
Introduction
Post translational modifications (PTMs) on histone as well as non-histone proteins regulate gene expression profiles through chromatin structure alterations. These events often dictate key cellular events such as mitosis, meiosis, DNA damage response, gene expression, cell cycle, cell signaling pathways, energy, and metabolic pathways. PTMs such as phosphorylation, N-terminal acetylation, methylation, sumoylation, ubiquitination, propionylation, butyrylation, carbonylation, neddylation, proline isomerization, glycosylation, citrullination and poly ADP ribosylation regulate diverse protein functions [1, 2]. Epigenetic modifications in chromatin’s structure affect gene expression, such that the loosely packed form of DNA, which is called euchromatin, shows higher transcriptional activity; in contrast, the tightly packed form of DNA, heterochromatin, presents lower transcriptional activity [3]. Chromatin remodeling complexes can alter the nucleosome structure thereby modifying DNA accessibility [4]. Chromatin-modifying complexes can be divided into the adenosine triphosphate (ATP)-dependent complex and covalent histone modifications. Various ATP-dependent nucleosome-remodeling complexes have been reported in the literature, suggesting different mechanisms of action involved in histone sliding, ejection or the incorporation of histone variants [5]. The ATP dependent chromatin remodeling complex is divided into four subfamilies, 1) switch/sucrose non-fermentable (SWI/SNF), 2) chromo domain helicase DNA-binding (CHD), 3) imitation switch (ISWI), 4) INO80 [6]. In addition, several subtypes of chromatin remodelers have been identified that are involved in DNA damage, apoptosis and developmental pathways [7, 8]. A range of enzymes including histone lysine acetyltransferases (KATs), histone lysine deacetylases (KDACs), histone lysine methyltransferases (KMTs) and histone lysine demethylases (KDMs) regulate histone modifications [9-11]. Recent studies have shown that long noncoding RNAs are also involved in histone-modifying activities through histone H3 lysine 4 trimethylation [12]. Acetylation of several proteins is often found and regulated a plethora of cellular functions further emphasizing the importance of this PTM. Mass spectrometry analysis of cellular proteins revealed that acetylation occurred across different compartments of cells and was not confined to the nucleus of the cell [13]. PhosphoSitePlus(®) (PSP, http://www.phosphosite.org/), a knowledgebase dedicated to mammalian PTMs, reports that over 330,000 non-redundant PTMs, including phospho, acetyl, ubiquityl and methyl groups [14, 15].
Aberrant histone modifications are known to play a key role in the pathogenesis of several human diseases such as cancer, inflammatory and neurodegenerative diseases. Cancer-specific telomerase reverse transcriptase gene (TERT) promoter mutations (-146C>T and -124C>T) have been linked to reactivation of the epigenetically silenced TERT. TERT promoter mutations in human embryonic stem cells and their differentiation into fibroblasts and neural progenitor cells revealed that these mutations were capable of overcoming epigenetic silencing [16-20]. The reduction of H3K4me3 and H3K9ac levels at mutant TERT promoters was similarly observed in BRAF-mutant, TERT (−146C > T) or TERT (−124C > T) melanoma cell lines, UACC257, A375, WM793, and Malme-3M, following si-BRAF treatment [20]. These data collectively demonstrate the critical role of RAS-ERK signaling in regulating the active chromatin state of mutant TERT promoters in BRAF/NRAS-mutant human melanoma [20]. Various cytokines such as interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), lipopolysaccharides and other stimulants can promote histone acetylation. KATs activate inflammatory genes, whereas KDACs repress the inflammatory gene expression. Promoters of pro-inflammatory cytokines (such as IL-1, IL-2, IL-8, and IL-12) are rapidly acetylated and become transcriptionally active. KDACs regulate transcription of both pro-inflammatory and anti-inflammatory cytokines via their co-repressor complexes and transcription factors such as forkhead box P3 (FOXP3), signal transducer and activator of transcription (STATs), GATAs, zinc finger E-Box binding homeobox 1 (ZEB1), and nuclear factor - κB (NF-κB [21, 22]. Furthermore H3tre11 is a specific substrate for tumor specific pyruvate kinase M2 (PKM2) in EGF mediated transcription initiation and H3K9 acetylation, thus leading to tumor cell proliferation [23]. In addition to nuclear function, histones also act like an endogenous signal when they locate at the extra-nuclear space. As a response to stress, immune cells, cerebellar neurons, Schwann cells, and microglia present H2A, H2B, H3, H4, and H1 on their cell surface or cytoplasm. The levels of circulating histones as well as nucleosomes are increased in cancer, inflammation and infection, which suggest that histones could be potentially useful as biomarkers and therapeutic targets for these diseases [24-27].
Epigenetic Changes in Tumor Suppressor Gene and Oncogene
Tumor suppressor gene p53
The tumor suppressor gene p53 is the most widely studied gene that, under normal physiological conditions, regulates cell cycle, senescence, and differentiation, and induces apoptosis [28]. p53 is often mutated, leading to uncontrolled cell proliferation in a variety of cancers [29]. One of the most frequently observed post-translational modifications is acetylation. Acetylation regulates both the loss of function and gain of function of p53 such as enhancing DNA binding and preventing non-specific binding by the p53 C terminal domain [30]. p53 is acetylated by p300 at 5 lysine residues at the C-terminal regulatory region [31]. Ultraviolet light, ionizing radiation and activated Ras oncogene and other stress factors induce acetylation of p53 [32, 33]. Interestingly, acetylation of p53 blocks its ubiquitination. PCAF (P300/CBP-associated factor) acetylation of p53 at K320 prevents ubiquitination by E4F1 [34]. Remarkably, both acetylation and prevention of ubiquitination activate p53. Ubiquitination of K370, K372, K373, K381, and K382 activates p53 nuclear translocation and its subsequent proteosomal degradation [35]. Therefore, the acetylation event stabilizes p53 and actively promotes its nuclear localization. Notably, p300 and PCAF acetylation sites are not mutated, except K120, which is mutated in cancers and is linked to cell cycle control and apoptosis mechanisms [28, 36-38]. TIP60, and MOF (males absent on the first)-two members of the MYST family- acetylate K120 and are located within the DNA binding domain of p53 [36, 37]. K120 acetylation by TIP60 and MOF induces apoptosis with minimal to no effect on cell cycle genes. Acetylation of p53 neutralizes the positive charge on the lysine residue thereby impairing its ability to form hydrogen bonds [35]. Acetylation also creates docking sites for transcriptional coactivators. For example, acetylation of K382 increases p53 affinity to the CBP bromodomain [39, 40]. In contrast, deacetylation of both K373 and K382 enhances interaction with tandem bromodomains of TAF1, a TFIID subunit [40]. PC4 is a transcription factor that is acetylated by p300. It has been shown that PC4 interaction with p53 leads to acetylation of both the proteins and enhances the p53 DNA binding ability and expresses p53 regulated gene products, especially during DNA damage [41, 42].
Oncogene cMYC
One of the most commonly activated oncogenes in cancer cells is Myc, which regulates the complex inflammatory response [33, 43]. In B-cells, activation of Myc rapidly induces the synthesis and release of IL1-β [36]. The pleiotropic effects of oncogenes include creating a pro-tumor microenvironment and a persistent, constitutive activation of pro-inflammatory transcription factors [44-46]. The involvement of human c-Myc in cancer progression is shown in multiple cancer types, including Burkitt’s lymphoma, multiple myeloma and T cell leukemia [47]. cMyc is deregulated and abnormally overexpressed in a variety of cancers [43]. Wasylishen AR et al., 2014 showed that Myc signaling is regulated by six lysine residues located at the highly conserved Myc homology box IV. These lysine residues negatively regulate Myc induced transformation and tumorigenesis, and thus serve as potential targets for cancer therapy [48]. This lysine rich region has previously been associated with Myc acetylation [49, 50]. Myc also serves as a substrate for KATs which modify different lysines. The following lysines, 143, 148, 157, 275, 317, 323, 371, and 417, are acetylated either in response to co-expression of mGCN5 or p300 or by in vitro acetylation with p300 [51]. Other studies indicated that knocking down JMJD1A, a histone demethylase inhibited the proliferation and survival of prostate cancer cells. . This study unveils the prime role of JMJD1A in upregulating c-Myc expression by AR (androgen receptor) dependent mechanism via H3K9 demethylation activity and controlling post transcriptional activity of c-Myc [52]
Epigenetic Origin of Sporadic Cancers
Genetic and environmental inter-relationships play a vital role in the development of sporadic forms of cancers and are often associated with genetic alterations in these tumors [53]. For instance, breast cancer and its subtypes can be differentiated based on their genomic and epigenetic aberrations. DNA methylation is often observed in both normal as well as in cancerous tissues. Several hundred genes are either hypermethylated or hypomethylated in breast cancer [54]. Epigenetic silencing or deletion of BRCA1 is commonly observed in sporadic breast cancer [55]. Familial CRC occurs in 15% of cases, hereditary in 5% of cases, and sporadic in almost 75-80% of cases. Several somatic mutations have been in identified in the tyrosine phosphatome, and PTPRG, PTPRT, PTPN3, PTPN13, and PTPN14 were all associated with loss of function [53]. Genetic and environmental factors play key roles in the susceptibility of individuals to sporadic forms of CRC resulting in loss of protein function.
Histone Phosphorylation in Cancer
Histone phosphorylation is one of the PTM events that take place during DNA damage, activation of transcription and chromatin remodeling/compaction during cell division and apoptosis, and other nuclear processes. Histone phosphorylation is regulated by various kinases and phosphatases [56]. These kinases and phosphatases can potentially act on PTM sites on any of the four nucleosomal histone tails. Traditionally, phosphorylation occurs on tyrosine, serine, and threonine residues [57, 58]. Several phosphorylation sites have been identified in histone tails that regulate nuclear processes. During DNA damage, phosphorylation of core histone H2A(X) is a key event; serine 139 gets phosphorylated on the H2A(X) variant histone, commonly referred to as γH2A(X) [59-63]. This phosphorylation occurs during cell cycle and DNA damage response, and is mediated by protein kinases yTel1 and yMec1 (ATM and ATR in mammals) in yeast [64, 65]. Importantly, histone phosphorylation is associated with transcriptional regulation and gene expression, especially genes that regulate cell cycle and proliferation [60]. Phosphorylation of serine (Ser) 10 and 28 on H3, and serine 32 on H2B have been associated with activation of epidermal growth factor (EGF) mediated gene transcription. This phenomenon has also been observed upon exposure to ultraviolet B radiation, whereH3ser10p and H2Bser32p upregulated the expression of proto-oncogenes such as c-myc, c-fos, c-jun [66-68]. Furthermore, H3ser28p regulated the activation of c-fos and α-globin [69]. Phosphorylation of H3 at ser10, 28 and threonine (Thr) 6 and 11 has been shown to occur upon androgen stimulation and DNA damage. [56, 70-73]. Taken together, a complex crosstalk between phosphorylation events leads to gene expression, and cell proliferation. H2Bser32p occurs ubiquitously in normal human cells; however, it is also extensively phosphorylated by RSK2 kinase (an AGC kinase of the RSK family of kinases) in skin cancer cells [67, 74]. Several protein kinases have been associated with phosphorylation of histones such as RSK2, MSK1/2, and mixed lineage kinase-like mitogen-activated protein triple kinase alpha (MLTK)-α [56]. In addition, androgen stimulation protein kinase C-beta (PKCβ) and protein kinase C related kinase 1 (PRK1) phosphorylate H3tre6 and 11 respectively in prostate cancer cells [70, 71]. Histone tyrosine (tyr) phosphorylation is also associated with transcription initiation. Janus kinase 2 (JAK2) has also been shown to phosphorylate H3tyr41, thereby disrupting chromatin binding by heterochromatin protein 1α (HP1α). HP1α has been shown to directly interact with H3 through its chromo-shadow domain. Thus, loss of HP1α from chromatin leads to constitutive activation of the JAK2 signaling pathway including oncogene imo2, leading to oncogenesis [75]. Several studies indicated that Aurora B was overexpressed in a variety of human cancers particularly in colorectal and breast cancer, and critical for H3 phosphorylation and for accurate chromosome segregation [76-79]. Remarkably, H2AX phosphorylation increases several fold upon DNA fragmentation and apoptosis [80, 81]. Mst1 kinase has been shown to phosphorylate H2AX. Overexpression of Mst1-induced apoptosis of HELA cells is associated with H2AXser139p [82] (Figure 1) (Table 1).
Table 1: Transcriptional and cellular role of novel histone modifications in cancers
Post-translational Modifications |
Histone residues modified |
Role in cell activity and transcription |
Enzymes |
References |
Phosphorylation(serine, threonine, tyrosine) |
H3 (Ser10, 28; Thr 3, 6, 11,45 and Tyr41) H2B (Ser32) Histone variant H2A(X) |
Activation DNA damage response |
ATM, ATR, RSK2, PKM2, Aurora B kinase. |
|
Lysine ubiquitination |
H2A (K119) H2B (K120) |
Transcription regulation |
E2A ubiquitinase |
|
p53 acetylation and ubiquitination |
K370; K372; K373; K381; K382; K120 |
Proteasomal degradation; apoptosis |
p300/PCAF; TIP60; MOZ; |
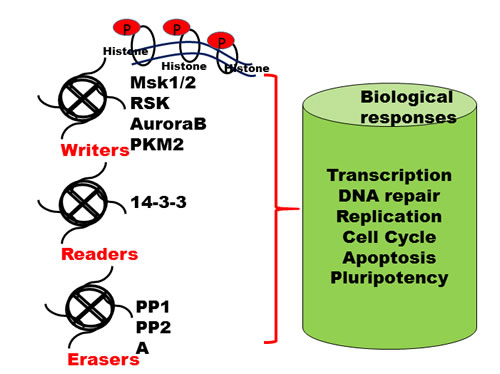
Figure 1: Writers, readers and erasers of histone marks. Reversible PTMs addition, identification and removal of these modifications on histone tails regulate various biological processes, including transcription, DNA replication and DNA repair and are highly dynamic in nature. The "writers" such as histone protein kinases add specific PTMs on specific amino acid residues on core histone tails. These marks are identified by specific protein domains called "readers" such as 14-3-3 proteins. The PTM marks are removed by active enzymes known as "erasers" such as phosphatases; Abbreviations: RSK, ribosomal protein S6 kinase A3; AuroraB, serine/threonine protein kinase Aurora B; PKM2, pyruvate kinase M2; Msk1/2, Mitogen- and stress-activated protein kinase 1 and 2, PP, protein phosphatases.
Histone Poly-ADP Ribosylation in Cancer
Poly-ADP ribosylation is involved in the development and progression of cancer thorough diverse pathways. Involvement of mono and/or poly-ADP-ribosylation has been shown to be involved in carcinogenesis and has been widely implicated in many cancers including hepatocellular carcinomas, lymphomas, endometrial, oral and colon cancers [83-88]. Poly-ADP-ribose-polymerase (PARP) and poly-ADP-ribose-glycohydrolase (PARG) function as DNA repair enzymes that play prominent roles in the base-excision repair and double strand break repair pathways [89]. The primary prerequisite in DNA repair of heterochromatic DNA is chromatin relaxation to allow DNA repair enzymes access to DNA lesion sites. Several studies have demonstrated that PARP-1 mediated poly-ribosylation is an essential component of different repair pathways such as base excision repair (BER) [90], nucleotide excision repair [91], homologous recombination [92], and non-homologous end joining [93]. PARP-1 also has a role in epigenetic regulations that promote oncogenesis [85]. Cohen-Armon et al., 2007 showed that phosphorylated ERK2 activates PARP-1 and Elk1, thereby promoting CBP/p300 dependent acetylation and transcription of Elk1 target genes [94]. In another study, CDK1 dependent phosphorylation of PARP-1 displaced histone H1 from chromatin, thus accelerating progestin-mediated breast cancer cell proliferation [95]. PARP-1 mediated PARylation of the lysine demethylase, KDM5B, inhibited binding to its target proteins and maintained the H3K4me3 methylation status in MCF7 breast cancer cells [96]. The transient ectopic overexpression of CCCTC-binding factor (CTCF) induces the PARP-1/CTCF (CCCTC binding factor) complex that suppresses DNA methyltransferase 1 (DNMT1)-mediated DNA methylation-dependent silencing and chromatin compaction [97, 98]. Furthermore, PARP-1 deficiency induces differentiation of embryonic stem cells to syncytiotrophoblastic giant cells and produces tumors phenotypically similar to teratocarcinoma [99, 100].
Histone Ubiquitination and Transcriptional Regulation
Epigenetic modifications are regulated by PTMs, including ubiquitination, and can directly modulate either histones or non-histone proteins such as transcription factors or their co-factors. The balance between ubiquitination and deubiquitination is important for cellular homeostasis, and any imbalance in this equation often leads to tumorigenesis. Ubiquitin is a 76-amino acid polypeptide containing seven lysine (K) residues with poly-ubiquitin chains linked through the K6, K11, K27, K29, K33, K48, and K63 residues Ubiquitin contains. The process of ubiquitination (attachment of ubiquitin to a histone protein) is an ATP dependent process activated by an E1 enzyme, followed by an E2 class enzyme allowing E3 ubiquitin ligase to ubiquitinate target proteins directly or indirectly [101-103]. The target protein can either be mono or poly-ubiquinated and the consequence depends on the type of chains formed [104]. Distinct mono and poly-ubiquitination are known to regulate target protein activity [105]. For instance, K6 and K48 poly-ubiquitin chains regulate proteosomal degradation, while K11 targets proteins degradation via endoplasmic reticulum mediated d pathways. K11 is also involved in regulation of cell cycle progression while K29 is involved in protein degradation by lysomes [106, 107]. Histone ubiquitination is implicated in several conserved cellular processes such as DNA repair, transcriptional regulation, and genome stability. Unlike other proteins, histone poly-ubiquitination does not mark histones for degradation by 26s proteasomes, but rather is associated with transcriptional regulation [108]. One major PTM is histone H2A ubiquitination, which plays a role in transcriptional silencing by polycomb proteins and genome maintenance. H2A is the most abundant histone fraction to be mono-ubiquitinated at K119 compared to H2B at K120. These ubiquitination marks can be deubiquitinated by MYSM1 (2A-DUB) [109]. All the four core histones H2A, H2B, H3, and H4 become mono-ubiquitinated upon DNA damage and are known ubiquitin substrates in vivo [106, 108]. H2B is usually monoubiquitinated while H2A can be mono or poly-ubiquitinated. Ubiquitination of the C-terminal tail of H2B promotes transcription elongation, disrupts chromatin structure and nucleosomes, and significantly increases transcript length [110, 111]. H2B monoubiquitination (H2Bub1) is a key PTM that plays an important role in both transcriptional activation and tumor suppression [112, 113]. Aberrant H2Bub1 ubiquitination and deubiquitination is the primary driver of tumorigenesis and directly impact upon chromatin structure beyond the single nucleosome level [114, 115]. H2B ubiquitination is regulated by ubiquitin-conjugating enzyme E2A (UBE2A or RAD6A), and the RNF20/RNF40 ubiquitin ligase complex. Aberrant expression of these enzymes often leads to the development and progression of several tumor types such as seen with hypermethylation of RNF20 promoters in breast tumor samples. Maintenance of H2B monoubiquitination is dependent on tumor suppressor cell division cycle 73 (CDC73). Mutation of CDC73 results in corresponding loss of H2B monoubiquitination in both in vitro and in vivo. Mutated or abnormally regulated CDC73 has been reported in parathyroid, renal, breast, gastric, and colorectal tumors, as well as in the germline of patients with the familial disorder-hyperparathyroidism jaw tumor syndrome [116]. Thus, altered expression levels of RNF20 contribute to the deferred H2Bub1 levels, leading to cancer development and progression. H2Bub1’s role in tumor suppression was correlated with decreased abundance in malignant breast cancer samples compared to normal and benign samples [117]. Aberrant expression and over-activation of deubiquitination enzymes also affect the global abundance of H2Bub1 [115, 118, 119]. Specific mutations such as non-synonymous single nucleotide polymorphisms, amplifications, and homozygous deletions in deubiquitinase USP22 have been reported in a variety of tumor types [108]. In a study by Zhang et al., USP22 was upregulated in breast cancer samples obtained from patients with aggressive phenotypes compared to benign tumors, and correlated with a decrease in H2Bub1 levels [120]. Furthermore, USP22 overexpression correlated with lymph node metastasis and breast cancer reoccurrence, and was an indicator of poor prognosis [120, 121].
Histone Sumoylation in Cancer
Similar to other reversible post-translational modifications, sumoylation of histones is characterized by covalent addition of small ubiquitin-like modifier (SUMO) to a target protein. It is one of the most essential post translational modifications and plays a key role in various physiological and pathological processes. SUMO exists in four isoforms named SUMO-1, -2, -3, -4 [122] which are 12 kDa in size, and share a structural similarity with ubiquitin [123-126]. Sumoylation of protein does not lead to proteosomal degradation; in contrast it is required for protein stability, transcription regulation of glucocorticoid receptor, Myb, CAAT/enhancer binding protein (C/EBP) and SP3. Sumoylation is a negative regulator and attenuates transcriptional activity [127, 128]. Desumoylation is mediated by specific cysteine-protease Sentrin-specific protease (SENP) which removes the sumoylation marks on proteins [129]. Several transcription factors like activating protein 2 (AP-2), androgen receptor (AR), c-Jun, c/EBP, c-Myb, CREB, GATA-2, heat shock factor (HSF1), interferon regulatory factor (IRF-1), p53, Sp3, STAT1, have been demonstrated to be SUMOylated in cancer [130-132]. Emerging evidence indicated that sumoylation is also known to regulate the enzymatic activity of histone modifying enzymes. Several histone modifying enzymes such as HDAC1, HDAC2, HDAC4, SIRT1, EZH2 and KDM5B are known to be modified by SUMO thereby linking SUMOylation to epigenetic regulation [133-137].
Histone Glycosylation in Cancer
Several other PTMs of histone have been identified which are associated with the regulation of histone function and chromatin remodeling. Core histones H2A, H2B, H3 and H4 were found to be glycosylated to O-linked N-acetylglucosamine (O-GluNAc) modification on serine and threonine residues such as H2A at T101, H2B at S36, H3S10 and T32 and H4 at S47 [138-142]. This modification is mediated by two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAc hydrolase (OGA). Both histones and cytosine demethylases are modified by O-GlcNAc in a variety of cancer cells [139, 141-143]. Over expression of OGT crosstalk with other histone PTM such as H3K9 acetylation, H3S10 phosphorylation, and H3R17/K27 methylation suggesting that O-GlcNAc signaling might regulate chromatin dynamics by affecting other histone marks [140, 141, 144]. Other studies have demonstrated the role of histone glycosylation in normal and cancer cells. This study indicated the early reduction of OGlcNAcylation level at H3 on serine 10 and 28 in S phase whereas elevated levels was observed in G2/M phase. However, the function and mechanism of histone O-GlcNAcylation and other histone modifications such as propionylation, butyrylation, carbonylation, citrullination and proline isomerization still remain to be elucidated.
Histone Neddylation in Cancer
Similar to the ubiquitin pathway, ubiquitin-like protein NEDD8 has been identified as targeting proteins for degradation via the neddylation pathway [145]. NEDD8 is 80% homologus to ubiquitin and modulates ubiquitin induced protein degradation. Unlike ubiquitin, NEDD8 conjugated proteins are more abundant in the nucleus than in the cytoplasm [146, 147]. The most commonly investigated protein substrates of neddylation are Cullin-RING ligases. These are conjugated to NEDD8 via its C-terminal Gly-76 by NEDD8 [147-149] and deneddylated by COP9 signalosome (CSN) [150] Interestingly, it was observed that the cellular stress condition leads to drastic increase in neddylation. However the reason for this phenomenon is still unclear. Several key molecules involved in tumorigenesis are known to be regulated by neddylation and ubiquitination. In a majority of sporadic renal cancers, mutation in VHL protein is often observed and this is a substrate for NEDD8 [150-152]. In high grade neuroendocrine tumors, high levels of neddylated culin-1 is associated with tumor progression [153]. Squamous Cell Carcinoma-related Oncogene SCCRO (also known as DCUN1D1) is a part of E3 ligase complex (Cul-1-ROC1-SCCRO) is associated with the development of primary of squamous cell carcinoma of the lung with malignant phenotypes [154, 155]. Several other substrates of neddylation have also been observed in cancers. The RNA binding protein Hu antigen R (HuR) regulates cell dedifferentiation, proliferation and survival is neddylated in hepatocellular and colorectal carcinoma [156]. Other studies indicated that nedyylation of p53 by FBXO11, a member of the F-box protein family suppressed the function of p53 in lung cancer models [157]. In gastrointestinal cancers, neddylation of NF-κB upstream kinase IKKγ leads to activation of NF-κB [158]. In several cancers Mdm2, a E3 ubiquitin protein ligase that is involved in the ubiquitination and degradation of tumor suppressor protein p53 is also neddylated and promotes conjugation of NEDD8 to p53 [159].
Conclusion and perspectives
Modifications in the tail regions and globular domains of histones can produce multifaceted consequences to cellular process such as transcription and replication, or even lead to malignant transformations. Several important histone modifiers have been identified and have been extensively discussed in this review. Histone acetylation is a mark associated with an active chromatin state. PTMs of histones such as histone acetyltransferases and deacetylases have been identified in the initiation, progression, and metastasis of cancers and some of these events are indicators of poor prognosis. Several novel histone post-translational modifications have been a subject of intense study for their role in tumor suppression, and also some exhibiting oncogenic functions. In addition, phosphorylation of histone by histone kinases, poly-ADP ribosylation, and histone ubiquitination have been implicated in the activation of multi-step and diverse pathways in transcription regulation and in the development of cancer. Therefore, histone modifications can be drivers of transcriptional change with relevant functions, rather than being mere by-products of PTMs or markers of the chromatin state. More work is needed to untangle the role of PTMs occurring at the same residue with contrasting functions. Several new epigenetic targets have also been identified with vast potential in epigenetic-specific therapy. Recent advances in epigenetics research have identified novel modifications in the core histones that have not yet been fully characterized. Overall, additional work is required to decipher the role of novel histone acylation, butyrylation, propionylation, and crotonylation, which will provide insight and extend our knowledge regarding chromatin compaction, transcription regulation, and the development of a variety of diseases including hematological malignancies, organ specific cancers, tumor metastasis, DNA repair, cellular reprogramming, differentiation, and pluripotency.
Acknowledgments and funding
This work was supported by NUHS Basic seed grant [T1-BSRG 2015-02] and Ministry of Education Tier 1 grant to G. Sethi. APK was supported by grants from National Medical Research Council of Singapore, NCIS Yong Siew Yoon Research Grant through donations from the Yong Loo Lin Trust and by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centers of Excellence initiative to Cancer Science Institute of Singapore, National University of Singapore.
ConflictS of Interest
There is no conflict of interest.
References
1. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128:693-705.
2. Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010; 142:682-685.
3. Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009; 10:161-172.
4. Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr Opin Genet Dev. 2003; 13:136-142.
5. Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017; 18:407-422.
6. Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006; 34:2887-2905.
7. Lessard JA, Crabtree GR. Chromatin regulatory mechanisms in pluripotency. Annu Rev Cell Dev Biol. 2010; 26:503-532.
8. Bao Y, Shen X. SnapShot: chromatin remodeling complexes. Cell. 2007; 129:632.
9. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015; 16:178-189.
10. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014; 15:703-708.
11. Shanmugam MK, Sethi G. Role of Epigenetics in Inflammation-Associated Diseases. In: Kundu TK, Editors. Epigenetics: Development and Disease. London: Springer Dordrecht; 2012. pp. 627-658.
12. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011; 472:120-124.
13. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325:834-840.
14. Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012; 40:D261-270.
15. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015; 43:D512-520.
16. Akincilar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, Tergaonkar V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016; 6:1276-1291.
17. Akincilar SC, Low KC, Liu CY, Yan TD, Oji A, Ikawa M, Li S, Tergaonkar V. Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett. 2015; 589:974-984.
18. Akincilar SC, Unal B, Tergaonkar V. Reactivation of telomerase in cancer. Cell Mol Life Sci. 2016; 73:1659-1670.
19. Khattar E, Kumar P, Liu CY, Akincilar SC, Raju A, Lakshmanan M, Maury JJ, Qiang Y, Li S, Tan EY, Hui KM, Shi M, Loh YH, Tergaonkar V. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J Clin Invest. 2016; 126:4045-4060.
20. Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc Natl Acad Sci U S A. 2016; 113:14402-14407.
21. Cantley MD, Haynes DR. Epigenetic regulation of inflammation: progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology. 2013; 21:301-307.
22. Tong L, Tergaonkar V. Rho protein GTPases and their interactions with NFkappaB: crossroads of inflammation and matrix biology. Biosci Rep. 2014; 34.
23. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012; 150:685-696.
24. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl). 2014; 92:465-472.
25. Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis. 2014; 5:e1370.
26. Kaypee S, Sudarshan D, Shanmugam MK, Mukherjee D, Sethi G, Kundu TK. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol Ther. 2016.
27. Shanmugam MK, Arfuso F, Sethi G. Epigenetic Modifications and Their Potential Role in Tumorigenesis. In: Dincer Y, Editor. Epigenetics: Mechanisms and Clinical Perspectives. New York: Nova Science Publishers, Inc.; 2016. pp. 31-82.
28. Soussi T, Ishioka C, Claustres M, Beroud C. Locus-specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer. 2006; 6:83-90.
29. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007; 8(4):275-283.
30. Friedler A, Veprintsev DB, Freund SM, von Glos KI, Fersht AR. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure. 2005; 13:629-636.
31. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997; 90595-606.
32. Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998; 12:2831-2841.
33. Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo J. 2001; 20:1331-1340.
34. Le Cam L, Linares LK, Paul C, Julien E, Lacroix M, Hatchi E, Triboulet R, Bossis G, Shmueli A, Rodriguez MS, Coux O, Sardet C. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell. 2006; 127:775-788.
35. Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003; 15:164-171.
36. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006; 24:841-851.
37. Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006; 24:827-839.
38. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006; 6:909-923.
39. Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, Sanchez R, Zeleznik-Le NJ, Ronai Z, Zhou MM. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell. 2004; 13:251-263.
40. Li AG, Piluso LG, Cai X, Gadd BJ, Ladurner AG, Liu X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol Cell. 2007; 28:408-421.
41. Banerjee S, Kumar BR, Kundu TK. General transcriptional coactivator PC4 activates p53 function. Mol Cell Biol. 2004; 24:2052-2062.
42. Batta K, Kundu TK. Activation of p53 function by human transcriptional coactivator PC4: role of protein-protein interaction, DNA bending, and posttranslational modifications. Mol Cell Biol. 2007; 27:7603-7614.
43. Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, Li Y, Franzoso G, Li S, Guccione E, Tergaonkar V. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest. 2015; 125:2109-2122.
44. Croce CM. Oncogenes and cancer. N Engl J Med. 2008; 358:502-511. 358/5/502
45. Borrello MG, Degl’Innocenti D, Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 2008; 267:262-270.
46. Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev. 2010; 20:65-71.
47. Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001; 20:5595-5610.
48. Wasylishen AR, Kalkat M, Kim SS, Pandyra A, Chan PK, Oliveri S, Sedivy E, Konforte D, Bros C, Raught B, Penn LZ. MYC activity is negatively regulated by a C-terminal lysine cluster. Oncogene. 2014; 33:1066-1072.
49. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, Blobel GA, McMahon SB. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol. 2004; 24:10826-10834.
50. Zhang K, Faiola F, Martinez E. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem Biophys Res Commun. 2005; 336:274-280.
51. Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006; 281:34725-34729.
52. Fan L, Peng G, Sahgal N, Fazli L, Gleave M, Zhang Y, Hussain A, Qi J. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene. 2016; 35:2441-2452.
53. Laczmanska I, Sasiadek MM. Tyrosine phosphatases as a superfamily of tumor suppressors in colorectal cancer. Acta Biochim Pol. 2011; 58:467-470.
54. Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, Nebdal D, Daviaud C, Gut I, Bukholm I, Naume B, Borresen-Dale AL, Tost J, Kristensen V. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol Oncol. 2011; 5:61-76.
55. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006; 8:R38.
56. Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012; 7:1098-1108.
57. Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007; 14:1025-1040.
58. Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011; 21:564-578.
59. van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005; 6:757-765.
60. Rossetto D, Truman AW, Kron SJ, Cote J. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res. 2010; 16:4543-4552.
61. Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin Ther Targets. 2010; 14:577-584.
62. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273:5858-5868.
63. Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003; 5:675-679.
64. Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000; 408:1001-1004.
65. Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004; 14:1703-1711.
66. Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, Davie JR. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999; 274:24914-24920.
67. Lau AT, Lee SY, Xu YM, Zheng D, Cho YY, Zhu F, Kim HG, Li SQ, Zhang Z, Bode AM, Dong Z. Phosphorylation of histone H2B serine 32 is linked to cell transformation. J Biol Chem. 2011; 286:26628-26637.
68. Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, Dong Z. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Res. 2005; 65:5818-5827.
69. Lau PN, Cheung P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc Natl Acad Sci U S A. 2011; 108:2801-2806.
70. Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Muller JM, Greschik H, Kirfel J, Ji S, Kunowska N, Beisenherz-Huss C, Gunther T, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010; 464:792-796.
71. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005; 437:436-439.
72. Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008; 132:221-232.
73. Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell. 2000; 5:917-926.
74. Cho YY, Yao K, Pugliese A, Malakhova ML, Bode AM, Dong Z. A regulatory mechanism for RSK2 NH(2)-terminal kinase activity. Cancer Res. 2009; 69:4398-4406.
75. Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009; 461:819-822.
76. Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000; 102:279-291.
77. Gopalan G, Chan CS, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J Cell Biol. 1997; 138:643-656.
78. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. Embo J. 1998; 17:3052-3065.
79. Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, Suzuki F, Terada Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998; 58:4811-4816.
80. Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000; 275:9390-9395.
81. Mukherjee B, Kessinger C, Kobayashi J, Chen BP, Chen DJ, Chatterjee A, Burma S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst). 2006; 5:575-590.
82. Wen W, Zhu F, Zhang J, Keum YS, Zykova T, Yao K, Peng C, Zheng D, Cho YY, Ma WY, Bode AM, Dong Z. MST1 promotes apoptosis through phosphorylation of histone H2AX. J Biol Chem. 2010; 285:39108-39116.
83. Tentori L, Leonetti C, Scarsella M, Muzi A, Mazzon E, Vergati M, Forini O, Lapidus R, Xu W, Dorio AS, Zhang J, Cuzzocrea S, Graziani G. Inhibition of poly(ADP-ribose) polymerase prevents irinotecan-induced intestinal damage and enhances irinotecan/temozolomide efficacy against colon carcinoma. FASEB J. 2006; 20:1709-1711.
84. Masutani M, Nakagama H, Sugimura T. Poly(ADP-ribose) and carcinogenesis. Genes Chromosomes Cancer. 2003; 38:339-348.
85. Masutani M, Fujimori H. Poly(ADP-ribosyl)ation in carcinogenesis. Mol Aspects Med. 2013; 34:1202-1216.
86. Idogawa M, Yamada T, Honda K, Sato S, Imai K, Hirohashi S. Poly(ADP-ribose) polymerase-1 is a component of the oncogenic T-cell factor-4/beta-catenin complex. Gastroenterology. 2005; 128:1919-1936.
87. Shimizu S, Nomura F, Tomonaga T, Sunaga M, Noda M, Ebara M, Saisho H. Expression of poly(ADP-ribose) polymerase in human hepatocellular carcinoma and analysis of biopsy specimens obtained under sonographic guidance. Oncol Rep. 2004; 12:821-825.
88. Das BR. Increased ADP-ribosylation of histones in oral cancer. Cancer Lett. 1993; 73:29-34.
89. Hottiger MO. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu Rev Biochem. 2015; 84:227-263.
90. Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Menissier-de Murcia J, de Murcia G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999; 81:69-75.
91. Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012; 199:235-249.
92. Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. Embo J. 2009; 28:2601-2615.
93. Spagnolo L, Barbeau J, Curtin NJ, Morris EP, Pearl LH. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012; 40:4168-4177.
94. Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz-Nezer S, Yao Z, Seger R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell. 2007; 25:297-308.
95. Wright RH, Castellano G, Bonet J, Le Dily F, Font-Mateu J, Ballare C, Nacht AS, Soronellas D, Oliva B, Beato M. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 2012; 26:1972-1983.
96. Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell. 2010; 39:736-749.
97. Guastafierro T, Cecchinelli B, Zampieri M, Reale A, Riggio G, Sthandier O, Zupi G, Calabrese L, Caiafa P. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J Biol Chem. 2008; 283:21873-21880.
98. Reale A, Matteis GD, Galleazzi G, Zampieri M, Caiafa P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene. 2005; 24:13-19.
99. Nozaki T, Masutani M, Watanabe M, Ochiya T, Hasegawa F, Nakagama H, Suzuki H, Sugimura T. Syncytiotrophoblastic giant cells in teratocarcinoma-like tumors derived from Parp-disrupted mouse embryonic stem cells. Proc Natl Acad Sci U S A. 1999; 96:13345-13350.
100. Ogino H, Nozaki T, Gunji A, Maeda M, Suzuki H, Ohta T, Murakami Y, Nakagama H, Sugimura T, Masutani M. Loss of Parp-1 affects gene expression profile in a genome-wide manner in ES cells and liver cells. BMC Genomics. 2007; 8:41.
101. Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A. 1980; 77:1365-1368.
102. Blumenfeld N, Gonen H, Mayer A, Smith CE, Siegel NR, Schwartz AL, Ciechanover A. Purification and characterization of a novel species of ubiquitin-carrier protein, E2, that is involved in degradation of non-”N-end rule” protein substrates. J Biol Chem. 1994; 269:9574-9581.
103. Villamil MA, Liang Q, Zhuang Z. The WD40-repeat protein-containing deubiquitinase complex: catalysis, regulation, and potential for therapeutic intervention. Cell Biochem Biophys. 2013; 67:111-126.
104. Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009; 37:937-953.
105. Martinez-Forero I, Rouzaut A, Palazon A, Dubrot J, Melero I. Lysine 63 polyubiquitination in immunotherapy and in cancer-promoting inflammation. Clin Cancer Res. 2009; 15:6751-6757.
106. McClurg UL, Robson CN. Deubiquitinating enzymes as oncotargets. Oncotarget. 2015; 6:9657-9668. https://doi.org/10.18632/oncotarget.3922
107. Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012; 13:508-523.
108. Thompson LL, Guppy BJ, Sawchuk L, Davie JR, McManus KJ. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013; 32:363-376.
109. Zhu P, Zhou W, Wang J, Puc J, Ohgi KA, Erdjument-Bromage H, Tempst P, Glass CK, Rosenfeld MG. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell. 2007; 27:609-621.
110. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007; 128:707-719.
111. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003; 301:1090-1093.
112. Davie JR, Murphy LC. Level of ubiquitinated histone H2B in chromatin is coupled to ongoing transcription. Biochemistry. 1990; 29:4752-4757.
113. Davie JR, Murphy LC. Inhibition of transcription selectively reduces the level of ubiquitinated histone H2B in chromatin. Biochem Biophys Res Commun. 1994; 203:344-350.
114. Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011; 7:113-119.
115. Johnsen SA. The enigmatic role of H2Bub1 in cancer. FEBS Lett. 2012; 586:1592-1601.
116. Hahn MA, Dickson KA, Jackson S, Clarkson A, Gill AJ, Marsh DJ. The tumor suppressor CDC73 interacts with the ring finger proteins RNF20 and RNF40 and is required for the maintenance of histone 2B monoubiquitination. Hum Mol Genet. 2012; 21:559-568.
117. Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, Hintermair C, Eick D, Kremmer E, Simons M, Beissbarth T, Johnsen SA. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011; 71:5739-5753.
118. Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S, Suzuki E, Le Guezennec X, Stunnenberg HG, Krasnov A, Georgieva SG, Schule R, Takeyama K, et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell. 2008; 29:92-101.
119. Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL, McMahon SB. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008; 29:102-111.
120. Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, Pang D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol. 2011; 137:1245-1253.
121. Liu YL, Yang YM, Xu H, Dong XS. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J Gastroenterol Hepatol. 2010; 25:1800-1805.
122. Bettermann K, Benesch M, Weis S, Haybaeck J. SUMOylation in carcinogenesis. Cancer Lett. 2012; 316:113-125.
123. Bayer P, Arndt A, Metzger S, Mahajan R, Melchior F, Jaenicke R, Becker J. Structure determination of the small ubiquitin-related modifier SUMO-1. J Mol Biol. 1998; 280:275-286.
124. Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002; 108:345-356.
125. Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997; 88:97-107.
126. Mahajan R, Gerace L, Melchior F. Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. J Cell Biol. 1998; 140(2):259-270.
127. Gill G. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev. 2003; 13:108-113.
128. Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004; 18:2046-2059.
129. Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009; 284:8223-8227.
130. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007; 8:947-956.
131. Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010; 11:861-871.
132. Lomeli H, Vazquez M. Emerging roles of the SUMO pathway in development. Cell Mol Life Sci. 2011; 68:4045-4064.
133. Brandl A, Heinzel T, Kramer OH. Histone deacetylases: salesmen and customers in the post-translational modification market. Biol Cell. 2009; 101:193-205.
134. Ouyang J, Gill G. SUMO engages multiple corepressors to regulate chromatin structure and transcription. Epigenetics. 2009; 4:440-444.
135. Brandl A, Wagner T, Uhlig KM, Knauer SK, Stauber RH, Melchior F, Schneider G, Heinzel T, Kramer OH. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J Mol Cell Biol. 2012; 4:284-293.
136. Citro S, Jaffray E, Hay RT, Seiser C, Chiocca S. A role for paralog-specific sumoylation in histone deacetylase 1 stability. J Mol Cell Biol. 2013; 5:416-427.
137. Riising EM, Boggio R, Chiocca S, Helin K, Pasini D. The polycomb repressive complex 2 is a potential target of SUMO modifications. PLoS One. 2008; 3:e2704.
138. Fong JJ, Nguyen BL, Bridger R, Medrano EE, Wells L, Pan S, Sifers RN. beta-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J Biol Chem. 2012; 287:12195-12203.
139. Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011; 480:557-560.
140. Sakabe K, Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J Biol Chem. 2010; 285(45):34460-34468. 10.1074/jbc.M110.158170
141. Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A. 2010; 107:19915-19920.
142. Zhang S, Roche K, Nasheuer HP, Lowndes NF. Modification of histones by sugar beta-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem. 2011; 286:37483-37495.
143. Forma E, Jozwiak P, Brys M, Krzeslak A. The potential role of O-GlcNAc modification in cancer epigenetics. Cell Mol Biol Lett. 2014; 19:438-460.
144. Singh JP, Zhang K, Wu J, Yang X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 2015; 356:244-250.
145. Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992; 185:1155-1161.
146. Kumar S, Yoshida Y, Noda M. Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem Biophys Res Commun. 1993; 195:393-399.
147. Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997; 272:28557-28562.
148. Hori T, Osaka F, Chiba T, Miyamoto C, Okabayashi K, Shimbara N, Kato S, Tanaka K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999; 18:6829-6834.
149. Liakopoulos D, Busgen T, Brychzy A, Jentsch S, Pause A. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc Natl Acad Sci U S A. 1999; 96:5510-5515.
150. Duncan K, Schafer G, Vava A, Parker MI, Zerbini LF. Targeting neddylation in cancer therapy. Future Oncol. 2012; 8:1461-1470.
151. Leung SK, Ohh M. Playing Tag with HIF: The VHL Story. J Biomed Biotechnol. 2002; 2:131-135.
152. Ohh M, Kaelin WG Jr. VHL and kidney cancer. Methods Mol Biol. 2003; 222:167-183.
153. Salon C, Brambilla E, Brambilla C, Lantuejoul S, Gazzeri S, Eymin B. Altered pattern of Cul-1 protein expression and neddylation in human lung tumours: relationships with CAND1 and cyclin E protein levels. J Pathol. 2007; 213:303-310.
154. Kim AY, Bommelje CC, Lee BE, Yonekawa Y, Choi L, Morris LG, Huang G, Kaufman A, Ryan RJ, Hao B, Ramanathan Y, Singh B. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J Biol Chem. 2008; 283:33211-33220.
155. Sarkaria I, Oc P, Talbot SG, Reddy PG, Ngai I, Maghami E, Patel KN, Lee B, Yonekawa Y, Dudas M, Kaufman A, Ryan R, Ghossein R, et al. Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 2006; 66:9437-9444.
156. Embade N, Fernandez-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutierrez de Juan V, Woodhoo A, Martinez-Lopez N, Rodriguez-Iruretagoyena B, Bustamante FJ, de la Hoz AB, Carracedo A, Xirodimas DP, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology. 2012; 55:1237-1248.
157. Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem. 2007; 282:1797-1804.
158. Noguchi K, Okumura F, Takahashi N, Kataoka A, Kamiyama T, Todo S, Hatakeyama S. TRIM40 promotes neddylation of IKKgamma and is downregulated in gastrointestinal cancers. Carcinogenesis. 2011; 32:995-1004.
159. Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004; 118:83-97.