the RAF/MEK/ERK and PI3K/AKT/mTOR as the two pathways that have been studied most in detail. Sorafenib is a multitarget TKI which also inhibits BRAF protein, while trametinib and selumetinib acts against MEK protein. On the other hand, buparlisib and ridaforolimus have been used as PI3K and mTOR inhibitors, respectively. Independently, to these two best characterized pathways, the research focused on the inhibition of other targets. For example, ganetespib, defactinib and abemaciclib act against HSP90, FAK and CDK4, respectively. The goal of these drugs is to stop the tumorigenesis promoted by mutant KRAS. Abbreviations: RTK: receptor tyrosine kinase; Hsp90: heat shock protein 90; GTP: guanosine triphosphate; GDP: guanosine diphosphate; KRAS: Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; BRAF: v-Raf murine sarcoma viral oncogene homolog B; MEK: mitogen-activated protein kinase kinase; ERK: extracellular signal-related kinase; MSK1: mitogen- and stress-activated protein kinase; c-MYC: v-Myc avian myelocytomatosis Viral Oncogene Homolog; CDK2/4: cyclin-dependent kinase 2/4; E2F: E2 transcription factor; Rb: retinoblastoma protein; FAK: focal adhesion kinase; PI3K: phosphoinositide-3-kinase; AKT: v-Akt murine thymoma viral oncogene homolog 1; mTOR: mammalian target of rapamycin.
RAF/MEK/ERK pathway inhibitors
Sorafenib is a multi-tyrosine kinase inhibitor against vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR) and Proto-Oncogene Tyrosine-Protein Kinase (KIT), competing with ATP in the hydrophobic pocket, leading to the inhibition of MAPK pathway [64]. Smit and colleagues reported in an early phase trial 4 partial responses and 6 stabilisations of diseasein 10 NSCLC patients, harboring the KRAS mutation treated with sorafenib after failure of chemotherapy [65]. In a phase II study, among thirty-seven patients with advanced NSCLC treated with sorafenib, KRAS mutations were found in 11 patients that obtained 60% of disease control rate (DCR) compared to 71% in the KRAS wild type tumors (p = 0.69) [66]. The phase II BATTLE trial enrolled patients with advanced NSCLC who had been previously treated with chemotherapy and subsequently experienced a disease relapse [15]. The trial enrolled 255 patients, randomized to one of 4 treatment arms (erlotinib, vandetanib, erlotinib plus bexarotene, or sorafenib) according to their baseline biomarker profile analysis, among which KRAS mutations. The stratified biomarker analysis showed that sorafenib had a higher DCR at 8 weeks in KRAS mutant patients, although the difference was not statistically significant (61% vs. 32%, p = 0.11) [15]. Interestingly, Ihle and colleagues pointed out that different types of KRAS mutations may predict for different outcomes: tumors with p.G12C and p.G12V mutations had the worse PFS compared with other codon 12 KRAS mutations or KRAS wild-type (median PFS was 1.84 months vs. 3.55 months vs. 1.95 months, respectively, p = 0.046). Moreover, the negative impact of KRAS p. G12C and p. G12V mutations on PFS was particularly evident in patients treated with sorafenib (p = 0.026) [67]. The MISSION trial failed in his primary endpoint, and highlighted that there were no differencies in PFS in mutant or not mutant patients for KRAS when treated with sorafenib [16]. Also in the first stage of the BATTLE II trial patients treated with sorafenib (29.5% were KRAS mutant), did not show a difference in DCR compared to wild type [68]. Moreover, several trials evaluated sorafenib in combination with erlotinib for NSCLC patients without any promising results (Figure 3) [69, 70].
Monotherapy and combinations with MEK inhibitors
Selumetinib is a potent, selective, and ATP-uncompetitive inhibitor of MEK1-2 kinases able to inhibit proliferation of KRAS mutant in NSCLC cell lines [71]. Due to its potency, selumetinib has been tested as either monotherapy or in combination with cytotoxic agents to target KRAS mutant NSCLC patients. In the CUSTOM trial selumetinib was studied in patients with KRAS, NRAS, HRAS, or BRAF mutations. Only one PR was observed in 9 patients treated with selumetinib with a median PFS time of 2.3 months, and a median OS of 6.5 months [72].
Trametinib is a reversible and highly selective allosteric inhibitor of MEK1 and MEK2 kinase activity and has been developed and studied in BRAF-mutant metastatic melanoma [73, 74]. In early phase of clinical development, trametinib has been given to 30 NSCLC patients obtaining only two PR, however, both patients were carrier of a KRAS mutation [75]. Following these results, trametinib was compared to docetaxel in a phase II trial in stage IV KRAS mutant NSCLC [76]. Unfortunately, trametinib did not show its superiority and the study was prematurely terminated.
Overall, these results suggest that the presence of a compensatory signaling pathway makes MEK inhibition not sufficient to achieve a significant antitumor effect. On the other hand, the efficacy of a combined treatment between trametinib or selumetinib and cytotoxic agents in NSCLC cells represented a strong rationale to use this drug combination as a potent strategy to improve treatment response in NSCLC patients with KRAS mutations (Figure 3) [77].
In preclinical models, docetaxel in combination with selumetinib showed an important inhibition of tumor growth and regression [78]. A randomized phase II clinical trial evaluated the combination of docetaxel with selumetinib or placebo, in patients with KRAS-mutant NSCLC that progressed after first-line chemotherapy [79]. Interestingly, patients with KRAS p.G12C or p.G12V mutations treated with selumetinib plus docetaxel showed greater improvement in OS, PFS and ORR compared with the other KRAS mutations [80]. Unfortunately, selumetinib in addiction to docetaxel failed to improve overall survival, progression free survival and overall response rate compared to docetaxel alone in the phase III clinical trial SELECT-1 for KRAS mutant NSCLCs [81].
Trametinib, has been evaluated in a phase I trial in combination with docetaxel or pemetrexed in advanced NSCLC patients with or without KRAS mutations [17]. Both combinations showed activity in KRAS mutant and wild-type tumors. Primary endpoint was the objective response rate (ORR). A confirmed PR was observed in 10 of the 47 patients with NSCLC who received trametinib plus docetaxel (21%). The ORR was 18% (four PRs in 22 patients) in those with KRAS wild-type NSCLC and 24% (six PRs in 25 patients) in those with KRAS-mutant NSCLC. Of the 42 patients with NSCLC treated with trametinib plus pemetrexed, six (14%) had a PR; the ORR was 17% (four of 23) in patients with KRAS-mutated NSCLC versus 11% (two of 19) in KRAS wild-type NSCLC [17]
As far as it is known, KRAS mutant NSCLCs do not respond to EGFR-TKIs treatment [82]. The combination of selumetinib with erlotinib was evaluated in randomized phase II trial and compared with selumetinib alone in KRAS mutant tumors or with erlotinib alone in KRAS wild type tumors [83]. In 41 KRAS mutant tumors, the PFS was 4.0 months (95% CI 2.9–7.8 months) for selumetinib alone and 2.3 months (95% CI 2.0–4.6 months) for the combination; the ORR was 10% and 0%, respectively. This study failed to show improvement in objective response rate or PFS with combination therapy of selumetinib and erlotinib over monotherapy in KRAS mutant and KRAS wild-type advanced NSCLC. Therefore, the authors suggested that further study of selumetinib with erlotinib is not warranted in NSCLC [83].
CDK inhibitors
Cyclin-dependent kinases (CDKs) are critical proteins for cell cycle’s. Proliferation stimuli, including those through KRAS, converge on the complex CDK4/6 cyclin-D, a critical regulator of the transition between phase G1 and S [84].
CDK4 has been shown necessary for tumor progression in a KRAS-induced lung adenocarcinoma model [85]. Abemaciclib, a CDK4 inhibitor, have shown efficacy in various xenograft models for human cancer including NSCLC [86]. Forty-nine pretreated NSCLC patients were enrolled in a phase I trial to evaluate the safety and the clinical activity of abemaciclib. KRAS mutations were present in 26 patients, while 19 were wild-type (and 4 with unknown KRAS status). Results were encouraging, with a DCR of 54% and 37% in KRAS mutant and wild type tumors, respectively [87]. In another trial, Patnaik et al. enrolled 68 pre-treated NSCLC patients for treatment with abemaciclib, including 29 patients harboring KRAS mutation. Even if responses were uncommon, the majority of tumor regression occurred in KRAS-mutant patients, with a stable disease as result [12]. The phase III trial JUNIPER is currently evaluating abemaciclib versus erlotinib in pretreated patients with NSCLC harboring KRAS mutations (Figure 3) [88].
Hsp90 inhibitors
Heat Shock Proteins (HSPs) are adenosine triphosphate (ATP)–dependent chaperones with an important role for the cell response to stress and for maintaining cellular homeostasis. The ubiquitously expressed HSP90 has been studied intensively because of its involvement in the folding, stability and function of several oncogenic driver proteins. Interestingly, HSP90 chaperone is necessary for the maturation of proteins involved in KRAS downstream pathways, such as mTOR and MEK [89]. Hence, this chaperone could be an attractive therapeutic target for KRAS mutant NSCLC, and several HSP90 inhibitors have been developed for the treatment of cancer. KRAS mutant NSCLC cell lines were sensitive to some HSP90 inhibitors, such as tanespimycin, alvespimycin or ganetespib [90, 91]. Ganetespib has been tested in a phase II study, in stage IIIB/IV NSCLC patients [92]. Unfortunately, this trial failed to demonstrate a significant activity of ganetespib with respect to KRAS mutant NSCLC [92]. Preclinical evidences suggest a synergistic effect of taxanes and ganetespib [93] and in the randomized phase II trial GALAXY-1, docetaxel was administered with or without ganetespib in pretreated NSCLC patients [13]. However, in the KRAS mutant population, the combination did not improve the PFS neither the OS [13], and these data were confirmed in the phase III trial GALAXY-2 (Figure 3) [94].
Immunotherapy for KRAS-mutated NSCLC patients
Recently, the attention has turned into the discovery of drugs able to interfere with specific immune checkpoints, among which programmed death-1 receptor (PD-1) and its ligand (PD-L1) have been the most studied in NSCLC. PD-1 and PD-L1 are expressed by activated immune cell types, including T-cells, B-cells, dendritic cells, and their interaction negatively regulates immune activity in peripheral tissues in response to infection or tumor progression [95]. Several studies have shown that PD-1/PD-L1 pathway is manipulated by cancer microenvironment, in particular PD-L1 is commonly up-regulated in NSCLC and PD-1 is expressed on the majority of tumor-infiltrating immune cells [96, 97].
FDA has recently approved nivolumab an anti-PD-1 antibody, for metastatic NSCLC progressed after prior platinum-based chemotherapy [98]. Subsequently, pembrolizumab, another anti-PD-1 antibody, was approved as a second-line for NSCLC patients whose tumors exhibit >50% of PD-L1 expression [99] and as first-line treatment in patients PD-L1 positive, with no EGFR or ALK genomic tumor aberrations [100, 101].
However, PD-L1 is clearly not an easy-to-handle biomarker. In fact, different assays are used to determine its expression levels, with different cut-off of PD-L1 positivity making difficult the definition of a strong cut-off value for PD-L1 positivity in NSCLC.
Interestingly, several data report that a high mutational burden is a positive predictive biomarker of response to immunotherapy [102]. For this reason, an increasing number of studies are exploring the incidence of PD-L1 expression with other genetic alterations [103, 104]. The relationship between the KRAS status and PD-1/PD-L1 expression is currently not enough defined in the subset of NSCLC tumors, with also different results [105–108]. However, the subgroup analysis of the CheckMate 057 trial reported an increased effect in terms of OS in favor of nivolumab for KRAS mutant patients, compared to those with not detected or not reported status [98]. In a retrospective study, the expression of PD-1 and PD-L1 has proved to be heterogeneous within KRAS mutant NSCLC, suggesting PD-L1 expression is not genetically driven by KRAS mutation [109]. D’Incecco et colleagues hypothesized that PD-1/PD-L1 expression could differ according to the molecular phenotype of the tumor [106]. Results of their study confirmed that patients harboring KRAS mutations had higher levels of PD-1 expression when compared to the KRAS wild-type population. By contrast, PD-L1-positive tumors showed driver mutations, as EGFR mutations and ALK rearrangements [106]. Conversely, a recent study found a statistical significance between PD-1 expression and KRAS status (p = 0.043) and a higher mutation rate in patients with lower PD-1 expression (8 out of 10 KRAS mutant patients); whereas PD-L1 expression was higher in patients harboring EGFR mutations or with wild-type KRAS status [110]. Moreover, according to the study by Zhang et al, mutational status of KRAS correlated with PD-L1 expression was not significantly associated with relapse-free survival and OS [107]. Scheel and colleagues demonstrated a strong association between mutations in KRAS with PD-L1 expression in adenocarcinoma specimens (OR = 2.5; p = 0.018) [111].
Recently, according to findings from the phase III OAK trial, atezolizumab, an anti-PD-L1 antibody, improved survival compared with docetaxel in NSCLC patients following the failure of platinum-based chemotherapy, regardless of PD-L1 expression or histology. Even if the number of patients with KRAS mutations was small, the benefit was consistent also across this subgroup [112].
Concerning the association between KRAS mutations and the response to anti-PD-1 antibodies, results of published studies are still limited and controversial.
To conclude, the results for immune checkpoint inhibitor in NSCLC are encouraging, however, a multitude of questions related to predictive biomarkers to anti-PD-1/anti-PD-L1 therapies remains unanswered.
A new hypothesis suggests that the extreme mutational heterogeneity of a tumor, such as in KRAS mutant NSCLC [113], may play a critical key role in the tumor susceptibility to checkpoints blockers by springing an intense immune response against neoantigens and, consequently, improving the response to immune checkpoint-targeting therapies [114]. Even if this concept would be difficult to introduce into daily clinical practice, it is a charming possibility that should be evaluated in future clinical trials.
KRAS and TKIs resistance
In a recent study on cell-free circulating tumor DNA (cftDNA), Del Re and colleagues evaluated the appearance of KRAS mutations in EGFR positive NSCLC patients progressed after a TKI regimen. Patient that developed a KRAS mutation showed a worse survival compared to KRAS wild-type patients, suggesting a role of KRAS mutations also in acquired resistance to anti-EGFR TKI [37]. Guibert and colleagues obtained similar results, highlighting the correlation between the presence of KRAS mutations at codon 12 with a poor response to therapy [115]. Chabon and colleagues observed resistance mechanisms in 46% of 43 NSCLC patients treated with the third-generation epidermal growth factor receptor (EGFR) inhibitor rociletinib. Again, three patients showed acquiring activating mutations in KRAS following treatment with rociletinib [116]. In two recent published case reports was reported the concomitant presence of EGFR, KRAS mutations and the c-ROS oncogene 1 (ROS1) rearrangement in one NSCLC patient and the concomitant presence of ALK rearrangement and KRAS mutations in other two. Interestingly, these patients experienced a rapid disease progression and primary resistance to crizotinib [36, 117]. Moreover, in 9 out of 16 NSCLC patients, KRAS mutations p.G12D or p.G12V appeared in cftDNA at the time of resistance to ALK-TKIs (crizotinib or ceritinib) and 3 of them presented simultaneously ALK mutations [118].
CONCLUSIONS
Several preclinical and clinical investigations have been launched with the hope to better understand the biologic world that surround the KRAS gene, its potential prognostic and predictive role and, importantly, to look for effective treatments for NSCLC patients harboring KRAS mutations. However, KRAS targeting seems to be a real challenge to overcome, a direct KRAS-targeting is probably not efficacious because it is able to activate multiple mechanisms of escape under the selective pressure of treatments. One of the best pharmacological rational approach could be based on a combination of treatments, in order to silence more that one driver at the same time. However, the toxicity-side in this case plays an important role and needs to be strongly considered.
Tumor heterogeneity increases the complexity of the system [119]. Acquired mutations, including KRAS, can affect tumor growth due to the evolution of sub-clones that evolve through the selection of advantageous driver, neutral “passenger” or deleterious mutations [120]. This dynamic diversity is the most important mechanism of acquired resistance to treatments.
Therefore, the landscape is becoming complex considering that KRAS mutations can concomitantly occur with two or more driver alterations in the same tumor.
Although we are fully aware of the laborious path that leads the scientific community to face this challenge, we are confident that some of the attempts discussed in this contest could be an effective treatment for KRAS mutant NSCLC patients, and we strongly believe that in this landscape a multidisciplinary approach can help in the management of this complex disease [121].
Author contributions
M. Del Re, E. Rofi, G. Restante conceived and wrote the article. S. Crucitta, E. Arrigoni, S. Fogli reviewed the manuscript. M. Di Maio, I. Petrini, R. Danesi critically reviewed the data and wrote the article.
ACKNOWLEDGMENTS
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
CONFLICTS OF INTEREST
None.
FUNDING
This article was funded by grants from the Italian Association for Cancer Research (AIRC, Milano, Italy), Istituto Toscano Tumori (ITT, Florence, Italy), Fondazione Cassa Risparmio Di Lucca (Lucca, Italy), Fondazione Berlucchi (2014) and from the Italian Ministry Of Instruction, University and Research (MIUR, Rome) to R Danesi.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, Chen H, Fujimoto J, Kugler K, Franklin WA, Iafrate AJ, Ladanyi M, Kris MG, et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol. 2015; 10:768–777.
3. Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, Justice R, Keegan P, Pazdur R. U.S. Food and Drug Administration approval summary: Erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014; 19:774–779.
4. Lo Russo G, Proto C, Garassino MC. Afatinib in the treatment of squamous non-small cell lung cancer: a new frontier or an old mistake? Transl Lung Cancer Res. 2016; 5:110–114.
5. Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, Liu Q, Zhao P, Zhao H, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res. 2015; 21:2436–2439.
6. Kazandjian D, Blumenthal GM, Chen HY, He K, Patel M, Justice R, Keegan P, Pazdur R. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014; 19:e5–11.
7. Gettinger SN, Bazhenova LA, Langer CJ, Salgia R, Gold KA, Rosell R, Shaw AT, Weiss GJ, Tugnait M, Narasimhan NI, Dorer DJ, Kerstein D, Rivera VM, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016; 17:1683–1696.
8. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, Ou SI, Perol M, Dziadziuszko R, Rosell R, Zeaiter A, Mitry E, Golding S, et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2017; 377:829–838.
9. Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, Xie Y, Gazdar AF, Mori M, Minna JD. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011; 10:336–346.
10. Adjei AA, Mauer A, Bruzek L, Marks RS, Hillman S, Geyer S, Hanson LJ, Wright JJ, Erlichman C, Kaufmann SH, Vokes EE. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003; 21:1760–1766.
11. Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W, Ginsberg MS. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011; 6:1435–1437.
12. Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW, Hilton JF, Nasir A, Beckmann RP, Schade AE, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016; 6:740–753.
13. Ramalingam S, Goss G, Rosell R, Schmid-Bindert G, Zaric B, Andric Z, Bondarenko I, Komov D, Ceric T, Khuri F, Samarzija M, Felip E, Ciuleanu T, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann Oncol. 2015; 26:1741–1748.
14. Han JY, Lee SH, Lee GK, Yun T, Lee YJ, Hwang KH, Kim JY, Kim HT. Phase I/II study of gefitinib (Iressa((R))) and vorinostat (IVORI) in previously treated patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2015; 75:475–483.
15. Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR Jr, Tsao A, Stewart DJ, Hicks ME, Erasmus J Jr, Gupta S, Alden CM, Liu S, Tang X, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011; 1:44–53.
16. Paz-Ares L, Hirsh V, Zhang L, de Marinis F, Yang JC, Wakelee HA, Seto T, Wu YL, Novello S, Juhasz E, Aren O, Sun Y, Schmelter T, et al. Monotherapy Administration of Sorafenib in Patients With Non-Small Cell Lung Cancer (MISSION) Trial: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib in Patients with Relapsed or Refractory Predominantly Nonsquamous Non-Small-Cell Lung Cancer after 2 or 3 Previous Treatment Regimens. J Thorac Oncol. 2015; 10:1745–1753.
17. Gandara DR, Leighl N, Delord JP, Barlesi F, Bennouna J, Zalcman G, Infante JR, Reckamp KL, Kelly K, Shepherd FA, Mazieres J, Janku F, Gardner OS, et al. A Phase 1/1b Study Evaluating Trametinib Plus Docetaxel or Pemetrexed in Patients With Advanced Non-Small Cell Lung Cancer. J Thorac Oncol. 2017; 12:556–566.
18. McGrath JP, Capon DJ, Smith DH, Chen EY, Seeburg PH, Goeddel DV, Levinson AD. Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature. 1983; 304:501–506.
19. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007; 129:865–877.
20. Westcott PM, To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer. 2013; 32:63–70.
21. Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP. Protein Kinase Ciota Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell. 2016; 29:367–378.
22. Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013; 3:112–123.
23. Uekita T, Fujii S, Miyazawa Y, Iwakawa R, Narisawa-Saito M, Nakashima K, Tsuta K, Tsuda H, Kiyono T, Yokota J, Sakai R. Oncogenic Ras/ERK signaling activates CDCP1 to promote tumor invasion and metastasis. Mol Cancer Res. 2014; 12:1449–1459.
24. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017; 116:415–423.
25. Kempf E, Rousseau B, Besse B, Paz-Ares L. KRAS oncogene in lung cancer: focus on molecularly driven clinical trials. European respiratory review. 2016; 25:71–76.
26. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of oncology. 2013; 24:2371–2376.
27. Heo SJ, Jung I, Lee CK, Kim JH, Lim SM, Moon YW, Shim HS, Jeong J, Kim HR, Cho BC. A randomized phase II trial of ERCC1 and RRM1 mRNA expression-based chemotherapy versus docetaxel/carboplatin in advanced non-small cell lung cancer. Cancer chemotherapy and pharmacology. 2016; 77:539–548.
28. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proceedings of the American Thoracic Society. 2009; 6:201–205.
29. Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, Li J, Chen Q. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010; 69:272–278.
30. Jo YM, Park TI, Lee HY, Jeong JY, Lee WK. Prognostic Significance of Aquaporin 5 Expression in Non-small Cell Lung Cancer. Journal of pathology and translational medicine. 2016; 50:122–128.
31. Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, Scanni A, Broggini M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Annals of oncology. 2011; 22:235–237.
32. Lee JW, Lee SM, Yun M, Cho A. Prognostic Value of Volumetric Parameters on Staging and Posttreatment FDG PET/CT in Patients With Stage IV Non-Small Cell Lung Cancer. Clinical nuclear medicine. 2016; 41:347–353.
33. Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochimica et biophysica acta. 2007; 1773:1177–1195.
34. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, Iafrate AJ, Arcila ME, Ladanyi M, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clinical cancer research. 2013; 19:4273–4281.
35. Lee T, Lee B, Choi YL, Han J, Ahn MJ, Um SW. Non-small Cell Lung Cancer with Concomitant EGFR, KRAS, and ALK Mutation: Clinicopathologic Features of 12 Cases. Journal of pathology and translational medicine. 2016; 50:197–203.
36. Mengoli MC, Barbieri F, Bertolini F, Tiseo M, Rossi G. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: Report of two cases and review of the literature. Lung Cancer. 2016; 93:55–58.
37. Del Re M, Tiseo M, Bordi P, D’Incecco A, Camerini A, Petrini I, Lucchesi M, Inno A, Spada D, Vasile E, Citi V, Malpeli G, Testa E, et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget. 2016; 8:13611–13619. https://doi.org/10.18632/oncotarget.6957.
38. Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L, Puccetti M, Capelli L, Calistri D, Verlicchi A, Gamboni A, Papi M, Mariotti M, et al. Nonsquamous, Non-Small-Cell Lung Cancer Patients Who Carry a Double Mutation of EGFR, EML4-ALK or KRAS: Frequency, Clinical-Pathological Characteristics, and Response to Therapy. Clinical lung cancer. 2016; 17:384–390.
39. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, Zhang J, Giri U, Gudikote J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015; 5:860–877.
40. Maida Y, Kyo S, Kanaya T, Wang Z, Yatabe N, Tanaka M, Nakamura M, Ohmichi M, Gotoh N, Murakami S, Inoue M. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene. 2002; 21:4071–4079.
41. Liu W, Yin Y, Wang J, Shi B, Zhang L, Qian D, Li C, Zhang H, Wang S, Zhu J, Gao L, Zhang Q, Jia B, et al. Kras mutations increase telomerase activity and targeting telomerase is a promising therapeutic strategy for Kras-mutant NSCLC. Oncotarget. 2016; 8:179–190. https://doi.org/10.18632/oncotarget.10162.
42. Chiappori AA, Kolevska T, Spigel DR, Hager S, Rarick M, Gadgeel S, Blais N, Von Pawel J, Hart L, Reck M, Bassett E, Burington B, Schiller JH. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Annals of oncology. 2015; 26:354–362.
43. Zhang YA, Nemunaitis J, Scanlon KJ, Tong AW. Anti-tumorigenic effect of a K-ras ribozyme against human lung cancer cell line heterotransplants in nude mice. Gene therapy. 2000; 7:2041–2050.
44. Srikar R, Suresh D, Zambre A, Taylor K, Chapman S, Leevy M, Upendran A, Kannan R. Targeted nanoconjugate co-delivering siRNA and tyrosine kinase inhibitor to KRAS mutant NSCLC dissociates GAB1-SHP2 post oncogene knockdown. Scientific Reports. 2016; 6:30245.
45. Lu Y, Liu L, Wang Y, Li F, Zhang J, Ye M, Zhao H, Zhang X, Zhang M, Zhao J, Yan B, Yang A, Feng H, et al. siRNA delivered by EGFR-specific scFv sensitizes EGFR-TKI-resistant human lung cancer cells. Biomaterials. 2016; 76:196–207.
46. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. Journal of clinical oncology. 2009; 27:5459–5468.
47. Kurtze I, Sonnemann J, Beck JF. KRAS-mutated non-small cell lung cancer cells are responsive to either co-treatment with erlotinib or gefitinib and histone deacetylase inhibitors or single treatment with lapatinib. Oncology reports. 2011; 25:1021–1029.
48. Greve G, Schiffmann I, Pfeifer D, Pantic M, Schuler J, Lubbert M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer. 2015; 15:947.
49. Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005; 1756:127–144.
50. Kim ES, Kies MS, Fossella FV, Glisson BS, Zaknoen S, Statkevich P, Munden RF, Summey C, Pisters KM, Papadimitrakopoulou V, Tighiouart M, Rogatko A, Khuri FR. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer. 2005; 104:561–569.
51. Khuri FR, Glisson BS, Kim ES, Statkevich P, Thall PF, Meyers ML, Herbst RS, Munden RF, Tendler C, Zhu Y, Bangert S, Thompson E, Lu C, et al. Phase I study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in solid tumors. Clinical cancer research. 2004; 10:2968–2976.
52. End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, Venet M, Sanz G, Poignet H, Skrzat S, Devine A, Wouters W, Bowden C. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer research. 2001; 61:131–137.
53. Gunning WT, Kramer PM, Lubet RA, Steele VE, End DW, Wouters W, Pereira MA. Chemoprevention of benzo(a)pyrene-induced lung tumors in mice by the farnesyltransferase inhibitor R115777. Clinical cancer research. 2003; 9:1927–1930.
54. Zhang J, Park D, Shin DM, Deng X. Targeting KRAS-mutant non-small cell lung cancer: challenges and opportunities. Acta biochimica et biophysica Sinica. 2016; 48:11–16.
55. Zundelevich A, Elad-Sfadia G, Haklai R, Kloog Y. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Molecular cancer therapeutics. 2007; 6:1765–1773.
56. Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015; 4:165–176.
57. Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, Wang H, Unger M, Testa JR. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004; 25:2053–2059.
58. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nature reviews Molecular cell biology. 2010; 11:329–341.
59. Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011; 2:261–274. https://doi.org/10.1177/1947601911408079.
60. Green S, Trejo CL, McMahon M. PIK3CA(H1047R) Accelerates and Enhances KRAS(G12D)-Driven Lung Tumorigenesis. Cancer research. 2015; 75:5378–5391.
61. Vansteenkiste JF, Canon JL, Braud FD, Grossi F, De Pas T, Gray JE, Su WC, Felip E, Yoshioka H, Gridelli C, Dy GK, Thongprasert S, Reck M, et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J Thorac Oncol. 2015; 10:1319–1327.
62. Wislez M, Spencer ML, Izzo JG, Juroske DM, Balhara K, Cody DD, Price RE, Hittelman WN, Wistuba II, Kurie JM. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer research. 2005; 65:3226–3235.
63. Martin P, Leighl NB, Tsao MS, Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013; 8:530–542.
64. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer research. 2004; 64:7099–7109.
65. Smit EF, Dingemans AM, Thunnissen FB, Hochstenbach MM, van Suylen RJ, Postmus PE. Sorafenib in patients with advanced non-small cell lung cancer that harbor K-ras mutations: a brief report. Journal of thoracic oncology. 2010; 5:719–720.
66. Kelly RJ, Rajan A, Force J, Lopez-Chavez A, Keen C, Cao L, Yu Y, Choyke P, Turkbey B, Raffeld M, Xi L, Steinberg SM, Wright JJ, et al. Evaluation of KRAS mutations, angiogenic biomarkers, and DCE-MRI in patients with advanced non-small-cell lung cancer receiving sorafenib. Clinical cancer research. 2011; 17:1190–1199.
67. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, Diao L, Coombes KR, Chen L, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. Journal of the National Cancer Institute. 2012; 104:228–239.
68. Papadimitrakopoulou V, Lee JJ, Wistuba II, Tsao AS, Fossella FV, Kalhor N, Gupta S, Byers LA, Izzo JG, Gettinger SN, Goldberg SB, Tang X, Miller VA, et al. The BATTLE-2 Study: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology. 2016 Aug 1. [Epub ahead of print].
69. Lind JS, Dingemans AM, Groen HJ, Thunnissen FB, Bekers O, Heideman DA, Honeywell RJ, Giovannetti E, Peters GJ, Postmus PE, van Suylen RJ, Smit EF. A multicenter phase II study of erlotinib and sorafenib in chemotherapy-naive patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010; 16:3078–3087.
70. Spigel DR, Burris HA 3rd, Greco FA, Shipley DL, Friedman EK, Waterhouse DM, Whorf RC, Mitchell RB, Daniel DB, Zangmeister J, Bass JD, Hainsworth JD. Randomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2011; 29:2582–2589.
71. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Molecular cancer therapeutics. 2007; 6:2209–2219.
72. Lopez-Chavez A, Thomas A, Rajan A, Raffeld M, Morrow B, Kelly R, Carter CA, Guha U, Killian K, Lau CC, Abdullaev Z, Xi L, Pack S, et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: a biomarker-derived, multiarm, multihistology phase II basket trial. Journal of clinical oncology. 2015; 33:1000–1007.
73. Goldman JW, Garon EB. Targeting MEK for the treatment of non-small-cell lung cancer. Journal of thoracic oncology. 2012; 7:S377–378.
74. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D, Laquerre SG. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clinical cancer research. 2011; 17:989–1000.
75. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, Peddareddigari VG, Le NT, Hart L, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13:773–781.
76. Blumenschein GR Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, Dunphy F, Udud K, Ahn MJ, Hanna NH, Kim JH, Mazieres J, Kim SW, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol. 2015; 26:894–901.
77. Holt SV, Logie A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. British journal of cancer. 2012; 106:858–866.
78. Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, Gao P, Woo MS, Xu C, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012; 483:613–617.
79. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crino L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The Lancet Oncology. 2013; 14:38–47.
80. Janne PA, Smith I, McWalter G, Mann H, Dougherty B, Walker J, Orr MC, Hodgson DR, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios CH, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. British journal of cancer. 2015; 113:199–203.
81. Bernabe R, Patrao A, Carter L, Blackhall F, Dean E. Selumetinib in the treatment of non-small-cell lung cancer. Future Oncol. 2016; 12:2545–2560.
82. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clinical cancer research. 2007; 13:2890–2896.
83. Carter CA, Rajan A, Keen C, Szabo E, Khozin S, Thomas A, Brzezniak C, Guha U, Doyle LA, Steinberg SM, Xi L, Raffeld M, Tomita Y, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann Oncol. 2016; 27:693–699.
84. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews Drug discovery. 2015; 14:130–146.
85. Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, Fang M, Tao S, Green CL, Khavari PA. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nature medicine. 2002; 8:1105–1114.
86. Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS, Neubauer BL, Young J, Chan EM, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investigational new drugs. 2014; 32:825–837.
87. Goldman JW, Gandhi L, Patnaik A, Rosen L, Hilton JF, Papadopoulos K, Tolaney SM, Beeram M, Rasco DW, Myrand SP, Beckmann RP, Kulanthaivel P, Frenzel M, et al. Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with non-small cell lung cancer. Journal of Clinical Oncology. 2014; 32:8026–8026.
88. Goldman JW, Shi P, Reck M, Paz-Ares L, Koustenis A, Hurt KC. Treatment Rationale and Study Design for the JUNIPER Study: A Randomized Phase III Study of Abemaciclib With Best Supportive Care Versus Erlotinib With Best Supportive Care in Patients With Stage IV Non-Small-Cell Lung Cancer With a Detectable KRAS Mutation Whose Disease Has Progressed After Platinum-Based Chemotherapy. Clinical lung cancer. 2016; 17:80–84.
89. Chatterjee S, Bhattacharya S, Socinski MA, Burns TF. HSP90 inhibitors in lung cancer: promise still unfulfilled. Clinical advances in hematology & oncology. 2016; 14:346–356.
90. Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, Shimamura T, Rauh D, Mermel C, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. The Journal of clinical investigation. 2009; 119:1727–1740.
91. Acquaviva J, Smith DL, Sang J, Friedland JC, He S, Sequeira M, Zhang C, Wada Y, Proia DA. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Molecular cancer therapeutics. 2012; 11:2633–2643.
92. Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical cancer research. 2013; 19:3068–3077.
93. Proia DA, Sang J, He S, Smith DL, Sequeira M, Zhang C, Liu Y, Ye S, Zhou D, Blackman RK, Foley KP, Koya K, Wada Y. Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Investigational new drugs. 2012; 30:2201–2209.
94. Hendriks LEL, Dingemans AC. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin Investig Drugs. 2017; 26:541–550.
95. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012; 366:2443–2454.
96. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Advances in immunology. 2006; 90:51–81.
97. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012; 12:252–264.
98. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015; 373:1627–1639.
99. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England journal of medicine. 2015; 372:2018–2028.
100. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. The New England journal of medicine. 2016; 375:1823–1833.
101. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016; 387:1540–1550.
102. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351:1463–1469.
103. Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A, Reibel JB, Walton Z, Ji H, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer cell. 2014; 25:590–604.
104. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery. 2013; 3:1355–1363.
105. Cooper WA, Tran T, Vilain RE, Madore J, Selinger CI, Kohonen-Corish M, Yip P, Yu B, O’Toole SA, McCaughan BC, Yearley JH, Horvath LG, Kao S, et al. PD-L1 expression is a favorable prognostic factor in early stage non-small cell carcinoma. Lung Cancer. 2015; 89:181–188.
106. D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, Tibaldi C, Minuti G, Salvini J, Coppi E, Chella A, Fontanini G, Filice ME, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. British journal of cancer. 2015; 112:95–102.
107. Zhang Y, Wang L, Li Y, Pan Y, Wang R, Hu H, Li H, Luo X, Ye T, Sun Y, Chen H. Protein expression of programmed death 1 ligand 1 and ligand 2 independently predict poor prognosis in surgically resected lung adenocarcinoma. OncoTargets and therapy. 2014; 7:567–573.
108. Yang CY, Lin MW, Chang YL, Wu CT, Yang PC. Programmed cell death-ligand 1 expression in surgically resected stage I pulmonary adenocarcinoma and its correlation with driver mutations and clinical outcomes. Eur J Cancer. 2014; 50:1361–1369.
109. Calles A, Liao X, Sholl LM, Rodig SJ, Freeman GJ, Butaney M, Lydon C, Dahlberg SE, Hodi FS, Oxnard GR, Jackman DM, Janne PA. Expression of PD-1 and Its Ligands, PD-L1 and PD-L2, in Smokers and Never Smokers with KRAS-Mutant Lung Cancer. Journal of thoracic oncology. 2015; 10:1726–1735.
110. Ji M, Liu Y, Li Q, Li X, Ning Z, Zhao W, Shi H, Jiang J, Wu C. PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/KRAS mutations. Cancer biology & therapy. 2016; 17:407–413.
111. Scheel AH, Ansen S, Schultheis AM, Scheffler M, Fischer RN, Michels S, Hellmich M, George J, Zander T, Brockmann M, Stoelben E, Groen H, Timens W, et al. PD-L1 expression in non-small cell lung cancer: Correlations with genetic alterations. Oncoimmunology. 2016; 5:e1131379.
112. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, Cortinovis DL, Leach J, Polikoff J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017; 389:255–265.
113. Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, Fredlund E, Quigley DA, Adams DJ, Balmain A. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015; 517:489–492.
114. Champiat S, Ferte C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology. 2014; 3:e27817.
115. Guibert N, Pradines A, Farella M, Casanova A, Gouin S, Keller L, Favre G, Mazieres J. Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS-mutated lung adenocarcinoma. Lung Cancer. 2016; 100:1–4.
116. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, Harding TC, Durkin KA, Otterson GA, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nature communications. 2016; 7:11815.
117. Ju L, Han M, Zhao C, Li X. EGFR, KRAS, ROS1 variants coexist in a lung adenocarcinoma patient. Lung Cancer. 2016; 95:94–97.
118. Bordi P, Del Re M, Danesi R, Tiseo M. 2PD Monitoring of secondary drug resistance mutations in circulating tumor DNA of patients with advanced ALK positive NSCLC. Journal of thoracic oncology. 2016; 11:S57.
119. Ilie M, Hofman V, Long E, Bordone O, Selva E, Washetine K, Marquette CH, Hofman P. Current challenges for detection of circulating tumor cells and cell-free circulating nucleic acids, and their characterization in non-small cell lung carcinoma patients. What is the best blood substrate for personalized medicine? Annals of translational medicine. 2014; 2:107.
120. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481:306–313.
121. Grech G, Zhan X, Yoo BC, Bubnov R, Hagan S, Danesi R, Vittadini G, Desiderio DM. EPMA position paper in cancer: current overview and future perspectives. EPMA J. 2015; 6:9.