
INTRODUCTION
Prostate cancer (PCa) is one of the leading causes of cancer-related death for men in North America and Europe [1]. Prostate-specific antigen (PSA) analysis, biopsy, as well as the Gleason score, are diagnostic tools that have improved the diagnosis and management of PCa [2]. Among treated PCa patients, pathological parameters can predict the outcome of patients. For example, serum PSA, biopsy, and the Gleason score are well-known predictors of biological outcome following primary therapy for PCa. However, outcome prediction has shifted from pathological parameters to biological molecules. Molecular biomarkers, such as the expression of specific protein-coding and non-coding genes have now greatly improved the accuracy of outcome prediction for patients after treatment [3].
Specific functions are correlated or predictive of pathological parameters that are characteristic of tumor progression. For example, adhesion-related genes are correlated with Gleason score. Specifically, in human prostate adenocarcinomas, the down-regulation of the adhesion molecule CD44 standard (CD44s) [4] and E-cadherin (CDH1) [5, 6] was reported to be associated with metastasis and high Gleason score. Cell cycle genes are also biomarkers that can predict the risk of clinicopathological outcomes [7] such as biochemical recurrence rate after prostatectomy therapy [8, 9]. More recently, the deregulation of ncRNAs has been associated with cell proliferation and survival of PCa [10]. Therefore, it is of great importance to determine the biological functions that are important for pathological features and to identify the corresponding novel biomarkers relevant for clinicopathological parameters, in particular, non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and lncRNAs.
In this study, we examined gene modules and their corresponding biological functions that are significantly linked to clinicopathological parameters. We found potent prognostic markers, including lncRNAs that were identified based on their association with survival time. We also used WGCNA to look for gene sets with similar biological function based on the TCGA dataset for PCa. Six gene modules were identified, one of which is related to patient survival time. Enrichment analysis revealed the genes in the survival time-related module are significantly associated with the regulation of protein translation. We further identified dysregulated lncRNAs involved in protein translation with prognostic potential in PCa and dissected the roles of lncRNAs and miRNAs via target predictions and co-expression networks.
RESULTS
Identification of gene modules using WGCNA
Extending the survival time is the final goal for patients suffering from PCa. In this follow up analysis of the TCGA PRAD dataset, WGCNA was used to create co-expressed gene networks associated with survival time. Only genes with appreciable expression levels (FPKM>1) in more than half of the PCa patients were subjected to analysis. Power 22 was selected as the soft threshold to identify co-expression gene modules (for details, see the Materials and Methods section). Seven gene-network modules were identified and color-coded. Since the “grey” module is reserved for unassigned genes, we focused on the other six modules instead. As shown in Figure 1, the turquoise, blue and brown modules are the top 3 modules which contained the highest number of genes. The turquoise module contained 532 genes, while the blue and brown had 523 and 305 genes, respectively.
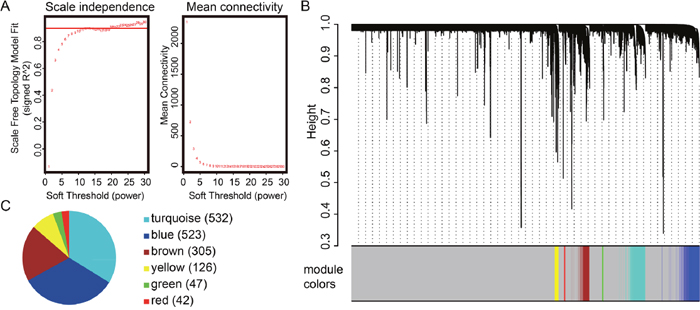
Figure 1: Gene modules detected using the weighted correlation network analysis (WGCNA). (A) Scale-free topology index and mean connectivity were used to determine the soft threshold. (B) Clustering dendrogram of genes. The dissimilarity of genes is based on topological overlap. The genes are assigned to different modules and are identified using different colors. (C) Number of genes in each module identified from WGCNA. The numbers in the bracket represent the number of genes in each module. The modules containing the most number of genes are the turquoise module, blue module and brown module.
Linking modules to pathological parameters
We further evaluated the relationship between these modules and the pathological parameters by calculating the correlation value of the eigengenes of each module (for a detailed definition, see the Materials and Methods section) with the clinical information obtained from the patients. The turquoise module was marginally significantly associated with survival time (p=0.07, Figure 2). The green module was associated with clinical parameters including Gleason score (p=4e-17), most PSA (p=9e-27) and lymph nodes according to haematoxylin and eosin (HE) staining (p=1e-06). Functional enrichment analysis based on KEGG pathways and biological process of Gene Ontology (GO) revealed the genes in the green module are involved in the GO term “cell cycle”. This observation is consistent with a previous report which also found cell cycle genes are correlated with PSA and the Gleason score [7]. The yellow module was also associated with Gleason score and lymph nodes according to HE examination. And as expected, the genes in this module were shown to participate in immune and defense responses. Finally, the brown module was negatively associated with Gleason score and lymph nodes according to HE examination. The genes assigned to this module significantly participate in focal adhesion pathways as well as the biological processes of muscle contraction and cell adhesion, consistent with previous studies on adhesion genes such as CD44 [4], CDH1 [5, 6] and GJA1 [11].
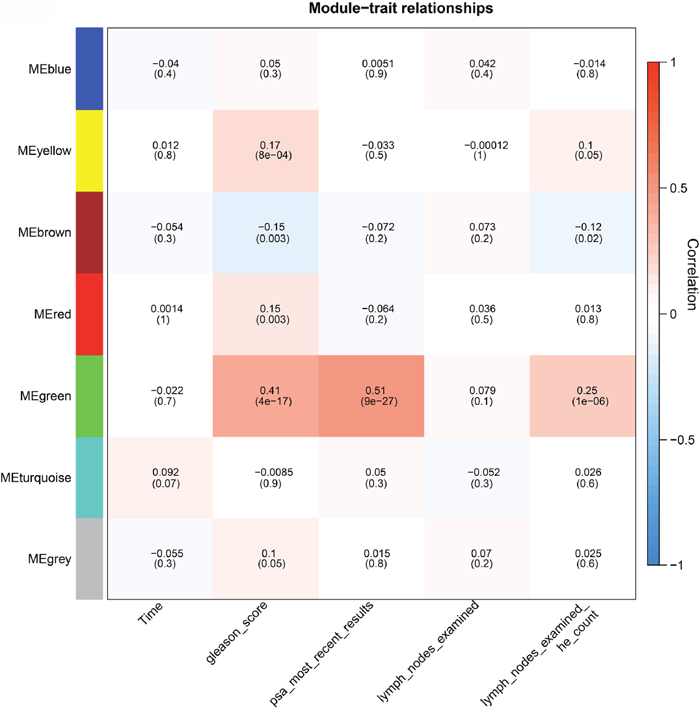
Figure 2: Module-trait associations. Each row corresponds to a module eigengene, and each column corresponds to a pathoclinical parameter. The module eigengene is defined as the first principal component of a given module and considered a representative of the gene expression profiles in a module. Each grid contains the correlation value, calculated based on eigengene expression and clinical traits. The corresponding p-value is the Student asymptotic p-value for the correlation. The grid is color-coded by correlation according to the color bar of the correlation.
The turquoise module is correlated with survival time and associated with RNA-processing and protein translation
In the present study, we used survival time as one of the sample traits. There are two methods that are commonly used to identify prognostic markers. One method is at the gene module level, according to the correlation between the survival time and eigengene for each module. The other method is at the single gene level according to the correlation value (for details, see the Materials and Methods section) of gene expression and survival time.
With the most number of genes, the turquoise module was marginally significantly associated with survival time (p=0.07). Functional enrichment analysis revealed the genes in this module are associated with RNA processing and translation (Supplementary Table 1). As shown in Figure 3A, there are primarily 3 clusters of genes representing different biological functions in the turquoise module. The cluster with the purple background represents a translation-related function, while the clusters with the gold and sea green background are associated with mitochondrial-related processes and RNA processing, respectively. Further functional enrichment analysis revealed the genes in the turquoise module are significantly involved in the ribosome pathway and neurodegenerative diseases (Figure 3B).
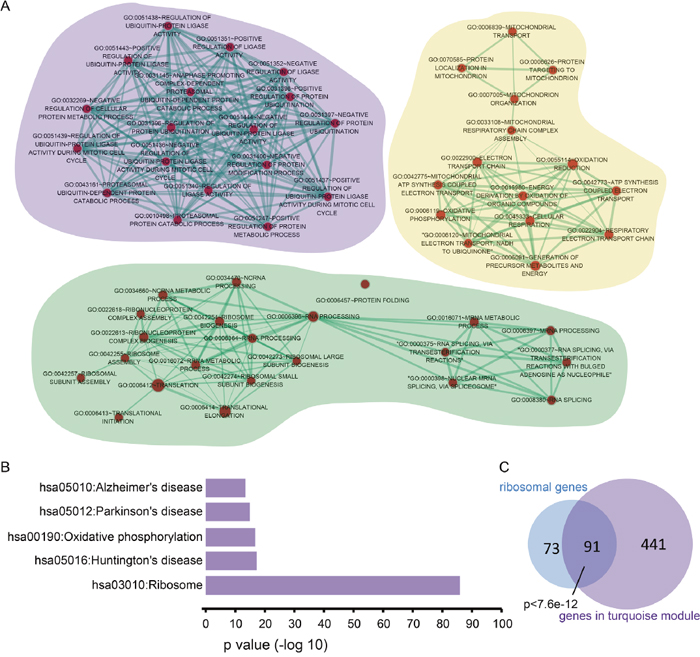
Figure 3: Genes in the turquoise module are involved in translation via ribosomal protein-coding genes. (A) Enrichment analysis was performed for genes in the turquoise module. For the Gene Ontology BP terms, the Cytoscape app, Enrichment Map was used to identify the most correlated terms for genes in the turquoise module. One node represents one biological process. The node size increased with number of genes. The thickness of the edges between two terms is proportional to the similarity coefficient of the associated terms. (B) The enriched KEGG pathway of the genes in the turquoise module. (C) The overlapped genes between ribosomal protein-coding genes and those in the turquoise module. The p-value was calculated using the hypergeometric test.
In addition to the above functional enrichment analyses, we also performed a hypergeometric test on the turquoise module. The hypergeometric test is a widely-used method to identify the function of gene sets based on overlapping genes with known functions [12]. A gene family comprises a set of similar genes with similar biochemical functions. The HUGO gene nomenclature committee (HGNC) contains the members of each gene family. According to HGNC, the ribosomal protein family is comprised of 164 genes encoding for ribosomal proteins, including L ribosomal proteins (RPL), S ribosomal proteins (RPS) and mitochondrial ribosomal proteins (MRPL, MRPS) [13]. As shown in Figure 3C, the genes in the turquoise module significantly overlapped with the HGNC ribosomal protein family (hypergeometric test, p<7.6e-12). Taken together, our results show the turquoise module is correlated with survival time and closely associated to RNA processing and protein translation.
FDZ7 and MEIS1 are good prognostic markers for PCa patient survival time in gleason score-related modules
Gene significance (GS) is a measure to quantify the correlation of individual genes with clinical information [14]. Similarly, for individual genes, module membership (MM) is a measure to evaluate the degree of correlation between the module eigengene and the expression level of a single gene [15]. In this study, survival time was used as the clinical information. At the single gene level, prognostic markers can be identified using the correlation of gene expression and survival time. Genes with high GS and MM are regarded as the most important components of the modules, which are remarkably correlated with survival time. Among the genes in modules which are notably linked to Gleason score, we identified genes associated with high GS and high MM.
Our current findings show there are four modules which are correlated with Gleason score: the yellow, red, green and brown modules (Figure 2). Considering the high soft threshold β =22, we used the cut-off GS>0.1 [15, 16] and MM>0.8 [17] to determine which genes are critical for survival time in these four modules. Only 9 genes (FZD7, PRTFDC1, FAXDC2, MEIS1, ST5, FBXL22, EOGT, and NPR2) in the brown module met the criteria. In the brown module, the co-expression network comprised of 296 nodes and 5698 edges with adjacency>0.02. Among the 9 genes identified, FZD7, FBXL22, and MEIS1 were the top 3 ranked genes based on the number of interacting genes.
Next, we examined the expression pattern of FZD7, FBXL22, and MEIS1 in several PCa cohorts to establish whether any of these genes could be potential biomarkers. FBXL22 is lowly expressed in the TCGA prostate cancer dataset (average FPKM=1.8, SD=1.2) and therefore may not be a good biomarker candidate. FZD7 is a member of the Frizzled receptor family and has been shown to be important in cancer development and progression by activating Wnt pathways [18]. Although FZD7 is up-regulated in multiple tumors, including colorectal cancer and breast cancer [18], we found FZD7 is down-regulated in PCa relative to normal tissue across multiple cohorts (Figure 4A-4D). Similar observations were also found in PCa cell lines (Figure 4E). Moreover, patients with high FZD7 expression have better disease-free survival rates (Figure 4F). MEIS1 is a novel AR co-repressor [19]. Similar to FZD7, we found MEIS1 is down-regulated in both prostate tumors (Figure 4G-4J) and PCa cell lines (Figure 4K), and a high MEIS1 expression is an indication of better overall survival for PCa patients (Figure 4L) [20]. Taken together, our GS and MM analysis have revealed FZD7 and MEIS1 as potentially new prognostic genes for PCa that are associated with good patient outcome.
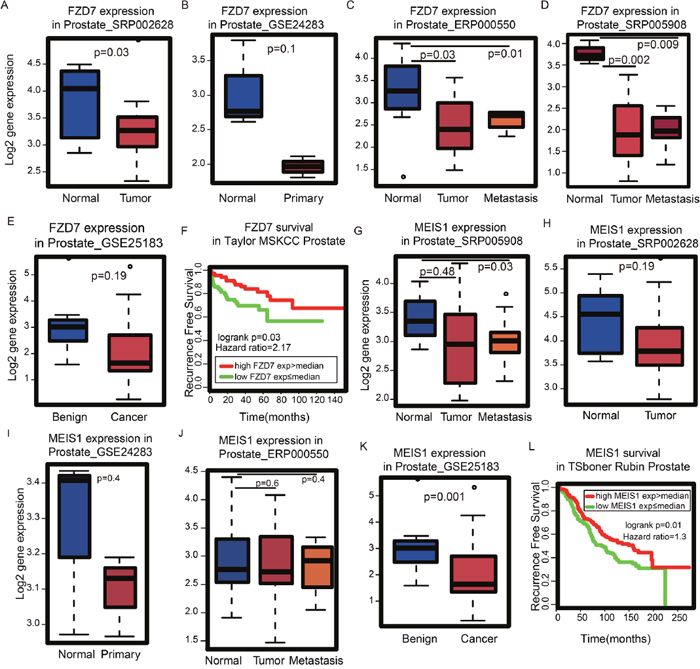
Figure 4: Identification of genes important for survival time in Gleason score-related modules. (A) FZD7 is down-regulated in tumor vs. normal in dataset SRP002628 from publication [PMID: 21571633], (B) GSE24283 from publication [PMID: 21261984], (C) ERP000550 from publication [PMID: 22349460], (D) SRP005908 from publication [PMID: 21036922] and (E) GSE25183 from publication [PMID: 21804560]. (F) According to the Kaplan-Meier plot, patients with high FZD7 expression have better survival probability using Taylor’s MSKCC dataset. (G) MEIS1 is down-regulated in tumor vs. normal in dataset SRP005908 from publication [PMID: 21036922], (H) SRP002628 from publication [PMID: 21571633], (I) GSE24283 from publication [PMID: 21261984], (J) ERP000550 from publication [PMID: 22349460] and (K) GSE25183 from publication [PMID: 21804560]. (L) According to the Kaplan-Meier plot, patients with high expression of MEIS1 expression have better survival probability using Tsboner Rubin’s dataset.
Identification of novel prognostic lncRNAs in PCa
The module that was most significantly associated with survival time was the turquoise module. Recently, a number of lncRNAs have been implicated in PCa biology. For example, PCAT-1, PRNCR1, and MALAT1 were shown to regulate the development and progression of PCa [21–23]. Therefore, we decided to see whether there are any potential prognostic lncRNAs in the turquoise module. Notably, we found four lncRNAs including NCBP2-AS2, LINC00116, GAS5, and ZFAS1. NCBP2-AS2 did not show any expression differences between normal and tumor tissues in PCa (data not shown), however, it has been reported to be up-regulated in lung squamous cell carcinoma compared to lung adenocarcinoma [24]. LINC00116 is up-regulated in PCa relative to normal tissue (data not shown), however, the function of LINC00116 has not been explored yet. For GAS5 and ZFAS1, both lncRNAs are also up-regulated in PCa relative to normal prostate tissues in the four datasets that we examined (Figure 5).
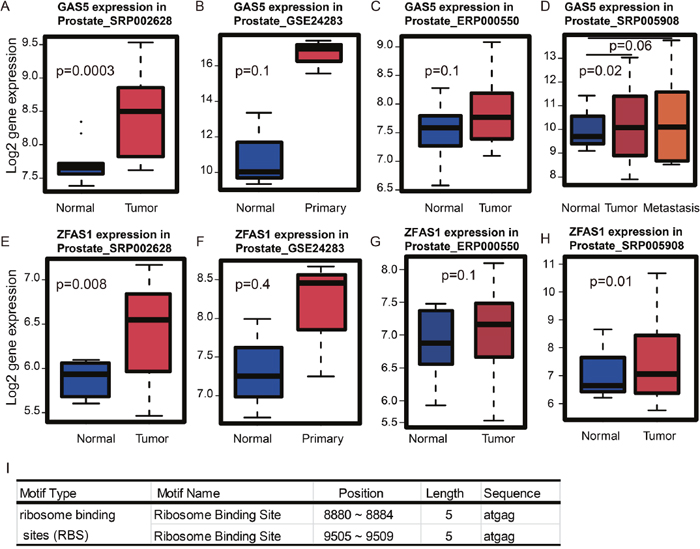
Figure 5: Cross-dataset expression of survival related lncRNAs. (A) GAS5 is up-regulated in tumors vs. normal tissues in dataset SRP002628 from publication [PMID: 21571633], (B) GSE24283 from publication [PMID: 21261984], (C) ERP000550 from publication [PMID: 22349460] and (D) SRP005908 from publication [PMID: 21036922]. (E) ZFAS1 is up-regulated in tumor vs. normal tissues in dataset SRP002628 from publication [PMID: 21571633], (F) GSE24283 from publication [PMID: 21261984], (G) ERP000550 from publication [PMID: 22349460]and (H) SRP005908 from publication [PMID: 21036922]. (I) RegRNA identified the ribosome-binding sites of ZFAS1.
As shown above, the genes in the turquoise module are highly associated with biological functions related to ribosomes. Therefore, we asked whether LINC00116, ZFAS1 or GAS5 could be directly involved in the translation process. To address this, we used the web tool, RegRNA, which looks for ribosome binding sites (RBS) in RNA sequences [25]. As shown in Figure 5I, ZFAS1 but not LINC00116 or GAS5 contains RBS. This finding suggests that ZFAS1 may be directly involved in ribosome-related translation.
Next, we assessed the prognostic potential of LINC00116, GAS5, and ZFAS1. For this, we performed survival analysis with a log-rank test to determine whether patients with high and low expression levels of these lncRNAs have significantly different survival rates. As shown in Figure 6A-6B, the high expression of GAS5 or ZFAS1 is correlated with a worse outcome in PCa. These results (Figure 6A-6B) are consistent with the data obtained from TANRIC (Supplementary Figure 1) [26]. In contrast, LINC00116 appears not to be a good predictor of patient outcome. Thus, we further focused on GAS5 and ZFAS1, which are up-regulated in PCa tissues relative to normal samples (Figure 5 and Figure 6C-6D). Since the turquoise module is associated with RNA-processing and protein translation (Figure 3), we examined the expression correlation between ribosomal genes, GAS5 and ZFAS1. In Pearson’s correlation coefficient (PCC) analysis, both ZFAS1 (0.5
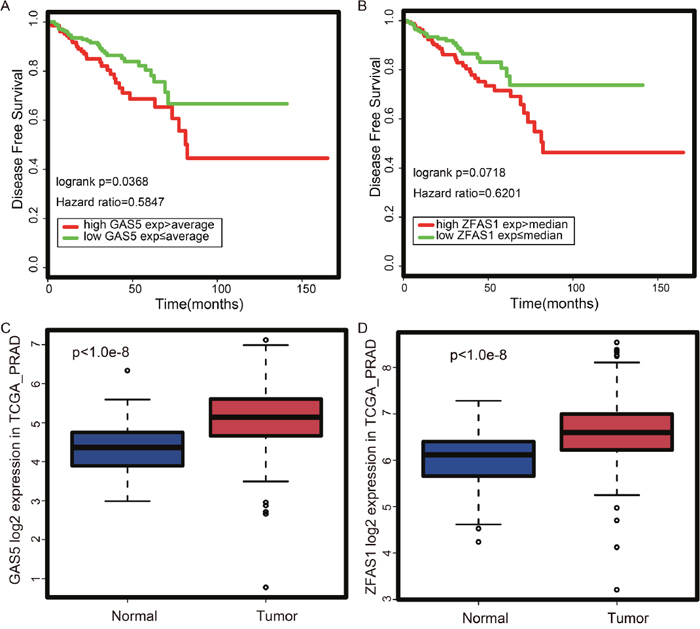
Figure 6: Expression and prognostic potential of GAS5 and ZFAS1 according to TCGA prostate cancer dataset. (A) According to the Kaplan-Meier plot, patients with high GAS5 expression have worse survival probability. (B) According to the Kaplan-Meier plot, patients with high ZFAS1 expression have worse survival probability. (C) GAS5 is up-regulated in tumor vs. normal tissues. (D) ZFAS1 is up-regulated in tumor vs. normal tissues.
The interaction network of miR-940 and lncRNAs in PCa
The reciprocity among miRNAs, lncRNAs, and protein-coding genes constitute an intricate interaction network, which is dysregulated in all types of human cancers [27]. To dissect this complex network, we began by exploring the role of miRNAs in the turquoise module. We found microRNA miR-940 in the turquoise module. MiR-940 is up-regulated in both primary and metastatic PCa patients (Figures 7A-7B). According to DIANA-miRPath [28], the targets of miR-940 are significantly enriched in “prostate cancer” (p=0.045). Moreover, miR-940 has been shown to suppress PCa migration and invasion by regulating the expression of MIEN1 [29]. To determine whether miR-940 could potentially regulate the expression of lncRNAs in the turquoise module, we used LncBase [30] which hosts a database of non-coding RNA targets of microRNA. Surprisingly, miR-940 has been experimentally validated by immunoprecipitation assays to interact with both GAS5 and ZFAS1 [31, 32]. Taken together, we speculate that the GAS5/ZFAS1/miR-940 axis plays key roles in PCa via the protein translation pathway. How these three factors influence the outcome of PCa patients remains elusive.
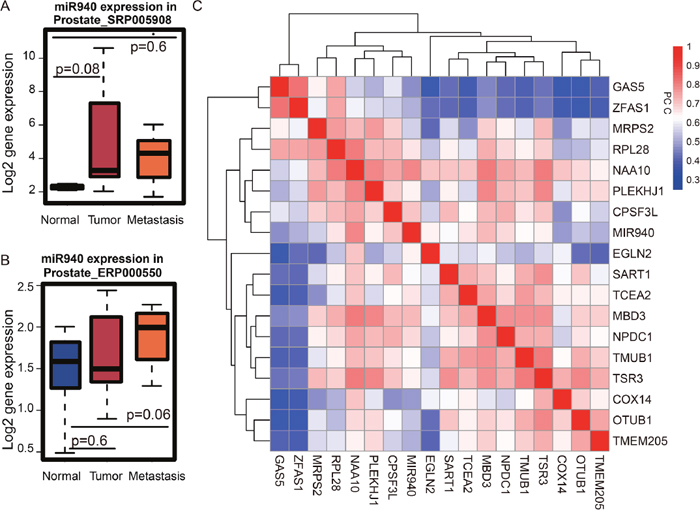
Figure 7: Expression of miRNA and its targets in the turquoise module. (A) MiR-940 is up-regulated in tumor vs. the normal in dataset SRP005908 from publication [PMID: 21261984], (B) ERP000550 from publication [PMID: 22349460]. (C) Based on the gene expression profile from TCGA prostate cancer, the expression similarity of the genes of interest is shown in the two-way clustering heat map. The interested genes include two lncRNAs (GAS5 and ZFAS1), one miRNA (miR-940) and target genes of miR-940 in the turquoise module. GAS5 and ZFAS1 are highly correlated with each other.
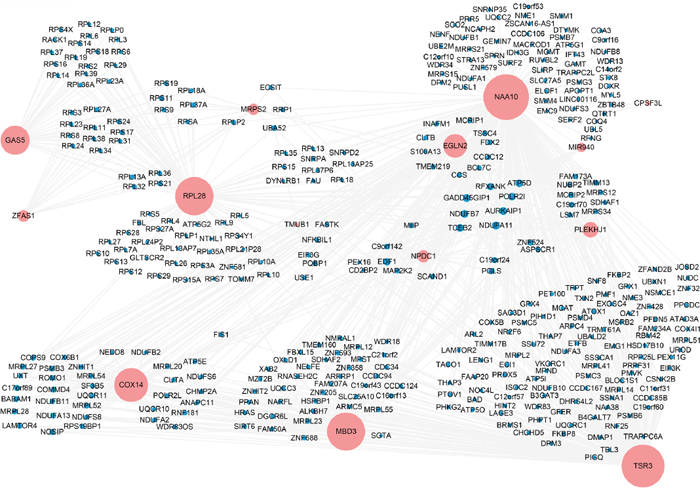
Figure 8: Co-expression network of GAS5, ZFAS1, miR-940 and target genes of miR-940 in the turquoise module. The co-expression network was constructed for the genes of interest which included two lncRNAs (GAS5 and ZFAS1), one miRNA (miR-940), target genes of miR-940 in the turquoise module and their co-expressed genes with correlation coefficients larger than 0.01. Node size is proportional to the number of co-expressed genes.
MiRNAs bind to partially complementary sequences of their target mRNAs, and many of these molecules have been widely implicated in various human diseases. Thus, to understand the relationship between miR-940 and its target genes in the turquoise module and how they are integrated as part of the GAS5/ZFAS1/miR-940 axis, we searched for mRNA targets of miR-940. For this, we used TarBase [33], a data warehouse that stores targets of miRNAs originating from both manual curation and experimental studies. From our search, we identified 15 gene targets of miR-940 belonging to the turquoise module, including COX14, CPSF3L, EGLN2, MBD3, MRPS2, NAA10, NPDC1, OTUB1, PLEKHJ1, RPL28, SART1, TCEA2, TMEM205, TMUB1, and TSR3. Next, to obtain additional information on how these genes are connected with miR-940 as well as GAS5 and ZFAS1, we obtained the expression information for these 15 genes along with miR-940, GAS5, and ZFAS1 and performed PCC analysis. Based on the correlation heat map for these genes, ZFAS1 and GAS5 are highly positively correlated with each other (PCC=0.83, p<1.0e-9, Figure 7C). Moreover, correlation analysis also revealed miR-940 is positively correlated with GAS5 (PCC=0.47) and ZFAS1 (PCC=0.49) (Figure 7C) which suggests miR-940 also likely to be involved in prostate cancer.
Finally, to further dissect the role of GAS5 and ZFAS1 in PCa, we created a gene co-expression subnetwork for the genes in the turquoise module, which included GAS5, ZFAS1, miR-940 and its targets from TarBase (Figure 8). As shown in the network, GAS5 and ZFAS1 share many co-expressed genes, including genes encoding both S and L ribosomal proteins. One of the target genes of miR-940 is NAA10 which interacts with the most genes in the network. Interestingly, both GAS5 and ZFAS1 are linked to MRPS2, a gene coding for mitochondrial ribosomal proteins which is also a target of miR-940 via NAA10. RPL28, as the third largest node in the network and a target of miR-940, interacts with GAS5 and ZFAS1 via other ribosomal protein-encoding genes. A simplified version of the gene co-expression network using the Cytoscape app, ThematicMap, can be found in Supplementary Figure 4. Again, this network map shows GAS5 and ZFAS1 are targeted by miR-940 via NAA10 and RPL28 and possibly other targets of miR-940 as well (genes in nodes 3 and 4, including ribosomal genes). In summary, our results show miR-940 indirectly targets GAS5 and ZFAS1 via its mRNA targets, including NAA10 and RPL28.
DISCUSSION
In this study, we performed co-expression gene network analysis on PCa patient RNA-seq samples and identified a gene module that correlated with patient survival time. In functional enrichment analysis, we showed the genes in this module are involved in translation and RNA-processing. Translation pathways have previously been implicated to predict patient survival in PCa. For example, higher expression of EIF4E, a family member of the eukaryotic translation initiation factor, is associated with worse outcome in PCa patients [34, 35]. Moreover, it is now known that RNA processing contributes to the generation of androgen receptor splice variants, the constitutive activation of which is associated with poor prognosis [36].
Due to its tissue-specific and cancer-specific expression, long non-coding RNAs are favorable candidates as diagnostic or prognostic biomarkers for cancer. Indeed, a number of lncRNAs have emerged as potential biomarkers for PCa. High SCHLAP1 expression in PCa has been reported to predict worse patient outcome [37–40]. The up-regulation of UCA1 [41] and NEAT1 [42] also indicates a poor prognosis in patients suffering from PCa. In contrast, a low PCAT29 expression has been shown as an indicator of a higher potential for recurrence [43]. Similarly, the down-regulation of PCAT14 [44, 45] and DRAIC [46] are both associated with poor prognosis of PCa. In this work, we identified several non-coding RNAs in the module that correlated with survival time, including GAS5, miR-940, and ZFAS1. Furthermore, our findings suggest that GAS5 and ZFAS1 are potential novel prognostic markers for PCa.
GAS5 expression and its clinical implication have been examined in many types of cancers. For example, GAS5 is down-regulated in breast cancer [47]. In squamous cell carcinoma of the head and neck, higher expression of GAS5 in patients indicates higher recurrence-free survival [48]. Functionally, GAS5 has been shown to bind to the DNA-binding domain of AR [49]. This is because part of the GAS5 sequence is similar to the glucocorticoid receptor responsive element [50]. Therefore, GAS5 can prevent the binding of AR to its target DNA sequences by sequestering the androgen/AR complex [49]. In PCa, GAS5 has been shown to promote apoptosis [51] and inhibit cell proliferation and cancer progression by targeting miR-103 and the mTOR pathway [52]. In the present study, our results suggest that GAS5 may also be involved in regulating protein translation in PCa and a high GAS5 expression is a predictor of worse disease-free survival.
The expression of ZFAS1, like GAS5, is also up-regulated in normal mammary glands compared to breast cancer tissues [53]. Our current results show that high ZFAS1 expression is an indicator of lower disease-free survival for PCa patients. This predictive power of ZFAS1 does not appear to be limited to PCa, as it has been reported for gliomas as well [54]. With regards to function, ZFAS1 has been shown to regulate cell proliferation and migration of ovarian cancer by targeting miR-150-5p [55], whereas, in gastric cancer, ZFAS1 was demonstrated to accelerate cell proliferation via repressing the expression of KLF2 and NKD2 [56]. In this study, we also showed ZFAS1 may have functions related to protein translation which has been previously reported for breast cancer [53].
Compared to GAS5 and ZFAS1, the role of miR-940 is less clear and appears to be different depending on the type of cancer. Moreover, in some cancer types, the finding has even been contradictory. In general, miR-940 has been reported as a tumor suppressor in many studies. In addition, it is highly expressed in normal tissues compared with tumors in nasopharyngeal carcinoma [4], breast cancer [4], pancreatic ductal adenocarcinoma [57], ovarian cancer [58], hepatocellular carcinoma and gastric cancer [59]. On the other hand, miR-940 has also been reported as an oncogene with higher expression in tumor compared to normal tissues in pancreatic cancer [60], oral tongue squamous cell carcinoma, cervical cancer [4] and gastric cancer [60]. Currently, studies of miR-940 in cancer are still sparse and contradictory. For example, in one study miR-940 was reported to act as an oncogene in gastric cancer by directly down-regulating ZNF24 expression [60]. But in another study also on gastric cancer, miR-940 was reported as a tumor suppressor [59]. Therefore, the difference in expression trend of miR-940 may lie in the different cohorts and cancer types. Further studies are needed to clarify these observations.
Here, we described the regulation between GAS5, ZFAS1, and miR-940 in PCa. Our findings suggest miR-940 directly targets NAA10 and indirectly targets ZFAS1 and GAS5 via MRPS2 and other ribosomal genes. In addition, we used WGCNA to detect gene modules that are significantly associated with pathoclinical parameters. We also identified prognostic biomarkers based on correlations between survival time and gene expression from both the single gene and gene module perspectives. Finally, we inferred the function of non-coding RNAs based on co-expressed genes. In conclusion, our work suggests that co-expression analysis of large-scale RNA-seq profiling can facilitate the identification and functional characterization of novel prognostic markers.
MATERIALS AND METHODS
Data acquisition
HTSeq-FPKM TCGA expression profiling was downloaded from https://gdc-portal.nci.nih.gov/projects/t for PCa, together with clinical data, including “BCR status”, “tumor status”, “Gleason score”, “pathologic N”, “pathologic T”, “psa_most_recent_results”, “lymph_nodes_examined”, “lymph_nodes_examined_he_count” and “residual tumor”. Clinical information was obtained from 380 patients, and the following analyses were performed on these patients.
Gene co-expression network construction and module identification
WGCNA was used to create gene co-expression networks and to identify gene modules [14, 61]. All transcripts expressed (FPKM>1) in at least half of the patients were included for WGCNA. First, a symmetric matrix of Pearson’s correlation was computed between all gene pairs. Second, the correlation matrix was raised to power β = 30 to obtain the adjacency matrix. Considering its characteristic of scale-free topology (R2 = 0.9), the power β=22 is selected to construct the adjacency network (Figure 1). The adjacency matrix was further transformed to a topological overlap matrix (TOM), which aims to evaluate the most strongly correlated genes. The matrix (1-TOM) was used for hierarchical clustering. In the hierarchical dendrogram, its branches are regarded as the gene modules, which are cut using branch cutting algorithms [62]
The gene significance (GS) of the ith gene can be defined: where is the expression profile of gene i and T is the sample trait. =22 (Figure 1A) is the power we used to find gene modules. For each module, the module eigengene was represented by the first principal component of the expression profile. Modules were merged together when the module eigengenes are highly correlated (correlation > 0.75). The module-trait relationships (Figure 2) exhibits the correlation of eigengene expression in a module () and clinical traits T (survival time, Gleason score, PSA and number of lymph nodes). The correlation value in each grid was calculated as with corresponding Student asymptotic p-value. The module membership (MM) quantifies the extent of similarity of a pair of gene and module. MM of each gene was calculated as . For more details, please refer to [63]. The correlation network of genes in the turquoise module was constructed based on adjacency threshold as 0.01.
Functional enrichment analysis
Fisher’s exact test was adopted to measure the gene enrichment in the annotation terms according to DAVID [64, 65]. When the Bonferroni-adjusted p≤ 0.05 was used, we assumed that the user gene lists were significantly enriched in this functional term. “Enrichment Map”, a Cytoscape plugin (http://cytoscape.org/) [66], was used to identify the function clusters for genes in the largest module (turquoise module), facilitating the interpretation of the enrichment terms.
Abbreviations
Long non-coding RNAs (lncRNAs), non-coding RNAs (ncRNAs), microRNAs (miRNAs), weighted correlation network analysis (WGCNA), Prostate cancer (PCa), prostate-specific antigen (PSA), cell cycle progression (CCP), hematoxylin and eosin (HE), HUGO gene nomenclature committee (HGNC), L ribosomal proteins (RPL), S ribosomal proteins (RPS), Gene significance (GS), module membership (MM), topological overlap matrix (TOM), ribosome binding sites (RBS) and Pearson’s correlation coefficient (PCC).
Author contributions
All authors contributed to the completion of this manuscript. XC and EC initiated the project. EC and CY provided funding, revised the draft and supervised the project. XC performed data acquisition, data processing and drafted the original manuscript. CY and SX generated the results visualization and revised the manuscript.
ACKNOWLEDGMENTS
This work was performed in part at the High-Performance Computing Cluster (HPCC), which is supported by Information and Communication Technology Office (ICTO) of the University of Macau. The authors would like to thank Jacky Chan Hoi Kei and William Pang from HPCC for support and assistance.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was financially supported in part by grants from the National Natural Science Foundation of China [Grant Nos. 61603099 and Grant Nos. U1201253], the Macau Science and Technology Development Fund [FDCT/023/2014/A1 and FDCT102/2015/A3]), a Multi-Year Research Grant [MYRG2015-00196-FHS] and a Start-up Research Grant [SRG2014-0000-FHS] from the University of Macau.
REFERENCES
1. Gannon PO, Lessard L, Stevens LM, Forest V, Begin LR, Minner S, Tennstedt P, Schlomm T, Mes-Masson AM, Saad F. Large-scale independent validation of the nuclear factor-kappa B p65 prognostic biomarker in prostate cancer. Eur J Cancer. 2013; 49: 2441-8. https://doi.org/10.1016/j.ejca.2013.02.026.
2. Formosa A, Markert EK, Lena AM, Italiano D, Finazzi-Agro E, Levine AJ, Bernardini S, Garabadgiu AV, Melino G, Candi E. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene. 2014; 33: 5173-82. https://doi.org/10.1038/onc.2013.451.
3. Tamayo P, Cho YJ, Tsherniak A, Greulich H, Ambrogio L, Schouten-van Meeteren N, Zhou T, Buxton A, Kool M, Meyerson M, Pomeroy SL, Mesirov JP. Predicting relapse in patients with medulloblastoma by integrating evidence from clinical and genomic features. J Clin Oncol. 2011; 29: 1415-23. https://doi.org/10.1200/JCO.2010.28.1675.
4. Aaltomaa S, Lipponen P, Ala-Opas M, Kosma VM. Expression and prognostic value of CD44 standard and variant v3 and v6 isoforms in prostate cancer. Eur Urol. 2001; 39: 138-44. doi: 52428.
5. Morton RA, Ewing CM, Nagafuchi A, Tsukita S, Isaacs WB. Reduction of E-cadherin levels and deletion of the alpha-catenin gene in human prostate cancer cells. Cancer Res. 1993; 53: 3585-90.
6. Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, Schaafsma HE, Debruyne FM, Isaacs WB. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992; 52: 5104-9.
7. Yamoah K, Johnson MH, Choeurng V, Faisal FA, Yousefi K, Haddad Z, Ross AE, Alshalafa M, Den R, Lal P, Feldman M, Dicker AP, Klein EA, et al. Novel biomarker signature that may predict aggressive disease in African American men with prostate cancer. J Clin Oncol. 2015; 33: 2789-96. https://doi.org/10.1200/JCO.2014.59.8912.
8. Freedland SJ, deGregorio F, Sacoolidge JC, Elshimali YI, Csathy GS, Dorey F, Reiter RE, Aronson WJ. Preoperative p27 status is an independent predictor of prostate specific antigen failure following radical prostatectomy. J Urol. 2003; 169: 1325-30. https://doi.org/10.1097/01.ju.0000054004.08958.f3.
9. Freedland SJ, Gerber L, Reid J, Welbourn W, Tikishvili E, Park J, Younus A, Gutin A, Sangale Z, Lanchbury JS, Salama JK, Stone S. Prognostic utility of cell cycle progression score in men with prostate cancer after primary external beam radiation therapy. Int J Radiat Oncol Biol Phys. 2013; 86: 848-53. https://doi.org/10.1016/j.ijrobp.2013.04.043.
10. Bolton EM, Tuzova AV, Walsh AL, Lynch T, Perry AS. Noncoding RNAs in prostate cancer: the long and the short of it. Clin Cancer Res. 2014; 20: 35-43. https://doi.org/10.1158/1078-0432.CCR-13-1989.
11. Xu N, Chen HJ, Chen SH, Xue XY, Chen H, Zheng QS, Wei Y, Li XD, Huang JB, Cai H, Sun XL. Reduced Connexin 43 expression is associated with tumor malignant behaviors and biochemical recurrence-free survival of prostate cancer. Oncotarget. 2016; 7: 67476-84. https://doi.org/10.18632/oncotarget.11231.
12. Hamoudi RA, Appert A, Ye H, Ruskone-Fourmestraux A, Streubel B, Chott A, Raderer M, Gong L, Wlodarska I, De Wolf-Peeters C, MacLennan KA, de Leval L, Isaacson PG, et al. Differential expression of NF-kappaB target genes in MALT lymphoma with and without chromosome translocation: insights into molecular mechanism. Leukemia. 2010; 24: 1487-97. https://doi.org/10.1038/leu.2010.118.
13. Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015; 43: D1079-85. https://doi.org/10.1093/nar/gku1071.
14. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9: 559. https://doi.org/10.1186/1471-2105-9-559.
15. Chen YC, Guo YF, He H, Lin X, Wang XF, Zhou R, Li WT, Pan DY, Shen J, Deng HW. Integrative analysis of genomics and transcriptome data to identify potential functional genes of BMDs in females. J Bone Miner Res. 2016; 31: 1041-9. https://doi.org/10.1002/jbmr.2781
16. Lou Y, Tian GY, Song Y, Liu YL, Chen YD, Shi JP, Yang J. Characterization of transcriptional modules related to fibrosing-NAFLD progression. Sci Rep. 2017; 7: 4748. https://doi.org/10.1038/s41598-017-05044-2.
17. Woodward NC, Levine MC, Haghani A, Shirmohammadi F, Saffari A, Sioutas C, Morgan TE, Finch CE. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J Neuroinflammation. 2017; 14: 84. https://doi.org/10.1186/s12974-017-0858-x.
18. King TD, Zhang W, Suto MJ, Li Y. Frizzled7 as an emerging target for cancer therapy. Cell Signal. 2012; 24: 846-51. https://doi.org/10.1016/j.cellsig.2011.12.009.
19. Cui L, Li M, Feng F, Yang Y, Hang X, Cui J, Gao J. MEIS1 functions as a potential AR negative regulator. Exp Cell Res. 2014; 328: 58-68. https://doi.org/10.1016/j.yexcr.2014.08.023.
20. Chen JL, Li J, Kiriluk KJ, Rosen AM, Paner GP, Antic T, Lussier YA, Vander Griend DJ. Deregulation of a Hox protein regulatory network spanning prostate cancer initiation and progression. Clin Cancer Res. 2012; 18: 4291-302. https://doi.org/10.1158/1078-0432.CCR-12-0373.
21. Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, Cao X, Jing X, Wang X, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011; 29: 742-9. https://doi.org/10.1038/nbt.1914.
22. Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, Evans CP, Rosenfeld MG. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013; 500: 598-602. https://doi.org/10.1038/nature12451.
23. Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ, Zhang J, Huang H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget. 2015; 6: 41045-55. https://doi.org/10.18632/oncotarget.5728.
24. Zhang HY, Yang W, Zheng FS, Wang YB, Lu JB. Long non-coding RNA SNHG1 regulates zinc finger E-box binding homeobox 1 expression by interacting with TAp63 and promotes cell metastasis and invasion in Lung squamous cell carcinoma. Biomed Pharmacother. 2017; 90: 650-8. https://doi.org/10.1016/j.biopha.2017.03.104.
25. Huang HY, Chien CH, Jen KH, Huang HD. RegRNA: an integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 2006; 34: W429-34. https://doi.org/10.1093/nar/gkl333.
26. Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN, Liang H. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015; 75: 3728-37. https://doi.org/10.1158/0008-5472.CAN-15-0273.
27. Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013; 12: 847-65. https://doi.org/10.1038/nrd4140.
28. Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, Hatzigeorgiou AG. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015; 43: W460-6. https://doi.org/10.1093/nar/gkv403.
29. Rajendiran S, Parwani AV, Hare RJ, Dasgupta S, Roby RK, Vishwanatha JK. MicroRNA-940 suppresses prostate cancer migration and invasion by regulating MIEN1. Mol Cancer. 2014; 13: 250. https://doi.org/10.1186/1476-4598-13-250.
30. Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, Zagganas K, Tsanakas P, Floros E, Dalamagas T, Hatzigeorgiou AG. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016; 44: D231-8. https://doi.org/10.1093/nar/gkv1270.
31. Balakrishnan I, Yang X, Brown J, Ramakrishnan A, Torok-Storb B, Kabos P, Hesselberth JR, Pillai MM. Genome-wide analysis of miRNA-mRNA interactions in marrow stromal cells. Stem Cells. 2014; 32: 662-73. https://doi.org/10.1002/stem.1531
32. Karginov FV, Hannon GJ. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013; 27: 1624-32. https://doi.org/10.1101/gad.215939.113.
33. Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D, Fevgas A, Dalamagas T, Hatzigeorgiou AG. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015; 43: D153-9. https://doi.org/10.1093/nar/gku1215.
34. Wang H, Ru Y, Sanchez-Carbayo M, Wang X, Kieft JS, Theodorescu D. Translation initiation factor eIF3b expression in human cancer and its role in tumor growth and lung colonization. Clin Cancer Res. 2013; 19: 2850-60.https://doi.org/10.1158/1078-0432.CCR-12-3084.
35. Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, Hurst BM, Deddens JA, Neubauer BL, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res. 2009; 69: 3866-73. https://doi.org/10.1158/0008-5472.CAN-08-3472.
36. Nakata D, Nakao S, Nakayama K, Araki S, Nakayama Y, Aparicio S, Hara T, Nakanishi A. The RNA helicase DDX39B and its paralog DDX39A regulate androgen receptor splice variant AR-V7 generation. Biochem Biophys Res Commun. 2017; 483: 271-6. https://doi.org/10.1016/j.bbrc.2016.12.153.
37. Mehra R, Shi Y, Udager AM, Prensner JR, Sahu A, Iyer MK, Siddiqui J, Cao X, Wei J, Jiang H, Feng FY, Chinnaiyan AM. A novel RNA in situ hybridization assay for the long noncoding RNA SChLAP1 predicts poor clinical outcome after radical prostatectomy in clinically localized prostate cancer. Neoplasia. 2014; 16: 1121-7. https://doi.org/10.1016/j.neo.2014.11.006.
38. Mehra R, Udager AM, Ahearn TU, Cao X, Feng FY, Loda M, Petimar JS, Kantoff P, Mucci LA, Chinnaiyan AM. Overexpression of the long non-coding RNA SChLAP1 independently predicts lethal prostate cancer. Eur Urol. 2016; 70: 549-52. https://doi.org/10.1016/j.eururo.2015.12.003.
39. Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, Jenkins RB, Triche TJ, Malik R, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013; 45: 1392-8. https://doi.org/10.1038/ng.2771.
40. Prensner JR, Zhao S, Erho N, Schipper M, Iyer MK, Dhanasekaran SM, Magi-Galluzzi C, Mehra R, Sahu A, Siddiqui J, Davicioni E, Den RB, Dicker AP, et al. RNA biomarkers associated with metastatic progression in prostate cancer: a multi-institutional high-throughput analysis of SChLAP1. Lancet Oncol. 2014; 15: 1469-80. https://doi.org/10.1016/S1470-2045(14)71113-1.
41. Fotouhi Ghiam A, Taeb S, Huang X, Huang V, Ray J, Scarcello S, Hoey C, Jahangiri S, Fokas E, Loblaw A, Bristow RG, Vesprini D, Boutros P, Liu SK. Long non-coding RNA urothelial carcinoma associated 1 (UCA1) mediates radiation response in prostate cancer. Oncotarget. 2017; 8: 4668-89. https://doi.org/10.18632/oncotarget.13576.
42. Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, MacDonald TY, Fontugne J, Erho N, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014; 5: 5383. https://doi.org/10.1038/ncomms6383.
43. Malik R, Patel L, Prensner JR, Shi Y, Iyer MK, Subramaniyan S, Carley A, Niknafs YS, Sahu A, Han S, Ma T, Liu M, Asangani IA, et al. The lncRNA PCAT29 inhibits oncogenic phenotypes in prostate cancer. Mol Cancer Res. 2014; 12: 1081-7. https://doi.org/10.1158/1541-7786.MCR-14-0257.
44. White NM, Zhao SG, Zhang J, Rozycki EB, Dang HX, McFadden SD, Eteleeb AM, Alshalalfa M, Vergara IA, Erho N, Arbeit JM, Karnes RJ, Den RB, et al. Multi-institutional analysis shows that low PCAT-14 expression associates with poor outcomes in prostate cancer. Eur Urol. 2017; 71: 257-66. https://doi.org/10.1016/j.eururo.2016.07.012.
45. Shukla S, Zhang X, Niknafs YS, Xiao L, Mehra R, Cieslik M, Ross A, Schaeffer E, Malik B, Guo S, Freier SM, Bui HH, Siddiqui J, et al. Identification and validation of PCAT14 as prognostic biomarker in prostate cancer. Neoplasia. 2016; 18: 489-99. https://doi.org/10.1016/j.neo.2016.07.001.
46. Sakurai K, Reon BJ, Anaya J, Dutta A. The lncRNA DRAIC/PCAT29 locus constitutes a tumor-suppressive nexus. Mol Cancer Res. 2015; 13: 828-38. https://doi.org/10.1158/1541-7786.MCR-15-0016-T.
47. Pickard MR, Williams GT. Regulation of apoptosis by long non-coding RNA GAS5 in breast cancer cells: implications for chemotherapy. Breast Cancer Res Treat. 2014; 145: 359-70. https://doi.org/10.1007/s10549-014-2974-y
48. Gee HE, Buffa FM, Camps C, Ramachandran A, Leek R, Taylor M, Patil M, Sheldon H, Betts G, Homer J, West C, Ragoussis J, Harris AL. The small-nucleolar RNAs commonly used for microRNA normalisation correlate with tumour pathology and prognosis. Br J Cancer. 2011; 104: 1168-77. https://doi.org/10.1038/sj.bjc.6606076.
49. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010; 3: ra8. https://doi.org/10.1126/scisignal.2000568.
50. Pickard MR, Williams GT. The hormone response element mimic sequence of GAS5 lncRNA is sufficient to induce apoptosis in breast cancer cells. Oncotarget. 2016; 7: 10104-16. https://doi.org/10.18632/oncotarget.7173.
51. Pickard MR, Mourtada-Maarabouni M, Williams GT. Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim Biophys Acta. 2013; 1832: 1613-23. https://doi.org/10.1016/j.bbadis.2013.05.005.
52. Yacqub-Usman K, Pickard MR, Williams GT. Reciprocal regulation of GAS5 lncRNA levels and mTOR inhibitor action in prostate cancer cells. Prostate. 2015; 75: 693-705. https://doi.org/10.1002/pros.22952
53. Hansji H, Leung EY, Baguley BC, Finlay GJ, Cameron-Smith D, Figueiredo VC, Askarian-Amiri ME. ZFAS1: a long noncoding RNA associated with ribosomes in breast cancer cells. Biol Direct. 2016; 11: 62. https://doi.org/10.1186/s13062-016-0165-y.
54. Lv QL, Chen SH, Zhang X, Sun B, Hu L, Qu Q, Huang YT, Wang GH, Liu YL, Zhang YY, Zhou HH. Upregulation of long noncoding RNA zinc finger antisense 1 enhances epithelial-mesenchymal transition in vitro and predicts poor prognosis in glioma. Tumour Biol. 2017; 39: 1010428317695022. https://doi.org/10.1177/1010428317695022
55. Xia B, Hou Y, Chen H, Yang S, Liu T, Lin M, Lou G. Long non-coding RNA ZFAS1 interacts with miR-150-5p to regulate Sp1 expression and ovarian cancer cell malignancy. Oncotarget. 2017; 8: 19534-46. https://doi.org/10.18632/oncotarget.14663.
56. Nie F, Yu X, Huang M, Wang Y, Xie M, Ma H, Wang Z, De W, Sun M. Long noncoding RNA ZFAS1 promotes gastric cancer cells proliferation by epigenetically repressing KLF2 and NKD2 expression. Oncotarget. 2016; 8: 38227-38. https://doi.org/10.18632/oncotarget.9611.
57. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11: R106. https://doi.org/10.1186/gb-2010-11-10-r106.
58. Arun G, Diermeier S, Akerman M, Chang KC, Wilkinson JE, Hearn S, Kim Y, MacLeod AR, Krainer AR, Norton L, Brogi E, Egeblad M, Spector DL. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016; 30: 34-51. https://doi.org/10.1101/gad.270959.115.
59. Bai Y, Dougherty L, Cheng L, Zhong GY, Xu K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genomics. 2015; 16: 612. https://doi.org/10.1186/s12864-015-1816-6.
60. Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, Tamez-Pena JG, Trevino V. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013; 8: e74250. https://doi.org/10.1371/journal.pone.0074250.
61. Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012.
62. Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008; 24: 719-20. https://doi.org/10.1093/bioinformatics/btm563.
63. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005; 4: Article17. https://doi.org/10.2202/1544-6115.1128.
64. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4: 44-57. https://doi.org/10.1038/nprot.2008.211.
65. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37: 1-13. https://doi.org/10.1093/nar/gkn923.
66. Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010; 5: e13984. https://doi.org/10.1371/journal.pone.0013984