
INTRODUCTION
Migraine is one of the most common neurological disorders in the world, affecting 14.2% of US adults [1]. Overall, migraineurs spend nearly 2.5 times more than non-migraineurs in direct healthcare costs, and recurrent episodes result in approximately 11 days of missed work that costs up to $10,000/case annually [2, 3]. Roughly two-thirds of migraineurs are female suggesting a potential role for estrogens in migraine pathophysiology [4], yet very few preclinical studies have used female subjects [5, 6].
According to the International Classification of Headache Disorders, two types of migraine exist: migraine with and without aura. Migraine with aura (MA), a focal neurological disturbance exhibiting predominantly visual symptoms, affects one-third of all migraineurs [7–9]; this phenomenon is closely associated with spreading depressions (SD). SD events are self-propagating waves of membrane depolarization that travel at rates of 2-5 mm/min followed by a negative shift of the direct current (DC) potential, reduction in EEG amplitude, and often, increases in cerebral blood flow [10–12]. In MA, cortical SD events (CSDs) are thought to occur during the aura phase of migraine and precede the headache phase in a subset of migraineurs [13]. CSD susceptibility (e.g., reduced CSD thresholds) and probabilities of migraine attack are enhanced by increases in cortical excitability [14–16] and result in an increased BOLD signal on fMRI [15, 17–20]. Female reproductive hormones, including estrogen and progesterone, are implicated in migraine development both clinically and pre-clinically [21–23]. Clinically, the onset of migraine attacks coincides with hormonal fluctuations such as those associated with puberty, some oral contraceptives, pregnancy, and menopause [22, 24–26]. Moreover, it is well accepted that migraine without aura can be triggered by steeply declining levels of ovarian hormones, whereas MA is exacerbated by increasing levels of female reproductive hormones [6, 21, 27–30]. Preclinical studies have shown that estrogens, acting through both nuclear α- and β-estrogen receptors as well as GPR30 [31, 32], increase cortical excitability and CSD susceptibility in brain slices [33]. In anesthetized rodent models of CSD, estrogen fluctuations reduced the CSD threshold and increased the frequency and velocity of CSD events [14, 34, 35]. Yet, questions of whether the anesthetic or invasiveness of intracortical recordings altering the CSD events have been raised [36, 37]. Non-invasive epidural EEGs have been used to determine brain activity in rats since 1979 [12, 38–41]. Over time, the implantation technique has been substantially improved upon to yield highly reproducible and stable recordings [12, 41] with successful epidural EEG recordings of CSD events by a number of groups in the context of migraine [42–44]. SD events in humans can be detected with traditional EEG leads on the cortical surface [45] or using scalp AC-EEG [46] intracortical and subdural recordings are only obtained during procedures with patients requiring intracranial pressure relief (i.e., TBI) or tissue removal [47–50].
To date, no studies have investigated the contribution of estrogens to SD induction and headache pain behavior in awake, freely moving, female rats. Here, we addressed this gap in knowledge and study the contribution of 17-β-estradiol to SD and headache pain using our non-anesthetized rodent model [43, 51, 52] including estrogen-receptor dependence and responsivity to sumatriptan, the leading abortive antimigraine. 17-β-estradiol administration induced SDs and behaviors associated with headache pain. Both SDs and pain behavior were inhibited by the non-selective estrogen receptor antagonist/GPR30 agonist ICI 182,780 and sumatriptan, confirming both estrogen receptor dependence and responsivity to the leading abortive antimigraine, respectively.
RESULTS
Serum estrogen (E2) levels
Fluctuations in estradiol serum concentrations are associated with CSD and migraine [6, 27]. We injected a supraphysiological dose of 17-β-estradiol (180μg/kg, ip) to maximize the rapid rise and fall of serum estradiol levels in order to capture multiple stages of estrogen activity, including a peak upswing to model pregnancy levels and downfall in levels to mimic sharp fluctuations in estrogen [53], or vehicle (castor oil, ip) 7 days post-OVX or in intact rats. Intact rats served as a control for ovariectomy; high dose estradiol did not effect cytology in any group. Serum samples were drawn before (t=0) and 30 min, 2, 4, 6, 12, and 24 hour after injection to quantify estrogen levels (Table 1). In naturally cycling female rats injected during the diestrus-2 phase of the estrogen cycle, basal serum estrogen levels were 3.53 ± 0.76 pg/mL (n=6). Vehicle injection did not significantly increase serum levels over 24 h as compared to baseline (Table 1, n=3). Administration of 17-β-estradiol (180μg/kg) significantly elevated serum estrogen levels (993.44 ± 498.3 pg/mL, n=3, p<0.01) after 30 min and remained significantly elevated for 4 hours (203.77 ± 50.29 pg/mL, n=3, p<0.0001) before returning to baseline levels after 24 hours (3.29 ± 0.01 pg/mL, n=3) in intact rats. We next determined if 17-β-estradiol administration similarly elevated serum estrogen levels in OVX rats. OVX rats had basal serum estrogen levels of <3.0 pg/mL (n=6) consistent with previous reports and below the limit of detection of our kit (Calbiotech, Spring Valley, CA). Acute administration of 17-β-estradiol elevated serum estrogen concentration that peaked 30 min post-injection (441.35 ± 47.10 pg/mL, n=3, p<0.0005) and remained significantly elevated for 6 hours (164.05 pg/mL ± 48.58, p=0.005). Within 24 h, estrogen levels returned to baseline (4.07 pg/mL ± 0.48, n=3). No significant fluctuation in serum 17-β-estradiol level was observed in vehicle-treated, OVX rats (n=3) at any time over the 24 h post-injection.
Table 1: Serum estradiol levels in intact and OVX animals following i.p. administration of 17-β-estradiol (180μg/kg) or vehicle
Surgery |
Treatment |
BL |
30 min |
2 h |
4 h |
6 h |
12 h |
24 h |
|---|---|---|---|---|---|---|---|---|
Intact |
Vehicle |
3.53 ± 0.76 |
10.24 ± 4.58 |
8.72 ± 2.64 |
9.12 ± 2.92 |
8.26 ± 2.39 |
8.16 ± 3.75 |
11.84 ± 5.19 |
17-β |
993.44 ± 498.30 |
857.50 ± 75.42 |
203.77 ± 50.29 |
103.51 ± 36.74 |
57.63 ± 43.83 |
3.29 ± 0.01 |
||
OVX |
Vehicle |
2.57 ± 0.32 |
< 3.0 |
< 3.0 |
< 3.0 |
< 3.0 |
< 3.0 |
< 3.0 |
17-β |
441.35 ± 47.12 |
204.45 ± 95.55 |
157.08 ± 77.78 |
164.05 ± 48.58 |
8.40 ± 2.5 |
4.07 ± 0.48 |
Data are mean ± SEM (pg/mL).
17-β-estradiol induces SD events
Elevated estradiol levels are associated with an increase in MA frequency but not in migraineurs without aura. Moreover, MA is typically associated with SD events. Therefore, we next asked if 17-β-estradiol induced SD events in awake, freely moving, female rats. Dural electroencephalogram traces were recorded for 12 hours following either 17-β-estradiol (180μg/kg) or vehicle administration, according to the timeline in Figure 1A. Although 1/8 of intact rats treated with vehicle experienced a single SD event, no intact animals treated with 17-β-estradiol had detectable SD events (n=7, Table 2). We also observed a single SD event in 1/10 vehicle-treated animals. In contrast, 6/10 OVX rats (60%) experienced SD events in 12 hours post administration of 17-β-estradiol compared to (Table 2). A total of 11 estrogen evoked SD events observed in 6 OVX rats spread mainly parietal to frontal (8/11); 3/11 traveled frontal to parietal. Overall, 17-β-estradiol SD events propagated at a velocity of 9.26 ± 1.15 mm/min. Negative DC-shifts had amplitudes of 1.30 ± 0.12 mV and durations of 65.01 ± 10.78 s (Table 2). Increases in the number of SDs induced was dependent on the interaction of the gonadal state and administration of exogenous 17-β-estradiol (2-way ANVOA, Interaction: F-value=8.318, p=.007, Figure 1D) suggesting that removal of the ovaries increases susceptibility to 17-β-estradiol-induced SD events.
Table 2: CSD characteristics amongst groups
Surgery |
Group |
% Animals with SD |
Speed of propagation (mm/min) |
Amplitude (mV) |
Duration (s) |
|---|---|---|---|---|---|
Intact |
Vehicle |
1/8 |
7.70 |
1.05 |
43.00 |
17-β |
0/7 |
N/A |
N/A |
N/A |
|
OVX |
Vehicle |
1/10 |
7.70 |
2.64 |
118.00 |
17-β |
6/10 |
9.26 ± 1.15 |
1.30 ± 0.12 |
65.01 ± 10.78 |
|
ICI |
0/6 |
N/A |
N/A |
N/A |
|
17-β/ICI |
0/6 |
N/A |
N/A |
N/A |
|
Suma |
0/6 |
N/A |
N/A |
N/A |
|
17-β/Suma |
0/6 |
N/A |
N/A |
N/A |
17-β, 17-β-estradiol; ICI, ICI 732,138; Suma, sumatriptan; OVX, ovariectomy; SD, spreading depression.
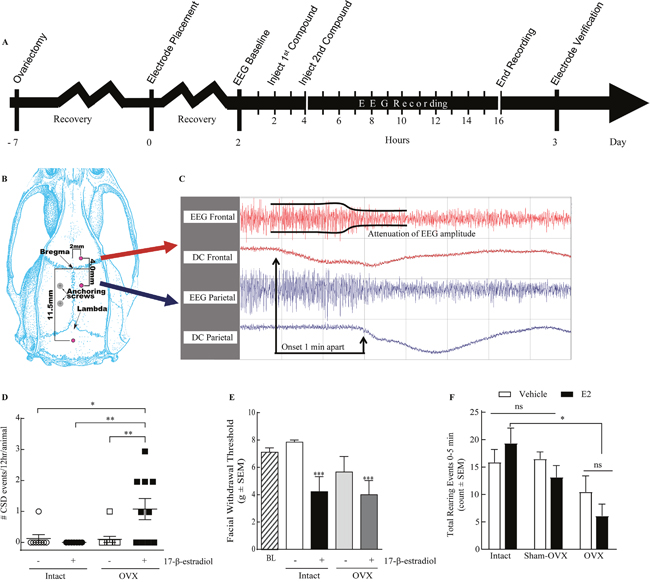
Figure 1: 17-β-estradiol administration induces SD events and pain behaviors in ovariectomized rats. (A) Schematic timeline of the procedures involved; electrode placement surgery is performed 1 week after ovariectomy and two days before dEEG recording. (B) Diagram of the electrode placement surgery. (C) A representative dEEG trace displaying a SD event with a propagation speed of 4mm/min. (D) 17-β-estradiol induced 1.1 ± 0.35 CSDs per animal (n=10) in ovariectomized rats, while none were elicited in intact rats (n=7). Vehicle had no significant effect on OVX (0.1 ± 0.1 SDs/animal, n=10) or intact (0.13 ± 0.13 CSDs per animal, n=8) rats over 12 hours. (E) Facial withdrawal threshold (FWT) is reduced by administration of 17-β-estradiol. Vehicle treated intact rats have a FWT of 7.85 ± 0.16 g (n=8), which is significantly reduced following estradiol administration (4.22 ± 1.11 g, n=8) or OVX and 17-β-estradiol administration 7 days later (3.99 ± 1.04 g, n=9). (F) Number of rearing events in 5 min following vehicle or 17-β-estradiol treatment in sham-operated animals was not significantly different from intact animals. 17-β-estradiol treated OVX animals (n=11) have significantly reduced rearing compared to estradiol treated intact animals (n=8). *p<0.05, **p<0.005 as compared to vehicle-treated, intact female rats (Kruskal-Wallis statistic = 26.8; Dunn’s multiple comparisons).
17-β-estradiol elicits pain behaviors after OVX
Induced SD events are not always associated with pain behaviors such as periorbital allodynia [52], and headache can occur independent from SD. Therefore, we evaluated pain behaviors in intact or OVX rats exposed to 17-β-estradiol or castor oil. Animals were evaluated for periorbital mechanical allodynia with calibrated von Frey filaments; baseline mechanical thresholds were 7.85 ± 0.15 g. Castor oil (vehicle) had no effect on facial sensitivity in naturally cycling (n=8) as compared to baseline values but administration of 17-β-estradiol significantly reduced mechanical threshold at 2 h in 5/8 intact rats (4.22 ± 1.11 g, p<0.05). Although OVX alone reduced facial withdrawal thresholds in 3/8 rats as compared to baseline values, overall hypersensitivity was not significantly different. 17-beta estradiol administration further reduced facial withdrawal thresholds in 6/9 OVX rats (OVX: 3.99 ± 1.03 g, p<0.005; Figure 1E). These data suggest that while loss of ovaries increases the percent of animals experiencing periorbital allodynia, administration of 17-β-estradiol induced periorbital allodynia regardless of gonadal state.
In addition to periorbital allodynia, suppression of exploratory behaviors was recently linked to preclinical migraine [54–56]. We determined if total times of exploration, ambulation, and fine movement, as well as rearing events, were decreased in rats 2 hours after exposure to high dose 17-β-estradiol or vehicle. Exploratory behaviors such as transitional exploratory events (i.e., chamber to chamber), ambulatory events, and fine movements were not significantly different between 17-β-estradiol and vehicle controls suggesting that these behaviors were not suppressed (p = 0.05, 0.12, and 0.25, respectively) 2h after administration of 17-β-estradiol. The total number of rearing events was assessed over 30 minutes, 2 h after vehicle or 17-β-estradiol administration. Rearing behavior was not significantly different between intact vehicle- and 17-β-estradiol- treated animals (52.73 ± 10.74, n=11 and 44.30 ± 6.21, n=10 respectively, p=0.85 Bonferroni). The total number of rearing events over a 30 min time course was significantly reduced in OVX vehicle treated rats (19.38 ± 5.23, n=8, p<0.05) and for OVX animals treated with 17-β-estradiol (9.857 ± 1.98, n=7, p<0.005 Bonferroni) compared to intact vehicle treated animals.
We next assessed if rearing reductions in OVX animals were due to exacerbation of post-surgical pain or a result of hormone fluctuations. A separate set of animals was generated in which sham-OVX surgeries were performed with rearing assessed before surgery and in the presence and absence of 17-β-estradiol (180 μg/kg). The total number of rearing events observed after sham-OVX + vehicle was not significantly different from intact, vehicle treated rats (Figure 1E, 64.5± 7.7, n=6 vs. 52.7 ± 10.7, p>0.99, Bonferroni). Likewise, administration of 17-β-estradiol to sham-OVX rats did not significantly reduce rearing over the 30 min observation period (33.5 ± 5.6, p>0.99, Bonferroni). Thus, suppressed rearing observed in OVX + 17-β-estradiol animals was not likely due to exacerbation of post-surgical pain.
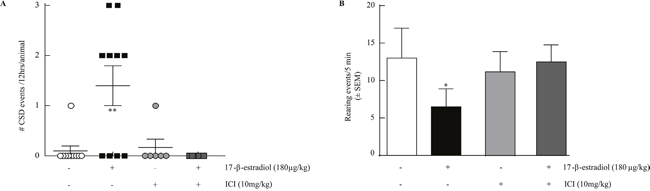
Figure 2: ICI 182,780 prevents estrogen induced SD and estrogen induced reduction in rearing. (A) 17-β-estradiol 180μg/kg induces SD events in OVX rats over 12 hours (1.4 ± 0.4 SDs, n=10) which is inhibited by ICI 182,780 10mg/kg, i.p. (0 ± 0 SDs, n=10). (B) 17-β-estradiol reduction of rearing behavior over 5 minutes (n=6) is inhibited by pretreatment with ICI 182,780 10mg/kg, i.p. (n=6). *p<0.05; **p<0.005 (One-way ANOVA).
To determine if reduced rearing correlated to exploring new environment, we assessed events in 5 min bins. Most rearing events occurred in the first 5 minutes (Figure 1F), consistent with exploring a new environment and subsided to very few (i.e., 0-2 events) over the 30 min regardless of treatment. Consistent with the total rearing data, both vehicle (8.25 ± 1.79, n=8, p<0.05; Figure 1C) and 17-β-estradiol (3.00 ± 0.65, n=7, p<0.005) treated OVX animals reared less over the first 10 min as compared to intact, vehicle-treated rats. No differences were seen between sham-OVX rats and intact rats at any time point. Suppression of rearing was dependent on gonadal state only. These data, together with periorbital allodynia, indicate that 17-β-estradiol induced distinct manifestations of pain behaviors in both intact and OVX rats.
17-β-estradiol evoked CSD events and suppressed rearing requires estrogen receptors
Focusing on the OVX rats, which had a significantly higher number of SDs and prominent pain behavior, we asked if estrogen receptor activation was required for 17-β-estradiol-induced SDs and suppression of rearing behaviors. We administered the estrogen antagonist ICI 182,780 (10 mg/kg, i.p.) 2 hr before 17-β-estradiol or vehicle. 17-β-estradiol evoked SD events were blocked in the presence of ICI 182,780 (Table 1, Figure 2A). Importantly, ICI 182,720 did not elicit SD events when administered alone. Similarly, pretreatment with ICI 182,780 significantly prevented 17-β-estradiol-induced suppression of rearing events in rats (12.5 ± 2.26, n=6, p<0.05; Figure 2B) within the 0-5 min period. Thus, 17-β-estradiol induced SD events and pain behaviors required activation of estrogen receptors in OVX rats.
Sumatriptan prevents 17-β-estradiol evoked CSD events and suppressed rearing
We assessed the ability of the abortive anti-migraine therapeutic sumatriptan (0.6mg/kg, i.p.) to prevent 17-β-estradiol SD events or rearing behaviors. Pre-treatment with sumatriptan 2 hours before 17-β-estradiol prevented the induction of SD events (n=6; Table 1, Figure 3A) and did not induce SDs on its own. Since the majority of rearing events occurred within the 0-5 min bin of the 30 min total observation period, we evaluated the effects of sumatriptan intervention within this time-period. Sumatriptan pretreatment increased the number of rears in 17-β-estradiol treated OVX animals (10.6 ± 1.95, n=10) back to that of OVX-vehicle treated rats (12.27 ± 3.14, n=6, p=0.21; Figure 3B).
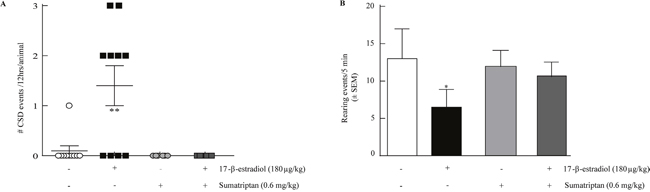
Figure 3: Sumatriptan prevents estrogen induced SD and estrogen induced reduction in rearing. (A) Sumatriptan 0.6mg/kg, i.p. (0 ± 0 SDs, n=6) inhibited 17-β-estradiol induced SD events in 12 hours with 2 h pretreatment. (B) 17-β-estradiol suppression of rearing behavior over 5 minutes (n=6) was inhibited by 2h pretreatment with sumatriptan 0.6mg/kg, i.p. (n=6). *p<0.05, **p<0.005 (One-way ANOVA).
DISCUSSION
Migraine with aura affects nearly 8% of migraineurs, the majority of whom are female [57–59]. In addition to the aura and headache, the patients suffer from a greatly diminished quality of life as evidenced by a greater absenteeism from work/school, less productivity, and less overall energy [60]. Increases in estrogen levels are implicated in the development of MA, but preclinical studies of this phenomenon in female subjects are few [6]. Here, we examined the role of 17-β-estradiol in the induction of SD events and headache-like pain, two characterizing features of MA, in non-anesthetized female rats. We show that exogenous application of 17-β-estradiol elicits SD events and corresponding pain behaviors in a gonadal state (intact versus OVX) dependent manner. These events were prevented with both estrogen receptor antagonist and administration of the anti-migraine therapeutic sumatriptan.
Women are 3 times more likely to suffer from migraine headaches than males with reports of higher pain levels and incidence of aura [5, 61–64]. Plasma estradiol and progesterone levels in non-migrainous women not taking hormonal birth control are lower throughout the menstrual cycle compared to migraineurs [65], implicating these hormones in migraine pathology. Interestingly, female migraineurs without aura taking combined oral contraceptives (i.e., those containing estrogen) reportedly have increased migraine attacks in between courses- when serum estrogen levels drop or are at the lowest point [66]; serum estradiol levels peak following administration of oral contraception between 100-120 pg/ml [67–69]. In contrast, in migraineurs with aura, contraceptives with estrogen are associated with increased frequency and severity of migraine attack as well as increased risk of ischemic stroke [70] and are thus contraindicated; conversely, progesterone-only pills are not [71]. These clinical findings highlight the idea that fluctuations in ovarian hormones (e.g., estrogen) can differentially affect migraineur sub-populations [62].
In the present study, we found that a single supraphysiological dose of 17-β-estradiol (180 μg/kg) increased serum concentrations of 17-β-estradiol in both intact and OVX rats for up to 6 hrs. Observed serum 17-β-estradiol levels were supraphysiological in animals mimicking reproductive (intact) and non-reproductive states (OVX) at values in line with patients with gonadotropinomas and other anterior pituitary tumors [72, 73]. This use of a bolus, supraphysiological dose allowed for assessment of SD events and pain behaviors across a sharp increase and decrease in estrogen levels During migraine without aura, these can be times of headache relief. However, elevated levels of estradiol in women with MA, are associated with increases in migraine frequency and severity [74] and induction of CSD events [6, 14, 15, 19, 75]. In our current investigation, SD events occurred 30 min after 17-β-estradiol administration in OVX rats suggesting that changing estrogen levels were required to induce SD events; this was dependent on gonadal state and administration of 17-β-estradiol. Interestingly, SDs persisted after estradiol levels had returned to baseline values in some rats (up to 12 hours) indicating an increased susceptibility to neuronal dysregulation after 17-β-estradiol. These observations align with work done by Sachs and colleagues showing acute estrogen alone was able to induce CSDs in rat neocortical slices [33], as well as studies showing that chronic estrogen can reduce the threshold for initiation of KCl-induced CSDs in anesthetized, OVX rats [34]. Likewise, our findings align with Chauvel et al., showing that OVX plays a role in SD susceptibility and frequency in non-anesthetized, female rats after 17-β-estradiol.
CSD events are linked to the aura that precedes migraine headache in 30% of migraineurs and are separate phenomena from headache-like pain behaviors [43]. In assessing headache pain behaviors, this investigation shows that 17-β-estradiol induces periorbital allodynia in both naturally cycling and OVX rats to similar degrees. However, 17-β-estradiol only suppressed spontaneous rearing behavior in OVX rats. Although OVX-vehicle treated rats reared less than intact rats at baseline, this was not an exacerbation of post-surgical pain [76, 77]. Our observations align with a recent study by An et al., showing that estrogen administered to OVX rats enhanced incision evoked tactile sensitivity [78] and suggests that estrogen fluctuation coupled to state of intactness may contribute to pain sensitivity. Given that periorbital allodynia (a reflex) and rearing (a spontaneous behavior that is suppressed during pain) represent two independent perceptions [79], it is plausible that the underlying mechanisms driving the two behaviors in the presence of E2 differ and account for the different presentations observed in intact and OVX rats. While we observed periorbital allodynia for all rats receiving 17-beta-estradiol, not all migraineurs experience this symptom [80]. Interestingly, presence of allodynia in patients corresponded with higher disability (suppression of normal behaviors); it is more prevalent in migraineurs with aura. These clinical observations may explain the dissociation of pain behaviors we observed (i.e., rearing behaviors only in OVX animals) and indicate a heterogeneous presentation of estradiol-induced head pain in rats.
Estradiol acts at nuclear α- and β-estrogen receptors as well as the membrane receptor GPR30 [31, 32]. These estrogen receptors are located throughout the craniofacial pain axis and the central nervous system [81–84]. Fluctuations in estrogen levels or total loss of endogenous estrogens can lead to changes in receptor expression in migraine relevant regions [82, 85–87]. We show that the non-selective estrogen receptor antagonist ICI 182,780 effectively prevented the induction of SD events and suppressed rearing behavior by 17-β-estradiol suggesting that estrogen receptors are required for these phenomena; identification of receptor sub-types responsible will require additional studies beyond the scope of this paper.
Triptans are first-line, abortive therapies for episodic migraine associated with high therapeutic gains and favorable side-effect profiles [88–91]. In the current study, we showed that pretreatment with sumatriptan prevented 17-β-estradiol induced SD events. In contrast to our observation, acute sumatriptan did not reduce cGMP- or KCl-induced CSD events in previous studies [92, 93]. Interestingly, sumatriptan effectively slowed propagation and reduced the amplitude/duration of retinal SD events in vitro [94, 95] suggesting that sumatriptan is capable of suppressing SD events. Differences in observations as reported within the migraine literature may reflect variability in subject sex, gonadal state (intact versus OVX), alertness (i.e., anesthetized/awake), model of SD induction (multiple/single events), and/or CNS penetration of sumatriptan under experimental conditions [89, 93, 96–99]. In addition to preventing SD induction, sumatriptan restored natural rearing behaviors to OVX, 17-β-estradiol treated rats suggesting that this behavior was sensitive to migraine therapeutics and relevant toclinical manifestations of pain [60, 91–93, 100, 101]. These data are consistent with reports that triptans need to be on-board during prodrome for effect in migraineurs with aura, and that triptans are ineffective when taken during the headache phase [88, 91, 102–106].
Limitations of this study
While we provide a novel model of hormone-mediated SD sensitivity, a few limitations to this study exist. First, although EEG recordings have been shown in animals and humans to represent a SD event, we do not have direct evidence using intracortical recordings that observed SD are cortical. In two animals (intact + vehicle and OVX+ vehicle), a single SD event was observed. While it is not possible to rule out induction of events in these animals due to endogenous factors such as the innate stress response or focal ischemia [107–109], no events occurred during the 2 h baseline recording nor at injection times suggesting that the stress associated with experimental execution was not responsible. Given that focal ischemia tends to produced negative shifts in DC potential of approximately 20mV in cortical recordings or 1-5mV shifts coupled with reduction in epidural EEG amplitudes during recordings [12], and our observed negative shifts were 1.05 and 2.64 mV, respectively, we cannot rule out a focal ischemic event triggering the observed SD events in these 2 rats. However, “energy–compromised” tissue (e.g., ischemic) induced events are typically evidenced by non-spreading depression or by peri-infarct depression where a negative shift in DC potential is not coupled to a reduction in ECoG amplitude [12]. Second, intact rats were assessed only in diestrus when estrogen levels are at their lowest [6, 110]; only OVX rats exhibit estradiol-induced SDs lending credence to use of intact rats during this phase. This provides a proof of concept that female sex hormones can induce highly reproducible SD. Third, we used a high dose of estradiol to induce SD events; this does not preclude the efficacy of lower doses in this model. Fourth, the acute administration and rapid onset of 17-β-estradiol in our model does not replicate most migraineurs but does allow for the induction and analysis of SD across estrogen peaks and falls.
MATERIALS AND METHODS
Animals
Female, Sprague Dawley rats (275-300g) purchased from Harlan (Indianapolis, IN) were housed in a climate controlled room on a regular 12 hour light/dark cycle with lights on at 7:00 am with food and water ad libitum. All procedures were performed during the 12-hour light cycle and according to the policies and recommendations of the International Association for the Study of Pain, the NIH guidelines for laboratory animals, and were approved by the IACUC of the University of Arizona.
Drugs
Ketamine/xylazine was purchased from Western Medical Supply (Arcadia, CA). 17-β-estradiol, ICI 182,780 and sumatriptan were purchased from Tocris (Ellisville, MO). 17-β-estradiol and ICI 182,780 were dissolved in castor oil to appropriate concentrations the day of experiments. Sumatriptan was dissolved in saline. Castor oil and sterile saline (0.9%) served as appropriate controls. All drugs were administered via intraperitoneal (i.p.) injections.
Removal of ovaries
Rats were anaesthetized with ketamine:xylazine (dose: 80:12 mg/kg, 1ml/kg) and ovariectomized [54]. The ovaries were removed via a bilateral side approach, whereby a 3-5 mm incision is made through the skin, fascia and muscle. Ovarian arteries were ligated and the ovary excised. Prophylactic gentamicin 8mg/mL, 1mL/kg was delivered i.p. following surgery. The rats recovered for 7 days before implantation of recording electrodes and/or any experiments (Figure 1A).
Vaginal smears
Estrous cycles of intact female rats were monitored by daily vaginal smears. The vaginal smears were interpreted as described by Goldman et al [110]. Briefly, vaginal openings were flushed with 200 μL of sterile saline. Fresh samples were evaluated for cytology at the same time daily for 8 days using a Zeiss Axioskop 40 (10x/0.3 numerical aperture EC Plan-Neofluar objective). CSD events, serum concentrations, and pain behaviors were assessed in the morning on diestrus day 2 by cytology.
Serum estrogen concentration
Serum concentrations of E2 before and at 30 min, 2, 4, 6, 12 and 24 hours post injection of either a dose of 17-β-estradiol (180 μg/kg, i.p., n=3/time-point) [55, 113] or vehicle (castor oil, 1ml/kg) were determined. Briefly, blood samples were drawn via intracardiac puncture under isoflurane anesthesia (5% induction, 2.5% maintenance in air- 2L/min); animals were then decapitated. Samples were allowed to clot for 30 min at RT then spun in microcentrifuge tubes at 10,000 RPM for 30 min at 4°C to separate serum from erythrocytes. Serum was collected, flash frozen in liquid nitrogen, and kept frozen at -80°C until day of concentration determination. An enzyme linked immunosorbent assay (ELISA) kit for rat estrogen (E2) was purchased (Calbiotech, Spring Valley, CA) and performed according to manufacturer’s instructions.
Implantation of recording electrodes
Silver chloride (AgCl) electrodes were prepared by flaming 0.25mm Ag wire (A-M Systems, Inc., Everett, WA) into spherical tips (1mm diameter) and subsequently coating the tips with chloride as previously reported [43]. Rats were anaesthetized with ketamine/xylazine, as above, 7 days post-ovariectomy. The rats were fixed to a stereotaxic frame (Stoelting) and three burr holes were drilled through the skull using a manual drill to allow placement of the AgCl recording electrodes. The frontal and parietal lead electrodes were placed in the right hemisphere 2 mm lateral and 1.5 mm anterior to bregma and 2 mm lateral and 2.5 mm posterior to bregma, respectively; the reference electrode was placed 7.5mm posterior to bregma (Figure 1B). Two screws (#MPX-080-3F-1M, Small Parts Inc., Miami Lakes, FL) were fastened into the left hemisphere of the skull separated by 2mm without going through the skull. The front screw served as a mounting support to assist in anchoring the multi-pin connector. The back screw served as a ground electrode, which was made by soldering silver wire onto the head of the screw. The four electrodes (three silver chloride electrodes and one ground electrode) were soldered into the bottom of a multi-pin connector (Continental Connector, Hatfield, PA) and the apparatus was fixed into place using dental cement.
Electrophysiological recordings
Forty-eight hours after dural electrode implantation, rats were placed in a recording chamber (40 cm L x 49 cm W x 37 cm H) and the multi-pin connector attached to an electro-cannular swivel (#CAY-675-6 commutator, Airflyte, Bayonne, NJ) mounted in the ceiling of the chamber. The swivel allowed rats to move freely about the chamber during the recording period. Animals habituated to the chamber for two hours prior to any pharmacological intervention to permit electrical recordings to stabilize. Only those rats with stable electrical recordings were included in experimental groups. Signals led to separate DC and AC amplifiers (Grass Model 15 amplifier system, 15A12 DC and 15A54 AC amplifiers, West Warick, RI) through insulated cables and collected with dEEG recording analysis software Gamma v.4.9 (Astro-Med, Inc. West Warick, RI). Following 17-β-estradiol or vehicle injection, the rats were observed for 12 hours without interruption. Subsequent to dEEG recordings, animals were sacrificed and cemented electrodes were carefully removed to ensure there was no damage to the dura. Data from rats with damaged dura were excluded from analysis.
Electrophysiological recording analysis
Following electrophysiological recording, the data were reviewed with Gamma Reviewer (Astro-Med, Inc. West Warick, RI) and analyzed for the SDs offline. SD events were determined by the DC shifts that were calculated from both the frontal and parietal electrodes that recorded the shifts. Cortical activity was defined as depressed during these events by measuring the significant reductions in both the power and amplitude of the dEEG tracings (i.e., recorded electrical activity was only considered an SD event when in both frontal and parietal electrodes: 1) the AC current was reduced by half; 2) the DC current exhibited a downward shift by a minimum of 1mV; and 3) for a minimal duration of 30 s [12, 41, 42, 44, 50]; Figure 1C). The velocity of SD propagation was determined by analyzing the difference in time of onset of DC current depression between the frontal and parietal electrodes and dividing into 4mm (the distance between the two electrodes).
Activity testing
Activity testing was conducted using behavioral techniques similar to those previously described [43, 51, 56, 114, 115]. Rats were acclimated to the testing room, but not to the activity recording chambers for 1 hour prior to injection. Rats were then administered an estrogen antagonist (ICI 182,780), sumatriptan, or vehicle 2 hours prior to 17-β-estradiol or vehicle injection. Two hours after the second injection, animals were placed in individual recording chambers (41 cm long X 41 cm wide X 39 cm high, Tru Scan Photobeam system: Coulbourn Instruments or 27.5 in long X 8.75 in wide X 13.125 in high, Place Preference system: San Diego Instruments). Each Tru Scan chamber has 2 photobeam sensor rings that measure all movements in 3 dimensions. One ring is located near the floor of the chamber measuring all horizontal movements including time, distance, and velocity of movements. The second ring is at the same fixed height across all chambers and records the vertical movements (i.e. rearing behavior) including the number of plane breaks and time the rat is vertical. Locomotor/exploratory activity was recorded and analyzed using Tru Scan software v.3.11 (Coulbourn Instruments). The Place Preference system records movement with a 4 X 16 photobeam array providing x,y movement, captured into PAS Software (San Diego Instruments) which displays the information as exploration, ambulation, and fine movements. Vertical rears were counted each time a rat stood on both hind paws without grooming.
Periorbital mechanical allodynia
Rats were acclimated to testing box 1 hour prior to evaluation of periorbital mechanical allodynia with von Frey filaments as previously described [43]. Behavioral responses were determined by applying calibrated von Frey filaments perpendicularly to the midline of the forehead at the level of the eyes with sufficient force to cause the filament to slightly bend while held for 5 seconds. A response was indicated by a sharp withdrawal of the head. Mechanical thresholds were evaluated at baseline, 6 days post OVX before drug administration, and 2 hours post 17-β-estradiol (180 μg/kg) or vehicle (castor oil, 1ml/kg) administration by an observer blinded to drug administration.
Statistical analysis
Prism software was employed to perform statistical analysis. All data are expressed as mean ± standard error of the mean (SEM). SD data was statistically compared using a one-way ANOVA and Student’s-Neuman-Keul’s post-hoc test. Statistical significance of periorbital allodynia was determined by repeated measure two-way ANOVA to analyze differences among treatment groups with a Bonferroni test applied post-hoc. In rearing experiments, data were expressed as mean ± SEM. A one-way ANOVA, Bonferroni post-hoc was used or an unpaired t-test with Welch’s (2 group) correction to determine statistical significance. Data were accepted as statistically significant when p ≤ 0.05.
CONCLUSION
While estrogen withdrawal is associated with induction of migraine without aura in humans [67], elevated estradiol likely contributes to MA. Here, utilizing awake adult-female rats measuring both pain behaviors and cortical spreading depression as a marker for MA, we link 17-β-estradiol levels [111, 112] to induction of CSD events and generation of pain behaviors in OVX rats. Our studies validate 17-β-estradiol as an exogenous stimulus that recapitulates several aspects of clinical migraine with aura including induction of pain behaviors and therapeutic responsiveness suggesting that both the direction, rate of change, and basal set point of estrogen levels may influence migraine pathogenesis [14, 43].
Abbreviations
Cortical spreading depression, CSD; direct current, DC; electroencephalogram, EEG; estradiol, E2; dural electroencephalogram, dEEG; hormone replacement therapy, HRT; migraine with aura, MA; ovariectomy, OVX; silver chloride, AgCl; spreading depression, SD.
Author contributions
AJS, TWV, and TML planned the experiments; AJS, KEC, MIM, and TML performed the experiments; AJS and TML primarily synthesized the manuscript; AJS, KEC, MIM, GD, TPD, TWV, and TML edited the manuscript and agreed to its final form.
ACKNOWLEDGMENTS
We would like to acknowledge Emily Galloway for her assistance in collecting data for the preparation of this manuscript. We thank the Drop-in statistical clinic athte University fo Arizona Colelge of Public Health with their assistance in statistical analyses.
CONFLICTS OF INTEREST
The authors have nothing to declare and no conflicts of interest.
FUNDING
This work was funded by University of Arizona Medical Pharmacology Departmental funding and NIH Grants 5 RO1 DA 11271-18 and 5 RO1 NS 42652-14.
REFERENCES
1. Burch RC, Loder S, Loder E, Smitherman TA. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache. 2015; 55: 21-34.
2. Messali A, Sanderson JC, Blumenfeld AM, Goadsby PJ, Buse DC, Varon SF, Stokes M, Lipton RB. Direct and indirect costs of chronic and episodic migraine in the United States: a web-based survey. Headache. 2013; 56: 306-322.
3. Hill K, Hoffman D, Welch N, Hart W, Hall JS, Hughes R, Rissi J. Truth and Consequences: Gambling, shifting, and hoping in Arizona Healthcare, Arizona Health Futures, (2009).
4. Marcus DA. Interrelationships of neurochemicals, estrogen, and recurring headache. Pain. 1995; 62:129-139.
5. Aegidius KL, Zwart JA, Hagen K, Dyb G, Holmen TL, Stovner LJ. Increased headache prevalence in female adolescents and adult women with early menarche. The Head-HUNT studies. Eur J Neurol. 2011; 18: 321-328.
6. Bolay H, Berman NE, Akcali D. Sex-related differences in animal models of migraine headache. Headache. 2011; 51: 891-904.
7. Lipton RB, Scher AI, Kolodner K, Liberman J, Steiner TJ, Stewart WF. Migraine in the United States: epidemiology and patterns of health care use. Neurology. 2002; 58: 885-894.
8. Ferrari MD. Migraine. Lancet. 1998; 351: 1043-1051.
9. Rasmussen BK, Olesen J. Migraine with aura and migraine without aura: an epidemiological study. Cephalalgia. 1992; 12: 221-228; discussion 186.
10. Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol. 1956; 19: 154-171.
11. Leo AP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944; 7: 359-390.
12. Hartings JA, Tortella FC, Rolli ML. AC electrocorticographic correlates of peri-infarct depolarizations during transient focal ischemia and reperfusion. J Cereb Blood Flow Metab. 2006; 26: 696-707.
13. Burgos-Vega C, Moy J, Dussor G. Meningeal afferent signaling and the pathophysiology of migraine. Prog Mol Biol Transl Sci. 2015; 131: 537-564.
14. Eikermann-Haerter K, Kudo C, Moskowitz MA. Cortical spreading depression and estrogen. Headache. 2007; 47: S79-S85.
15. Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001; 98: 4687-4692.
16. Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011; 31: 17-35.
17. Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015; 95: 953-993.
18. Aurora SK, Cao Y, Bowyer SM, Welch KM. The occipital cortex is hyperexcitable in migraine: experimental evidence. Headache. 1999; 39: 469-476.
19. Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009; 119: 99-109.
20. Ostergaard L, Dreier JP, Hadjikhani N, Jespersen SN, Dirnagl U, Dalkara T. Neurovascular coupling during cortical spreading depolarization and -depression. Stroke. 2015; 46: 1392-1401.
21. Sacco S, Ricci S, Degan D, Carolei A. Migraine in women: the role of hormones and their impact on vascular diseases. J Headache Pain. 2012; 13: 177-189.
22. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part I. Headache. 2006; 46: 3-23.
23. Silberstein SD. Headache and female hormones: what you need to know. Curr Opin Neurol. 2001; 14: 323-333.
24. Aegidius KL, Zwart JA, Hagen K, Schei B, Stovner LJ. Hormone replacement therapy and headache prevalence in postmenopausal women. The Head-HUNT study. Eur J Neurol. 2007; 14: 73-78.
25. Chai NC, Peterlin BL, Calhoun AH. Migraine and estrogen. Curr Opin Neurol. 2014; 27: 315-324.
26. Martin VT, Pavlovic J, Fanning KM, Buse DC, Reed ML, Lipton RB. Perimenopause and menopause are associated with high frequency headache in women with migraine: results of the American migraine prevalence and prevention study. Headache. 2016; 56: 292-305.
27. Borsook D, Erpelding N, Lebel A, Linnman C, Veggeberg R, Grant PE, Buettner C, Becerra L, Burstein R. Sex and the migraine brain. Neurobiol Dis. 2014; 68: 200-214.
28. Mattsson P. Hormonal factors in migraine: a population-based study of women aged 40 to 74 years. Headache. 2003; 43: 27-35.
29. Granella F, Sances G, Pucci E, Nappi RE, Ghiotto N, Napp G. Migraine with aura and reproductive life events: a case control study. Cephalalgia. 2000; 20: 701-707.
30. Melhado EM, Bigal ME, Galego AR, Galdezzani JP, Queiroz LP. Headache classification and aspects of reproductive life in young women. Arq Neuropsiquiatr. 2014; 72: 17-23.
31. Gu Q, Korach KS, Moss RL. Rapid action of 17beta-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen receptors. Endocrinology. 1999; 140: 660-666.
32. Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999; 13: 307-319.
33. Sachs M, Pape HC, Speckmann EJ, Gorji A. The effect of estrogen and progesterone on spreading depression in rat neocortical tissues. Neurobiol Dis. 2007; 25: 27-34.
34. Chauvel V, Schoenen J, Multon S. Influence of ovarian hormones on cortical spreading depression and its suppression by L-kynurenine in rat. PLoS One. 2013; 8: e82279.
35. Chauvel V, Vamos E, Pardutz A, Vecsei L, Schoenen J, Multon S. Effect of systemic kynurenine on cortical spreading depression and its modulation by sex hormones in rat. Exp Neurol. 2012; 236: 207-214.
36. Piper RD, Lambert GA. Inhalational anesthetics inhibit spreading depression: relevance to migraine. Cephalalgia. 1996; 16: 87-92.
37. Kudo C, Nozari A, Moskowitz MA, Ayata C. The impact of anesthetics and hyperoxia on cortical spreading depression. Exp Neurol. 2008; 212: 201-206.
38. Narebski J, Waczynska W, Tymicz J, Tegowska E. Reversible brain hyperthermia during audiogenic seizures in rats. Acta Physiol Pol. 1979; 30: 273-277.
39. Tortella FC, Robles L, Mosberg HI. Evidence for mu opioid receptor mediation of enkephalin-induced electroencephalographic seizures. J Pharmacol Exp Ther. 1987; 240: 571-577.
40. Tortella FC, Rose J, Robles L, Moreton JE, Hughes J, Hunter JC. EEG spectral analysis of the neuroprotective kappa opioids enadoline and PD117302. J Pharmacol Exp Ther. 1997; 282: 286-293.
41. Hartings JA, Rolli ML, Lu XC, Tortella FC. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci. 2003; 23: 11602-11610.
42. Adamek S, Vyskocil F. Potassium-selective microelectrode revealed difference in threshold potassium concentration for cortical spreading depression in female and male rat brain. Brain Res. 2011; 1370: 215-219.
43. Fioravanti B, Kasasbeh A, Edelmayer R, Skinner DP Jr, Hartings JA, Burklund RD, De Felice M, French ED, Dussor GO, Dodick DW, Porreca F, Vanderah TW. Evaluation of cutaneous allodynia following induction of cortical spreading depression in freely moving rats. Cephalalgia. 2011; 31: 1090-1100.
44. Accioly NE, Benevides Rde D, da Costa BL, Guedes RC. Ovariectomy in the developing rat decelerates cortical spreading depression in adult brain. Int J Dev Neurosci. 2012; 30: 405-410.
45. Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE. Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res. 1996; 740: 268-274.
46. Drenckhahn C, Winkler MK, Major S, Scheel M, Kang EJ, Pinczolits A, Grozea C, Hartings JA, Woitzik J, Dreier JP; COSBID study group. Correlates of spreading depolarization in human scalp electroencephalography. Brain. 2012; 135: 853-868.
47. Bogdanov VB, Middleton NA, Theriot JJ, Parker PD, Abdullah OM, Ju YS, Hartings JA, Brennan KC. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci. 2016; 36: 4733-4743.
48. Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006; 129: 778-790.
49. McLachlan RS, Girvin JP. Spreading depression of Leao in rodent and human cortex. Brain Res. 1994; 666: 133-136.
50. Hartings JA, Watanabe T, Dreier JP, Major S, Vendelbo L, Fabricius M. Recovery of slow potentials in AC-coupled electrocorticography: application to spreading depolarizations in rat and human cerebral cortex. J Neurophysiol. 2009; 102: 2563-2575.
51. Edelmayer RM, Le LN, Yan J, Wei X, Nassini R, Materazzi S, Preti D, Appendino G, Geppetti P, Dodick DW, Vanderah TW, Porreca F, Dussor G. Activation of TRPA1 on dural afferents: a potential mechanism of headache pain. Pain. 2012; 153: 1949-1958.
52. Edelmayer RM, Vanderah TW, Majuta L, Zhang ET, Fioravanti B, De Felice M, Chichorro JG, Ossipov MH, King T, Lai J, Kori SH, Nelsen AC, Cannon KE, et al. Medullary pain facilitating neurons mediate allodynia in headache-related pain. Ann Neurol. 2009; 65: 184-193.
53. Tam A, Morrish D, Wadsworth S, Dorscheid D, Man SF, Sin DD. The role of female hormones on lung function in chronic lung diseases. BMC Womens Health. 2011; 11: 24.
54. Lam KK, Hu CT, Ou TY, Yen MH, Chen HI. Effects of oestrogen replacement on steady and pulsatile haemodynamics in ovariectomized rats. Br J Pharmacol. 2002; 136: 811-818.
55. Becker JB, Arnold AP, Berkley KJ, Blaustein JD, Eckel LA, Hampson E, Herman JP, Marts S, Sadee W, Steiner M, Taylor J, Young E. Strategies and methods for research on sex differences in brain and behavior. Endocrinology. 2005; 146: 1650-1673.
56. Wallace VC, Blackbeard J, Segerdahl AR, Hasnie F, Pheby T, McMahon SB, Rice AS. Characterization of rodent models of HIV-gp120 and anti-retroviral-associated neuropathic pain. Brain. 2007; 130: 2688-2702.
57. Russell MB, Rasmussen BK, Thorvaldsen P, Olesen J. Prevalence and sex-ratio of the subtypes of migraine. Int J Epidemiol. 1995; 24: 612-618.
58. Burstein R, Noseda R, Borsook D. Migraine: multiple processes, complex pathophysiology. J Neurosci. 2015; 35: 6619-6629.
59. Faria V, Erpelding N, Lebel A, Johnson A, Wolff R, Fair D, Burstein R, Becerra L, Borsook D. The migraine brain in transition: girls vs boys. Pain. 2015; 156: 2212-2221.
60. Jhingran P, Davis SM, LaVange LM, Miller DW, Helms RW. MSQ: Migraine-Specific Quality-of-Life Questionnaire. Further investigation of the factor structure. Pharmacoeconomics. 1998; 13: 707-717.
61. Ruau D, Liu LY, Clark JD, Angst MS, Butte AJ. Sex differences in reported pain across 11,000 patients captured in electronic medical records. J Pain. 2012; 13: 228-234.
62. Shuster LT, Faubion SS, Sood R, Casey PM. Hormonal manipulation strategies in the management of menstrual migraine and other hormonally related headaches. Curr Neurol Neurosci Rep. 2011; 11: 131-138.
63. Mathew PG, Dun EC, Luo JJ. A cyclic pain: the pathophysiology and treatment of menstrual migraine. Obstet Gynecol Surv. 2013; 68: 130-140.
64. Misakian AL, Langer RD, Bensenor IM, Cook NR, Manson JE, Buring JE, Rexrode KM. Postmenopausal hormone therapy and migraine headache. J Womens Health (Larchmt). 2003; 12: 1027-1036.
65. Epstein MT, Hockaday JM, Hockaday TD. Migraine and reporoductive hormones throughout the menstrual cycle. Lancet. 1975; 1: 543-548.
66. Somerville BW. The role of estradiol withdrawal in the etiology of menstrual migraine. Neurology. 1972; 22: 355-365.
67. Edelman AB, Cherala G, Munar MY, McInnis M, Stanczyk FZ, Jensen JT. Correcting oral contraceptive pharmacokinetic alterations due to obesity: a randomized controlled trial. Contraception. 2014; 90: 550-556.
68. Cawello W, Rosenkranz B, Schmid B, Wierich W. Pharmacodynamic and pharmacokinetic evaluation of coadministration of lacosamide and an oral contraceptive (levonorgestrel plus ethinylestradiol) in healthy female volunteers. Epilepsia. 2013; 54: 530-536.
69. Edelman AB, Cherala G, Munar MY, Dubois B, McInnis M, Stanczyk FZ, Jensen JT. Prolonged monitoring of ethinyl estradiol and levonorgestrel levels confirms an altered pharmacokinetic profile in obese oral contraceptives users. Contraception. 2013; 87: 220-226.
70. MacGregor EA. Migraine and use of combined hormonal contraceptives: a clinical review. J Fam Plann Reprod Health Care. 2007; 33: 159-169.
71. Aegidius K, Zwart JA, Hagen K, Schei B, Stovner LJ. Oral contraceptives and increased headache prevalence: the Head-HUNT Study. Neurology. 2006; 66: 349-353.
72. Cooper O, Geller JL, Melmed S. Ovarian hyperstimulation syndrome caused by an FSH-secreting pituitary adenoma. Nat Clin Pract Endocrinol Metab. 2008; 4: 234-238.
73. Levy MJ, Matharu M, Goadsby PJ. Chronic headache and pituitary tumors. Curr Pain Headache Rep. 2008; 12: 74-78.
74. Brandes JL. The influence of estrogen on migraine: a systematic review. JAMA. 2006; 295: 1824-1830.
75. Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001; 50: 582-587.
76. Fan XL, Duan XB, Chen ZH, Li M, Xu JS, Ding GM. Lack of estrogen down-regulates CXCR4 expression on Treg cells and reduces Treg cell population in bone marrow in OVX mice. Cell Mol Biol (Noisy-le-grand). 2015; 61: 13-17.
77. Zeng QM, Liu DC, Zhang XC, Wu JP, Ding L, Meng DH, Zhang J. Estrogen deficiency inducing shifted cytokines profile in bone marrow stromal cells inhibits Treg cells function in OVX mice. Cell Mol Biol (Noisy-le-grand). 2015; 61: 64-68.
78. An G, Li W, Yan T, Li S. Estrogen rapidly enhances incisional pain of ovariectomized rats primarily through the G protein-coupled estrogen receptor. Int J Mol Sci. 2014; 15: 10479-10491.
79. Stevenson GW, Cormier J, Mercer H, Adams C, Dunbar C, Negus SS, Bilsky EJ. Targeting pain-depressed behaviors in preclinical assays of pain and analgesia: drug effects on acetic acid-depressed locomotor activity in ICR mice. Life Sci. 2009; 85: 309-315.
80. Lipton RB, Bigal ME, Ashina S, Burstein R, Silberstein S, Reed ML, Serrano D, Stewart WF; American Migraine Prevalence Prevention Advisory Group. Cutaneous allodynia in the migraine population. Ann Neurol. 2008; 63: 148-158.
81. Fenzi F, Rizzzuto N. Estrogen receptors localization in the spinal trigeminal nucleus: an immunohistochemical study in humans. Eur J Pain. 2011; 15: 1002-1007.
82. Puri J, Bellinger LL, Kramer PR. Estrogen in cycling rats alters gene expression in the temporomandibular joint, trigeminal ganglia and trigeminal subnucleus caudalis/upper cervical cord junction. J Cell Physiol. 2011; 226: 3169-3180.
83. Vanderhorst VG, Terasawa E, Ralston HJ 3rd. Estrogen receptor-alpha immunoreactive neurons in the brainstem and spinal cord of the female rhesus monkey: species-specific characteristics. Neurosci. 2009; 158: 798-810.
84. Wang MW, Kumar U, Dong XD, Cairns BE. Expression of NMDA and oestrogen receptors by trigeminal ganglion neurons that innervate the rat temporalis muscle. Chin J Dent Res. 2012; 15: 89-97.
85. Spary EJ, Chapman SE, Sinfield JK, Maqbool A, Kaye J, Batten TF. Novel G protein-coupled oestrogen receptor GPR30 shows changes in mRNA expression in the rat brain over the oestrous cycle. Neurosignals. 2013; 21: 14-27.
86. Mohamed MK, Abdel-Rahman AA. Effect of long-term ovariectomy and estrogen replacement on the expression of estrogen receptor gene in female rats. Eur J Endocrinol. 2000; 142: 307-314.
87. Liverman CS, Brown JW, Sandhir R, McCarson KE, Berman NE. Role of the oestrogen receptors GPR30 and ERalpha in peripheral sensitization: relevance to trigeminal pain disorders in women. Cephalalgia. 2009; 29: 729-741.
88. Gilmore B, Michael M. Treatment of acute migraine headache. Am Fam Physician. 2011; 83: 271-280.
89. Humphrey PP, Feniuk W, Perren MJ, Beresford IJ, Skingle M, Whalley ET. Serotonin and migraine. Ann N Y Acad Sci. 1990; 600: 587-598; discussion 598-600.
90. Smitherman TA, Burch R, Sheikh H, Loder E. The prevalence, impact, and treatment of migraine and severe headaches in the United States: a review of statistics from national surveillance studies. Headache. 2013; 53: 427-436.
91. Thorlund K, Mills EJ, Wu P, Ramos E, Chatterjee A, Druyts E, Goadsby PJ. Comparative efficacy of triptans for the abortive treatment of migraine: a multiple treatment comparison meta-analysis. Cephalalgia. 2014; 34: 258-267.
92. Bradley DP, Smith MI, Netsiri C, Smith JM, Bockhorst KH, Hall LD, Huang CL, Leslie RA, Parsons AA, James MF. Diffusion-weighted MRI used to detect in vivo modulation of cortical spreading depression: comparison of sumatriptan and tonabersat. Exp Neurol. 2001; 172: 342-353.
93. Read SJ, Hirst WD, Upton N, Parsons AA. Cortical spreading depression produces increased cGMP levels in cortex and brain stem that is inhibited by tonabersat (SB-220453) but not sumatriptan. Brain Res. 2001; 891: 69-77.
94. Wiedemann M, de Lima VM, Hanke W. Effects of antimigraine drugs on retinal spreading depression. Naunyn Schmiedebergs Arch Pharmacol. 1996; 353: 552-556.
95. Maranhao-Filho PA, Martins-Ferreira H, Vincent MB, Ribeiro LJ, Novis SA. Sumatriptan blocks spreading depression in isolated chick retina. Cephalalgia. 1997; 17: 822-825.
96. Moskowitz MA, Nozaki K, Kraig RP. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci. 1993; 13: 1167-1177.
97. Ingvardsen BK, Laursen H, Olsen UB, Hansen AJ. Possible mechanism of c-fos expression in trigeminal nucleus caudalis following cortical spreading depression. Pain. 1997; 72: 407-415.
98. Jhee SS, Shiovitz T, Crawford AW, Cutler NR. Pharmacokinetics and pharmacodynamics of the triptan antimigraine agents: a comparative review. Clin Pharmacokinet. 2001; 40: 189-205.
99. Kaube H, Hoskin KL, Goadsby PJ. Inhibition by sumatriptan of central trigeminal neurones only after blood-brain barrier disruption. Br J Pharmacol. 1993; 109: 788-792.
100. Rendas-Baum R, Bloudek LM, Maglinte GA, Varon SF. The psychometric properties of the Migraine-Specific Quality of Life Questionnaire version 2.1 (MSQ) in chronic migraine patients. Qual Life Res. 2013; 22: 1123-1133.
101. Kim SY, Park SP. The role of headache chronicity among predictors contributing to quality of life in patients with migraine: a hospital-based study. J Headache Pain. 2014; 15: 68.
102. Bigal ME, Krymchantowski AV, Ho T. Migraine in the triptan era: progresses achieved, lessons learned and future developments. Arq Neuropsiquiatr. 2009; 67: 559-569.
103. Bigal ME, Lipton RB. Excessive acute migraine medication use and migraine progression. Neurology. 2008; 71: 1821-1828.
104. Martelletti P. The therapeutic armamentarium in migraine is quite elderly. Expert Opin Drug Metab Toxicol. 2015; 11: 175-177.
105. Kowacs PA, Piovesan EJ, Tatsui CE, Lange MC, Ribas LC, Werneck LC. Prolonged migraine aura without headache arrested by sumatriptan. A case report with further considerations. Cephalalgia. 1999; 19: 241-242.
106. Bates D, Ashford E, Dawson R, Ensink FB, Gilhus NE, Olesen J, Pilgrim AJ, Shevlin P. Subcutaneous sumatriptan during the migraine aura. Sumatriptan Aura Study Group. Neurology. 1994; 44: 1587-1592.
107. Becerra L, Bishop J, Barmettler G, Xie Y, Navratilova E, Porreca F, Borsook D. Triptans disrupt brain networks and promote stress-induced CSD-like responses in cortical and subcortical areas. J Neurophysiol. 2016; 115: 208-217.
108. Green AL, Gu P, De Felice M, Dodick D, Ossipov MH, Porreca F. Increased susceptibility to cortical spreading depression in an animal model of medication-overuse headache. Cephalalgia. 2014; 34: 594-604.
109. Koponen S, Keinanen R, Roivainen R, Hirvonen T, Narhi M, Chan PH, Koistinaho J. Spreading depression induces expression of calcium-independent protein kinase C subspecies in ischaemia-sensitive cortical layers: regulation by N-methyl-D-aspartate receptors and glucocorticoids. Neuroscience. 1999; 93: 985-993.
110. Goldman JM, Murr AS, Cooper RL. The rodent estrous cycle: characterization of vaginal cytology and its utility in toxicological studies. Birth Defects Res B Dev Reprod Toxicol. 2007; 80: 84-97.
111. Stricker R, Eberhart R, Chevailler MC, Quinn FA, Bischof P, Stricker R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med. 2006; 44: 883-887.
112. Mishell DR Jr, Thorneycroft IH, Nakamura RM, Nagata Y, Stone SC. Serum estradiol in women ingesting combination oral contraceptive steroids. Am J Obstet Gynecol. 1972; 114: 923-928.
113. Vermeer LM, Gregory E, Winter MK, McCarson KE, Berman NE. Behavioral effects and mechanisms of migraine pathogenesis following estradiol exposure in a multibehavioral model of migraine in rat. Exp Neurol. 2015; 263: 8-16.
114. Matson DJ, Broom DC, Cortright DN. Locomotor activity in a novel environment as a test of inflammatory pain in rats. Methods Mol Biol. 2010; 617: 67-78.
115. Stucky NL, Gregory E, Winter MK, He YY, Hamilton ES, McCarson KE, Berman NE. Sex differences in behavior and expression of CGRP-related genes in a rodent model of chronic migraine. Headache. 2011; 51: 674-692.