
INTRODUCTION
Endometrial cancer (EC) is one of the three main tumors originating in the female genital system. Its incidence is higher than that of cervical cancer in many countries, and is located in the top of gynecologic malignant tumors [1, 2]. EC patients often present with abnormal vaginal bleeding; although many patients are diagnosed when the disease is still confined to the uterus, about 30% are diagnosed with EC in its later stage. Most clinical trials for the treatment of advanced and recurrent EC have shown limited benefits, and the mortality rate has increased dramatically over the past few years [3, 4].
Abnormal genetic and epigenetic alterations have been widely recognized as being associated with EC [5, 6]. However, genetic markers have not yet proven reliable for identifying the entire spectrum of the disease, especially sporadic EC. Aberrant DNA methylation is one of the most widely studied epigenetic modifications with a critical role in EC [7, 8]. Diagnostic markers based on gene methylation have recently been developed and have shown considerable promise for the detection of EC, while aberrant promoter methylation has been found to be an early and widespread alteration in endometrial tumorigenesis [9]. However, although specific gene methylation patterns have been widely used for the diagnosis of many different cancers, including EC, there is currently a lack of effective diagnostic biomarkers for EC, and novel, accurate markers are urgently needed.
Previous studies have investigated the use of aberrant gene methylation in tissue samples as potential diagnostic biomarkers for EC, with encouraging but variable results. Further studies are therefore needed to describe the associations between DNA methylation in different tumor suppressor genes and the clinicopathologic features of sporadic EC. Moreover, no systematic review or meta-analysis has yet been conducted to evaluate the diagnostic accuracy of the existing studies. We therefore conducted a comprehensive, systematic review and meta-analysis of eligible studies to resolve the inconsistent and ambiguous findings, and to clarify the diagnostic value of DNA methylation in EC. Furthermore, we aimed to identify the diagnostic accuracy of gene methylation markers to predict other clinical pathological outcomes of EC.
RESULTS
Study characteristics
The workflow of the systematic literature search is displayed in Figure 1. The primary search of PubMed and Web of Science identified 701 articles, of which 194 were duplicate articles. A total of 120 studies were initially obtained after filtering the titles, abstracts, and full texts. Of these, 98 studies were excluded based on the eligibility criteria: 11 studies were excluded based on the use of in vitro/ex vivo cell lines and human xenografts; 41 studies did not have healthy normal controls; 11 were not case-control studies; it was not possible to extract or calculate the diagnostic sensitivity and specificity of the methylation biomarkers in 30 studies; and five studies had small sample sizes (n ≤ 10). All the included studies focused on DNA methylation/hypermethylation in tissues. The systematic literature search thus finally yielded 22 studies including 1930 participants (1418 patients and 512 normal individuals). None of the patients had received preoperative chemotherapy, radiotherapy, or hormone therapy. The included studies were published between 2001 and 2016 and originated from nine regions (China, Czech Republic, Hong Kong, Japan, Netherland, Russia, Slovak Republic, Taiwan, and USA) (Table 1). The sample sizes of these studies ranged from 19–155 (median 88). Fourteen studies evaluated the diagnostic value of methylation of a single gene (Sasaki et al., 2001(1) [10]; Sasaki et al., 2001(2) [11]; Saito et al., 2003 [12]; Sasaki et al., 2003 [13]; Li et al., 2005 [14]; Pijnenborg et al., 2007 [17]; Yanokura et al., 2007 [18]; Tse et al., 2009 [20]; Varley et al., 2009 [21]; Yi et al., 2011 [22]; Kovalenko et al., 2013 [25]; Yang et al., 2013 [27]; Chmelarova et al., 2014 [28]; Dong et al., 2015 [30]), five studies evaluated multiple genes (Banno et al., 2006 [15]; Suehiro et al., 2008 [19]; Fiolka et al., 2013 [24]; Visnovsky et al., 2013 [26]; Sheng et al., 2016 [31]), and the other three studies evaluated both single and combined genes (Shih et al., 2006 [16]; Zhang et al., 2011 [23]; Chen et al., 2015 [29]). Seventeen studies measured the methylation patterns of the genes using methylationspecific polymerase chain reaction (MSP), three studies used combined bisulfite restriction analysis (COBRA), one used quantitative MSP (qMSP), and one used MSP and COBRA for two genes, respectively. Details of the DNA methylation biomarkers and their diagnostic powers are shown in Supplementary Table 1. All 22 selected publications were evaluated and checked by two reviewers. High levels of methodological quality (more than five stars) were observed according to the NOS scale.
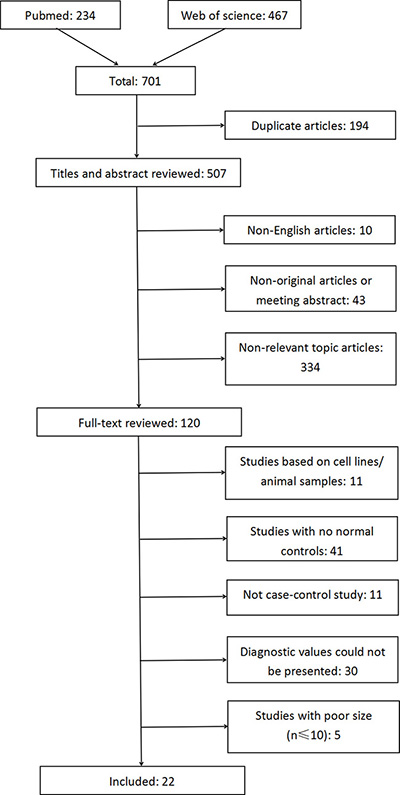
Figure 1: Flow diagram of the literature search process (search prior to 30/04/2017).
Table 1: The major characteristics of all included studies
No. |
Study |
Region |
Method |
Biomarker |
Case |
Control |
Alteration type |
|---|---|---|---|---|---|---|---|
1 |
Sasaki et al. (2001) [10] |
Japan |
MSP |
single |
83 |
33 |
hypermethylation |
2 |
Sasaki et al. (2001) [11] |
Japan |
MSP |
single |
88 |
46 |
methylation |
3 |
Saito et al. (2003) [12] |
Japan |
MSP |
single |
104 |
21 |
hypermethylation |
4 |
Sasaki et al. (2003) [13] |
Japan |
MSP |
single |
60 |
10 |
methylation |
5 |
Li et al. (2005) [14] |
Japan |
MSP |
single |
64 |
16 |
hypermethylation |
6 |
Banno et al. (2006) [15] |
Japan |
MSP |
multiple |
52 |
18 |
methylation |
7 |
Shih et al. (2006) [16] |
Taiwan |
MSP |
single/combined |
35 |
20 |
methylation |
8 |
pijnenborg et al. (2007) [17] |
Netherland |
MSP |
single |
95 |
27 |
methylation |
9 |
Yanokura et al. (2007) [18] |
Japan |
MSP |
single |
50 |
9 |
hypermethylation |
10 |
Suehiro et al. (2008) [19] |
Japan |
MSP/COBRA |
multiple |
106 |
27 |
hypermethylation |
11 |
Tse et al. (2009) [20] |
Hongkong |
COBRA |
single |
125 |
30 |
methylation |
12 |
Varley et al. (2009) [21] |
USA |
COBRA |
single |
14 |
5 |
methylation |
13 |
Yi et al. (2011) [22] |
China |
MSP |
single |
82 |
32 |
methylation |
14 |
Zhang et al. (2011) [23] |
China |
MSP |
single/combined |
35 |
22 |
methylation |
15 |
Fiolka et al. (2013) [24] |
Slovak |
MSP |
multiple |
41 |
20 |
methylation |
16 |
Kovalenko et al. (2013) [25] |
Russia |
COBRA |
single |
18 |
10 |
methylation |
17 |
Visnovsky et al. (2013) [26] |
Slovak |
MSP |
multiple |
50 |
35 |
methylation |
18 |
Yang et al. (2013) [27] |
China |
MSP |
single |
97 |
40 |
methylation |
19 |
Chmelarova et al. (2014) [28] |
Czech |
MSP |
single |
54 |
18 |
methylation |
20 |
Chen et al. (2015) [29] |
Taiwan |
qMSP |
single/combined |
26 |
18 |
methylation |
21 |
Dong et al. (2015) [30] |
China |
MSP |
single |
80 |
28 |
hypermethylation |
22 |
Sheng et al. (2016) [31] |
China |
MSP |
multiple |
59 |
27 |
methylation |
Meta-analysis of diagnostic value
We assessed the risk of bias for each study. The detailed evaluation criteria and results for each item are shown in Supplementary Table 2 and Supplementary Figure 1, and the risk of bias is summarized in Figure 2. The risk of bias was high or unclear in most included studies. Four studies stated that the sequences of participants were generated randomly. The diagnostic values for all the assessed methylation biomarkers were reported in 64% of studies, indicating no selective reporting. Eight studies were reported to be free of other biases and were defined as low risk.
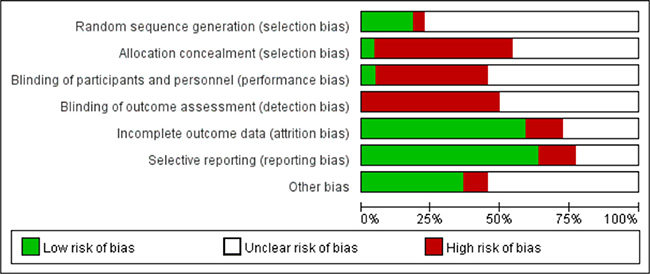
Figure 2: Risk of bias graph (reviewers’ judgements about each risk of bias item presented as percentages across enrolled studies).
Twenty-two studies were pooled for the meta-analysis of diagnostic accuracy. The pooled sensitivity and specificity were 0.93 (95% confidence interval [CI]: 0.91–0.94) and 0.48 (95% CI: 0.46–0.50), respectively (Figure 3). The PLR and NLR were 2.02 (95%CI: 1.77–2.29) and 0.15 (95% CI: 0.11–0.21), respectively, and the pooled DOR was 18.84 (95% CI: 12.01–29.54). Significant heterogeneity was observed in the diagnostic meta-analysis of these studies (sensitivity: I2 = 69.1%, p < 0.001; specificity: I2 = 86.7%, p < 0.001). There was no significant threshold effect according to Spearman correlation (ρ =−0.19, p = 0.15). Subgroup analysis (Table 2) was therefore carried out according to five different parameters: sample size (<85 vs. ≥85), alteration type (methylation vs. hypermethylation), race (Mongolian vs. Caucasian), detection methods (MSP vs. COBRA vs. qMSP), and genes (single vs. any one vs. both genes). Lower heterogeneities for sensitivity were only detected in the subgroups of COBRA (sensitivity: I2 = 44.8%, p = 0.143), qMSP (sensitivity: I2 = 0%, p = 0.547), any one gene (sensitivity: I2 = 0%, p = 0.920), and both genes (sensitivity: I2 = 0%, p = 1.0); while the specificity heterogeneity was reduced in the Caucasian (specificity: I2 = 39.8%, p = 0.092) and any one gene (specificity: I2 = 0%, p = 1.0) subgroup. Subgroup analysis for DOR revealed homologous trends in hypermethylation, COBRA, qMSP, and one or two genes (hypermethylation: I2 = 49.9%, p = 0.062; COBRA: I2 = 0%, p = 0.871; qMSP: I2 = 0%, p = 0.994; any one gene: I2 = 0%, p = 0.998; both genes: I2 = 32.6%, p = 0.157). These results suggested that the methylation-detection method and type of gene combination might contribute to the heterogeneity. The other measures of diagnostic value in the subgroups are summarized in Table 2. Meta-regression analysis based on those factors was also applied to explore the possible heterogeneity source, as shown in Supplementary Table 3. Only the detection method significantly changed the heterogeneity of the universal diagnostic value (p < 0.001). Taken together, we considered that the detection method and gene combination contributed to the heterogeneity source. Further, well-designed studies with different detection methods are therefore needed to clarify the diagnostic role of gene methylation in sporadic EC.
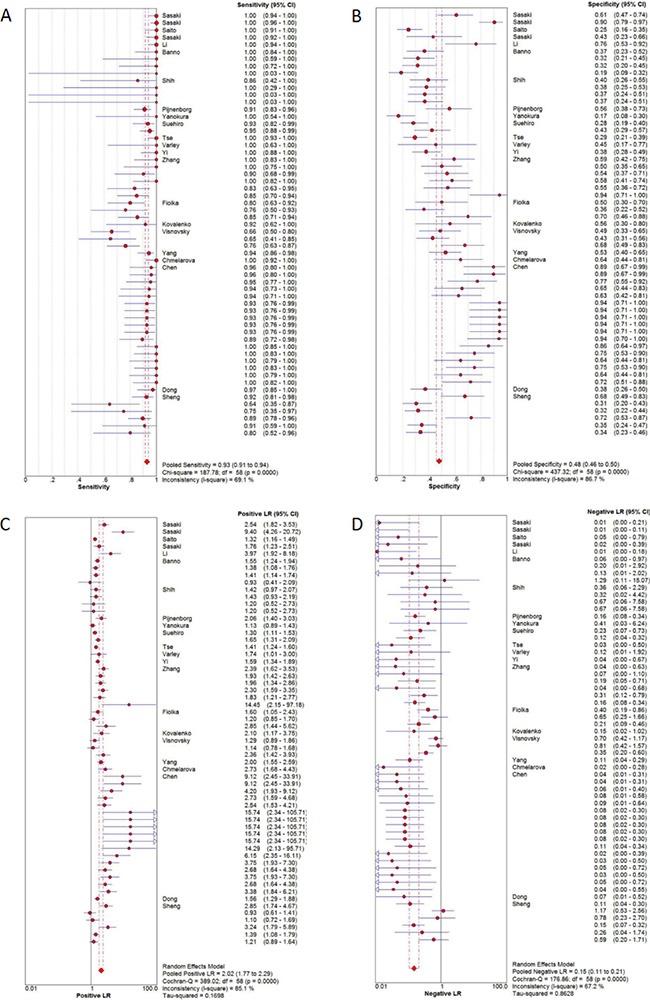
Figure 3: Diagnostic accuracy of forest plots. (A) Forest plots of pooled sensitivity. (B) Forest plots of pooled specificity. (C) Forest plots of pooled positive likelihood ratio. (D) Forest plots of pooled negative likelihood ratio.
Table 2: Subgroup analysis of diagnosis parameters
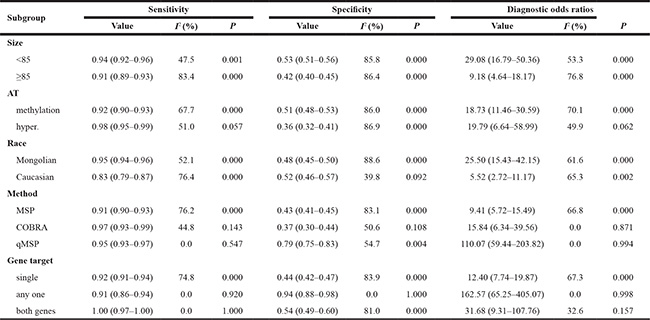
Abbreviation: AT = alteration type; I2 = inconsistency index; hyper. = hypermethylation; P = p value.
Association of DNA methylation with clinicopathological characteristics in patients with sporadic EC
We evaluated the association between DNA methylation and several clinicopathological features in patients with sporadic EC based on 13 different studies, including three involving multiple genes. We did not analyze age in relation to DNA methylation because there was no single cut-off value for age among the patients. Methylation status was significantly associated with lymph node metastasis (negative/positive, pooled odds ratio [OR]: 0.28, 95% CI: 0.15–0.52, p < 0.001) (Table 3), indicating that gene methylation was positively associated with the risk of lymph node metastasis in patients with sporadic EC, and thus potentially with a relatively poor prognosis. However, gene methylation status showed no significant relationship with body mass index (≤25.9/>25.9, pooled OR: 1.04, 95% CI: 0.44–2.47, p = 0.92), pathological type (endometrioid/other, pooled OR: 0.63, 95% CI: 0.21–1.88, p = 0.41), grade (G1/G2–3, pooled OR: 0.80, 95% CI: 0.41–1.54, p = 0.50), invasion (<1/2/≥1/2, pooled OR: 0.58, 95% CI: 0.19–1.76, p = 0.33), and stage (I–II/III–IV, pooled OR: −0.08, 95%CI: −0.20–0.04, p = 0.20).
Table 3: Meta-analysis of the association DNA methylation with clinicopathological features
Stratification |
No. of studies |
No. of patients |
Pooled OR |
95% CI |
P |
Heterogeneity |
|
|---|---|---|---|---|---|---|---|
I2 (%) |
P |
||||||
BMI (≤25.9/>25.9) |
5 |
130 |
1.04 |
0.44–2.47 |
0.92 |
0.0 |
0.50 |
Pathological type (endometrioid/others) |
3 |
228 |
0.63 |
0.21–1.88 |
0.41 |
0.0 |
0.95 |
Grade (G1/G2-3) |
18 |
877 |
0.80 |
0.41–1.54 |
0.50 |
65.0 |
<0.001 |
Invasion (<1/2/≥1/2) |
6 |
413 |
0.58 |
0.19–1.76 |
0.33 |
82.0 |
<0.001 |
Lymph metastasis (negative/positive) |
6 |
384 |
0.28 |
0.15–0.52 |
<0.001 |
0.0 |
0.85 |
Stage (I-II/III-IV) |
16 |
789 |
-0.08 |
–0.20–0.04 |
0.20 |
72.0 |
<0.001 |
Abbreviation: The implication of No. of studies and patients containing multiple and different genes detected in the same study; P = p value; OR=odds ratio; I2 = inconsistency index; 95% CI = 95% confidence intervals; BMI = body mass index; others = non-endometrioid; Invasion = myometrial invasion.
Sensitivity analysis and publication bias
Sensitivity analysis was conducted to evaluate the stability of the results, by removing one study at a time. However, this did not significantly affect the pooled OR or inconsistency index, indicating the stability of our meta-analysis. The funnel plot was relatively symmetrical and no single study fell outside the funnel (Figure 4), suggesting that there were no significant publication biases in this meta-analysis of gene methylation in patients with sporadic EC.
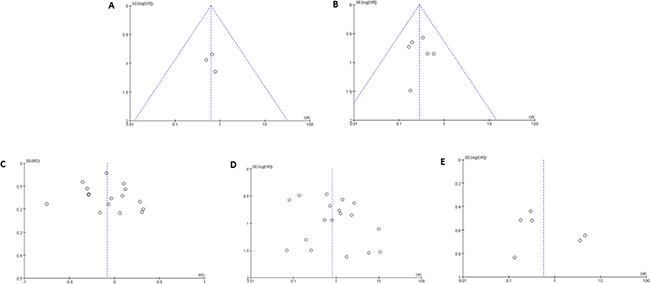
Figure 4: Funnel plots for the evaluation of publication bias. The funnel plots from 13 enrolled studies (including 20 different genes) comparing endometrioid and non-endometrioid EC (A), comparing negative and positive lymph metastasis EC (B), comparing early staged (I-II) and advanced EC (III-IV) (C), comparing grade 1 and grade 2-3 EC (D), and comparing EC with myometrial invasion <1/2 and ≥1/2 (E). X-axis:value of odds ratio (OR); Y-axis: standard errors (SE) multiply log scale of OR.
DISCUSSION
Surgical resection or biopsy specimens are used for diagnosing numerous cancers, and are considered to be the gold standard for clinical applications. Pathological results from these tissues can also provide important information in relation to clinical decision making. DNA methylation has previously demonstrated potential use as a biomarker for EC, showing distinctly different patterns between EC and normal tissues [32]. Alterations in DNA methylation in EC reflect transcription changes during endometrial carcinogenesis. According to a recent review, numerous studies have assessed abnormal promoter methylation of different genes in EC [33]. Although methylation studies in relation to EC remain at the preclinical stage, many results offer the potential for clinical use as diagnostic, prognostic, and therapeutic-response biomarkers, as well as targets of epigenetic therapies. However, although aberrant methylated regions of P16 and RASSF1A demonstrated sensitivity and specificity for the detection of EC [34, 35], aberrant methylation statuses of other genes, such as ER, PR, MLH1, MGMT, APC, and CDH1, have shown variable diagnostic accuracies for EC [33].
In the present study, we evaluated the diagnostic accuracy of methylated markers for sporadic EC, based on previously published studies. Overall, the pooled sensitivity and specificity of DNA methylation for EC diagnosis were 0.93 and 0.48, respectively, and the area under the SROC curve (AUSROC) was 0.88 in tissue samples. The presence of DNA methylation had a relatively high diagnostic ability (area under curve [AUC] = 0.8834) for the risk of sporadic EC (Figure 5), but a relatively low specificity (0.48) and high sensitivity (0.93). In terms of sensitivity, DNA methylation is chemically stable and has shown high sensitivity in other tumors [36]. The present evidence demonstrated that methylated genes were superior to protein biomarkers such as CA-125, either alone or in combination, which had diagnostic sensitivities of only 40%–80% [37, 38]. A clinical diagnosis of EC currently relies on a combination of ultrasound, magnetic resonance imaging, and CA-125, though none of these alone is completely satisfactory. CA-125 is often used as a biomarker for ovarian cancer, but has also been suggested as a good prognostic marker for EC [37]. The presence of circulating tumor DNA (ctDNA) has been considered to be highly specific for certain cancers, mainly because somatic mutations identified in tumor DNA are absent from normal DNA, whereas gene methylation may occur in normal as well as cancer DNA [39]. Our study confirmed that the detection of DNA methylation in clinical samples had a relatively low diagnostic specificity for EC (pooled specificity: 0.48, 95%CI: 0.46–0.50), suggesting that more specific methylated genes are required.
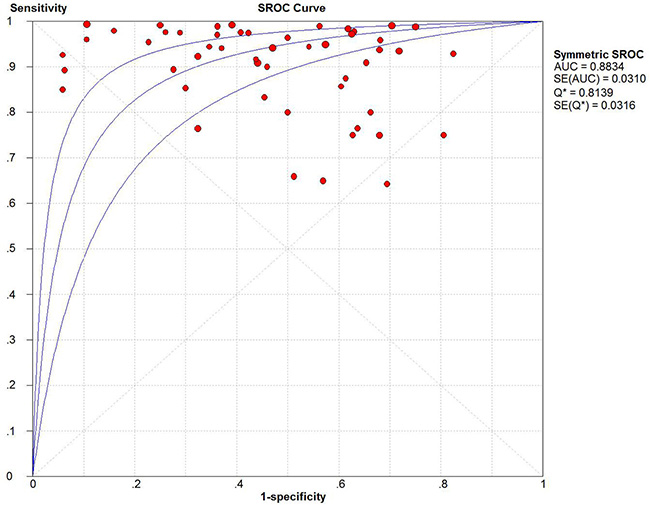
Figure 5: Summary receiver operating characteristic (SROC) plot with the associated 95% confidence region.
The current study included gene methylation or hypermethylation alterations, which could lead to aberrant silencing of tumor suppressor genes in most cases. Consistent with these types of gene alterations, the most common detection method for methylation was MSP, which has various advantages including relatively high sensitivity [40]. In our meta-analysis, MLH1, CDH1, and RASSF1A gene methylation were detected in more than one study. MLH1 was identified as the cause of abnormal DNA mismatch repair and microsatellite instability (MSI), and its dysfunction might be associated with Lynch syndrome (also known as hereditary nonpolyposis colorectal cancer syndrome, HNPCC) [41]. Although MSI has been detected in numerous patients with EC, its mutation frequency is extremely low, suggesting that it may arise as a result of aberrant methylation of promoter regions in MSI-positive EC patients [15]. Furthermore, the diagnostic value of aberrant DNA methylation for EC was not significantly influenced by the technique used, except possibly for qMSP, suggesting that this kind of assay may show greater specificity. A combination of gene methylation markers also showed potentially higher diagnostic accuracy in sporadic EC.
This preliminary study clearly indicated that detecting gene methylation patterns could offer a diagnostic test for sporadic EC with high sensitivity, but low specificity. The presence of DNA methylation in EC patients may predict positive lymph node metastasis and unfavorable survival. In general, there is lack of diagnostic and prognostic biomarkers for EC, and methylation markers may thus be useful for distinguishing between EC and non-malignant causes in women with abnormal vaginal bleeding. Further meta-analyses need to be conducted to address the use of accurate methylated targets and suitable detection techniques, while more prospective studies utilizing consistent and standardized methodologies are urgently required to resolve these problems.
This study had several limitations. Firstly, selection bias might have occurred due to enrichment of studies reporting positive results, and the relatively small sample sizes in some of the selected literature may also have led to bias. Furthermore, the included studies were mostly from East Asia, and the conclusions may therefore not be universally applicable. The use of different MSP primers and/or equipment, and the lack of a well-accepted methylated gene in sporadic EC might also have been potential sources of bias.
MATERIALS AND METHODS
Search strategy
We conducted a systematic literature search to identify studies assessing DNA methylation changes as biomarkers for the early diagnosis of sporadic EC. We searched the PubMed and Web of Science databases for all relevant English-language papers published prior to April 30, 2017, using the following combination of keywords: [tissue (or) sample (or) specimen (or) circulating cell free DNA (or) cfDNA (or) ctDNA (or) circulating tumor DNA (or) circulating tumor cell (or) CTC (or) blood (or) white blood cell (or) serum (or) plasma] (and) [endometrial (or) endometrioid (or) endometrium (or) endometria] (and) [neoplasm (or) cancer (or) tumor (or) carcinoma (or) adenocarcinoma (or) malignancy] (and) [methylation (or) methylated (or) hypermethylation (or) hypomethylation]. Two authors (YF) and (YW) consulted, and screened the titles and abstracts of the identified papers independently. All the studies were evaluated and discussed by the authors until a consensus of criteria was reached. The Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) statement [42] was applied as the template for the searching process.
Eligibility criteria
Duplicate articles were removed after combining the retrieved publications from the two databases. Initial screening was conducted by reviewing the titles and abstracts. Only full-text reports of original studies were included, and meeting abstracts, reviews, and editorials were excluded. Articles not focusing on DNA methylation changes in tissues and not in the context of sporadic EC detection/diagnosis were also excluded. After the first-round examination, we conducted a full-text review of the remaining articles and excluded studies based on the following exclusion criteria: studies without healthy normal individuals or controls (e.g., studies with only paired samples or benign disease were not considered); studies that were not case-control studies; studies for which sensitivity and specificity values for the diagnosis of sporadic EC were not reported or could not be calculated from the published data; studies with small sample sizes (n ≤ 10); and studies based on cell lines/animals rather than human clinical samples.
Data extraction and statistical analysis
Two investigators independently reviewed and evaluated the eligible studies according to the Newcastle-Ottawa scale (NOS) [43], and studies awarded five or more stars were considered as high-quality. The following data were then extracted by the two authors using a standardized form: first author, year of publication, region, sample size, detection techniques, biomarkers, alteration type, diagnostic sensitivity and specificity values, and risk of bias according to criteria from the Cochrane Collaboration tool (Cochrane handbook for systematic reviews of interventions version 5.1.0.). The following risk-of-bias items were evaluated using standardized methods: random sequencing generation, allocation concealment, blinding of patients and study personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other biases. Any disagreement was resolved by further discussion. If methylation values were not definitively reported, the information was extracted from available tables or figures as far as possible. The diagnostic value or accuracy of the methylated genes for sporadic EC was evaluated by analyzing diagnostic variables such as sensitivity, specificity, positive likelihood ratios (PLR) and negative likelihood ratios (NLR), diagnostic ratios (DOR), and summary receiver operating characteristic curves (SROC) using Meta-DiSc software [44]. A PLR >5.0 and NLR <0.2 were considered clinically significant. The DOR represented the increased risk of EC in patients with the methylated gene compared with those without. Sensitivity and publication bias were analyzed using Review Manager 5.3, and publication bias was presented using funnel plots. Substantial heterogeneity was considered to exist when I2 was >50%. In the event of heterogeneity among studies, the results were pooled using a random effect model; otherwise a fixed effect model was adopted.
Abbreviations
EC: endometrial cancer; MSP: methylation-specific PCR; COBRA: combined bisulfite restriction analysis; SROC: summary receiver operating characteristic curve; AUSROC: area under summary receiver operating characteristic curve; HNPCC: hereditary nonpolyposis colorectal cancer syndrome; MSI: microsatellite instability; ctDNA: circulating tumor DNA; AUC: area under curve; OR: odds ratio; SE: standard errors; BMI: body mass index; PLR: positive likelihood ratios; NLR: negative likelihood ratios; DOR: diagnostic ratios; CI: confidence intervals; NOS: Newcastle-Ottawa scale; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-analysis.
Author contributions
Conceived and designed this study: QW; Performed this study: YF, YW and SF; Analyzed the data: YF, YW and LY; Contributed analysis tools: SL and QF; Wrote the manuscript: YF and YW.
ACKNOWLEDGMENTS
We thank Yunwei Han to provide helps for data extraction and analysis.
CONFLICTS OF INTEREST
None of the authors has any commercial or other associations that might pose a conflicts of interest.
FUNDING
This study was supported by Research Foundation of Southwest Medical University for Youth (No. 0903-00030685), and by grants from National Natural Science Foundation of China (No. 81201784, 81201682) and Health and Family Planning Commission of Sichuan Province (No. 17PJ557).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics. CA Cancer J Clin. 2017; 67:7–30.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China. CA Cancer J Clin. 2016; 66:115–132.
3. Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet. 2016; 387:1094–1108.
4. Bradford LS, Rauh-Hain JA, Schorge J, Birrer MJ, Dizon DS. Advances in the management of recurrent endometrial cancer. Am J Clin Oncol. 2015; 38:206–212.
5. Silva JL, Paulino E, Dias MF, Melo AC. Endometrial cancer: redefining the molecular-targeted approach. Cancer Chemother Pharmacol. 2015; 76:1–11.
6. Bartosch C, Lopes JM, Jeronimo C. Epigenetics in endometrial carcinogenesis - part 2: histone modifications, chromatin remodeling and noncoding RNAs. Epigenomics. 2017; 9:873–892.
7. Li M, Sun Q, Wang X. Transcriptional landscape of human cancers. Oncotarget. 2017; 8:34534–34551. http://doi.org/10.18632/oncotarget.15837.
8. Llinas-Arias P, Esteller M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. 2017; 7:170152.
9. Caplakova V, Babusikova E, Blahovcova E, Balharek T, Zelieskova M, Hatok J. DNA Methylation Machinery in the Endometrium and Endometrial Cancer. Anticancer Res. 2016; 36:4407–4420.
10. Sasaki M, Dharia A, Oh BR, Tanaka Y, Fujimoto S, Dahiya R. Progesterone receptor B gene inactivation and CpG hypermethylation in human uterine endometrial cancer. Cancer Res. 2001; 61:97–102.
11. Sasaki M, Kotcherguina L, Dharia A, Fujimoto S, Dahiya R. Cytosine-phosphoguanine methylation of estrogen receptors in endometrial cancer. Cancer Res. 2001; 61:3262–3266.
12. Saito T, Nishimura M, Yamasaki H, Kudo R. Hypermethylation in promoter region of E-cadherin gene is associated with tumor dedifferention and myometrial invasion in endometrial carcinoma. Cancer. 2003; 97:1002–1009.
13. Sasaki M, Kaneuchi M, Sakuragi N, Dahiya R. Multiple promoters of catechol-O-methyltransferase gene are selectively inactivated by CpG hypermethylation in endometrial cancer. Cancer Res. 2003; 63:3101–3106.
14. Li R, Saito T, Tanaka R, Satohisa S, Adachi K, Horie M, Akashi Y, Kudo R. Hypermethylation in promoter region of retinoic acid receptor-beta gene and immunohistochemical findings on retinoic acid receptors in carcinogenesis of endometrium. Cancer Lett. 2005; 219:33–40.
15. Banno K, Yanokura M, Susumu N, Kawaguchi M, Hirao N, Hirasawa A, Tsukazaki K, Aoki D. Relationship of the aberrant DNA hypermethylation of cancer-related genes with carcinogenesis of endometrial cancer. Oncol Rep. 2006; 16:1189–1196.
16. Shih MC, Yeh KT, Tang KP, Chen JC, Chang JG. Promoter methylation in circadian genes of endometrial cancers detected by methylation-specific PCR. Mol Carcinog. 2006; 45:732–740.
17. Pijnenborg JM, Dam-de Veen GC, Kisters N, Delvoux B, van Engeland M, Herman JG, Groothuis PG. RASSF1A methylation and K-ras and B-raf mutations and recurrent endometrial cancer. Ann Oncol. 2007; 18:491–497.
18. Yanokura M, Banno K, Kawaguchi M, Hirao N, Hirasawa A, Susumu N, Tsukazaki K, Aoki D. Relationship of aberrant DNA hypermethylation of CHFR with sensitivity to taxanes in endometrial cancer. Oncol Rep. 2007; 17:41–48.
19. Suehiro Y, Okada T, Okada T, Anno K, Okayama N, Ueno K, Hiura M, Nakamura M, Kondo T, Oga A, Kawauchi S, Hirabayashi K, Numa F, et al. Aneuploidy predicts outcome in patients with endometrial carcinoma and is related to lack of CDH13 hypermethylation. Clin Cancer Res. 2008; 14:3354–3361.
20. Tse KY, Liu VW, Chan DW, Chiu PM, Tam KF, Chan KK, Liao XY, Cheung AN, Ngan HY. Epigenetic alteration of the metallothionein 1E gene in human endometrial carcinomas. Tumour Biol. 2009; 30:93–99.
21. Varley KE, Mutch DG, Edmonston TB, Goodfellow PJ, Mitra RD. Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Res. 2009; 37:4603–4612.
22. Yi TZ, Guo J, Zhou L, Chen X, Mi RR, Qu QX, Zheng JH, Zhai L. Prognostic value of E-cadherin expression and CDH1 promoter methylation in patients with endometrial carcinoma. Cancer Invest. 2011; 29:86–92.
23. Zhang QY, Yi DQ, Zhou L, Zhang DH, Zhou TM. Status and significance of CpG island methylator phenotype in endometrial cancer. Gynecol Obstet Invest. 2011; 72:183–191.
24. Fiolka R, Zubor P, Janusicova V, Visnovsky J, Mendelova A, Kajo K, Lasabova Z, Plank L, Danko J. Promoter hypermethylation of the tumor-suppressor genes RASSF1A, GSTP1 and CDH1 in endometrial cancer. Oncol Rep. 2013; 30:2878–2886.
25. Kovalenko TF, Sorokina AV, Ozolinya LA, Patrushev LI. Methylation of the pseudogene PTENP1 5’-terminal region in endometrial cancer and hyperplasia. Russian Journal Of Bioorganic Chemistry. 2013; 39:397–405.
26. Visnovsky J, Fiolka R, Kudela E, Slavik P, Krkoska M, Lasabova Z, Danko J. Hypermethylation of selected genes in endometrial carcinogenesis. Neuro Endocrinol Lett. 2013; 34:675–680.
27. Yang T, Qiu H, Bao W, Li B, Lu C, Du G, Luo X, Wang L, Wan X. Epigenetic inactivation of EFEMP1 is associated with tumor suppressive function in endometrial carcinoma. PLoS One. 2013; 8:e67458.
28. Chmelarova M, Kos S, Dvorakova E, Spacek J, Laco J, Ruszova E, Hrochova K, Palicka V. Importance of promoter methylation of GATA4 and TP53 genes in endometrioid carcinoma of endometrium. Clin Chem Lab Med. 2014; 52:1229–1234.
29. Chen YC, Tsao CM, Kuo CC, Yu MH, Lin YW, Yang CY, Li HJ, Yan MD, Wang TJ, Chou YC, Su HY. Quantitative DNA methylation analysis of selected genes in endometrial carcinogenesis. Taiwan J Obstet Gynecol. 2015; 54:572–579.
30. Dong R, Pu H, Wang Y, Yu J, Lian K, Mao C. TESTIN was commonly hypermethylated and involved in the epithelial-mesenchymal transition of endometrial cancer. APMIS. 2015; 123:394–400.
31. Sheng Y, Wang H, Liu D, Zhang C, Deng Y, Yang F, Zhang T, Zhang C. Methylation of tumor suppressor gene CDH13 and SHP1 promoters and their epigenetic regulation by the UHRF1/PRMT5 complex in endometrial carcinoma. Gynecol Oncol. 2016; 140:145–151.
32. Wentzensen N, Bakkum-Gamez JN, Killian JK, Sampson J, Guido R, Glass A, Adams L, Luhn P, Brinton LA, Rush B, d’Ambrosio L, Gunja M, Yang HP, et al. Discovery and validation of methylation markers for endometrial cancer. Int J Cancer. 2014; 135:1860–1868.
33. Bartosch C, Lopes JM, Jeronimo C. Epigenetics in endometrial carcinogenesis - part 1: DNA methylation. Epigenomics. 2017; 9:737–755.
34. Su L, Wang H, Miao J, Liang Y. Clinicopathological Significance and Potential Drug Target of CDKN2A/p16 in Endometrial Carcinoma. Sci Rep. 2015; 5:13238.
35. Pabalan N, Kunjantarachot A, Ruangpratheep C, Jarjanazi H, Christofolini DM, Barbosa CP, Bianco B. Potential of RASSF1A promoter methylation as biomarker for endometrial cancer: A systematic review and meta-analysis. Gynecol Oncol. 2017; 146:603–608.
36. Cheuk IW, Shin VY, Kwong A. Detection of Methylated Circulating DNA as Noninvasive Biomarkers for Breast Cancer Diagnosis. J Breast Cancer. 2017; 20:12–19.
37. Rizner TL. Discovery of biomarkers for endometrial cancer: current status and prospects. Expert Rev Mol Diagn. 2016; 16:1315–1336.
38. Knific T, Osredkar J, Smrkolj S, Tonin I, Vouk K, Blejec A, Frkovic Grazio S, Rizner TL. Novel algorithm including CA-125, HE4 and body mass index in the diagnosis of endometrial cancer. Gynecol Oncol. 2017; 147:126–132.
39. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579–586.
40. Huang Z, Bassil CF, Murphy SK. Methylation-specific PCR. Methods Mol Biol. 2013; 1049:75–82.
41. Chang L, Chang M, Chang HM, Chang F. Expending Role of Microsatellite Instability in Diagnosis and Treatment of Colorectal Cancers. J Gastrointest Cancer. 2017; 48:305–313.
42. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009; 6:e1000097.
43. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010; 25:603–605.
44. Zamora J, Abraira V, Muriel A, Khan K, Coomarasamy A. Meta-DiSc: a software for meta-analysis of test accuracy data. BMC Med Res Methodol. 2006; 6:31.