
INTRODUCTION
Familial myelodysplastic syndrome (MDS) and aplastic anemia (AA) are rarely reported in adults and then usually in the context of early-onset diseases. In MDS for example, germline (GL) RUNX1 mutations are associated with thrombocytopenia and subsequent clonal evolution [1, 2]. Similar findings have also been observed for GL CEBPA and GATA2 [3–7]. A patient’s family history may indicate inherited predisposition, but late onset, or incomplete penetrance often obscure assessment of hereditary factors and thus, aging or environmental exposures are implicated more often than genetic factors in the pathogenesis of sporadic MDS.
Fanconi Anemia (FA) is a prototypic inherited autosomal recessive bone marrow failure (BMF) syndrome with a highly variable penetrance [8]. Most of the mutations in FA gene pathway are found within three genes, FANCA, FANCC and FANCG [8]. FA cells exhibit hypersensitivity to DNA cross-linking agents, a laboratory marker of chromosomal instability [9] and FA patients frequently develop AA and MDS. While heterozygous carriers have been presumed to be asymptomatic in FA, various GL FA gene mutations have been suggested to increase susceptibility to breast cancer [10–13]. Dyskerin mutations are associated in autosomal dominant fashion with another hereditary BMF syndrome, dyskeratosis congenita (DC) [14], but mutations of other members of the telomerase complex, chiefly TERT and TERC, have been implicated in predisposition to both AA and MDS [15–18].
Based on the close association between AA and MDS as manifestations of congenital BMF syndromes, we hypothesized that both AA and heterozygous FA gene mutations as well as low penetrance telomerase gene alterations may represent risk factors for otherwise typical, sporadic adult MDS and AA. For that purpose we have systematically screened the open reading frames of FA and telomerase complex genes to identify and map both novel and established FA sequence alterations.
RESULTS
Identification of germline variants in Fanconi Anemia and telomerase complex genes
All coding frames of 14 FA and 5 telomerase genes (Supplementary Table 1) were sequenced in a cohort of patients with BMF (N = 489), including MDS/sAML (N = 305), Myeloproliferative neoplasms (N = 33), and AA/paroxysmal nocturnal hemoglobinuria (PNH) (N = 151; Table 1). For the purpose of this analysis, the candidate alterations were sub-classified to prioritize subsequent analytic steps (Supplementary Figure 1); tier-1 lesions included known disease-prone sequence alterations/mutations (OMIM [19]/Rockefeller Fanconi mutation database [20]), new nonsense or frameshift mutations, and exceedingly rare, individual, missense mutations (<0.01%, no reported homozygotes form in the control cohort), all of which are predicted as deleterious by >4/6 applied scoring algorithms (PolyPhen2 [21], PhyloP [22], SIFT [23], LRT [24], MutationAssesor and MutationTaster [25] within Annovar [26]). All tier-1 alterations were screened to exclude somatic lesions and technical artifacts. AML Tier-1 variants’ germline status was confirmed using CD3 derived DNA, where specimen was available. Tier-2 lesions were defined as variants with a high general population frequency of >0.01% and were not studied further. Instead, our subsequent analysis focused on tier-1 lesions. In total, we found 36 tier-1 gene mutations including 2 recurrent frameshift FANCG mutations. We also found 2 splicing variants (RAD51C and FANCA) and one stop gain variant (FANCC) (Figure 1, Supplementary Table 2). Only two variants among 36 reported were related to telomerase pathway and both were located on TERT gene. Among the genes tested, the most frequent SNVs were found in FANCA (N = 10) and BRCA2 (N = 6). Overall, carriers of heterozygous FANCA alterations showed an increased risk of developing MDS (OR 4.9, P < 0.001), patients with FANCG showed an increased risk of developing MDS (OR 5.9, P < 0.05) and patients carrying FANCE variants tended to show a stronger risk for development of MDS although not statistically significant (OR 4.9, P < 0.10). RAD51C was the only gene which was nominally associated with development of AA (OR 6.9, P < 0.05). When averaged rates of total number of FA gene variants were compared to a healthy cohort of samples from ExAC database, we could find statistically significant differences for selected genes only, with varying frequencies among different disease subgroups. FANCA, FANCE and FANCG gene were present at higher rates among MDS subcohort (Table 2). Overall there was a higher rate of FA variants among disease subgroups, when compared with healthy population, but when looked closely it was driven only by a handful of genes within that pathway (Table 2).
Table 1: Clinical characteristics of patients participating in the study
Patient characteristics |
% (n/range) |
|---|---|
Median age (years, median, range) |
65 (9–83) |
Sex (male) |
56% (254) |
Type of disease |
|
MDS |
48% (233) |
MDS low risk |
27% (130) |
MDS high risk |
14% (70) |
MDS/MPN |
7% (33) |
sAML |
15% (72) |
AA+AA/PNH+PNH |
31% (151) |
total |
489 |
Karyotype (MDS and AML only) |
|
Normal |
33% (114) |
del(5q),-5 |
10% (33) |
del(7q),-7 |
10% (33) |
-Y |
2% (8) |
del(20q),-20 |
6% (21) |
trisomy 8 |
4% (14) |
Complex (≥3) |
17% (57) |
Others |
21% (73) |
TOTAL |
344 |
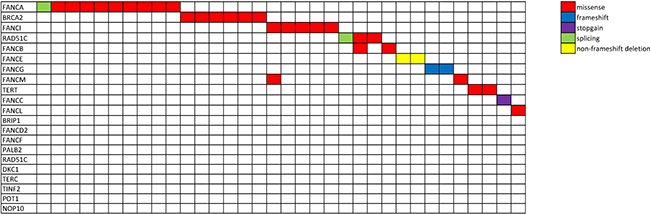
Figure 1: Mutational spectrum of patients harboring Fanconi Anemia and telomerase germline variants. Schematic representation of the cohort of patients carrying germline variants in Fanconi Anemia and telomerase complex genes. Each row represents a gene and each column represents an individual patient (N = 34). Two patients harbored two variants each. Each color encodes a different type of mutation (red, missense; blue, frameshift; purple, stopgain; green, splicing variant; yellow, non-frameshift deletion).
Table 2: Variant distribution among different patient population, compared with general, healthy population (ExAC)
Gene |
AA |
OR_AA |
Pval_AA |
MDS |
OR_MDS |
Pval_MDS |
AML |
OR_AML |
Pval_AML |
ExAC |
|---|---|---|---|---|---|---|---|---|---|---|
BRCA2 |
1/151 |
0.72 |
0.59683 |
3/233 |
1.41 |
0.47748 |
2/72 |
3.09 |
0.14243 |
306/33370 |
BRIP1 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
0.63722 |
0/72 |
N/A |
1.00000 |
174/33370 |
FANCA |
1/151 |
0.82 |
0.65464 |
9/233 |
4.93 |
0.00016 |
0/72 |
N/A |
1.00000 |
270/33370 |
FANCB |
2/151 |
2.45 |
0.20129 |
0/233 |
N/A |
0.64111 |
0/72 |
N/A |
1.00000 |
182/33370 |
FANCC |
0/151 |
N/A |
1.00000 |
1/233 |
2.21 |
0.36851 |
0/72 |
N/A |
1.00000 |
65/33370 |
FANCD2 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
0.63479 |
0/72 |
N/A |
1.00000 |
168/33370 |
FANCE |
0/151 |
N/A |
1.00000 |
2/233 |
5.00 |
0.06520 |
0/72 |
N/A |
1.00000 |
58/33370 |
FANCF |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
21/33370 |
FANCG |
0/151 |
N/A |
1.00000 |
2/233 |
5.89 |
0.04889 |
0/72 |
N/A |
1.00000 |
49/33370 |
FANCI |
1/151 |
0.90 |
0.69582 |
2/233 |
1.17 |
0.69043 |
1/72 |
1.90 |
0.41266 |
245/33370 |
FANCL |
0/151 |
N/A |
1.00000 |
1/233 |
1.79 |
0.43123 |
0/72 |
N/A |
1.00000 |
80/33370 |
FANCM |
2/151 |
2.04 |
0.26091 |
0/233 |
N/A |
0.41155 |
0/72 |
N/A |
1.00000 |
218/33370 |
PALB2 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
113/33370 |
RAD51C |
2/151 |
7.00 |
0.03583 |
1/233 |
2.24 |
0.36410 |
0/72 |
N/A |
1.00000 |
64/33370 |
FA_combined |
9/151 |
0.99 |
0.57945 |
21/233 |
1.46 |
0.07000 |
3/72 |
0.64 |
0.80130 |
2002/33370 |
DKC1 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
31/33370 |
NOP10 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
14/33370 |
POT1 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
59/33370 |
TERT |
0/151 |
N/A |
1.00000 |
1/233 |
2.52 |
0.33230 |
1/72 |
8.23 |
0.11760 |
57/33370 |
TERC |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
0/33370 |
TINF2 |
0/151 |
N/A |
1.00000 |
0/233 |
N/A |
1.00000 |
0/72 |
N/A |
1.00000 |
27/33370 |
TELO_combined |
0/151 |
N/A |
0.27713 |
1/233 |
0.38 |
0.52778 |
1/72 |
1.24 |
0.55638 |
374/33370 |
OR, Odds ratio; AA, aplastic anemia; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia, Fisher’s exact test used to calculate p-values.
When all coding frames of telomerase machinery genes (Supplementary Table 1) were analyzed, none of the 5 different variants in TERT previously reported to be pathogenic in hereditary AA [16] were found in our MDS and AA cohorts. However, combined, new 2 tier-1 TERT variants showed higher frequency in AML (P > 0.1; OR = 11), but without statistical significance, partially because of small sample size. Variants previously reported to be associated with AA (Ala1062Thr and His412Tyr) were either not present or not increased in their frequency in AA as compared to controls, a finding in contrast with previous reports [16–18] (Supplementary Table 3).
Characterization of clinical phenotypes and molecular spectrum of carriers of germline variants in Fanconi Anemia and telomerase complex genes
Subsequent analysis focused on clinical features of carriers of FA and telomerase variants. The presence of heterozygous GL mutations in FA was associated with a higher frequency of somatic deletion of chr5 (3.6 OR, P < .0.039) and deletion of chr7 (OR 2.3, P = 0.08). We also observed lower rates of normal cytogenetics, higher prevalence of complex karyotype and trisomy 8, but due to small sample size none of them reached statistical significance. FA carrier status among MDS/AML patients showed no impact on survival (Supplementary Figure 2), neither association with age or clinical laboratory parameters (Table 3). We also tested the association of individual GL variants with the occurrence of specific somatic mutations, but no specific association was observed (list of somatic mutations in Supplementary Figures 3 and 4). However, when analyzed as a group, FA variant carriers were associated with the absence of SF3B1 mutations and the presence of PRPF8 mutations (Supplementary Figure 3). Overall, frequencies of specific types of somatic mutations (frameshift vs. missense/nonsense) or the total number of observed mutations were not statistically different between FA carrier and non-carrier groups. Neither family history of leukemia nor higher rates of other cancers were found.
Table 3: Clinical characteristics of MDS patients with respect to the presence of FA germline variants
MDS WT FA (N = 188) |
MDS Mutant FA (N = 16) |
P value |
|
|---|---|---|---|
Age (median) |
65 |
65 |
1.000 |
Sex (Male, %) |
54% (102) |
56% (9) |
0.757 |
Cytogenetics |
|||
Normal |
37% (69) |
18% (3) |
0.180 |
del(5q),-5 |
7% (14) |
25% (4) |
0.039 |
del(7q),-7 |
11% (19) |
25% (4) |
0.087 |
-Y |
2% (3) |
0% (0) |
1.000 |
del(20q),-20 |
8% (15) |
6% (1) |
0.385 |
+8 |
4% (13) |
12% (2) |
0.501 |
Complex(≥3) |
18% (35) |
26% (4) |
0.491 |
others |
14% (27) |
32% (5) |
0.066 |
WBC (1,000/mm3) |
3.9 |
3.7 |
1.000 |
ANC (1,000/mm3) |
1.9 |
1.7 |
1.000 |
BM blast (%) |
4.6 |
5.0 |
1.000 |
Plt (1,000/mm3) |
115 |
124 |
0.720 |
OS (months) |
21 |
18 |
0.070 |
Somatic mutations |
1.4 |
1.6 |
0.828 |
Significant associations were put in bold.
DISCUSSION
To date, few hereditary factors have been described for seemingly sporadic adult MDS and other BMF syndromes. We hypothesized that GL alterations may be genetic predisposition traits that in a heterozygous configuration are associated with long disease anticipation. Classical FA is an autosomal recessive disease with various physical sequelae and presumed asymptomatic heterozygous carriers. Most reports do not associate the presence of heterozygous FA variants to the increased risk of cancer or development of BMF such as AA or MDS [27–30]. Telomerase gene mutations, previously reported in AA, appear to be autosomal dominant with variable penetrance.
Our study provides the first evidence that heterozygous FA gene carriers may have delayed onset and increased risk for the development of MDS and AML, implying that the normal alleles may not be entirely protective. The observed alterations included 1 recurrent nonsense heterozygous mutation (FANCC), 2 recurrent frameshift, heterozygous mutations (FANCG) and 1 splicing variant (FANCA). Interestingly, FA alterations were associated with both MDS and AML, consistent with the pathophysiologic overlap between MDS and AML. The functionally detrimental impact of tier-1 heterozygous alterations has been predicted using consistent results from an array of 6 prediction tools as well as public Fanconi Anemia databases and other resources. The molecular impact of some of the identified variants needs to be tested, since only one variant (e.g., FANCC p.R185X) was previously found in patients with FA (Rockefeller Fanconi mutation database [20]).
Consistent with our results, in another publication, relatives of FA carriers showed an increased rate of breaks per cell (using standard DEB and MMC testing), although the frequency of aberrant cells was lower [31] suggesting that the penetrance of the damaged allele is low, thus explaining the long latency needed to accumulate critical numbers of aberrant cells. Another interesting observation of our cohort is the enrichment of del5 and del7 alterations among FA carriers identified consistent with the high frequency of -7/del7q found among typical FA patients who develop MDS/AML.
Similar to DC associated with BMF telomeres shortening [32–34], telomerase complex genes have previously been implicated in some forms of idiopathic AA [16–18]. Previously, telomerase variant frequencies in AA were estimated at 1.5% [35] to 4.0% [16]. However, in our cohort, we identified roughly identical rates of tier-1 alterations in MDS, AML, and AA when compared to large control populations currently available in public databases (http://exac.broadinstitute.org/; date accessed [July 2016]). For example, His412Thr (rs34094720) was previously reported to be present [16] in 1% of AA patients and 0% of controls. Yet with today’s access to vast genetic databases we know that this variant is present in nearly ~2% of healthy, non-Finnish Europeans. Usage of an extended set of controls, genotyped in unbiased fashion, enabled finer disease-risk calculation and thus some of the former GWAS approaches may require careful reconsideration. While some of the variants described in AA (TERT His412Thr and Ala202Thr) may have shown a negative impact on telomere maintenance using in vitro assays, given the comparable frequencies of these variants in controls, previous studies might have captured disease-risk independent factors that could attribute to disease course/severity or play a risk factor role in the hematopoietic cell transplantation [36].
In sum, our study of the investigation of FA and telomerase gene alterations suggests that heterozygous carriers of certain FA genes may have increased risk of myeloid neoplasia but not AA and thus conversely, some of the seemingly sporadic cases of MDS may have FA-related genetic background.
MATERIALS AND METHODS
Patients
Tumor DNA was obtained from patient’s bone marrow and/or peripheral blood. Written informed consent for sample collection was obtained according to protocols approved by the Institutional Review Board of the Cleveland Clinic and in accordance with the Declaration of Helsinki. Disease diagnoses were assigned according to the World Health Organization (WHO) classification criteria. The clinical characteristics of patients investigated in this study are presented in Table 1.
Population frequencies for detected variants, functional annotation and variant prioritization
We used Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) [June, 2015]. All the variants from tested genes were extracted for further functional annotation and processing. Sequencing results were annotated using ANNOVAR, http://annovar.openbioinfromatics.org. Mutations that were predicted to be damaging/probably damaging by 4/5 algorithms (PhyloP, SIFT, PolyPhen2, LRT and MutationTaster) were prioritized for further analysis (tier1). This functional pre-processing analysis was also carried out on control population variants that were extracted from ExAC database. Ethnically matched population and the variants extracted that we analyzed were derived from ExAC database consisting 33370 normal, White/Caucassian individuals. For the purpose of this analysis, the candidate alterations were sub-classified to prioritize known disease-prone sequence alterations/mutations (OMIM/Rockefeller Fanconi mutation database)
Whole exome sequencing
Whole exome capture using SureSelect Human All Exon 50 Mb kit was used for targeted, exome capture according to the manufacture’s protocol (SureSelect®, Agilent Technology, Santa Clara, CA), according to the manufacturer’s protocols. The captured targets were subjected to massive sequencing using Illumina HiSeq 2000 (Illumina, INC., San Diego, CA). Generation of .bam files with its preprocessing and detection of somatic point mutations/insertions and deletions was according with GATK best practices.
Targeted sequencing
Targeted sequencing was completed using TruSeq Enrichment library (Illumina). Two panels were created, one for somatic mutations and one for GL variants. Somatic variants were reported previously [37]. List of genes targeted can be found in Supplementary Table 1. The enriched targets were subjected to massive sequencing using Illumina Miseq sequencer, with average depth of 500×. Only variants with minimum depth at least 20 and 8 positive, high quality reads were considered for further analysis. Alterations with Variant Allelic Frequency below 35% were removed being considered somatic variants.
Statistical analysis
For comparison between groups, with and without FA mutations, statistical analysis was performed using standard, unpaired, two sample t-test (for continuous, normally distributed variables) and Fisher’s Exact test (for discrete data). Reported p-values here are not adjusted for multiple comparisons. Odds ratio were calculated as follows: number of patients with damaging variant multiplied by a number of normal individuals without damaging variants and divided by number of normal individuals with damaging variant multiplied by number of patients without damaging variants.
Abbreviations
Chr: chromosome; RUNX1: Runt Related Transcription Factor 1; CEBPA: CCAAT/Enhancer Binding Protein Alpha; GATA2: GATA Binding Protein 2; FANC: Fanconi Anemia Complementation Group; TERT: Telomerase Reverse Transcriptase; TERC: Telomerase RNA Component; BRCA2: BRCA2: DNA Repair Associated.
Author contributions
B.P designed the research, analyzed the data and wrote the paper, H.M., M.A.S., SKB. assisted with the interpretation of the results and edited the manuscript; S.T., B.J.P, M.C. collected clinical data; C.H., B.D. performed experiments; J.P.M conceived the study, designed the experiments, and wrote the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by generous funds from EPE1509JM – Edward P. Evans Foundation and R01HL123904/R01HL118281/R01HL128425 - NHLBI.
REFERENCES
1. Preudhomme C, Renneville A, Bourdon V, Philippe N, Roche-Lestienne C, Boissel N, Dhedin N, André JM, Cornillet-Lefebvre P, Baruchel A, Mozziconacci MJ, Sobol H. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009; 113:5583–7. https://doi.org/10.1182/blood-2008-07-168260.
2. Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, Shigesada K, Ito Y, Benson KF, Raskind WH, Bossier C, Antonarakis SE, Israels S, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: Implications for mechanisms of pathogenesis. Blood. 2002; 99:1364–72. https://doi.org/10.1182/blood.V99.4.1364.
3. Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, Weissmann S, Dicker F, Kohlmann A, Schindela S, Kern W, Haferlach T, Schnittger S. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2014; 28:794–803. https://doi.org/10.1038/leu.2013.273.
4. Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CAJ, Wouters BJ, van der Poel-van de Luytgaarde SC, Damm F, Krauter J, Ganser A, Schlenk RF, Löwenberg B, Delwel R, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2010; 117:2469–75. Available from http://www.bloodjournal.org/content/117/8/2469.abstract.
5. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, Frucht DM, Vinh DC, Auth RD, Freeman AF, Olivier KN, Uzel G, Zerbe CS, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011; 118:2653–5. https://doi.org/10.1182/blood-2011-05-356352.
6. Hyde RK, Liu PP. GATA2 mutations lead to MDS and AML. Nat Genet. 2011; 43:926–7. https://doi.org/10.1038/ng.949.
7. Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, Murday VA, Hodgson S, Keenan R, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011; 43:929–31. https://doi.org/10.1038/ng.923.
8. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009; 668:4–10. https://doi.org/10.1016/j.mrfmmm.2009.01.013.
9. Kupfer GM. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J Biol Med. 2013; 86:491–7. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3848103&tool=pmcentrez&rendertype=abstract.
10. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010; 362:1909–19. https://doi.org/10.1056/NEJMra0809889.
11. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, et al. Breast-Cancer Risk in Families with Mutations in PALB2. N Engl J Med. 2014; 371:497–506. https://doi.org/10.1056/NEJMoa1400382.
12. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006; 38:1239–41. https://doi.org/10.1038/ng1902.
13. Offit K, Levran O, Mullaney B, Mah K, Nafa K, Batish SD, Diotti R, Schneider H, Deffenbaugh A, Scholl T, Proud VK, Robson M, Norton L, et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst. 2003; 95:1548–51. https://doi.org/10.1093/jnci/djg072.
14. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998; 19:32–8. https://doi.org/10.1038/ng0598-32.
15. Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, Young NS. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003; 102:916–8. https://doi.org/10.1182/blood-2003-01-0335.
16. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005; 352:1413–24. https://doi.org/10.1056/NEJMoa042980.
17. Calado RT, Regal JA, Hills M, Yewdell WT, Dalmazzo LF, Zago MA, Lansdorp PM, Hogge D, Chanock SJ, Estey EH, Falcão RP, Young NS. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci U S A. 2009; 106:1187–92. https://doi.org/10.1073/pnas.0807057106.
18. Calado RT, Cooper JN, Padilla-Nash HM, Sloand EM, Wu CO, Scheinberg P, Ried T, Young NS. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia. 2012; 26:700–7. https://doi.org/10.1038/leu.2011.272.
19. Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005; 33:D514–7. https://doi.org/10.1093/nar/gki033.
20. Cotton RGH, Auerbach AD, Beckmann JS, Blumenfeld OO, Brookes AJ, Brown AF, Carrera P, Cox DW, Gottlieb B, Greenblatt MS, Hilbert P, Lehvaslaiho H, Liang P, et al. Recommendations for locus-specific databases and their curation. Hum Mutat. 2008; 29:2–5. https://doi.org/10.1002/humu.20650.
21. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Current Protocols in Human Genetics. 2013; 7.20.1–7.20.41. https://doi.org/10.1002/0471142905.hg0720s76.
22. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005; 15:901–13. https://doi.org/10.1101/gr.3577405.
23. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073–81. https://doi.org/10.1038/nprot.2009.86.
24. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009; 19:1553–61. https://doi.org/10.1101/gr.092619.109.
25. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010; 7:575–6. https://doi.org/10.1038/nmeth0810-575.
26. Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:1–7. https://doi.org/10.1093/nar/gkq603.
27. Tischkowitz M, Easton DF, Ball J, Hodgson SV, Mathew CG. Cancer incidence in relatives of British Fanconi Anaemia patients. BMC Cancer. 2008; 8:257. https://doi.org/10.1186/1471-2407-8-257.
28. Seal S, Barfoot R, Jayatilake H, Smith P, Renwick A, Bascombe L, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N, Breast Cancer Susceptibility Collaboration. Evaluation of Fanconi Anemia genes in familial breast cancer predisposition. Cancer Res. 2003; 63:8596–9. Available from http://www.ncbi.nlm.nih.gov/pubmed/14695169.
29. Berwick M, Satagopan JM, Ben-Porat L, Carlson A, Mah K, Henry R, Diotti R, Milton K, Pujara K, Landers T, Batish SD, Morales J, Schindler D, et al. Genetic heterogeneity among fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007; 67:9591–6. https://doi.org/10.1158/0008-5472.CAN-07-1501.
30. Potter NU, Sarmousakis C, Li FP. Cancer in relatives of patients with aplastic anemia. Cancer Genet Cytogenet. 2015; 9:61–5. https://doi.org/10.1016/0165-4608(83)90025-0.
31. Fargo JH, Rochowski A, Giri N, Savage SA, Olson SB, Alter BP. Comparison of Chromosome Breakage in Non-Mosaic and Mosaic Patients with Fanconi Anemia, Relatives, and Patients with Other Inherited Bone Marrow Failure Syndromes. Cytogenet Genome Res. 2014; 144:15–27. Available from http://www.karger.com/DOI/10.1159/000366251.
32. Vulliamy TJ, Knight SW, Mason PJ, Dokal I. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol Dis. 2001; 27:353–7. https://doi.org/10.1006/bcmd.2001.0389.
33. Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, Dokal I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A. 2008; 105:8073–8. https://doi.org/10.1073/pnas.0800042105.
34. Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999; 402:551–5. https://doi.org/10.1038/990141.
35. Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, Chanock SJ, Boyer TD, Young NS. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology. 2011; 53:1600–7. https://doi.org/10.1002/hep.24173.
36. Gadalla SM, Wang T, Haagenson M, Spellman SR, Lee SJ, Williams KM, Wong JY, De Vivo I, Savage SA. Association Between Donor Leukocyte Telomere Length and Survival After Unrelated Allogeneic Hematopoietic Cell Transplantation for Severe Aplastic Anemia. JAMA. 2015; 313:594. https://doi.org/10.1001/jama.2015.7.
37. Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, Przychodzen B, Nagata Y, Meggendorfer M, Sanada M, Okuno Y, Hirsch C, Kuzmanovic T, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017; 49:204–12. https://doi.org/10.1038/ng.3742.